Introduction
Acute myeloid leukemia (AML) is a clonal disorder of
hematopoiesis characterized by the uncontrolled proliferation and
accumulation of immature and dysfunctional hematopoietic
progenitors accompanied by blockage in normal hematopoiesis. During
the last decade, chemotherapy has been widely used as a main
approach and preferred therapy for AML treatment. However, the
majority of adults diagnosed with AML are destined to relapse, and
the major cause of relapse and therapeutic failure in AML is
resistance to chemotherapy (1).
Many factors account for the occurrence of chemotherapeutic
multidrug resistance (MDR), including overexpression of drug
resistance-related proteins, alterations in drug targets, escape
from cell cycle checkpoints, altered pharmacokinetics, increased
drug efflux and stem cell development (2). Given the pressing need to improve
outcomes in AML patients, it is crucial to elucidate the mechanisms
of chemoresistance.
MicroRNAs (miRNAs, miRs) represent a new class of
small, non-coding endogenous RNAs that range in size from 19 to 25
nucleotides (nt) and can negatively regulate target gene expression
at the post-transcriptional level. Mature miRNAs are incorporated
into the RNA-induced silencing complex (RISC) to cause either
degradation or inhibition of translation by binding to the
3’-untranslated region (3’-UTR) of target mRNAs (3,4). Due
to their diverse functions in cell proliferation, apoptosis,
invasion, cell differentiation, cell cycle progression, and
hematopoiesis, overwhelming evidence has indicated the important
regulatory roles of miRNAs during carcinogenesis and
chemoresistance (5,6). Moreover, modulation of these
dysregulated miRNAs sensitizes cancer drug-resistant cells to
chemotherapy, suggesting the potential of miRNAs as targets for
anticancer drug resistance. For example, overexpression of
miR-331-5p and/or miR-27a can effectively increase
the drug sensitivity of leukemia DOX-resistant cells. Furthermore,
miR-331-5p and miR-27a were verified to target the
multidrug resistance 1 gene (MDR1), the most extensively studied
gene directly involved in drug resistance (7).
miR-181b belongs to the miR-181
family, which is known to be evolutionarily conserved among the
vertebrate lineage, with high homology (8). Functional research identified
miR-181b as a key regulator of restricting B cell
lymphomagenesis. miR-181b impairs the class switch
recombination (CSR) reaction and results in the downregulation of
activation-induced cytidine deaminase (AID) in activated B cells
(9). It is important to note that
the effects of miR-181b in chemoresistance vary according to
different tumor microenvironments. In hepatocellular carcinoma
(HCC) cells, miR-181b enhances resistance to the anticancer
drug doxorubicin (10). A markedly
enhanced expression of miR-181b was also shown in more
aggressive breast cancers and chemotherapy-resistant breast cancer
cells, and knockdown of miR-181b can be used to render
breast tumors more responsive to tamoxifen (11,12).
In contrast, forced miR-181b expression sensitizes human MDR
gastric cancer cells and lung cancer cells to chemotherapy-induced
apoptosis by directly targeting Bcl-2 protein (13). Also, miR-181b is
downregulated in chronic lymphocytic leukemia (CLL) compared to
normal controls (14–16), and its expression further decreases
during the progression of CLL; indeed, downregulation of
miR-181a and miR-181b was associated with shorter
overall survival (OS) and disease-free survival in CLL patients
(17). Nevertheless, whether
miR-181b is mechanistically associated with AML progression
and relapse remains unknown.
miR-451 is a positive regulator of late-stage
maturation of committed erythroid precursors (18). Many studies have shown that
miR-451 is widely dysregulated in human cancers and plays a
critical role in tumorigenesis and tumor progression (19,20).
In addition, miR-451 is involved in mediating the resistance
of breast cancer cells to the chemotherapeutic drug doxorubicin
through regulating MDR1 expression (21). Aberrant expression of
miR-486-5p is a frequent molecular event that has important
functions in human malignances (22,23);
however, evidence of biological roles for miR-451 and
miR-486 in relapsed/refractory AML has not yet been
reported.
Therefore, based on the important functions of
miRNAs in AML initiation and progression, we sought to investigate
roles of these 3 miRNAs in chemoresistance of AML. We provided
evidence that overexpression of miR-181b increased the drug
sensitivity of AML MDR cells by targeting high-mobility group box-1
protein (HMGB1) and myeloid cell leukemia-1 (Mcl-1). In conclusion,
the identification of miR-181b function highlights a new
approach for the development of drug resistance therapy in AML.
Materials and methods
Patient samples
Forty-three AML patients, including 31 newly
diagnosed AML patients who had not undergone any therapy or
treatment and 12 relapsed/refractory AML patients, were included in
our study. All relapsed/refractory AML cases either failed to
respond to initial chemotherapy or relapsed after initial complete
remission (CR). We excluded patients with inherently resistant AML
in the newly diagnosed AML group. Informed consent was obtained
from all patients in accordance with the Declaration of Helsinki
and with approval of the Medical Ethics Committee of Qilu Hospital,
Shandong University. Mononuclear cells (MNCs) from bone marrow
aspirates were isolated by density-gradient centrifugation with the
use of Ficoll-Paque Plus (Ficoll, Pharmacia LKB Biotechnology,
Piscataway, NY, USA). Among those AML patients, 3 matched-pair BM
samples were available both at the diagnosis time prior to
treatment and the relapsed/refractory state. Detailed clinical
information for the AML patients is summarized in Table I.
 | Table I.Characteristics of the 43 AML
patients. |
Table I.
Characteristics of the 43 AML
patients.
| Variables | Newly
diagnosed |
Relapsed/refractory |
|---|
| No. of
patients | 31 | 12 |
| Gender
(male/female) | 14/17 | 6/6 |
| Age: median years
(range) | 45 (14–80) | 47.5 (13–78) |
| WBC count
(109/l) median: (range) | 13.67
(1.28–259.8) | 7.55
(1.09–204.3) |
| Hemoglobin (g/dl)
median: (range) | 78 (43–125) | 84 (68–157) |
| Platelet count
(109/l) median: (range) | 40 (3–250) | 116.5 (11–236) |
| FAB
classification | | |
| No. of
patients | | |
| M0 | 1 | 0 |
| M1 | 2 | 0 |
| M2 | 5 | 2 |
| M3 | 5 | 3 |
| M4 | 8 | 2 |
| M5 | 10 | 5 |
| M6 | 0 | 0 |
Cell culture and transfection
The human leukemia cell lines K562 and HL-60 and
their multidrug-resistant counterparts, K562/A02 and HL-60/ADM,
were purchased from the Institute of Hematology and Blood Diseases
Hospital, Chinese Academy of Medical Sciences and Peking Union
Medical College (Tianjin, China). Cells were cultured in complete
medium (RPMI-1640 supplemented with 10% fetal bovine serum, 100
U/ml penicillin G, 100 μg/ml streptomycin, and 2 mM
L-glutamine), at 37°C in humidified air containing 5%
CO2 and were routinely subcultured every 2–3 days. In
addition, in order to maintain the MDR phenotype, doxorubicin was
added to the medium of drug-resistant cell lines until 2 weeks
before use in experiments.
The synthetic miR-181b mimic, miR-181b
inhibitor, and negative controls were purchased from GenePharma
(Shanghai, China). Short hairpin RNA targeting human HMGB1 was
synthesized from Ribobio (Guangzhou, China). Transfection of miRNAs
and short hairpin RNAs was performed with Lipofectamine 2000
reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the
manufacturer’s protocol.
Cell viability assay
Cells were seeded in 96-well culture plates at
density of 5×104 cells/ml and were treated with serial
dilutions of doxorubicin (DOX) or cytarabine (Ara-C) for 48 h. Ten
microliters of CCK8 (Beyotime, China) was added to each well, and
the cells were incubated at 37°C for 4 h. The absorbance in each
well was read at 450 nm by an automated microplate
spectrophotometer (Thermo Scientific, USA), with a reference
wavelength of 650 nm. Each sample was measured in triplicate, and
experiments were repeated 3 times.
Apoptosis assay
Apoptosis was detected using an Annexin V/FITC and
PI apoptosis detection kit (Invitrogen). Briefly, after treatment
with DOX (2.0 μg/ml) or Ara-C (1.0 μM) for 48 h,
2×105 cells were harvested, resuspended in 100 μl
flow cytometry binding buffer, and stained with 5 μl Annexin
V/FITC followed by 1 μl PI. Cells were then incubated in the
dark for 15 min at room temperature, and 400 μl binding
buffer was added. The cells were immediately measured by
FACSCalibur (Becton-Dickinson, CA, USA).
Quantitative real-time polymerase chain
reaction (qRT-PCR) analysis
Total RNA was extracted from all samples using
TRIzol reagent (Invitrogen). To detect miR-181b expression,
cDNA was reverse transcribed from total RNA using special stem-loop
primers and the mirVana reverse transcription kit (Ambion Inc.,
Austin, TX, USA), followed by qPCR using TaqMan primer/probe sets
from Ambion. U6 small nucleolar RNA was used as an internal control
for miRNAs. To detect Mcl-1 and HMGB1 expression, cDNA was
synthesized from ∼1 μg total RNA using the M-MLV RTase cDNA
Synthesis kit (Takara, Dalian, China) according to the
manufacturer's instructions. Quantitative PCR was conducted on an
Applied Biosystems 7900HT system (ABI Prism, Foster City, CA, USA)
with SYBR Green PCR Master Mix (Toyobo, Osaka, Japan). GAPDH was
used to normalize Mcl-1 and HMGB1 expression levels. Each sample
was measured in triplicate, and fold-changes in mRNA expression
levels were calculated using the comparative threshold cycle (Ct)
method. The sequences of primer pairs specific for each gene are
shown in Table II.
| Table II.Primer sets and genes included in
qPCR. |
Table II.
Primer sets and genes included in
qPCR.
| Name | Forward primer | Reverse primer |
|---|
| HMGB1 |
5′-GCACTCCCTCCATCTTTGGA-3′ |
5′-CAGCTCCGACAGATCCAGTTC-3′ |
| Mcl-1 |
5′-TGCTTCGGAAACTGGACATCA-3′ |
5′-TAGCCACAAAGGCACCAAAAG-3′ |
| β-actin |
5′-CACTGTGTTGGCGTACAGGT-3′ |
5′-TCATCACCATTGGCAATGAG-3′ |
Western blot analysis
Cells were lysed with RIPA buffer (0.15 mM NaCl,
0.05 mM Tris-HCl, pH 7.5, 1% Triton, 0.1% SDS, 0.1% sodium
deoxycholate, and 1% NP40) containing protease and phosphatase
inhibitors, and stored at −20°C. Samples (30–50 μg) were
separated by SDS-polyacrylamide gel electrophoresis and transferred
to polyvinylidene difluoride (PVDF) membranes. Membranes were
blocked with 5% non-fat dry milk in Tris-buffered saline containing
0.05% Tween-20 (TBST) for 1 h at room temperature, then incubated
with the following specific antibodies: rabbit anti-HMGB1, rabbit
anti-Mcl-1 (Epitomics, Burlingame, CA, USA), or anti-β-actin (Cell
Signaling Technology, New England BioLabs Inc., USA) for 1 h or
overnight at 4°C. After washing with TBST 3 times, membranes were
incubated with secondary antibodies (horseradish
peroxidase-conjugated anti-rabbit immunoglobulin; Santa Cruz
Biotechnology) for 1 h at room temperature. Protein bands were
visualized using an Anmobilon Western Chemiluminescent HRP
Substrate system (Millipore Corp., Billerica, MA, USA).
Dual luciferase activity assay
The 3’-UTRs of HMGB1 and Mcl-1 mRNAs were
PCR-amplified from human genomic DNA and inserted into the
SpeI and HindIII sites in the pMIR-Report vector
(Ambion Inc., Austin, TX, USA) downstream from the firefly
luciferase coding sequence. 293T cells were cotransfected with
pMIR-Report constructs, miR-181b mimic or scramble control
in combination with pRL-TK (Promega) using Lipofectamine 2000.
Firefly and Renilla luciferase activities were determined using the
dual luciferase reporter assay system (Promega) according to the
manufacturer’s instructions.
Statistical analysis
Data are expressed as means ± standard errors of at
least 3 independent experiments. Student’s t-test and one-way
analysis of variance were used to determine significance between
groups. Statistical analysis was carried out using SPSS software
(version 17.0). Differences with P-values of <0.05 were
considered statistically significant.
Results
MiRNA expression in bone marrow blasts
from AML patients and cell lines
To investigate whether miRNA participated in drug
resistance in AML, we analyzed the expression of 3 miRNAs
(miR-181b, miR-451 and miR-486) in bone marrow
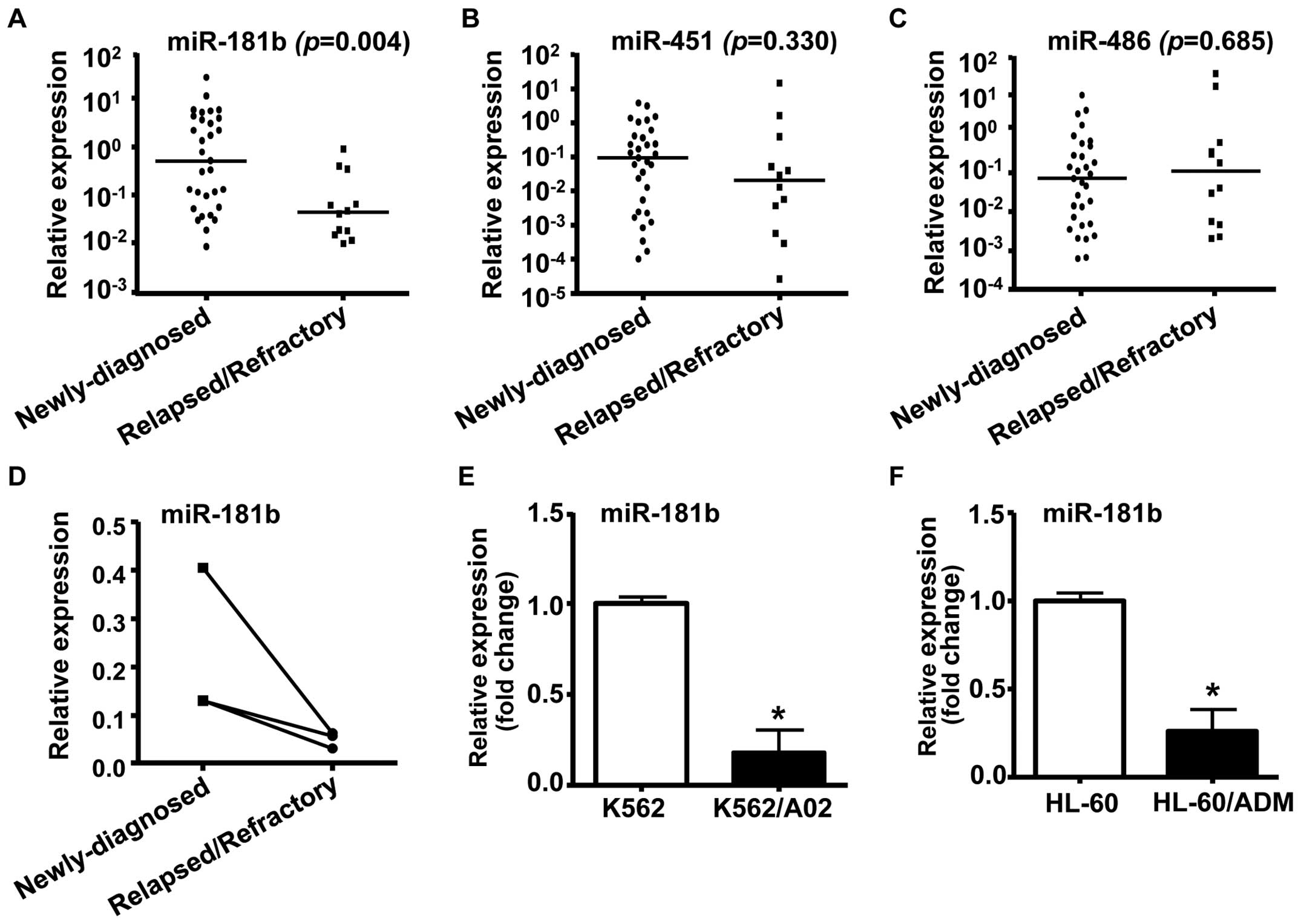
samples from AML patients. The results showed that miR-181b
was downregulated in AML samples from relapsed/refractory patients
in comparison with those of newly diagnosed AML patients (Fig. 1A, P<0.01). However, qRT-PCR
assay revealed that there were no differences in miR-451 or
miR-486 levels between the 2 groups of AML patients
(Fig. 1B and C). BM samples from 3
paired AML patients were analyzed for miR-181b levels at
both the newly diagnosed and relapsed/refractory state. We found a
high level of miR-181b at diagnosis, whereas a significant
decrease in miR-181b expression in the relapsed/refractory
state (Fig. 1D).
Next, we performed quantitative RT-PCR to compare
miR-181b expression between human MDR leukemia cells and
their parental drug-sensitive cells. As shown in Fig. 1E and F, the expression of
miR-181b was decreased significantly in human MDR leukemia
cell lines K562/A02 and HL-60/ADM, as compared to their parental
cell lines K562 and HL-60, respectively. These results suggested
that miR-181b may be involved in the development of drug
resistance and disease progression in AML.
The levels of miR-181b in newly diagnosed AML
patients were then split into two classes (high and low
expressions, according to the median expression in all samples).
miR-181b expression showed a negative correlation with
treatment response in our enrolled cases, in which low expression
of miR-181b was observed more frequently in poor prognosis
subset (8/11, 72.7%) than in good prognosis subset (7/20, 35%).
These results confirm that low miR-181b expression can act
as a prognostic factor associated with poor outcome of AML
patients.
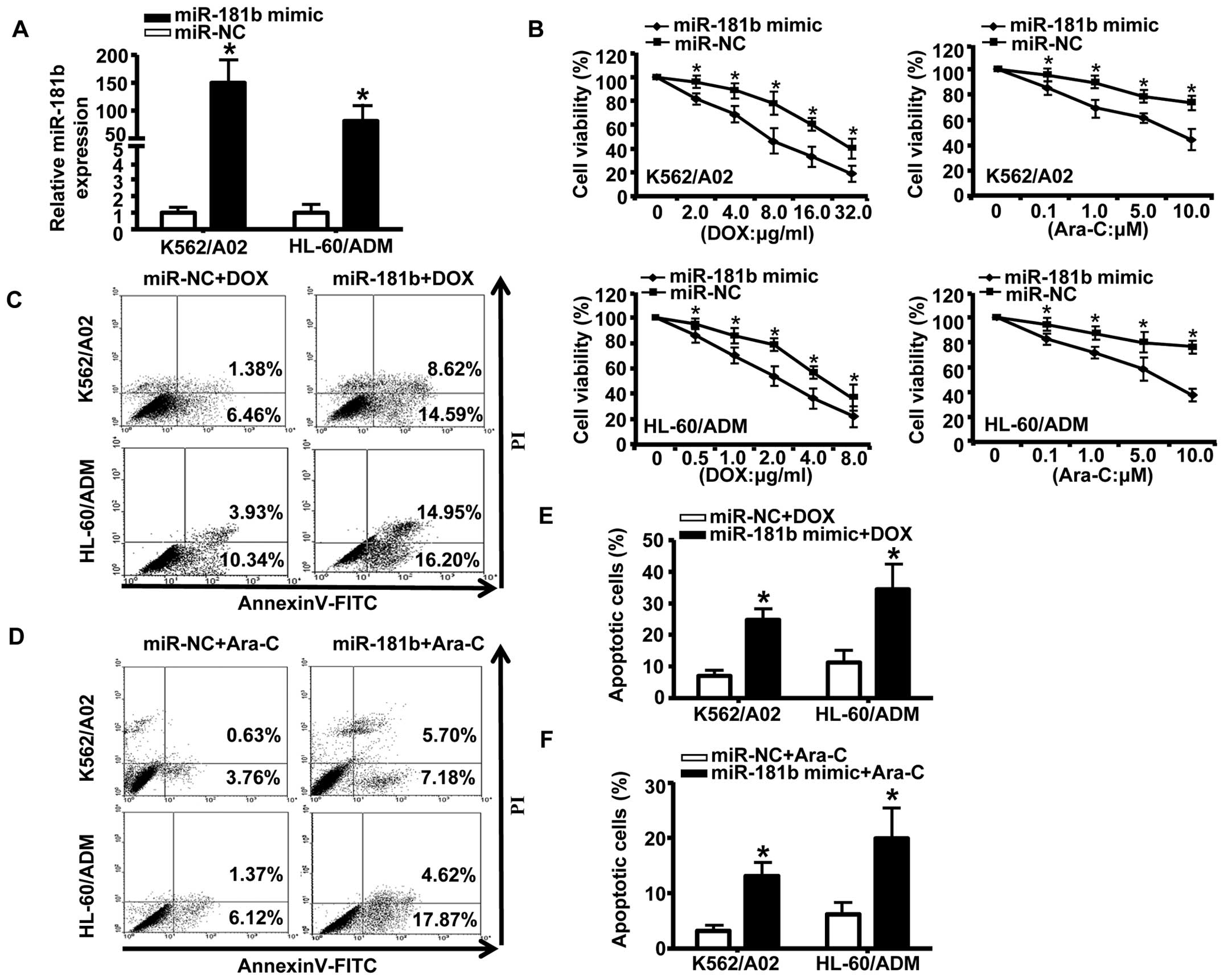
Forced miR-181b expression sensitizes
K562/A02 and HL-60/ADM cells to chemotherapeutic agents
To further explore the effects of miR-181b on
chemoresistance in AML, we transiently transfected K562/A02 and
HL-60/ADM cells with miR-181b mimic or a negative control.
Quantitative RT-PCR confirmed that miR-181b mimic
effectively enhanced the expression of miR-181b (Fig. 2A). Following transfection of
drug-resistant cells with the mimic of miR-181b, we treated
the cells with a series of concentrations of DOX or Ara-C for 48 h.
As shown in Fig. 2B, transfection
with the miR-181b mimic significantly inhibited cell growth
compared to transfection with the negative control. We next
analyzed the effects of miR-181b on apoptosis in AML by flow
cytometry. The results showed that ectopic expression of
miR-181b markedly increased chemotherapy-inducing apoptosis
(as measured by the percentage of Annexin V-FITC-positive cells) in
AML drug-resistant cells (Fig.
2C–F). Taken together, these data indicated that the forced
expression of miR-181b increased the drug sensitivity of AML
MDR cells to chemotherapy and promoted apoptosis.
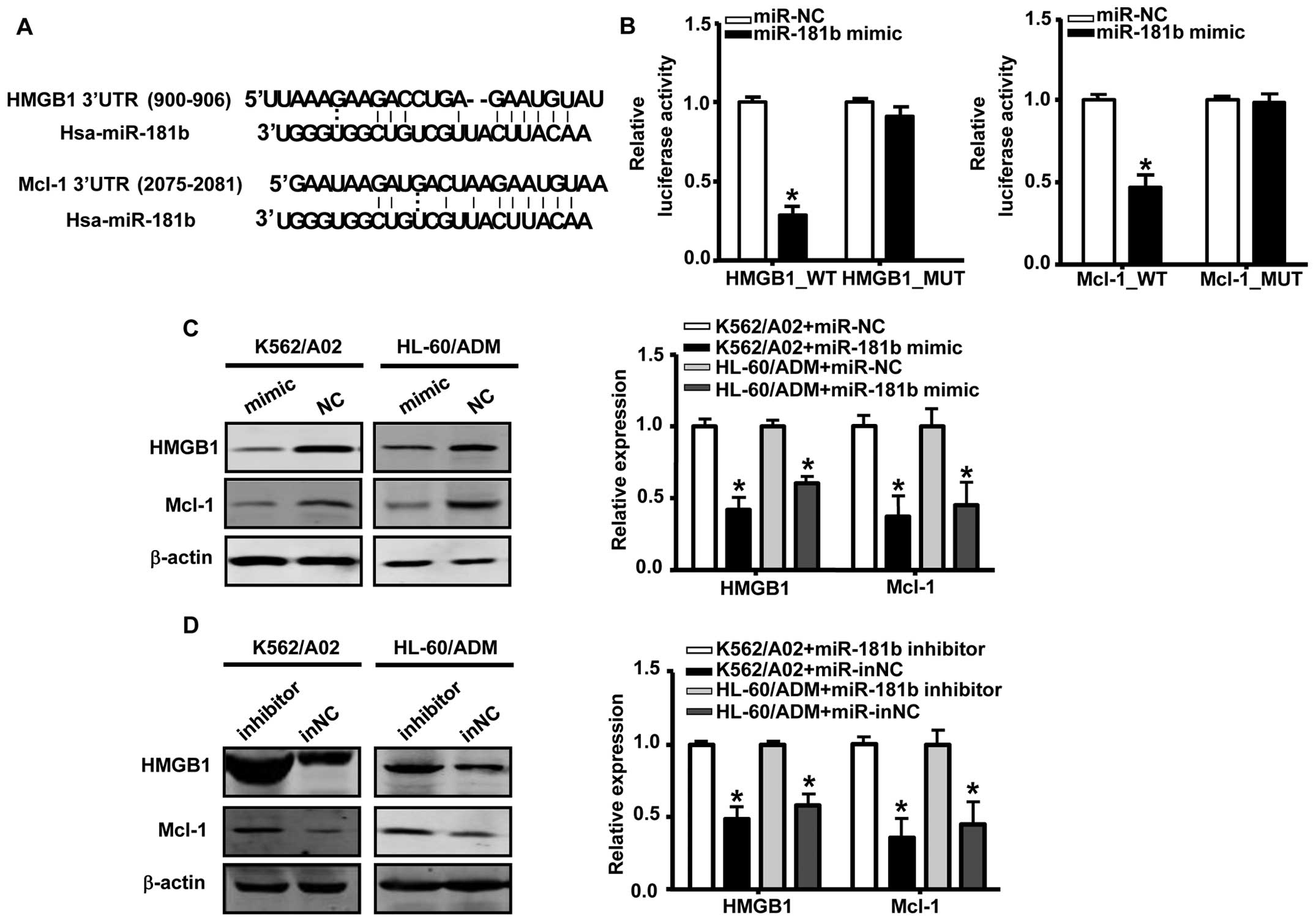
HMGB1 and Mcl-1 were identified as
targets of miR-181b
The database Target Scan Human 6.2 was used to
predict candidate targets of miR-181b. We identified HMGB1
and Mcl-1 as potential targets of miR-181b; these targets
contain putative binding sites in the 3’-UTR that match with the
‘seed’ sequence of miR-181b (Fig. 3A). To validate these interactions,
we constructed luciferase reporter vectors carrying wild-type or
mutated HMGB1 or Mcl-1 3’-UTR target sites and cotransfected these
vectors with the miR-181b mimic into 293T cells. As
illustrated in Fig. 3B,
transfection with the miR-181b mimic significantly decreases
luciferase activity, whereas mutation of the 3’-UTR binding sites
of HMGB1 or Mcl-1 in the reporter vector abrogated this effect,
indicating that miR-181b directly interacted with the 3’-UTR
of HMGB1 and Mcl-1.
In order to verify whether miR-181b affected
endogenous levels of HMGB1 and Mcl-1 in AML, we analyzed HMGB1 and
Mcl-1 expression after transfection with the miR-181b mimic
or inhibitor for 48 h. The results revealed that the ectopic
expression of miR-181b in K562/A02 and HL-60/ADM cells
robustly suppressed endogenous HMGB1 and Mcl-1 expression both at
mRNA and protein levels (Fig. 3C).
Conversely, knockdown of miR-181b by miR-181b
inhibitor markedly increased the expression of both HMGB1 and Mcl-1
(Fig. 3D). These results
demonstrated that HMGB1 and Mcl-1 were direct targets of
miR-181b in AML MDR cells.
Restoration of miR-181b increased the
drug sensitivity of AML MDR cells by targeting HMGB1 and Mcl-1
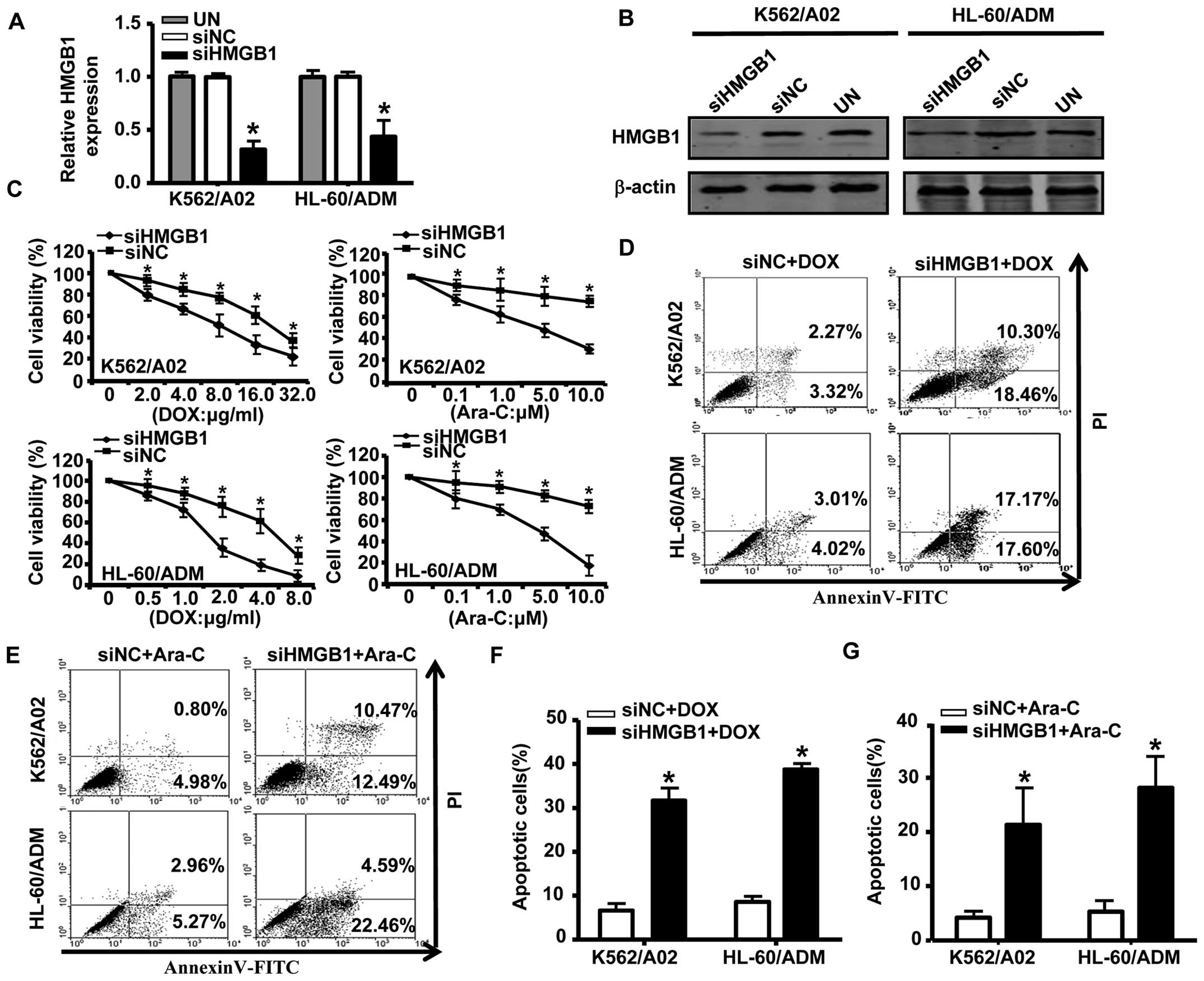
To further elucidate the role of HMGB1 in drug
resistance in AML, we transfected K562/A02 and HL-60/ADM cells with
HMGB1 siRNA; the effectiveness of the siRNAs designed to silence
HMGB1 in cells is shown in Fig. 4A and
B. Compared with negative controls, knockdown of HMGB1
dramatically decreased survival of K562/A02 and HL-60/ADM cells
exposed to different concentrations of DOX or Ara-C (Fig. 4C). Annexin V/PI analysis showed
that the proportion of apoptotic cells was significantly higher in
HMGB1 siRNA-transfected cells compared to cells transfected with
negative control siRNA (Fig.
4D–G). We previously reported that downregulation of Mcl-1 via
RNA interference sensitized MDR leukemia cells to chemotherapy and
induced apoptosis (24). Thus, our
results suggested that downregulation of Mcl-1 and HMGB1 was one
pathway through which miR-181b increased drug sensitivity in
AML MDR cells.
Overexpression of HMGB1 in
relapsed/refractory AML patients
After verifying that HMGB1 was a target of
miR-181b, we then sought to elucidate its role in AML. We
first investigated HMGB1 expression by quantitative RT-PCR in BM
cells obtained from 31 newly diagnosed AML patients and 12 patients
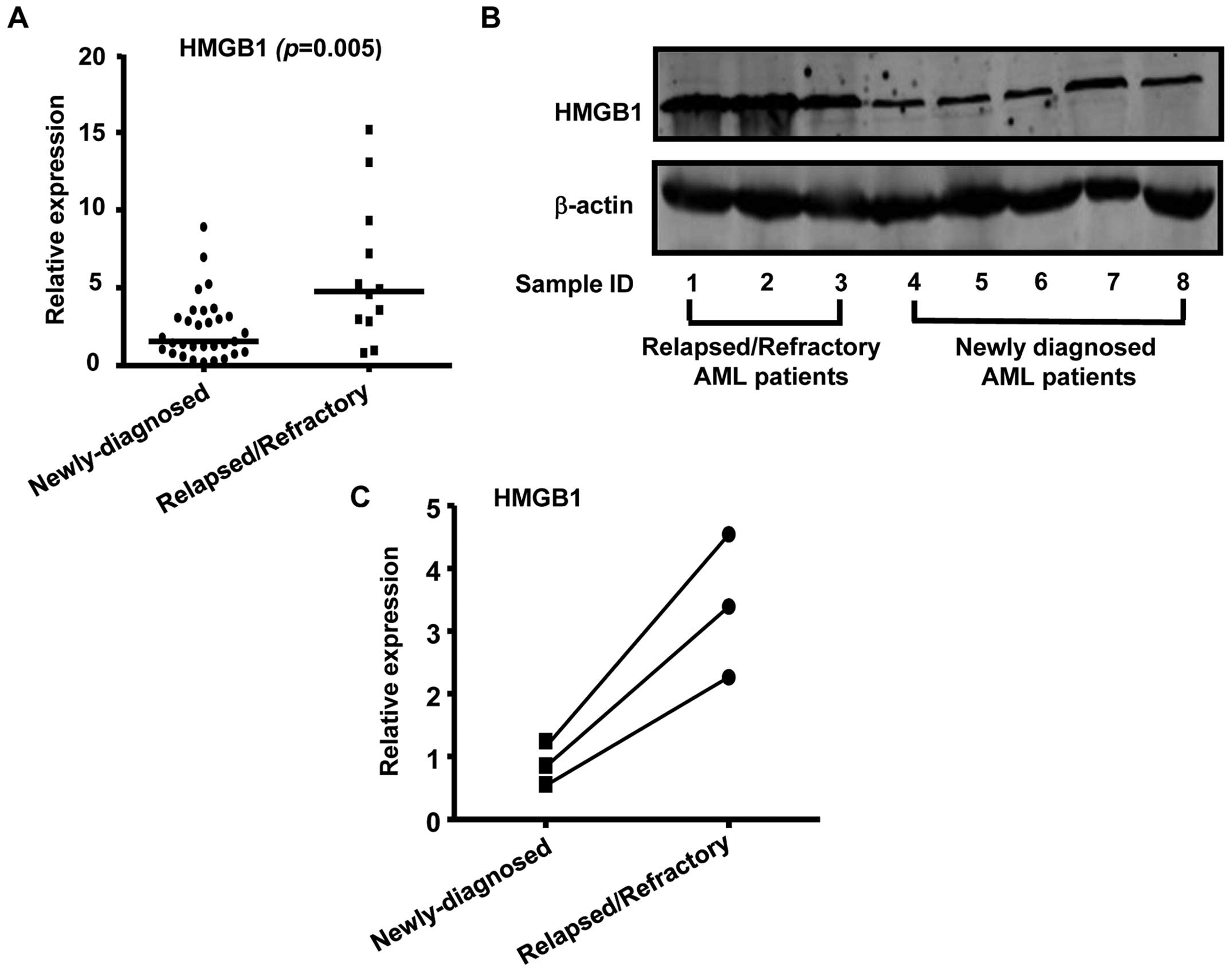
with relapsed/refractory leukemia. As shown in Fig. 5A, HMGB1 expression was
significantly increased in relapsed/refractory AML patients
compared to newly diagnosed AML patients. Consistent with the
real-time RT-PCR data, western blot analysis showed that HMGB1
protein levels were upregulated in relapsed/refractory AML patients
than in newly diagnosed AML patients (Fig. 5B). We also noted an increase in the
expression level of HMGB1 in sequential samples obtained from 3
paired AML patients and found that these HMGB1 levels were
inversely correlated with miR-181b expression levels
(Fig. 5C). In conclusion, our data
supported that the HMGB1 gene was aberrantly expressed in AML and
was required for the development and progression of multidug
resistance in AML.
Discussion
Expression and function analyses have unraveled the
close relationship between aberrant miR-181b expression and
the pathogenesis, diagnosis, and prognosis of AML. It has been
demonstrated that expression of miR-181b is associated with
lower CR rates and shorter relapse-free survival (RFS) and OS in
adult patients with de novo AML (25). Multivariable analysis has revealed
that increased expression of miR-181a and miR-181b is
also significantly associated with favorable outcomes in
cytogenetically abnormal AML with CEBPA mutations and
cytogenetically normal AML patients (26–29).
However, an obvious increase of miR-181b-5p was observed in
AML serum samples and that higher expression levels of
miR-181b-5p in serum are correlated with a poorer OS
(30). Possible explanation for
the different roles of miR-181b in serum and tissues of AML
patients could be the different origins and AML patient samples
used. These results also indicated that miR-181b may be
controlled by complex regulatory pathways in AML. miR-181a,
another important member of the miR-181 family, was
downregulated in the chemoresistant leukemia cell lines K562/A02
and HL-60/Ara-C compared to the parental K562 and HL-60 cells, and
restoration of miR-181a expression could sensitize K562/A02
and HL-60/Ara-C cell lines to chemotherapeutic agents by targeting
Bcl-2 (31,32). However, the role of miR-181b
in the development of chemoresistance in AML cells is still
unknown. The expression data reported in this study showed that
among the selected miRNAs, only miR-181b was differentially
expressed in relapsed/refractory AML patients and newly diagnosed
AML patients. Consistent with the results in AML patient samples,
miR-181 expression was lower in drug-resistant versus
parental drug-sensitive AML cell lines. Additionally, in BM samples
collected from 3 patients both at the diagnosis, prior to treatment
and after relapse, we also noted decreases in the expression levels
of miR-181b in sequential samples. Although this cutoff
point needs to be validated in an extended patient cohort, the
current results suggested that lower expression of miR-181b
contributed to disease aggressiveness in AML. Furthermore, we
verified that both AML drug-resistant cell lines K562/A02 and
HL-60/ADM exhibited greatly enhanced sensitivity to DOX or Ara-C
after transfection with the miR-181b mimic. These results
suggested that miR-181b may play an important role in the
development and maintenance of MDR in AML.
HMGB1, a highly conserved DNA-binding protein, is
ubiquitously expressed in the nuclei and cytoplasm of almost all
eukaryotic cells. Within the nucleus, HMGB1 stabilizes nucleosome
formation, assists in DNA mismatch repair, replication, and
recombination, and regulates the transcription of many genes.
Extracellular HMGB1 was identified as a prototypical
damage-associated molecular pattern molecule (DAMP) that is
released both actively and passively from cells in response to
infection or injury. Once released, HMGB1 can act as a chemokine or
cytokine by ligation with specific receptors, including the
receptor for advanced glycation end products (RAGE) and toll-like
receptors (TLRs)-2, -4 and -9 (33). Recent studies demonstrated that the
high expression of HMGB1 is tightly associated with unlimited
replicative potential, angiogenesis, apoptosis, inflammation,
invasion, and metastasis in cancer (34). Serum levels of HMGB1 are
significantly higher in children with acute lymphoid leukemia (ALL)
in the initial treatment group compared with healthy controls and
the complete remission group (35). In addition to being involved in
pathogenesis of leukemia, HMGB1 can be released from leukemia cell
lines after chemotherapy-induced cytotoxicity and can promote
chemotherapy resistance by inducing autophagy in leukemia cells
(36). In this study, we showed
that HMGB1 expression was significantly increased in
relapsed/refractory AML patients compared to newly diagnosed
patients. Inhibition of HMGB1 using siRNA enhanced drug sensitivity
in leukemia cells, and this result was consistent with that in a
previous study by Xie et al (37). Our study also identified HMGB1 as a
direct and functional target of miR-181b. In addition, an
obvious increase in HMGB1 levels and an inverse correlation with
miR-181b expression were also observed in blasts from the
same AML patient. Thus, based on these results, HMGB1 appears to
constitute a novel, powerful therapeutic target for AML
patients.
Another well-distinguished target of miR-181b
in our study was Mcl-1, an anti-apoptotic member of the Bcl-2
family. Mcl-1 contains 3 BH domains and has a very short half-life.
Functionally, Mcl-1 acts at mitochondria by binding to and
sequestering a subset of BH3-only pro-apoptotic Bcl-2 family
members, including Bak, Bax, Bim, Bid, Bik, Noxa and Puma, thereby
preventing the release of cytochrome c into the cytoplasm (38). The high expression of Mcl-1 in a
wide variety of cancers is being intensively studied. Indeed,
numerous reports have documented that overexpression of Mcl-1
protects cancer cells from apoptosis, representing a significant
barrier to the efficacy of chemotherapeutic agents (39). Additionally, elevated expression of
Mcl-1 was shown to correlate with leukemic relapse in AML patients
(40), and recent studies have
shown that Mcl-1 is upregulated in FMS-like tyrosine
kinase-3-internal tandem duplication (FLT3-ITD)-positive AML cell
lines and primary MNCs from AML patients. Mcl-1 is an essential
effector of FLT3-ITD-mediated drug resistance, and suppression of
endogenous Mcl-1 sensitizes FLT3-ITD-positive leukemias to
cytotoxic therapies (41). We have
previously reported that newly diagnosed or relapsed/refractory
leukemia patients express higher Mcl-1 levels than patients that
are in complete remission. Consistent with this, knockdown of Mcl-1
sensitizes MDR leukemia cells to chemotherapy and induces apoptosis
(24). In the present study, we
demonstrated that miR-181b directly regulated Mcl-1
expression post-transcriptionally in AML drug-resistant cell lines,
suggesting that downregulation of Mcl-1 is one of the major
mechanisms through which miR-181b promoted drug sensitivity in AML
MDR cells. In CLL, Mcl-1 has also been identified as a target of
miR-181b and miR-181a, and increased Mcl-1 protein
levels have been shown to be inversely correlated with decreased
miR-181b and miR-181a expression (15,42).
It is generally accepted that resistance to apoptosis is the main
mechanism of drug resistance. The mitochondrial apoptotic pathway
is tightly regulated by the Bcl-2 family. Suppression of HMGB1 by
siRNA in K562/A02 leukemia cells promotes ADM-induced Smac/DIABLO
release from the mitochondria to the cytoplasm, increasing the
activation of caspase-3 (37). In
addition, a recent study showed that autophagy-mediated HMGB1
release antagonizes vincristine-induced apoptosis in gastric cancer
cells via transcriptional regulation of Mcl-1 and that
HMGB1-mediated upregulation of Mcl-1 transcription is dependent on
RAGE (43). Further in-depth
studies are needed to investigate the interactions of HMGB1 and
Mcl-1 in the regulation of AML drug resistance.
In conclusion, the present study showed that
miR-181b functioned as a tumor suppressor in AML
chemoresistance. The abnormally decreased expression of
miR-181b was responsible for the occurrence of drug
resistance in some AML patients. Forced expression of
miR-181b could enhance drug sensitivity and apoptosis in AML
MDR cells at least partially though direct suppression of its
target genes, HMGB1 and Mcl-1. Because the biological effects and
regulatory networks of miR-181b in AML are more complex than
was once recognized, further studies are needed to confirm these
results in an extended patient cohort. However, our data implied
that ectopic implantation of miR-181b alone or in
conjunction with other anticancer agents may be a promising
strategy to combat MDR in AML.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (81070422,
30871088, 81070407, 81000223), SRFDP of Educational Ministry
(20100131110060), Medical and Health Science Technology Development
of Shandong Province, China (2013WS0229).
References
|
1.
|
Estey EH: Treatment of relapsed and
refractory acute myelogenous leukemia. Leukemia. 14:476–479. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Chen Y, Tang Y, Guo C, Wang J, Boral D and
Nie D: Nuclear receptors in the multidrug resistance through the
regulation of drug-metabolizing enzymes and drug transporters.
Biochem Pharmacol. 83:1112–1126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Garzon R, Marcucci G and Croce CM:
Targeting microRNAs in cancer: rationale, strategies and
challenges. Nat Rev Drug Discov. 9:775–789. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Nagano H, Tomimaru Y, Eguchi H, et al:
MicroRNA-29a induces resistance to gemcitabine through the
Wnt/beta-catenin signaling pathway in pancreatic cancer cells. Int
J Oncol. 43:1066–1072. 2013.PubMed/NCBI
|
|
6.
|
Croce C: Introduction to the role of
microRNAs in cancer diagnosis, prognosis, and treatment. Cancer J.
18:213–214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Feng DD, Zhang H, Zhang P, et al:
Down-regulated miR-331-5p and miR-27a are associated with
chemotherapy resistance and relapse in leukaemia. J Cell Mol Med.
15:2164–2175. 2011. View Article : Google Scholar
|
|
8.
|
Li QJ, Chau J, Ebert PJ, et al: miR-181a
is an intrinsic modulator of T cell sensitivity and selection.
Cell. 129:147–161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
de Yebenes VG, Belver L, Pisano DG, et al:
miR-181b negatively regulates activation-induced cytidine deaminase
in B cells. J Exp Med. 205:2199–2206. 2008.PubMed/NCBI
|
|
10.
|
Wang B, Hsu SH, Majumder S, et al:
TGFbeta-mediated upregulation of hepatic miR-181b promotes
hepatocarcinogenesis by targeting TIMP3. Oncogene. 29:1787–1797.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Bisso A, Faleschini M, Zampa F, et al:
Oncogenic miR-181a/b affect the DNA damage response in aggressive
breast cancer. Cell Cycle. 12:1679–1687. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Lu Y, Roy S, Nuovo G, et al:
Anti-microRNA-222 (anti-miR-222) and -181B suppress growth of
tamoxifen-resistant xenografts in mouse by targeting TIMP3 protein
and modulating mitogenic signal. J Biol Chem. 286:42292–42302.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Zhu W, Shan X, Wang T, Shu Y and Liu P:
miR-181b modulates multidrug resistance by targeting BCL2 in human
cancer cell lines. Int J Cancer. 127:2520–2529. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Marton S, Garcia MR, Robello C, et al:
Small RNAs analysis in CLL reveals a deregulation of miRNA
expression and novel miRNA candidates of putative relevance in CLL
pathogenesis. Leukemia. 22:330–338. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Visone R, Veronese A, Rassenti LZ, et al:
miR-181b is a biomarker of disease progression in chronic
lymphocytic leukemia. Blood. 118:3072–3079. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Li S, Moffett HF, Lu J, et al: MicroRNA
expression profiling identifies activated B cell status in chronic
lymphocytic leukemia cells. PLoS One. 6:e169562011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Visone R, Veronese A, Balatti V and Croce
CM: MiR-181b: new perspective to evaluate disease progression in
chronic lymphocytic leukemia. Oncotarget. 3:195–202.
2012.PubMed/NCBI
|
|
18.
|
Dore LC, Amigo JD, Dos Santos CO, et al: A
GATA-1-regulated microRNA locus essential for erythropoiesis. Proc
Natl Acad Sci USA. 105:3333–3338. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Wang R, Wang ZX, Yang JS, Pan X, De W and
Chen LB: MicroRNA-451 functions as a tumor suppressor in human
non-small cell lung cancer by targeting ras-related protein 14
(RAB14). Oncogene. 30:2644–2658. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Bian HB, Pan X, Yang JS, Wang ZX and De W:
Upregulation of microRNA-451 increases cisplatin sensitivity of
non-small cell lung cancer cell line (A549). J Exp Clin Cancer Res.
30:202011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Kovalchuk O, Filkowski J, Meservy J, et
al: Involvement of microRNA-451 in resistance of the MCF-7 breast
cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther.
7:2152–2159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Wang J, Tian X, Han R, et al:
Downregulation of miR-486-5p contributes to tumor progression and
metastasis by targeting protumorigenic ARHGAP5 in lung cancer.
Oncogene. 33:1181–1189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ragusa M, Majorana A, Statello L, et al:
Specific alterations of microRNA transcriptome and global network
structure in colorectal carcinoma after cetuximab treatment. Mol
Cancer Ther. 9:3396–3409. 2010. View Article : Google Scholar
|
|
24.
|
Ji M, Li J, Yu H, et al: Simultaneous
targeting of MCL1 and ABCB1 as a novel strategy to overcome drug
resistance in human leukaemia. Br J Haematol. 145:648–656. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Xiang L, Li M, Liu Y, et al: The clinical
characteristics and prognostic significance of MN1 gene and
MN1-associated microRNA expression in adult patients with de novo
acute myeloid leukemia. Ann Hematol. 92:1063–1069. 2013. View Article : Google Scholar
|
|
26.
|
Li Z, Huang H, Li Y, et al: Up-regulation
of a HOXA-PBX3 homeobox-gene signature following down-regulation of
miR-181 is associated with adverse prognosis in patients with
cytogenetically abnormal AML. Blood. 119:2314–2324. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Garzon R, Garofalo M, Martelli MP, et al:
Distinctive microRNA signature of acute myeloid leukemia bearing
cytoplasmic mutated nucleophosmin. Proc Natl Acad Sci USA.
105:3945–3950. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Marcucci G, Maharry K, Radmacher MD, et
al: Prognostic significance of, and gene and microRNA expression
signatures associated with, CEBPA mutations in cytogenetically
normal acute myeloid leukemia with high-risk molecular features: a
Cancer and Leukemia Group B Study. J Clin Oncol. 26:5078–5087.
2008. View Article : Google Scholar
|
|
29.
|
Marcucci G, Radmacher MD, Maharry K, et
al: MicroRNA expression in cytogenetically normal acute myeloid
leukemia. N Engl J Med. 358:1919–1928. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Zhi F, Cao X, Xie X, et al: Identification
of circulating microRNAs as potential biomarkers for detecting
acute myeloid leukemia. PLoS One. 8:e567182013. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Li H, Hui L and Xu W: miR-181a sensitizes
a multidrug-resistant leukemia cell line K562/A02 to daunorubicin
by targeting BCL-2. Acta Biochim Biophys Sin (Shanghai).
44:269–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Bai H, Cao Z, Deng C, Zhou L and Wang C:
miR-181a sensitizes resistant leukaemia HL-60/Ara-C cells to Ara-C
by inducing apoptosis. J Cancer Res Clin Oncol. 138:595–602. 2012.
View Article : Google Scholar
|
|
33.
|
Andersson U and Tracey KJ: HMGB1 is a
therapeutic target for sterile inflammation and infection. Annu Rev
Immunol. 29:139–162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Tang D, Kang R, Zeh HJ III and Lotze MT:
High-mobility group box 1 and cancer. Biochim Biophys Acta.
1799:131–140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Kang R, Tang DL, Cao LZ, Yu Y, Zhang GY
and Xiao XZ: High mobility group box 1 is increased in children
with acute lymphocytic leukemia and stimulates the release of tumor
necrosis factor-alpha in leukemic cell. Zhonghua Er Ke Za Zhi.
45:329–333. 2007.(In Chinese).
|
|
36.
|
Liu L, Yang M, Kang R, et al:
HMGB1-induced autophagy promotes chemotherapy resistance in
leukemia cells. Leukemia. 25:23–31. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Xie M, Kang R, Yu Y, et al: Enhancive
effect of HMGB1 gene silence on adriamycin-induced apoptosis in
K562/A02 drug resistance leukemia cells. Zhonghua Xue Ye Xue Za
Zhi. 29:549–552. 2008.(In Chinese).
|
|
38.
|
Kozopas KM, Yang T, Buchan HL, Zhou P and
Craig RW: MCL1, a gene expressed in programmed myeloid cell
differentiation, has sequence similarity to BCL2. Proc Natl Acad
Sci USA. 90:3516–3520. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Placzek WJ, Wei J, Kitada S, Zhai D, Reed
JC and Pellecchia M: A survey of the anti-apoptotic Bcl-2 subfamily
expression in cancer types provides a platform to predict the
efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis.
1:e402010. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Kaufmann SH, Karp JE, Svingen PA, et al:
Elevated expression of the apoptotic regulator Mcl-1 at the time of
leukemic relapse. Blood. 91:991–1000. 1998.PubMed/NCBI
|
|
41.
|
Kasper S, Breitenbuecher F, Heidel F, et
al: Targeting MCL-1 sensitizes FLT3-ITD-positive leukemias to
cytotoxic therapies. Blood Cancer J. 2:e602012. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Zhu DX, Zhu W, Fang C, et al: miR-181a/b
significantly enhances drug sensitivity in chronic lymphocytic
leukemia cells via targeting multiple anti-apoptosis genes.
Carcinogenesis. 33:1294–1301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Zhan Z, Li Q, Wu P, et al:
Autophagy-mediated HMGB1 release antagonizes apoptosis of gastric
cancer cells induced by vincristine via transcriptional regulation
of Mcl-1. Autophagy. 8:109–121. 2012. View Article : Google Scholar : PubMed/NCBI
|