Introduction
One of the major problems associated with the
treatment of cancer is the development of resistance to
chemotherapeutic drugs (1,2). The most common mechanisms that
produce drug resistance in cancer cells include: i) altered cell
cycle check points; ii) induction of emergency response genes; iii)
alterations in membrane lipids; iv) compartmentalization; v)
inhibition of apoptosis; vi) altered drug targets; vii) decreased
uptake and viii) increased efflux of drugs (2–6). One
type of resistance that is common and highly problematic is
multidrug resistance (MDR). MDR occurs when cancer cells become
resistant to structurally and mechanistically distinct classes of
chemotherapeutic compounds (2,7,8). A
common mediator of MDR in cancer cells is the family of specific
transmembrane, energy-dependent transporters known as ATP-binding
cassette (ABC) transporters (9).
The ABC transporter family is divided into seven subfamilies, ABCA
through ABCG (10,11). Currently, 48 members of the ABC
transporter family have been isolated and identified (1,11).
Mechanistically, the catalytic cycle of the transporter involves
two ATPs. The first molecule of ATP is hydrolyzed by ATPase,
producing a structural modification of the trans-membrane domains
that flips the inner membrane leaf to the outside of cell membrane,
thereby removing or effluxing the compound. The second ATP is
hydrolyzed to restore the transporter back to its original high
affinity state for substrate transport (12). The breast cancer resistance protein
(BCRP, also called ABCG2) produces MDR in a broad range of human
cancers (13,14). ABCG2, a 72 kDa protein, is known as
a half transporter that effluxes or extrudes molecules with
amphiphilic characteristics (15).
The substrates of ABCG2 include sulfated hormone metabolites,
methotrexate, mitoxantrone (MX), topotecan and irinotecan (16). ABCG2 is a widely distributed
transporter that is present mainly in the plasma membrane, and is
highly expressed in the placental syncytiotrophoblasts, apical
surface of small intestines, colon epithelium, liver canalicular
membrane, luminal surfaces of microvessel endothelium of human
brain and in the veins and capillaries of blood vessels (17–20).
Its wide distribution and expression suggests that it is involved
in protecting the fetus and adult against endogenous and exogenous
toxins (21). ABCG2 is also
abundantly expressed in the placenta and is also called ABCP1 (ABC
transporter expressed in placenta) (22). It is expressed in colon cancer
cells resistant to MX, thereby giving ABCG2 the name MX resistant
protein (MXR) (23). Mutations in
the ABCG2 gene produce distinct substrate preferences within the
mutant and wild-type variants. For example, a mutation at position
482 is the most important mutation for the determination of
substrate specificity (24). The
amino acid arginine (Arg or R) is located on the carboxy terminal
of the third transmembrane segment of the membrane spanning domain,
where substrate binding occurs probably due to the formation of
salt bridges (15). These
mutations cause conformational changes and alter the drug binding
and efflux capacity of the transporter (25–27).
The replacement of Arg with threonine (Thr or T) or glycine (Gly or
G) at position 482 produces changes in the substrate profiles among
the variants (15,28). Indeed, the multidrug efflux pump
ABCG2 has been implicated as the cause of the ‘side population’
which helps define adult stem cells of various tissues and tumors,
including placental trophoblasts, neural stem cells or progenitors
and hematopoietic progenitors (29,30).
Numerous studies over the past 3 decades have shown
that MDR in cancer cells can be attenuated or even reversed by the
inhibitors of ABC transporters (26,31–34).
However, many of these ABC transporter inhibitors, at concentration
that reversed MDR, also produced unacceptable toxicity as well as
problematic pharmacokinetic interactions (35). These limitations prompted the
development of a number of new compounds that are more potent and
selective. Recently, we have reported that several tyrosine kinase
inhibitors (TKIs), including tivozanib (36), imatinib (37), nilotinib (38), lapatinib (32) and erlotinib (39), can reverse ABC transporter mediated
MDR. However, none of these MDR inhibitors have been used
clinically in combination with conventional anti-neoplastic
drugs.
Masitinib, a novel phenyl aminothiazole derivative,
is a TKI used in the management of various diseases including
multiple sclerosis (40,41), asthma (42,43),
rheumatoid arthritis (44,45) and neoplasmic conditions such as
gastro-intestinal stromal tumor, and pancreatic cancer (46–49).
In a phase II trial, masitinib significantly increased the overall
survival rate and progression-free survival in patients with
locally advanced or metastatic gastro-intestinal stromal tumor
(48). Masitinib, in combination
with gemcitabine, significantly increased the media
time-to-progression in patients with advanced pancreatic cancer
compared to gemcitabine-treated patients (48,49).
Currently, no studies have examined the effect of masitinib on
ABCG2-mediated MDR. Therefore, in this study, we examined the
effect of masitinib on MDR to various antineoplastic drugs in
HEK293 and H460 cells overexpressing ABCG2 (14,50,51).
Materials and methods
Reagents
[3H]-MX (4 Ci/mmol) was purchased from
Moravek Biochemicals, Inc. (Brea, CA) Dulbecco’s modified Eagle’s
medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin
and trypsin 0.25% were purchased from HyClone (Waltham, MA). A
monoclonal antibody against GAPDH was purchased from Cell Signaling
Technologies (Beverly, MA). The antibody BXP-21 against ABCG2 was
obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Masitinib was purchased from LC Laboratories (Woburn, MA). MX, SN38
and cisplatin were purchased from Tocris Bioscience (Ellisville,
MO). 3-(4,5-Dimethylthiazol-yl)-2,5-diphenyltetrazolium bromide
(MTT), dimethyl sulfoxide (DMSO) and doxorubicin were obtained from
Sigma-Aldrich Chemical Co. (St. Louis, MO). Nilotinib was obtained
from Selleck Chemicals (Houston, TX).
Cell lines
The HEK293/pcDNA3.1 (empty vector), wild-type
HEK293/ABCG2-482-R2, mutant HEK293/ABCG2-482-G2 and mutant
HEK293/ABCG2-482-T7 cells were established by transfecting HEK293
with either the pcDNA3.1 or vectors containing the full length
ABCG2 containing either arginine (R), glycine (G), or threonine (T)
at amino acid 482, respectively. The cells were cultured in a
medium containing 2 mg/ml of G418 (52). The parental human non-small cell
lung cancer H460 cells were grown in DMEM, supplemented with 5%
heat-inactivated FBS. Resistant H460/MX20 cells were cultured in
the above-mentioned medium with the addition of 20 nM MX. All the
above cell lines were kindly provided by Dr Susan E. Bates and Dr
Robert W. Robey (NCI, NIH, Bethesda, MD).
Cell sensitivity by tetrazolium dye
assay
A modified
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay was performed to detect the viability of the cells to
anticancer drugs in vitro (53). The cell numbers seeded into the
96-well plates were 5,000/well for HEK293/pcDNA3.1,
HEK293/ABCG2-482-R2, HEK293/ABCG2-482-T7 and HEK293/ABCG2-482-G2
cells and 6,000/well for H460 and H460/MX20. The MTT assay was run
in triplicate and the compounds tested included MX (0.001 to 1
μM), SN38 (0.001 to 1 μM), doxorubicin (0.003 to 3
μM), cisplatin (0.1 to 100 μM), masitinib (1.25 and
2.5 μM), nilotinib (2.5 μM). After seeding cells in
180 μl of medium in 96-well plates and incubation for 24 h
at 37°C, 20 μl of the appropriate anticancer drug at various
concentrations was added (20 μl of a fixed concentration of
test compound for reversal experiments were added 1 h prior to
adding the anticancer drugs). Subsequently, cells were incubated
with anticancer drugs (in DMEM supplemented with 10% fetal bovine
serum) at 37°C for 72 h. After 72 h, 20 μl MTT (4 mg/ml) was
added to each well. The plates were incubated at 37°C for another 4
h. The MTT/medium was removed from each well, and 100 μl of
DMSO was added to each well. The absorbance was read at 570 nm
using an Opsys microplate reader (Dynex Technologies, Chantilly,
VA). The degree of resistance was calculated by dividing the
IC50 for the MDR cells by that of the parental sensitive
cells. The degree of the reversal of MDR was calculated by dividing
the IC50 for cells with the anticancer drug in the
absence of masitinib or other reversal compounds by that obtained
in the presence of masitinib or other reversal compounds.
[3H]-MX accumulation
assay
The HEK293/pcDNA3.1, HEK293/ABCG2-482-R2,
HEK293/ABCG2-482-T7, HEK293/ABCG2-482-G2, H460 and H460/MX20 cell
lines were harvested at 80% confluency in T75 flasks for this
experiment. All cell lines were trypsinized with 0.25% trypsin
after observing their confluency in T75 flasks under a microscope
and cell count was done using a hemocytometer. Approximately
6×106 cells were incubated at 37°C in DMEM supplemented
with 10% FBS with and without masitinib concentrations of 1.25 and
2.5 μM for 2 h. Subsequently, HEK293/pcDNA3.1,
HEK293/ABCG2-482-R2, HEK293/ABCG2-482-T7, HEK293/ABCG2-482-G2, H460
and H460/MX20 cells were incubated with 0.01 μM
[3H]-MX for 2 h. Following incubation, the medium was
removed and the cells were rinsed three times with cold phosphate
buffer saline (PBS). The cells were lysed by adding 200 μl
of lysis buffer and transferred to scintillation vials. Each sample
was placed in scintillation fluid and radioactivity was measured in
a Packard TriCarb® 1900CA liquid scintillation analyzer
from Packard Instrument Company, Inc (Downers Grove, IL).
[3H]-MX efflux assay
To measure [3H]-MX efflux, cells were
prepared using the procedure discussed for the drug accumulation
experiment and then incubated in fresh medium at 37°C at various
times (0, 30, 60 and 120 min) in the presence or absence of the
test compounds. After washing three times with ice-cold PBS, the
cells were lysed by adding 200 μl lysis buffer and
transferred to scintillation vials. Each sample was placed in
scintillation fluid and radioactivity was measured using a Packard
TriCarb 1900CA liquid scintillation analyzer from Packard
Instrument Company, Inc.
Preparation of cell lysates
Approximately 6×105 cells were harvested
and suspended in PBS, followed by centrifugation at 2,000 rpm for 2
min, and the cells were washed twice with the PBS. Lysate buffer
and 1% aprotinin were added to the suspension followed by
vortexing. The resuspended cells were kept on ice for 30 min
followed by centrifugation at 12,000 rpm for 20 min. The
supernatant was separated and was stored at −80°C for the
experiment. Protein concentrations in the vesicles were determined
using the bicinchonic acid (BCATM) based protein assay
(Thermo Scientific, Rockford, IL).
Immunoblot analysis
Equal amounts of total cell lysates (40 μg
protein) were resolved by sodium dodecyl sulfate polycrylamide gel
electrophoresis and electrophoretically transferred onto
polyvinylidene fluoride (PVDF) membranes. After incubation in a
blocking solution (5% skim milk) of TBST buffer (10 mM Tris-HCl, pH
8.0, 150 mM NaCl, and 0.1% Tween-20) for 1 h at room temperature,
the membranes were immunoblotted overnight with primary monoclonal
antibodies against ABCG2 at 1:200 dilution or GAPDH at 1:1,000 at
4°C, and were then incubated for 3 h at room temperature with
horseradish peroxide (HRP)-conjugated secondary antibody (1:1,000
dilution). The protein-antibody complex was detected by enhanced
chemiluminescence detection system (Amersham, Piscataway, NJ). The
protein expression was quantified by Scion Image Software (Scion
Corp., Frederick, MD).
Molecular modeling of ABCG2
The structure of masitinib was built using the
fragment dictionary of Maestro v9.0. The energy was minimized by a
Macromodel program v9.7 (Schrödinger, Inc., New York, NY) using the
OPLSAA force field with the steepest descent followed by a
truncated Newton conjugate gradient protocol. The low-energy 3D
structures of masitinib were generated by LigPrep v2.3 and the
parameters were defined based on different protonation states at
physiological pH ± 2.0, and all possible tautomers and ring
conformations. The ligand structures obtained from the LigPrep v2.3
run were further used for generating 100 ligand conformations for
each protonated structure using the default parameters of mixed
torsional/low-mode sampling function. The conformations were
filtered with a maximum relative energy difference of 5 kcal/mol to
exclude redundant conformers. The output conformational search
(Csearch) file containing 100 unique conformers of masitinib were
used as input for docking simulations into each binding site of the
human ABCG2 transporter.
A homology model of ABCG2 was built using the mouse
apoprotein (PDB ID: 3G5U) as a template (54). To identify drug binding sites on
ABCG2 homology model, we generated various grids based on the
following residues as centroids, for example, Arg482 (grid 1),
Asn629 (grid 2), Arg383 (grid 3) and Leu241 along with Gly83 (grid
4). The choice of these residues was based on their involvement in
ABCG2 function as determined through mutational experiments
(52,55). The grid 2 generated using Asn629 as
the centroid was found to have the best docking score; hence,
docking discussion was based on binding mode of masitinib at this
site. Glide v5.0 docking protocol was followed with the default
functions (Schrödinger, Inc.). The top scoring masitinib
conformation at Asn629 site of ABCG2 was used for graphical
analysis. All computations were carried out on a Dell Precision
470n dual processor with the Linus OS (Red Hat Enterprise WS 4.0)
(56).
Statistical analysis
Differences of the parameters between two cell
groups were analyzed by two tailed Student’s unpaired t-test. The
a priori significance level was set at p<0.05.
Results
Masitinib significantly enhances the
sensitivity of cells over-expressing ABCG2 to antineoplastic
drugs
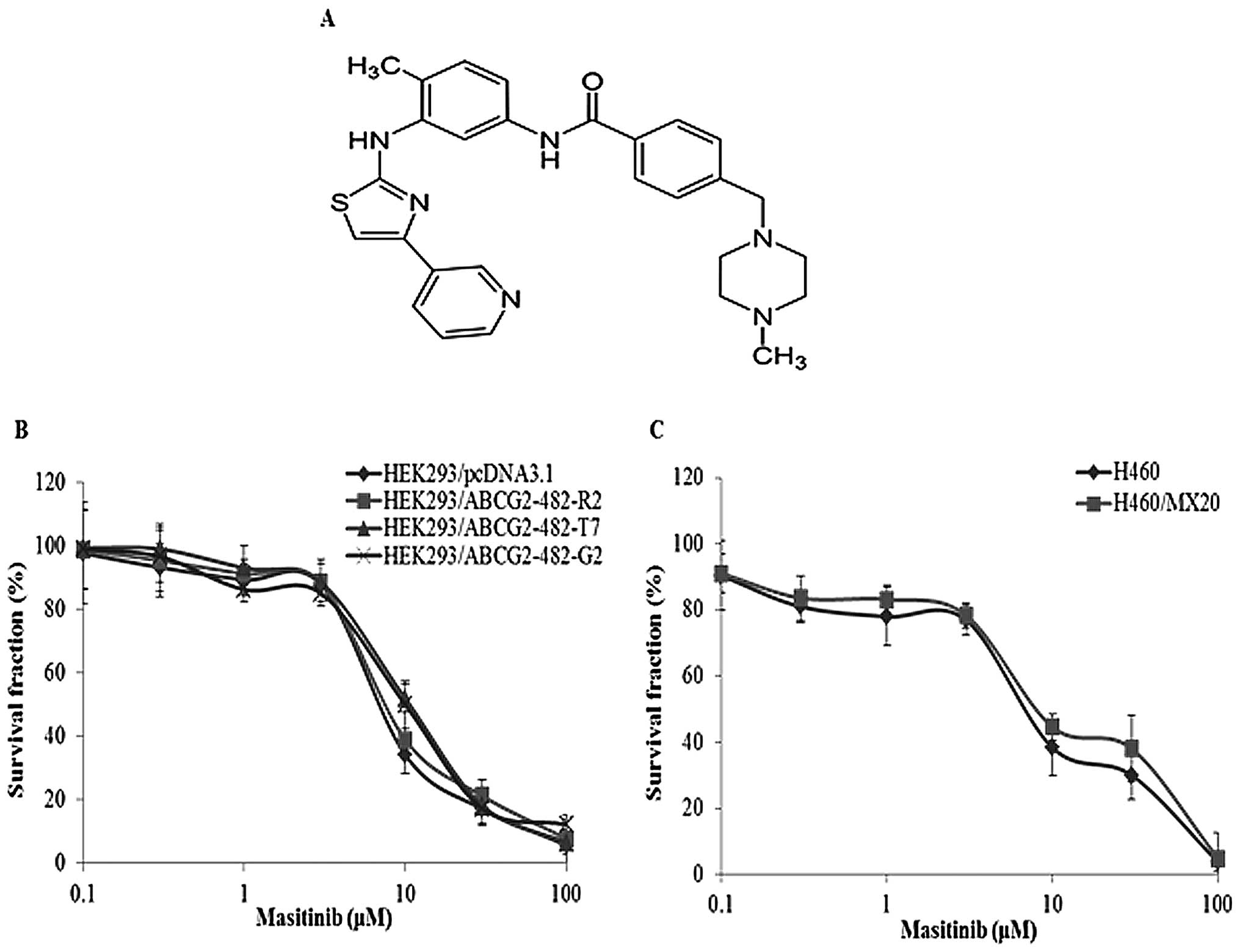
Cytotoxicity assays were performed to determine the
non-toxic concentration of masitinib for the reversal studies
(Fig. 1). It has been established
that mutations at position 482 in ABCG2 can alter substrate and
antagonist specificity of ABCG2 (15). Therefore, in the present study,
both wild-type (R482) and two mutant forms (R482T and R482G) of
ABCG2 were used. Masitinib, at 1.25 and 2.5 μM, produced a
concentration-dependent decrease in ABCG2-mediated resistance to
MX, SN38 and doxorubicin as indicated by the decrease in the
IC50 values (Table I).
We also used nilotinib (2.5 μM) as a positive control and as
previously reported (38), it
significantly decreased the resistance of wild-type
HEK293/ABCG2-482-R2, mutant HEK293/ABCG2-482-T7 and mutant
HEK293/ABCG2-482-G2 to MX, SN38 and doxorubicin respectively, as
compared to HEK293/pcDNA3.1 cells (Table I). In addition, masitinib did not
significantly alter the IC50 values for cisplatin, which
is not a substrate of ABCG2 (Table
I). These results suggest that masitinib enhances the
sensitivity of ABCG2 substrates in both wild-type and R482T/G
mutant ABCG2 overexpressing cells, i.e. it selectively reverses
MDR.
 | Table I.The effect of masitinib and nilotinib
on the survival of HEK293/pcDNA3.1, HEK293/ABCG2-482-R2,
HEK293/ABCG2-482-T7 and HEK293/ABCG2-482-G2 cells in the presence
of MX, SN38, doxorubicin and cisplatin. |
Table I.
The effect of masitinib and nilotinib
on the survival of HEK293/pcDNA3.1, HEK293/ABCG2-482-R2,
HEK293/ABCG2-482-T7 and HEK293/ABCG2-482-G2 cells in the presence
of MX, SN38, doxorubicin and cisplatin.
|
HEK293/pcDNA3.1 |
HEK293/ABCG2-482-R2 |
HEK293/ABCG2-482-T7 |
HEK293/ABCG2-482-G2 |
|---|
|
|
|
|
|---|
| Compounds |
IC50±SDa (nM) | FRb | IC50±SD
(nM) | FR | IC50±SD
(nM) | FR | IC50±SD
(nM) | FR |
|---|
| MX | 20.39±2.1 | 1.0 | 172.82±6.6 | 8.5 | 534.38±18.9 | 26.2 | 588.62±28.8 | 28.8 |
| +Masitinib 1.25
μM | 19.68±2.3 | 0.9 | 114.75±6.3d | 5.6 | 332.19±38.7d | 16.3 | 401.06±11.8d | 19.6 |
| +Masitinib 2.5
μM | 18.24±3.0 | 0.9 | 37.94±2.5d | 1.9 | 45.15±1.8d | 2.2 | 44.59±3.6d | 2.2 |
| +Nilotinib 2.5
μM | 16.24±3.6 | 0.8 | 18.5±1.5d | 0.9 | 26.68±2.9d | 1.3 | 28.46±1.5d | 1.4 |
| SN38 | 3.25±0.3 | 1.0 | 39.13±1.0 | 12.0 | 79.47±3.9 | 24.4 | 98.0±5.2 | 30.1 |
| +Masitinib 1.25
μM | 2.59±0.1c | 0.8 | 12.13±1.8d | 3.7 | 69.4±0.7d | 21.3 | 73.14±9.4d | 22.4 |
| +Masitinib 2.5
μM | 2.31±0.2c | 0.7 | 3.54±0.6d | 1.1 | 10.77±1.6d | 3.3 | 12.16±2.6d | 3.7 |
| +Nilotinib 2.5
μM | 1.98±0.2c | 0.6 | 2.25±0.5d | 0.7 | 3.14±0.2d | 0.9 | 6.07±0.6d | 1.9 |
| Doxorubicin | 25.28±3.2 | 1.0 | 139.51±1.5 | 5.5 | 212.58±29.5 | 8.4 | 306.96±12.3 | 12.1 |
| +Masitinib 1.25
μM | 23.75±2.6 | 0.9 | 90.35±4.7d | 3.6 | 115.2±11.3d | 4.5 | 266.07±9.7d | 10.5 |
| +Masitinib 2.5
μM | 21.41±2.0 | 0.8 | 41.43±2.8d | 1.6 | 61.18±4.4d | 2.4 | 84.23±10.2d | 3.3 |
| +Nilotinib 2.5
μM | 21.09±3.0 | 0.8 | 22.16±3.2d | 0.9 | 34.91±6.7d | 1.4 | 65.72±16.8d | 2.5 |
| Cisplatin | 2,813.9±102.6 | 1.0 | 2,339.1±98.3 | 0.8 | 1,895.0±487.1 | 0.7 | 1,925.5±128.1 | 0.7 |
| +Masitinib 2.5
μM | 2,778.7±231.0 | 1.0 | 2,265.7±84.1 | 0.8 | 2,052.0±224.8 | 0.7 | 1,958.7±240.7 | 0.7 |
| +Nilotinib 2.5
μM | 2,836.4±74.9 | 1.0 | 2,719.9±186.1 | 0.9 | 2,542.81±294.9 | 0.9 | 2,600.4±353.0 | 0.9 |
Masitinib reversed ABCG2-mediated resistance to MX
in transfected cell lines. Consequently, we determined whether
masitinib could also reverse MX resistance in an ABCG2
overexpressing H460/MX20 lung cancer cell line that specifically
confers resistance to MX. Masitinib, at 1.25 and 2.5 μM, in
combination with MX, SN38 or doxorubicin, significantly decreased
the resistance of H460/MX20 cell line as compared to the parental
H460 cell line. Nilotinib, 2.5 μM, was used as a positive
control and the results showed that it significantly decreased the
resistance of H460/MX20 cells compared to that of controls.
However, neither nilotinib nor masitinib (2.5 μM)
significantly altered the IC50 value of cisplatin (which
is not an ABCG2 substrate) in H460 and H460/MX20 cells (Table II).
| Table II.The effect of masitinib and nilotinib
on the survival of H460 and H460/MX20 cells to MX, SN38,
doxorubicin and cisplatin. |
Table II.
The effect of masitinib and nilotinib
on the survival of H460 and H460/MX20 cells to MX, SN38,
doxorubicin and cisplatin.
| H460 | H460/MX20 |
|---|
|
|
|---|
| Compounds |
IC50±SDa (nM) | FRb | IC50±SD
(nM) | FR |
|---|
| MX | 41.91±3.0 | 1.0 | 3700.2±143.7 | 88.2 |
| +Masitinib 1.25
μM | 33.45±2.1 | 0.8 | 203.1±9.3c | 4.8 |
| +Masitinib 2.5
μM | 31.0±0.5 | 0.7 | 61.47±2.2c | 1.4 |
| +Nilotinib 2.5
μM | 28.7±0.8 | 0.7 | 46.55±1.0c | 1.1 |
| SN38 | 20.77±1.4 | 1.0 | 1,414.7±191.5 | 68.0 |
| +Masitinib 1.25
μM | 20.3±1.6 | 1.0 | 622.3±75.5c | 30.0 |
| +Masitinib 2.5
μM | 17.62±0.8 | 0.8 | 80.84±5.1c | 3.9 |
| +Nilotinib 2.5
μM | 15.74±1.1 | 0.7 | 39.61±3.3c | 1.9 |
| Doxorubicin | 26.04±0.8 | 1.0 | 986.7±23.1 | 37.9 |
| +Masitinib 1.25
μM | 25.08±1.5 | 1.0 | 218.2±11.2c | 8.4 |
| +Masitinib 2.5
μM | 25.43±1.1 | 1.0 | 52.61±1.2c | 2.0 |
| +Nilotinib 2.5
μM | 24.32±0.8 | 0.9 | 32.64±1.9c | 1.2 |
| Cisplatin | 2,839.32±43.1 | 1.0 | 2,783.54±32.1 | 1.0 |
| +Masitinib 2.5
μM | 2,532.54±35.2 | 0.9 | 2,343.23±12.3 | 1.2 |
| +Nilotinib 2.5
μM | 2,742.55±23.1 | 1.0 | 2,711.98±53.7 | 1.0 |
Masitinib significantly increases the
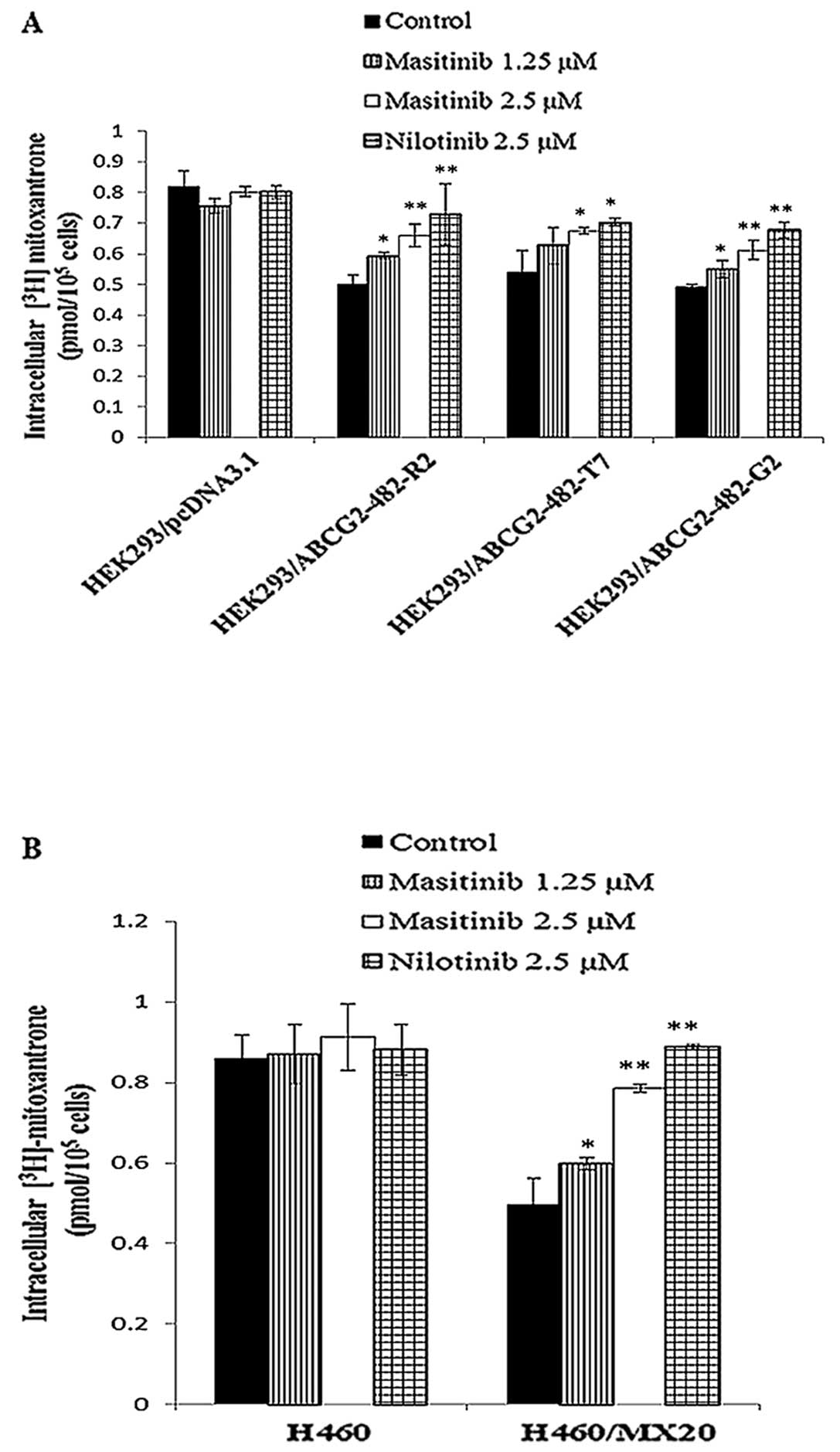
intracellular accumulation of [3H]-MX
In order to determine the mechanism by which
masitinib attenuates ABCG2-mediated MDR, we measured the effect of
masitinib on the intracellular accumulation of [3H]-MX,
a known substrate of ABCG2. The incubation of wild-type
HEK293/ABCG2-482-R2, mutant HEK293/ABCG2-482-T7, mutant
HEK293/ABCG2-482-G2, and H460/MX20 cells with 1.25 or 2.5 μM
masitinib significantly increased the intracellular accumulation
[3H]-MX in a concentration-dependent manner as compared
to the parental HEK293/pcDNA3.1 and H460 cell lines (Fig. 2). Nilotinib (2.5 μM), an
inhibitor of ABCG2 (38), also
significantly increased the accumulation of [3H]-MX.
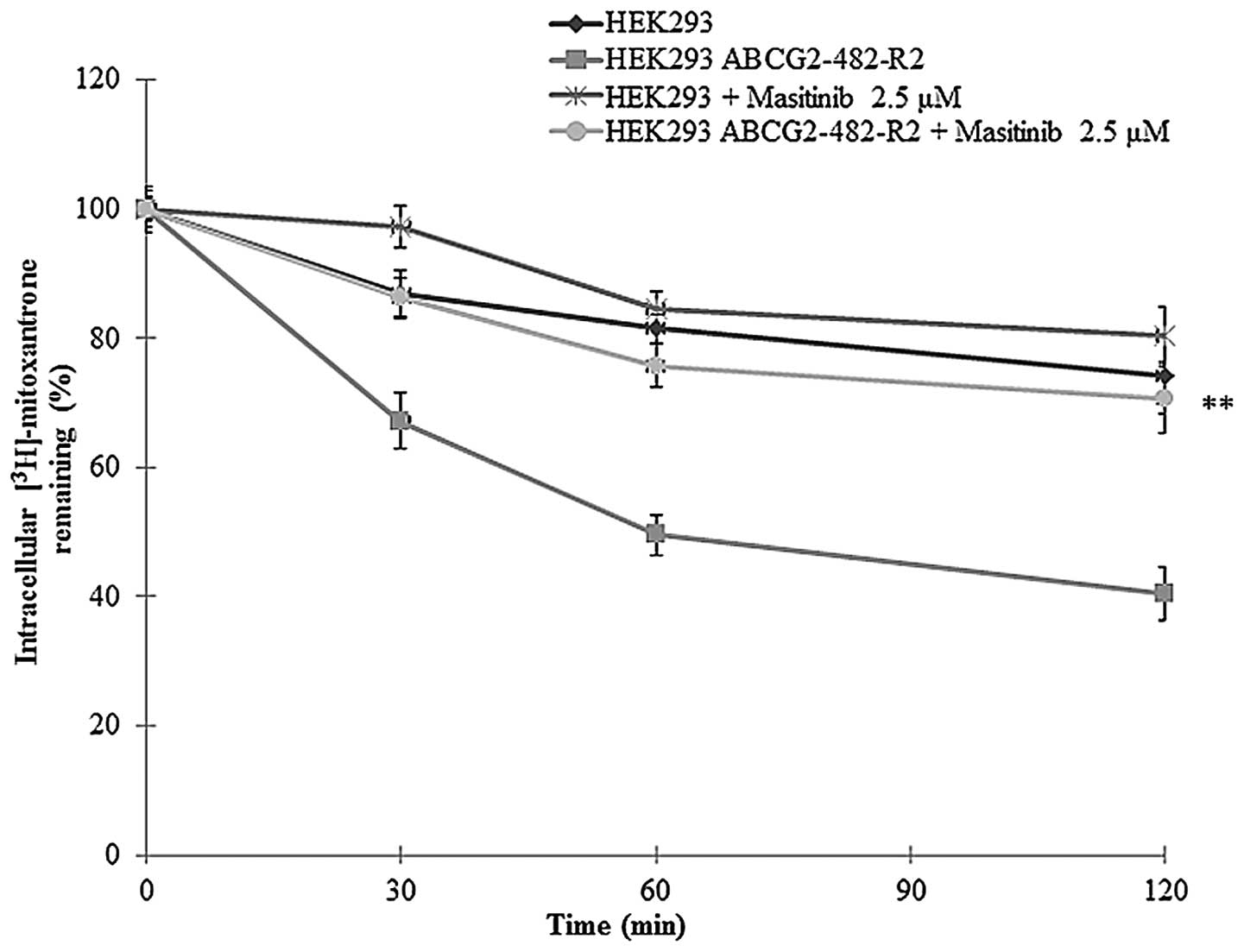
Masitinib significantly decreases the
cellular efflux of [3H]-MX
In these experiments, we determined the amount of
[3H]-MX present in the cells following incubation with
masitinib. The amount of [3H]-MX present in the
HEK293/ABCG2-482-R2 cell line was lower compared to HEK293/pcDNA3.1
cells due to the active efflux of [3H]-MX by the MDR
transporter ABCG2. However, in the presence of masitinib (2.5
μM), after 0, 30, 60 and 120 min the efflux of
[3H]-MX was significantly reduced (Fig. 3).
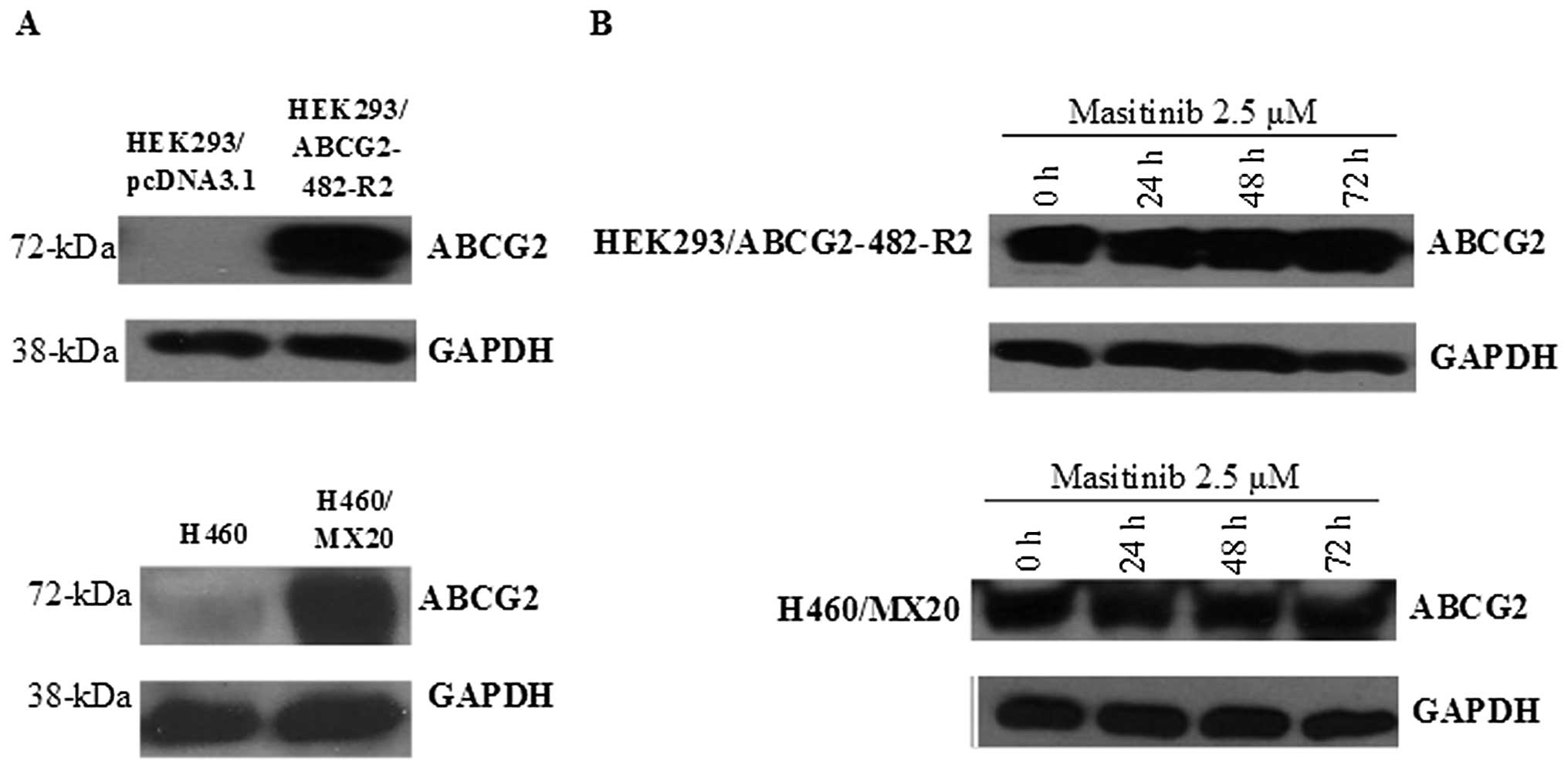
Masitinib does not alter the expression
of ABCG2 protein levels
Immunoblot analysis indicated a band with a
molecular weight of approximately 72-kDa in the HEK293/ABCG2-482-R2
and H460/MX20 cell lysates, suggesting the presence of the ABCG2
protein. However, this band was not present in the HEK293/pcDNA3.1
and H460 parental cell lines, indicating the absence of the ABCG2
protein in these cell lines (Fig.
4A).
In order to confirm that the masitinib-induced
reversal of MDR was not due to a decrease in the expression of the
ABCG2 protein, we measured the expression levels of ABCG2 in the
cell lysates after incubation with masitinib (2.5 μM) for 0,
24, 48 or 72 h. There was no significant change in the expression
levels of the ABCG2 protein in HEK293/ABCG2-482-R2 and H460/MX20
cells (Fig. 4B). These findings
suggest that the reversal of MDR by masitinib was not due to a
decrease in ABCG2 protein expression.
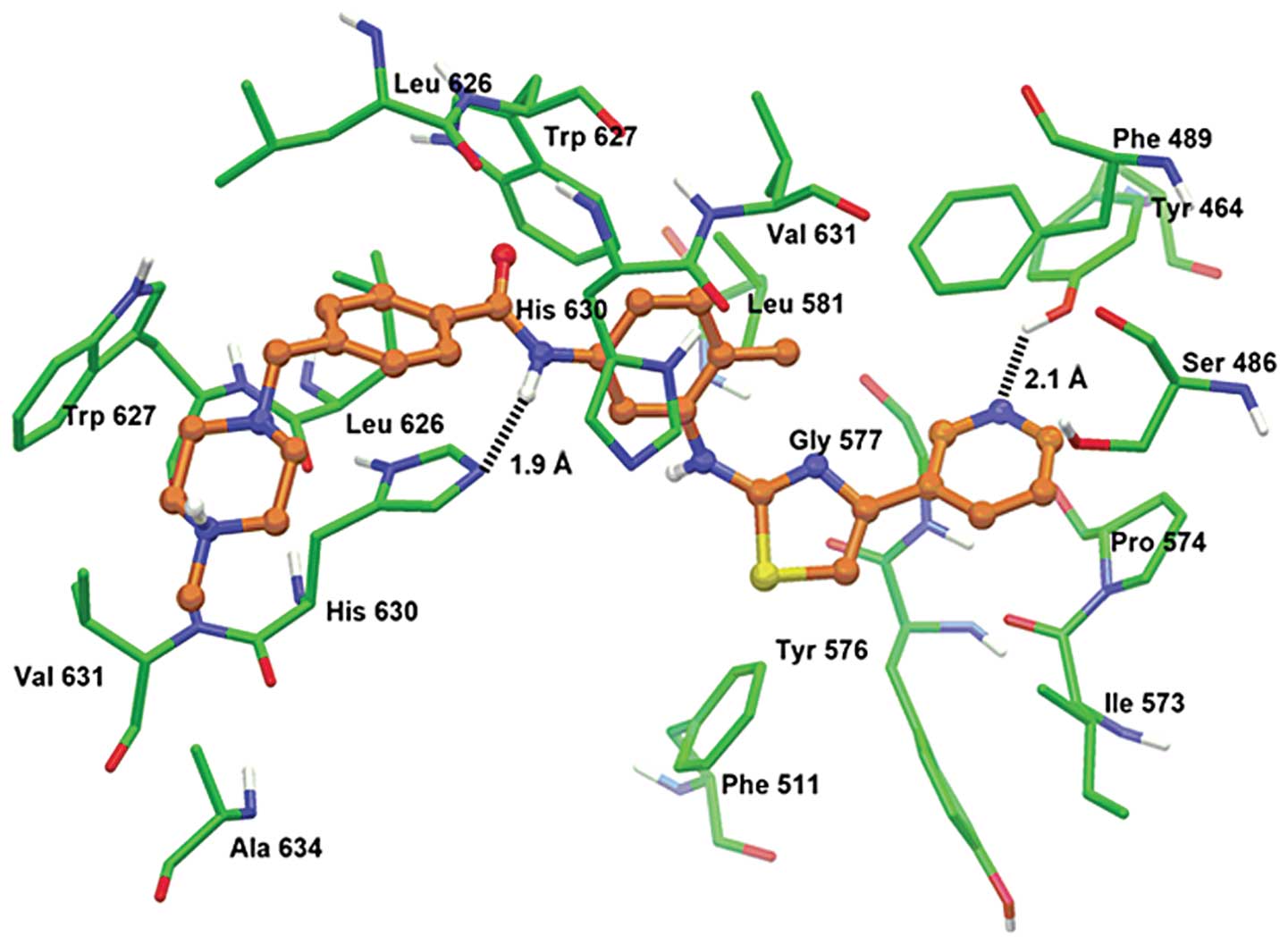
Model for binding of masitinib to
ABCG2
The XP-Glide predicted docked model of masitinib at
Asn629 centroid-based grid of human ABCG2 is shown in Fig. 5. The (4-methyl-piperazin-1-yl)
methyl-benzamide group formed hydrophobic interactions with the
side chains of two copies of Leu626, Trp627 and His630 along with
Val631 and Ala634. The amide group of the benzamide was involved in
a hydrogen bonding interaction with the imidazole ring nitrogen of
His630 (NH•••N-His630, 1.9 Å). The tolyl group was stabilized
through hydrophobic contacts with the side chains of Phe489,
Trp627, His630 and Val631. The thiazole ring and pyridine ring
formed hydrophobic interactions with Tyr464, Phe489, Phe511,
Ile573, Pro574, Tyr576 and Gly577. In addition, the pyridine ring
nitrogen atom formed a hydrogen bond with the hydroxy group of
Tyr464 (N•••HO-Tyr464, 2.1 Å). Docking studies were performed at
various grid-based sites of human ABCG2 homology model.
Discussion
One major finding of this study was that masitinib
(1.25 and 2.5 μM) significantly enhanced the sensitivity of
HEK293/ABCG2-482-R2, HEK293/ABCG2-482-T7 and HEK293/ABCG2-482-G2
cells overexpressing the ABCG2 transporter to MX, SN38 and
doxorubicin, which are substrates for the ABCG2 transporter
(57–59). Specifically, masitinib produced a
significant decrease in the IC50 values of the substrate
drugs for the ABCG2 transporter in the MTT assay. In contrast,
masitinib did not significantly alter the IC50 values
for the aforementioned substrate drugs in parental HEK293/pcDNA3.1
or in H460 cancer cells which do not overexpress ABCG2 transporter.
Furthermore, masitinib did not alter the sensitivity of
HEK293/ABCG2-482-R2, HEK293/ABCG2-482-T7 and HEK293/ABCG2-482-G2
cell lines to cisplatin, a drug that is not a substrate for the
ABCG2 transporter. These results suggest that masitinib
significantly reverses MDR mediated by the overexpression of the
ABCG2 transporter.
In order to gain insight into the mechanism of
action of masitinib, we also assessed the effect of masitinib on i)
the intracellular accumulation of [3H]-MX and ii) the
efflux of [3H]-MX in wild-type HEK293/ABCG2-482-R2,
mutant HEK293/ABCG2-482-T7, mutant HEK293/ABCG2-482-G2 and
H460/MX20 cells. Masitinib produced a concentration-dependent
increase in the response to the substrate drugs in the
aforementioned cell lines but not in parental HEK293/pcDNA3.1 and
H460 cell lines.
Masitinib significantly reverses MDR mediated by the
overexpression of the ABCG2 transporter. Previously, it has been
reported that small TKIs, including lapatinib (32), gefitinib (60), sunitinib (61) and apatinib (62) can reverse MDR in cell lines by
inhibiting the efflux of the substrate drugs from the cells. In
this in vitro study, masitinib (2.5 μM) did not
significantly alter the expression of the ABCG2 protein in
HEK293/ABCG2-482-R2 or H460/MX20 cells. This suggests that reversal
of MDR by masitinib is unlikely due to its decreasing the
expression of the ABCG2 protein. This finding does not exclude the
possibility that masitinib is preventing the translocation of the
ABCG2 protein to the cell membrane (data not shown). To understand
molecular interactions of masitinib, docking studies were performed
at various grid-based sites of human ABCG2 homology model.
According to its hydrophobic character (Clog P-value = 5.1), the
inhibition of ABCG2 by masitinib may be explained by its
significant distribution within the biomembrane, where it is
extracted by the ABCG2 transporter. In addition, pharmacophoric
features such as hydrophobic groups and/or aromatic ring center
(phenyl ring, piperazine ring, pyridine ring and thiazole ring),
hydrogen bond donor (-NH-) and hydrogen bond acceptor (pyridine
nitrogen) have been reported critical for ABCG2 inhibition
(63).
Our finding that masitinib reverses MDR and
resensitizes cells by inhibiting the activity of the ABCG2
transporter may have clinical application. For example, there is a
significant positive correlation between the overexpression of the
ABCG2 transporter and MDR in many types of cells, including
non-small cell lung cancer cells, thyroid and breast cancer cells
as well as hematological malignancies (64–71).
The presence of ABCG2 in esophageal squamous cell carcinoma and
advanced non-small cell lung cancer is significantly correlated
with a decreased survival (72–74).
The ABCG2 transporter is present in certain populations (side
population phenotype) of cancer stem cells and normal primitive
stem cells and its presence increases the likelihood of resistance
to various antineoplastic drugs (64,66,75–79).
Overall, it is possible that masitinib, in combination with
antineoplastics that are ABCG2 substrates, may be used in the
treatment of certain MDR cancers. The validation of this hypothesis
will require testing masitinib in clinical trials.
Collectively, the results of this in vitro
study indicated that masitinib, at concentrations that were
non-toxic to HEK293/pcDNA3.1 and H460 cells, significantly
increased the toxic effects of substrate antineoplastic drugs in
cells that overexpressed the ABCG2 transporter. This effect was
most likely due to the masitinib inhibition of the efflux activity
of the efflux transporter. Masitinib also reversed MDR in H460/MX20
lung cancer cells as indicated by their resensitization to MX, SN38
or doxorubicin. Our current results, provided they can be
clinically translated, suggest that masitinib, in combination with
other antineoplastics, may be efficacious in treating MDR cancers
due to the overexpression of ABCG2 transporter.
Acknowledgements
This study was supported by funds from
NIH (no. 1R15CA143701) and St. John’s University Research Seed
Grant (no. 579-1110-7002) to Z.-S.C. We are thankful to Drs Susan
E. Bates and Robert W. Robey (NIH) for ABCG2-transfected cell lines
and mitoxantrone selected H460/MX20. We thank Dr Mark F. Rosenberg
(University of Manchester, Manchester, UK) and Dr Zsolt Bikádi
(Virtua Drug Ltd., Budapest, Hungary) for providing coordinates of
ABCG2 homology model.
References
|
1.
|
Wu CP, Calcagno AM and Ambudkar SV:
Reversal of ABC drug transporter-mediated multidrug resistance in
cancer cells: evaluation of current strategies. Curr Mol Pharmacol.
1:93–105. 2008. View Article : Google Scholar
|
|
2.
|
Gottesman MM: Mechanisms of cancer drug
resistance. Annu Rev Med. 53:615–627. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Quintieri L, Fantin M and Vizler C:
Identification of molecular determinants of tumor sensitivity and
resistance to anticancer drugs. Adv Exp Med Biol. 593:95–104. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Liu YY, Han TY, Giuliano AE and Cabot MC:
Ceramide glycosylation potentiates cellular multidrug resistance.
FASEB J. 15:719–730. 2001. View Article : Google Scholar
|
|
5.
|
Lowe SW, Ruley HE, Jacks T and Housman DE:
p53-dependent apoptosis modulates the cytotoxicity of anticancer
agents. Cell. 74:957–967. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Synold TW, Dussault I and Forman BM: The
orphan nuclear receptor SXR coordinately regulates drug metabolism
and efflux. Nat Med. 7:584–590. 2001. View
Article : Google Scholar
|
|
7.
|
Deeley RG, Westlake C and Cole SP:
Transmembrane transport of endo- and xenobiotics by mammalian
ATP-binding cassette multidrug resistance proteins. Physiol Rev.
86:849–899. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bradbury PA and Middleton MR: DNA repair
pathways in drug resistance in melanoma. Anticancer Drugs.
15:421–426. 2004. View Article : Google Scholar
|
|
9.
|
Ambudkar SV, Kim IW, Xia D and Sauna ZE:
The A-loop, a novel conserved aromatic acid subdomain upstream of
the Walker A motif in ABC transporters, is critical for ATP
binding. FEBS Lett. 580:1049–1055. 2006. View Article : Google Scholar
|
|
10.
|
Liu FS: Mechanisms of chemotherapeutic
drug resistance in cancer therapy - a quick review. Taiwan J Obstet
Gynecol. 48:239–244. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Gillet JP, Efferth T and Remacle J:
Chemotherapy-induced resistance by ATP-binding cassette transporter
genes. Biochim Biophys Acta. 1775:237–262. 2007.PubMed/NCBI
|
|
12.
|
Linton KJ and Higgins CF: Structure and
function of ABC transporters: the ATP switch provides flexible
control. Pflugers Arch. 453:555–567. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Sodani K, Patel A, Kathawala RJ and Chen
ZS: Multidrug resistance associated proteins in multidrug
resistance. Chin J Cancer. 31:58–72. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Tiwari AK, Sodani K, Dai CL, Ashby CR Jr
and Chen ZS: Revisiting the ABCs of multidrug resistance in cancer
chemotherapy. Curr Pharm Biotechnol. 12:570–594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Mao Q and Unadkat JD: Role of the breast
cancer resistance protein (ABCG2) in drug transport. AAPS J.
7:E118–E133. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Suzuki M, Suzuki H, Sugimoto Y and
Sugiyama Y: ABCG2 transports sulfated conjugates of steroids and
xenobiotics. J Biol Chem. 278:22644–22649. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Rocchi E, Khodjakov A, Volk EL, et al: The
product of the ABC half-transporter gene ABCG2 (BCRP/MXR/ABCP) is
expressed in the plasma membrane. Biochem Biophys Res Commun.
271:42–46. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Maliepaard M, Scheffer GL, Faneyte IF, et
al: Subcellular localization and distribution of the breast cancer
resistance protein transporter in normal human tissues. Cancer Res.
61:3458–3464. 2001.PubMed/NCBI
|
|
19.
|
Cooray HC, Blackmore CG, Maskell L and
Barrand MA: Localisation of breast cancer resistance protein in
microvessel endothelium of human brain. Neuroreport. 13:2059–2063.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Doyle LA, Yang W, Abruzzo LV, et al: A
multidrug resistance transporter from human MCF-7 breast cancer
cells. Proc Natl Acad Sci USA. 95:15665–15670. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Schinkel AH and Jonker JW: Mammalian drug
efflux transporters of the ATP binding cassette (ABC) family: an
overview. Adv Drug Deliv Rev. 55:3–29. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Dean M and Allikmets R: Complete
characterization of the human ABC gene family. J Bioenerg Biomembr.
33:475–479. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Miyake K, Mickley L, Litman T, et al:
Molecular cloning of cDNAs which are highly overexpressed in
mitoxantrone-resistant cells: demonstration of homology to ABC
transport genes. Cancer Res. 59:8–13. 1999.
|
|
24.
|
Chen ZS, Robey RW, Belinsky MG, et al:
Transport of methotrexate, methotrexate polyglutamates, and
17beta-estradiol 17-(beta-D-glucuronide) by ABCG2: effects of
acquired mutations at R482 on methotrexate transport. Cancer Res.
63:4048–4054. 2003.PubMed/NCBI
|
|
25.
|
Honjo Y, Hrycyna CA, Yan QW, et al:
Acquired mutations in the MXR/BCRP/ABCP gene alter substrate
specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res.
61:6635–6639. 2001.PubMed/NCBI
|
|
26.
|
Dai CL, Liang YJ, Wang YS, et al:
Sensitization of ABCG2-overexpressing cells to conventional
chemotherapeutic agent by sunitinib was associated with inhibiting
the function of ABCG2. Cancer Lett. 279:74–83. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Pozza A, Perez-Victoria JM, Sardo A,
Ahmed-Belkacem A and Di Pietro A: Purification of breast cancer
resistance protein ABCG2 and role of arginine-482. Cell Mol Life
Sci. 63:1912–1922. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Ejendal KF and Hrycyna CA: Multidrug
resistance and cancer: the role of the human ABC transporter ABCG2.
Curr Protein Pept Sci. 3:503–511. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Padmanabhan R, Chen KG, Gillet JP, et al:
Regulation and expression of the ATP-binding cassette transporter
ABCG2 in human embryonic stem cells. Stem Cells. 30:2175–2187.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Evseenko DA, Paxton JW and Keelan JA:
Independent regulation of apical and basolateral drug transporter
expression and function in placental trophoblasts by cytokines,
steroids, and growth factors. Drug Metab Dispos. 35:595–601. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Chen ZS, Aoki S, Komatsu M, et al:
Reversal of drug resistance mediated by multidrug resistance
protein (MRP) 1 by dual effects of agosterol A on MRP1 function.
Int J Cancer. 93:107–113. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Dai CL, Tiwari AK, Wu CP, et al: Lapatinib
(Tykerb, GW572016) reverses multidrug resistance in cancer cells by
inhibiting the activity of ATP-binding cassette subfamily B member
1 and G member 2. Cancer Res. 68:7905–7914. 2008. View Article : Google Scholar
|
|
33.
|
Kathawala RJ, Wang YJ, Ashby CR Jr and
Chen ZS: Recent advances regarding the role of ABC subfamily C
member 10 (ABCC10) in the efflux of antitumor drugs. Chin J Cancer.
Oct 9–2013.(Epub ahead of print).
|
|
34.
|
Deng W, Dai CL, Chen JJ, et al: Tandutinib
(MLN518) reverses multidrug resistance by inhibiting the efflux
activity of the multidrug resistance protein 7 (ABCC10). Oncol Rep.
29:2479–2485. 2013.PubMed/NCBI
|
|
35.
|
Szakacs G, Paterson JK, Ludwig JA,
Booth-Genthe C and Gottesman MM: Targeting multidrug resistance in
cancer. Nat Rev Drug Discov. 5:219–234. 2006. View Article : Google Scholar
|
|
36.
|
Yang D, Kathawala RJ, Chufan EE, et al:
Tivozanib reverses multidrug resistance mediated by ABCB1
(P-glycoprotein) and ABCG2 (BCRP). Future Oncol. Dec 3–2013.(Epub
ahead of print).
|
|
37.
|
Shen T, Kuang YH, Ashby CR, et al:
Imatinib and nilotinib reverse multidrug resistance in cancer cells
by inhibiting the efflux activity of the MRP7 (ABCC10). PLoS One.
4:e75202009. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Tiwari AK, Sodani K, Wang SR, et al:
Nilotinib (AMN107, Tasigna) reverses multidrug resistance by
inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR
transporters. Biochem Pharmacol. 78:153–161. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Shi Z, Peng XX, Kim IW, et al: Erlotinib
(Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B
member 1 and ATP-binding cassette subfamily G member 2-mediated
drug resistance. Cancer Res. 67:11012–11020. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Vermersch P, Benrabah R, Schmidt N, et al:
Masitinib treatment in patients with progressive multiple
sclerosis: a randomized pilot study. BMC Neurol. 12:362012.
View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Rommer PS and Stuve O: Management of
secondary progressive multiple sclerosis: prophylactic
treatment-past, present, and future aspects. Curr Treat Options
Neurol. 15:241–258. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Humbert M, de Blay F, Garcia G, et al:
Masitinib, a c-kit/PDGF receptor tyrosine kinase inhibitor,
improves disease control in severe corticosteroid-dependent
asthmatics. Allergy. 64:1194–1201. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Lee-Fowler TM, Guntur V, Dodam J, Cohn LA,
DeClue AE and Reinero CR: The tyrosine kinase inhibitor masitinib
blunts airway inflammation and improves associated lung mechanics
in a feline model of chronic allergic asthma. Int Arch Allergy
Immunol. 158:369–374. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Tebib J, Mariette X, Bourgeois P, et al:
Masitinib in the treatment of active rheumatoid arthritis: results
of a multicentre, open-label, dose-ranging, phase 2a study.
Arthritis Res Ther. 11:R952009. View
Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Walker UA: More about masitinib. Arthritis
Res Ther. 11:1202009. View
Article : Google Scholar
|
|
46.
|
Georgin-Lavialle S, Lhermitte L, Suarez F,
et al: Mast cell leukemia: identification of a new c-Kit mutation,
dup(501–502) and response to masitinib, a c-Kit tyrosine kinase
inhibitor. Eur J Haematol. 89:47–52. 2012.PubMed/NCBI
|
|
47.
|
Paul C, Sans B, Suarez F, et al: Masitinib
for the treatment of systemic and cutaneous mastocytosis with
handicap: a phase 2a study. Am J Hematol. 85:921–925. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Le Cesne A, Blay JY, Bui BN, et al: Phase
II study of oral masitinib mesilate in imatinib-naive patients with
locally advanced or metastatic gastro-intestinal stromal tumour
(GIST). Eur J Cancer. 46:1344–1351. 2010.PubMed/NCBI
|
|
49.
|
Mitry E, Hammel P, Deplanque G, et al:
Safety and activity of masitinib in combination with gemcitabine in
patients with advanced pancreatic cancer. Cancer Chemother
Pharmacol. 66:395–403. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Gottesman MM, Fojo T and Bates SE:
Multidrug resistance in cancer: role of ATP-dependent transporters.
Nat Rev Cancer. 2:48–58. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Kruh GD, Guo Y, Hopper-Borge E, Belinsky
MG and Chen ZS: ABCC10, ABCC11, and ABCC12. Pflugers Arch.
453:675–684. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Robey RW, Honjo Y, Morisaki K, et al:
Mutations at amino-acid 482 in the ABCG2 gene affect substrate and
antagonist specificity. Br J Cancer. 89:1971–1978. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Carmichael J, DeGraff WG, Gazdar AF, Minna
JD and Mitchell JB: Evaluation of a tetrazolium-based semiautomated
colorimetric assay: assessment of chemosensitivity testing. Cancer
Res. 47:936–942. 1987.PubMed/NCBI
|
|
54.
|
Hazai E and Bikadi Z: Homology modeling of
breast cancer resistance protein (ABCG2). J Struct Biol. 162:63–74.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Alqawi O, Bates S and Georges E:
Arginine482 to threonine mutation in the breast cancer resistance
protein ABCG2 inhibits rhodamine 123 transport while increasing
binding. Biochem J. 382:711–716. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Sun YL, Kathawala RJ, Singh S, et al:
Zafirlukast antagonizes ATP-binding cassette subfamily G member
2-mediated multidrug resistance. Anticancer Drugs. 23:865–873.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Litman T, Brangi M, Hudson E, et al: The
multidrug-resistant phenotype associated with overexpression of the
new ABC half-transporter, MXR (ABCG2). J Cell Sci. 113:2011–2021.
2000.PubMed/NCBI
|
|
58.
|
Schellens JH, Maliepaard M, Scheper RJ, et
al: Transport of topoisomerase I inhibitors by the breast cancer
resistance protein. Potential clinical implications. Ann NY Acad
Sci. 922:188–194. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Jonker JW, Smit JW, Brinkhuis RF, et al:
Role of breast cancer resistance protein in the bioavailability and
fetal penetration of topotecan. J Natl Cancer Inst. 92:1651–1656.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Nakamura Y, Oka M and Soda H: Gefitinib
(‘Iressa’, ZD1839), an epidermal growth factor receptor tyrosine
kinase inhibitor, reverses breast cancer resistance
protein/ABCG2-mediated drug resistance. Cancer Res. 65:1541–1546.
2005.
|
|
61.
|
Shukla S, Robey RW, Bates SE and Ambudkar
SV: Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine
kinase inhibitor, blocks function of the ATP-binding cassette (ABC)
transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos.
37:359–365. 2009. View Article : Google Scholar
|
|
62.
|
Mi YJ, Liang YJ, Huang HB, et al: Apatinib
(YN968D1) reverses multidrug resistance by inhibiting the efflux
function of multiple ATP-binding cassette transporters. Cancer Res.
70:7981–7991. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Nicolle E, Boumendjel A, Macalou S, et al:
QSAR analysis and molecular modeling of ABCG2-specific inhibitors.
Adv Drug Deliv Rev. 61:34–46. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
An Y and Ongkeko WM: ABCG2: the key to
chemoresistance in cancer stem cells? Expert Opin Drug Metab
Toxicol. 5:1529–1542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Chen YJ, Huang WC, Wei YL, et al: Elevated
BCRP/ABCG2 expression confers acquired resistance to gefitinib in
wild-type EGFR-expressing cells. PLoS One. 6:e214282011. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Natarajan K, Xie Y, Baer MR and Ross DD:
Role of breast cancer resistance protein (BCRP/ABCG2) in cancer
drug resistance. Biochem Pharmacol. 83:1084–1103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67.
|
Kerr ID, Haider AJ and Gelissen IC: The
ABCG family of membrane-associated transporters: you don’t have to
be big to be mighty. Br J Pharmacol. 164:1767–1779. 2011.
|
|
68.
|
Robey RW, Medina-Perez WY, Nishiyama K, et
al: Overexpression of the ATP-binding cassette half-transporter,
ABCG2 (Mxr/BCrp/ABCP1), in flavopiridol-resistant human breast
cancer cells. Clin Cancer Res. 7:145–152. 2001.PubMed/NCBI
|
|
69.
|
Robey RW, Ierano C, Zhan Z and Bates SE:
The challenge of exploiting ABCG2 in the clinic. Curr Pharm
Biotechnol. 12:595–608. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70.
|
Woodward OM, Kottgen A and Kottgen M: ABCG
transporters and disease. FEBS J. 278:3215–3225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
Xu J, Peng H and Zhang JT: Human multidrug
transporter ABCG2, a target for sensitizing drug resistance in
cancer chemotherapy. Curr Med Chem. 14:689–701. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72.
|
Hang D, Dong HC, Ning T, Dong B, Hou DL
and Xu WG: Prognostic value of the stem cell markers CD133 and
ABCG2 expression in esophageal squamous cell carcinoma. Dis
Esophagus. 25:638–644. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73.
|
Li J, Li ZN, Du YJ, Li XQ, Bao QL and Chen
P: Expression of MRP1, BCRP, LRP, and ERCC1 in advanced
non-small-cell lung cancer: correlation with response to
chemotherapy and survival. Clin Lung Cancer. 10:414–421. 2009.
View Article : Google Scholar
|
|
74.
|
Tsunoda S, Okumura T, Ito T, et al: ABCG2
expression is an independent unfavorable prognostic factor in
esophageal squamous cell carcinoma. Oncology. 71:251–258. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
75.
|
Bunting KD: ABC transporters as phenotypic
markers and functional regulators of stem cells. Stem Cells.
20:11–20. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
76.
|
Kim M, Turnquist H, Jackson J, et al: The
multidrug resistance transporter ABCG2 (breast cancer resistance
protein 1) effluxes Hoechst 33342 and is overexpressed in
hematopoietic stem cells. Clin Cancer Res. 8:22–28. 2002.PubMed/NCBI
|
|
77.
|
Kusuhara H and Sugiyama Y: ATP-binding
cassette, subfamily G (ABCG family). Pflugers Arch. 453:735–744.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
78.
|
Ding XW, Wu JH and Jiang CP: ABCG2: a
potential marker of stem cells and novel target in stem cell and
cancer therapy. Life Sci. 86:631–637. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
79.
|
Sung JM, Cho HJ, Yi H, et al:
Characterization of a stem cell population in lung cancer A549
cells. Biochem Biophys Res Commun. 371:163–167. 2008. View Article : Google Scholar : PubMed/NCBI
|