Introduction
Cadmium is a toxic metal, classified as a human
carcinogen by the International Agency for Research on Cancer
(1). There are probable
associations between long-term environmental exposure to cadmium
and an increased risk of mortality from stomach, esophageal and
lung cancers (2). However, the
molecular and cellular mechanisms underlying cadmium-mediated
tumorigenic effects in tissue remain unclear. Studies using in
vitro cell culture and in vivo animal models have
revealed some of the mechanisms underlying cadmium carcinogenesis.
Cadmium may induce genotoxic effects, such as
8-ox-7,8-dihydro-2′-deoxyguanosine adducts, DNA strand breaks,
chromosomal aberration, and gene mutations, in a variety of in
vitro cell culture and animal experimental systems (3). Most of the genotoxic events induced
by cadmium are mediated through the generation of reactive oxygen
species (ROS) (4). Cadmium also
activates gene expression of c-myc and c-Jun, inhibits tumor
suppressor genes, such as p53 and p27, and accelerates the
proliferation of cells that are already stimulated with organic
carcinogens (5,6).
Cadmium has been reported to activate
mitogen-activated protein kinase (MAPKs) signaling. In mammals,
MAPKs consist of extracellular signal-regulated kinases (ERK),
c-Jun N-terminal kinases (JNK), and p38 MAPK. Cadmium activates ERK
signaling and elevates the expression of a key downstream
proangiogenic molecule, hypoxia-inducible factor-1 (HIF-1), in
immortalized lung epithelial cells (7). The JNK pathway has also been reported
to be involved in the acquisition of apoptotic resistance in
cadmium carcinogenesis, contributing to both tumor initiation and
malignant progression (8).
Additionally, cadmium has been reported to induce diverse
modulation of the transcription patterns of p38 MAPK isoform genes
and the accumulation of related protein products in breast cancer
cells. However, the roles of MAPK signals in cadmium-mediated cell
invasion and migration have not yet been explored.
The process of cancer cell invasion and migration is
multifactorial, requiring the coordinated action of cell-secreted
proteolytic enzymes and their inhibitors (9). A recent study has shown that cadmium
treatment increased the potential of cells to invade and migrate
(10). Urokinase-type plasminogen
activator (uPA), its inhibitors, and the uPA receptor (uPAR) form a
complex proteolytic system that has been implicated in cancer
invasion and metastasis. As a serine protease, uPA has the ability
to convert inactive plasminogen to active plasmin by binding to its
receptor, uPAR. The uPA-uPAR interaction can affect independently
cell motility, integrin function, and gene expression (11). uPAR expression has been shown to
play important roles in the invasion and metastasis of a number of
cancers, such as gastric (12),
prostate (13) and breast cancers
(14). In addition to mediating
proteolysis, this receptor also appears to mediate cell signaling,
proliferation, and survival, and these observations have suggested
novel ways to target uPAR. To examine the role and mechanisms of
cadmium in regulating invasion and migration, we investigated the
effects of cadmium on mitogen-activated protein kinase signaling
and the downstream transcription factors, NF-κB and AP-1, of uPAR
in gastric cancer AGS cells.
Materials and methods
Cell culture and reagents
Human gastric cancer AGS cells were purchased from
the American Type Culture Collection (Manassas, VA, USA), and MKN28
and SNU638 cells were obtained from the Korean Cell Line Bank
(Seoul, Korea). The cells were incubated in RPMI-1640 supplemented
with 10% fetal bovine serum (FBS), 100 IU/ml penicillin and 100
mg/ml streptomycin in a humidified atmosphere containing 5%
CO2 incubator at 37°C. To determine the effects of
cadmium (Sigma, St. Louis, MO, USA) on uPAR expression, cells were
harvested at various intervals and the levels of uPAR mRNA were
measured by RT-PCR analysis. To examine the effects of cadmium on
ERK1/2, JNK, and p38 MAPK activation, cells were harvested at
various intervals and phosphorylated and total protein levels were
determined by western blot analysis. To examine the role of
specific signaling pathways in uPAR induction by cadmium, cells
were pretreated with 25 μM PD98059 (a MEK inhibitor, New England
Biolabs Inc., Beverly, MA, USA), 10 μM SP600125 (a c-Jun-N-terminal
kinase inhibitor, Calbiochem, San Diego, CA, USA), 5 μM SB203580 (a
specific p38 MAPK inhibitor; Calbiochem), 20 μM Bay11-7082 (NF-κB
inhibitor, Calbiochem), or curcumin (AP-1 inhibitor, Sigma) for 1 h
prior to cadmium treatment. The levels of uPAR mRNA were then
measured by northern blot and RT-PCR analysis.
RT-PCR
Cells were incubated overnight in medium containing
1% FBS and then treated with specific inhibitors for 1 h prior to
cadmium treatment for 4 h. After the incubation, total cellular RNA
was isolated from the cells using the TRIzol reagent (Invitrogen,
Carlsbad, CA, USA). Total RNA (1 μg) was used for the first-strand
cDNA synthesis using random primers and Superscript reverse
transcriptase (Invitrogen). The cDNA was subjected to PCR
amplification with the primer sets for glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) and uPAR. The specific primers sequences were
GAPDH sense, 5′-TTG TTG CCA TCA ATG ACC CC-3′; GAPDH antisense,
5′-TGA CAA AGT GGT CGT TGA GG-3′ (836 bp) and uPAR sense, 5′-CAC
GAT CGT GCG CTT GTG GG-3′, and uPAR antisense, 5′-TGT TCT TCA GGG
CTG CGG CA-3′ (285 bp). The PCR conditions were 30 cycles of
denaturation at 95°C for 20 sec, annealing at 53°C for 30 sec, and
extension at 72°C for 40 sec. The products were electrophoresed in
1.5% agarose gel containing ethidium bromide.
Measurement of uPAR promoter
activity
The transcriptional regulation of uPAR was examined
by the transient transfection of a uPAR promoter luciferase
reporter construct (pGL3-uPAR). The plasmid pGL3-uPAR promoter
(15) was provided by Dr Y. Wang
(Australian National University). AGS cells (5×105) were
seeded and grown until they reached 60-70% confluence, then pRL-TK
(an internal control plasmid containing the herpes simplex
thymidine kinase promoter linked to the constitutively active
Renilla luciferase reporter gene) and pGL3-uPAR were cotransfected
into the cells using FuGENE (Boehringer-Mannheim, Mannheim,
Germany) according to the manufacturer’s protocol. pRL-TK was
transfected as an internal control. Cells were incubated in the
transfection medium for 20 h and treated with 0-20 μM cadmium for 4
h. The effects of signaling inhibitors on uPAR promoter activity
were determined by pretreating cells with the inhibitors for 1 h
prior to the addition of cadmium. The cotransfection studies were
performed in the presence or absence of the AP-1 decoy
oligodeoxynucleotides (ODNs) or MEK-1 (pMCL-K97M), I-κBα, I-κBβ, or
NF-κB-inducing kinase (NIK). The phosphorothioate double-stranded
ODNs with sequences against the AP-1 binding site (5′-CAC TCA GAA
GTC ACT TC-3′ and 3′-GAA GTG ACT TCT GAG CTG-5′) were prepared
(Genotech, St. Louis, MO, USA) and annealed (AP-1 decoy ODNs). The
expression vector encoding the inactive MEK-1 (pMCL-K97M) was a
gift from Dr N.G. Ahn (University of Colorado). The dominant
negative mutants of I-κBα and I-κBβ and NIK were provided by Dr
D.W. Ballard (Vanderbilt University, Nashville) and Dr W.C. Greene
(University of California), respectively. The importance of NF-κB
and AP-1 during the induction of uPAR by cadmium was examined by
transfecting AGS cells with pGL3-uPAR, in which the NF-κB and AP-1
sites had been mutated. After incubation, the cells were harvested
and lysed with passive lysis buffer (Dual-Luciferase Reporter Assay
system; Promega, Madison, WI, USA), and luciferase activity was
measured using a luminometer according to the manufacturer’s
protocol.
Western blot analysis
Cells pretreated with 0-20 μM cadmium for various
periods were washed in phosphate-buffered saline (PBS), detached
using trypsin-EDTA buffer, and stored at -70°C until needed. The
protein was extracted with RIPA buffer (1% NP-40, 0.5% sodium
deoxycholate, 0.1% sodium dodecyl sulfate) and protease inhibitors
(aprotinin, leupeptin, phenylmethanesulfonyl fluoride, benzamidine,
trypsin inhibitor, sodium orthovanadate). Then, 50 μg of the
protein was separated by 10% SDS-PAGE and transferred to PVDF
membranes. The membranes were blocked in a PBS solution containing
5% non-fat dry milk, incubated with primary antibody in blocking
solution overnight at 4°C, and washed three times with 0.1%
Tween-20 in Tris-buffered saline (TBST) at 10-min intervals.
Horseradish peroxidase-conjugated secondary antibody (Amersham,
Arlington Heights, IL, USA) was used to detect the immunoreactive
proteins by chemiluminescence. The following antibodies were used:
anti-uPAR (American Diagnostica, Greenwich, CT, USA), anti-phospho
p44/42 MAPK (ERK-1/2) (Cell Signaling Technology, Danvers, MA,
USA), anti-phospho-JNK (Cell Signaling Technology), and
anti-phospho-p38 MAPK (Cell Signaling Technology). Total protein
levels were assayed by washing the blotted membrane with a
stripping solution consisting of 100 mM 2-mercaptoethanol, 2% SDS,
and 62.5 mM Tris-HCl, pH 6.7, for 30 min at 50°C, and the membrane
was then reprobed with the anti-β-actin (Sigma-Aldrich),
anti-ERK-1/2 (Cell Signaling Technology), anti-JNK-2 (Cell
Signaling Technology), or antip38 MAPK (Cell Signaling Technology)
monoclonal antibody.
Extraction of nuclear proteins
AGS cells at 80–90% confluence were incubated
overnight in medium containing 5% FBS and then treated with 0–30 μM
cadmium. The cells were then resuspended in 500 μl cold buffer A
[50 mM Tris, pH 7.4, 150 mM NaCl, 0.2 mM EDTA, 3% (v/v) glycerol,
and 1.5 mM MgCl2]. After the cells had been allowed to
swell for 5 min on ice, they were lysed with 500 μl of buffer B
(identical to buffer A except containing 0.05% Nonidet P-40). The
homogenate was gently layered onto an equal volume cushion of
buffer C [10 mM Tris, pH 7.4, 25% (v/v) glycerol, and 1.5 mM
MgCl2] and centrifuged (12,000 × g, 5 min). The white
nuclear pellet was resuspended in 75 μl of a cold high-salt lysis
buffer (20 mM HEPES, pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM DTT, and
1 mM PMSF). This suspension was agitated for 30 min at 40°C and
then microcentrifuged (15 min, 4°C). The resulting supernatant was
stored in aliquots at −80°C.
Electrophoretic mobility shift assay
(EMSA)
EMSA was carried out using a Gel Shift assay system
(Promega). Briefly, oligonucleotides containing the consensus
sequences of AP-1 (5′-CGC TTG ATG AGT CAG CCG GAA-3′) and NF-κB
(5′-AGT TGA GGG GAC TTT CCC AGG-3′) were endlabeled with
[γ-32P] adenosine triphosphate (3000 μCi/mmol; Amersham
Pharmacia Biotech., Buckinghamshire, UK) using T4 polynucleotide
kinase, purified with Microspin G-25 columns (Sigma-Aldrich) and
used as the probe for EMSA. The nuclear extract proteins (6 μg)
were pre-incubated with the binding buffer [10 mM Tris-HCl, pH 7.5,
50 mM NaCl, 0.5 mM EDTA, 1 mM MgCl2, 0.5 mM DTT, 4%
(v/v) glycerol, and 0.05 mg/ml poly(dI-dC)] for 5 min and then
incubated with the radiolabeled probe for 15 min at 37°C. Each
sample was electrophoresed in a 5% non-denaturing polyacrylamide
gel in 0.5X TBE buffer. The gel was then dried and subjected to
autoradiography.
Transient transfection of AP-1 and NF-κB
reporters
The AP-1 and NF-κB reporter constructs were
purchased from Clontech (Palo Alto, CA, USA). Once the cells had
reached 60–70% confluence, they were washed with RPMI-1640 and
incubated in medium without serum or antibiotics for 18 h. The
cells were then transfected with AP-1 and NF-κB reporters in the
pGL3 vector using Lipofectamine 2000 (Invitrogen).
Reporter-transfected cells were treated with 0–30 μM cadmium for 4
h. After incubation, the luciferase activity was measured using a
luminometer.
Matrigel invasion assay
A cell invasion assay was performed using BioCoat
Matrigel invasion chambers (Becton-Dickinson, Bedford, MA, USA)
with 10% FBS as the chemoattractant in the lower chamber. AGS cells
(105) in 300 μl were added to each chamber with cadmium
and allowed to invade the Matrigel for 24 h. The non-invading cells
on the upper surface of each membrane were removed from the
chamber, and the invading cells on the lower surface of each
membrane were stained with the Quick-Diff stain kit
(Becton-Dickinson, Franklin Lakes, NJ, USA). After two washes with
water, the chambers were allowed to air-dry. The number of invading
cells was counted using a phase-contrast microscope. To determine
the effects of anti-uPAR antibody and inhibitors of ERK-1/2, NF-κB
and AP-1 on cadmium-induced cell invasion, AGS cells were
preincubated with a neutralizing antibody to uPAR or non-specific
IgG, inhibitors of ERK-1/2, NF-κB and AP-1 for 1 h, and added to 20
μM cadmium for 4 h.
Statistical analyses
Data are shown as means ± SD, and represent the
means of at least three separate experiments each performed in
triplicate. Differences between data sets were determined using
Student’s t-test. Differences described as significant in the text
correspond to P<0.05.
Results
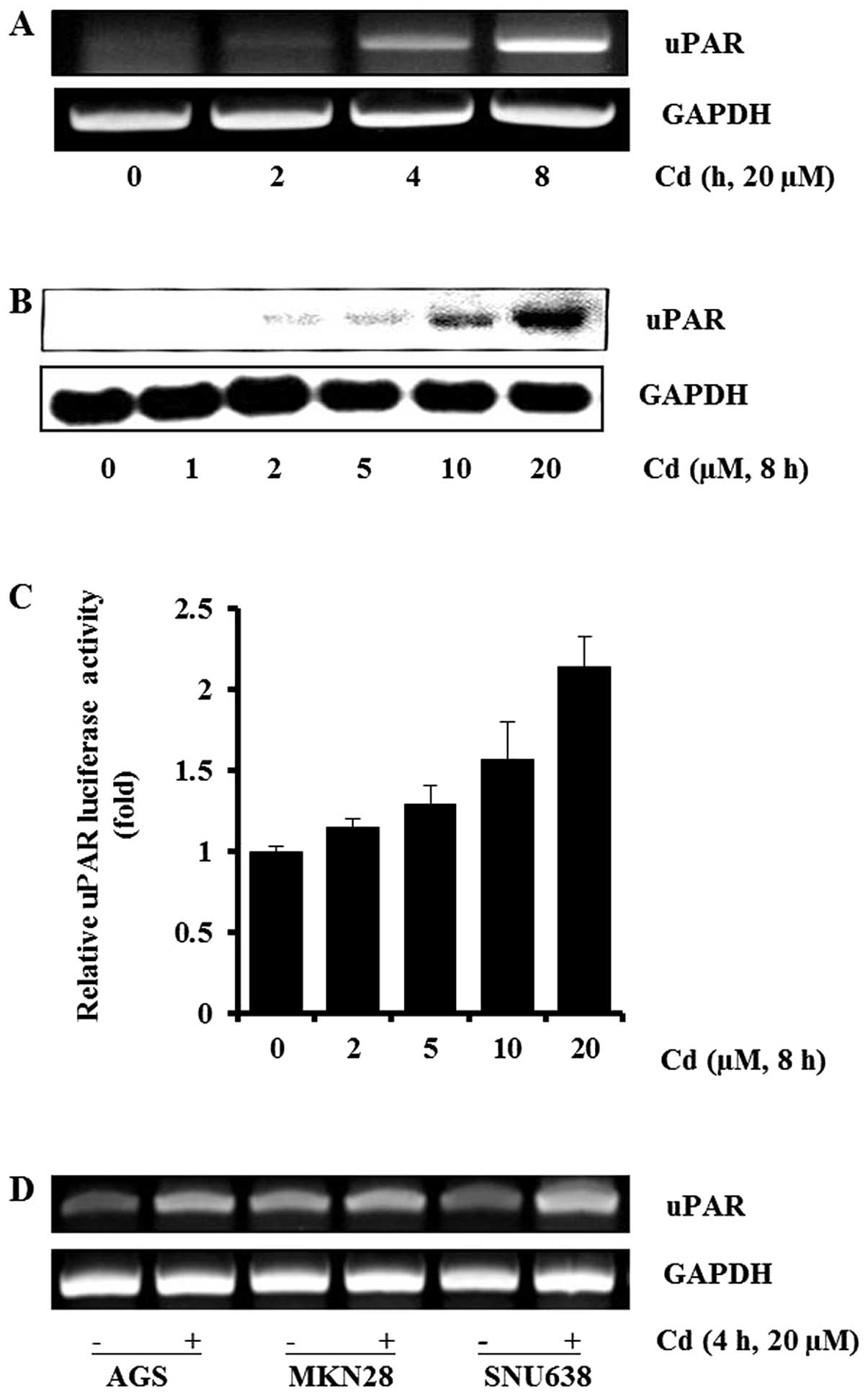
Effects of cadmium on uPAR expression in
gastric cancer cells
To determine the effects of cadmium on uPAR
expression in gastric cancer AGS cells, cells were treated with
cadmium and the expression of uPAR were measured by RT-PCR and
western blot analyses. As shown in Fig. 1A, cadmium induced uPAR mRNA
expression in a time-dependent manner in AGS cells. The uPAR mRNA
expression increased appreciably 4 h after addition of cadmium to
the cells. We also found that cadmium induced the uPAR protein in a
dose-dependent manner at 0–20 μM (Fig.
1B). Next, we sought to examine the effect of cadmium on
transcriptional regulation of the uPAR gene. AGS cells were
transiently transfected with the promoter-reporter construct
(pGL3-uPAR) containing the human uPAR promoter and the luciferase
gene. The AGS cells transfected with pGL3-uPAR showed an increase
in promoter activity with cadmium exposure in a dose-dependent
manner (Fig. 1C). Cadmium at the
concentrations used in this experiment did not affect cell
viability (data not shown). Gastric cancer AGS, MKN28 and SNU638
cells were used to explore whether cadmium was able to induce uPAR
expression in various gastric cancer cells. As shown in Fig. 1D, cadmium induced uPAR expression
in AGS, MKN28, and SNU638. Collectively, these results demonstrate
that cadmium upregulated the expression of the uPAR gene in human
gastric cancer cells.
Involvement of MAPK in cadmium-induced
uPAR mRNA expression
To determine the signaling pathways involved in uPAR
induction by cadmium, AGS cells exposed to cadmium for various
periods were examined for levels of phospho- and total ERK-1/2,
JNK, and p38 MAPK. Cadmium treatment resulted in marked increases
in ERK-1/2, JNK, and p38 MAPK phosphorylation within 30 min and the
increased levels were maintained for 60–90 min. The levels of total
ERK-1/2, JNK2, and p38 MAPK were not significantly altered after
cadmium treatment (Fig. 2A).
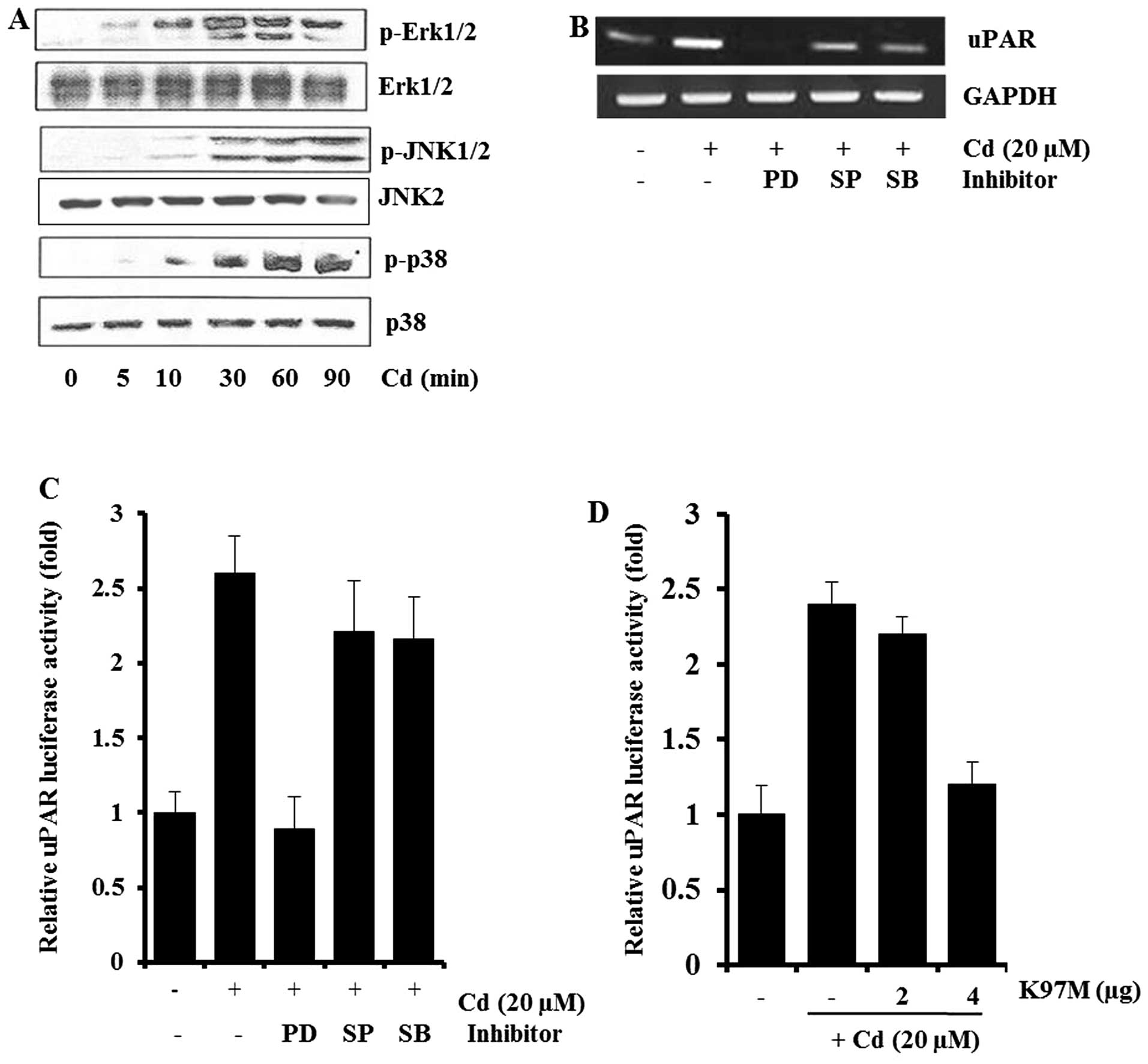 | Figure 2Involvement of MAPK in cadmium-induced
uPAR expression in AGS cells. AGS cells were incubated with 20 μM
cadmium for 0–90 min, and cell lysates were analyzed for
phospho-Erk1/2, phospho-JNK1/2, and phospho-p38 MAPK by western
blot analysis (A). AGS cells pretreated with 25 μM PD98059 (PD), 10
μM SP600125 (SP), or 5 μM SB203580 (SB) for 1 h were incubated with
20 μM cadmium for 4 h. After incubation, uPAR mRNA in the cell
lysates were determined by RT-PCR (B). Cells transiently
transfected with pGL3-uPAR after being pretreated with PD98059
(PD), SP600125 (SP), or SB203580 (SB) were incubated with 20 μM
cadmium for 4 h. After incubation, the cells were lysed, and
luciferase activity was measured using a luminometer (C). An
expression vector encoding a mutated MEK-1 (K97M) was cotransfected
with pGL3-uPAR into AGS cells. After incubation with 20 μM cadmium
for 4 h, luciferase activities were determined using a luminometer
(D). The data represent means ± standard deviations from triplicate
measurements. |
To further examine the specific roles of ERK-1/2,
JNK, and p38 MAPK in cadmium-induced uPAR expression, AGS cells
were pretreated with 25 μM PD98059 (MEK inhibitor), 10 μM SP600125
(JNK inhibitor), or 5 μM SB203580 (p38 MAPK inhibitor) before
cadmium treatment. As shown in Fig.
2B, the levels of uPAR in AGS cells pretreated with MAPK
inhibitors decreased. In particular, the MEK inhibitor PD98059
almost completely blocked cadmium-induced uPAR mRNA expression, by
RT-PCR analysis. When the transfected cells were pretreated with 25
μM PD98059 (MEK inhibitor) for 1 h before cadmium treatment, the
induction of uPAR promoter activity was inhibited markedly
(Fig. 2C). Consistent with the
results of Fig. 2B and C, when the
dominant-negative mutant of MEK-1 (K97M) was cotransfected with
pGL-uPAR in AGS cells, induction of the uPAR promoter activity by
cadmium was inhibited dose-dependently (Fig. 2D). These results suggest that
ERK-1/2 signaling pathways are involved in the cadmium-induced
activation of uPAR transcription.
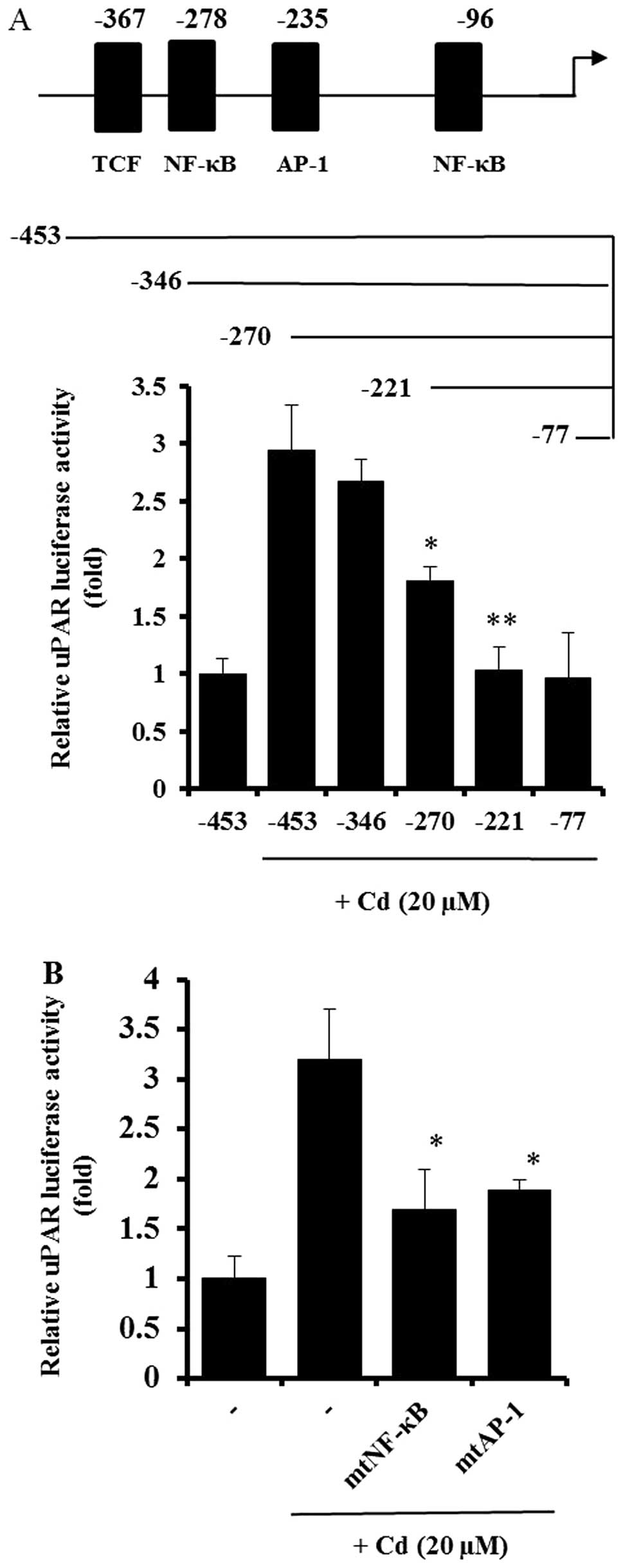
Effect of cadmium on the activation of
transcription factors during uPAR induction
As shown in Figs. 1
and 2, cadmium treatment increased
the activity of the uPAR promoter in AGS cells. A deletion study
was performed to explore the uPAR promoter and its specific
sequence-activity characteristics. Deletion of the upstream (5′)
region of position −346 bp had little effect on cadmium-induced
uPAR promoter activation. In contrast, elimination of the region
between positions −346 and −270 bp resulted in a substantial
decrease in promoter activity, and another cadmium-inducible
element was identified between nucleotides −270 and −221 (Fig. 3A). The uPAR gene fragments spanning
positions −346 to −270 bp and −270 to −221 bp contain DNA-protein
interaction sites for the transcription factors NF-κB (−278) and
AP-1 (−235), respectively. To study the importance of the NF-κB and
AP-1 sites in uPAR induction by cadmium, AGS cells were transfected
with site-specific mutant forms of the uPAR promoter linked to the
luciferase gene. As shown in Fig.
3B, mutation of either the NF-κB or AP-1 binding site
significantly decreased uPAR promoter activity, suggesting that
both NF-κB and AP-1 are important in uPAR upregulation by cadmium.
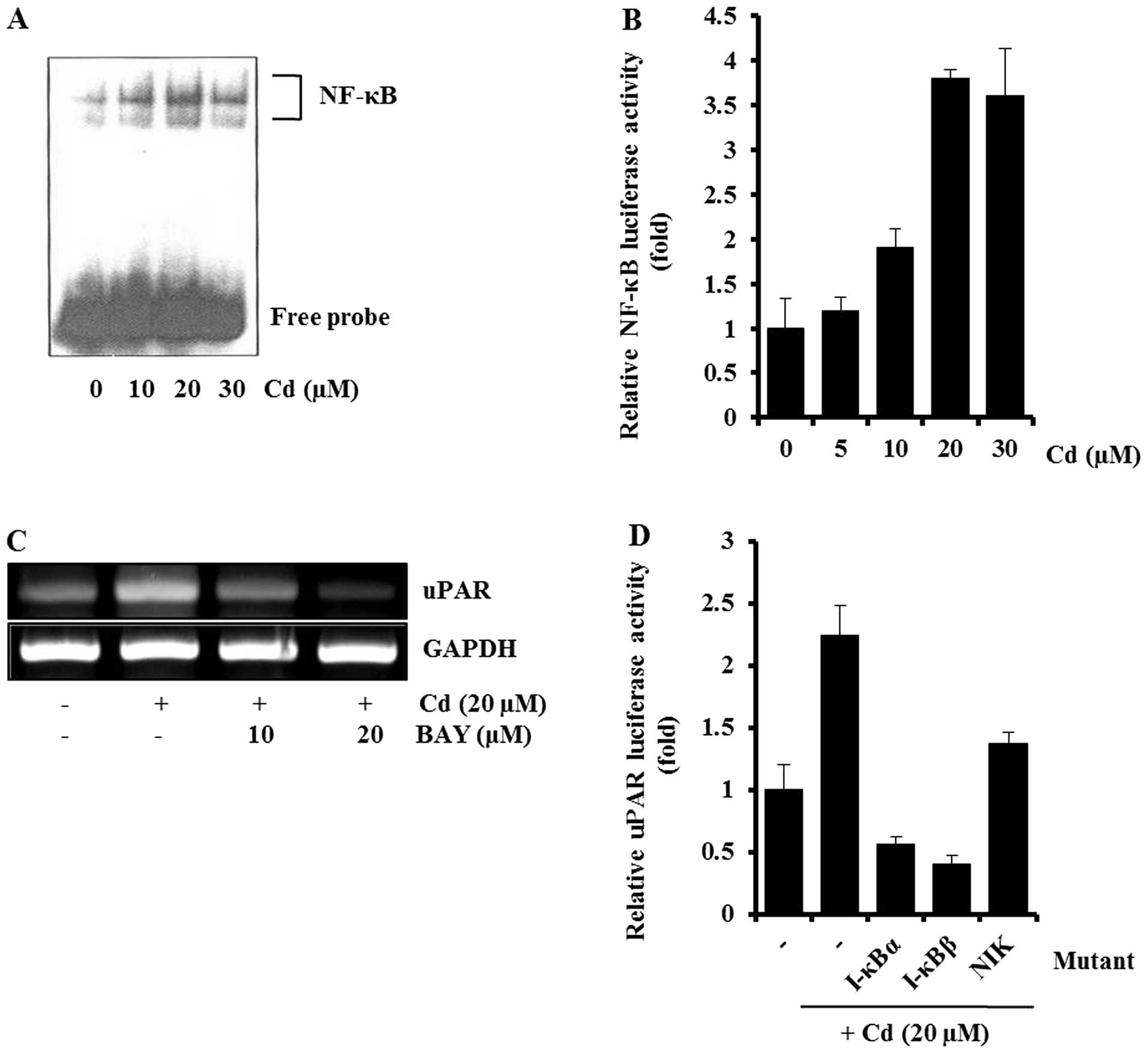
This was further supported by EMSA and inhibitor studies. In EMSA,
cadmium treatment caused a marked increase in the amount of NF-κB
that could form a complex with the radiolabeled oligonucleotide
probe (Fig. 4A). Consistent with
the EMSA result, cadmium treatment caused an increase in
NF-κB-dependent transcriptional activity (Fig. 4B), and Bay11-7082 (NF-κB inhibitor)
partially blocked the cadmium-induced uPAR expression, by RT-PCR
(Fig. 4C). The involvement of
NF-κB in the induction of uPAR by cadmium was confirmed by
cotransfecting AGS cells with a uPAR promoter reporter and
dominant-negative mutant forms of NF-κB-related molecules. As shown
in Fig. 4D, the expression of
dominant-negative mutant forms of NIK, I-κBα, or I-κBβ resulted in
a decrease in the cadmium-induced uPAR promoter activity.
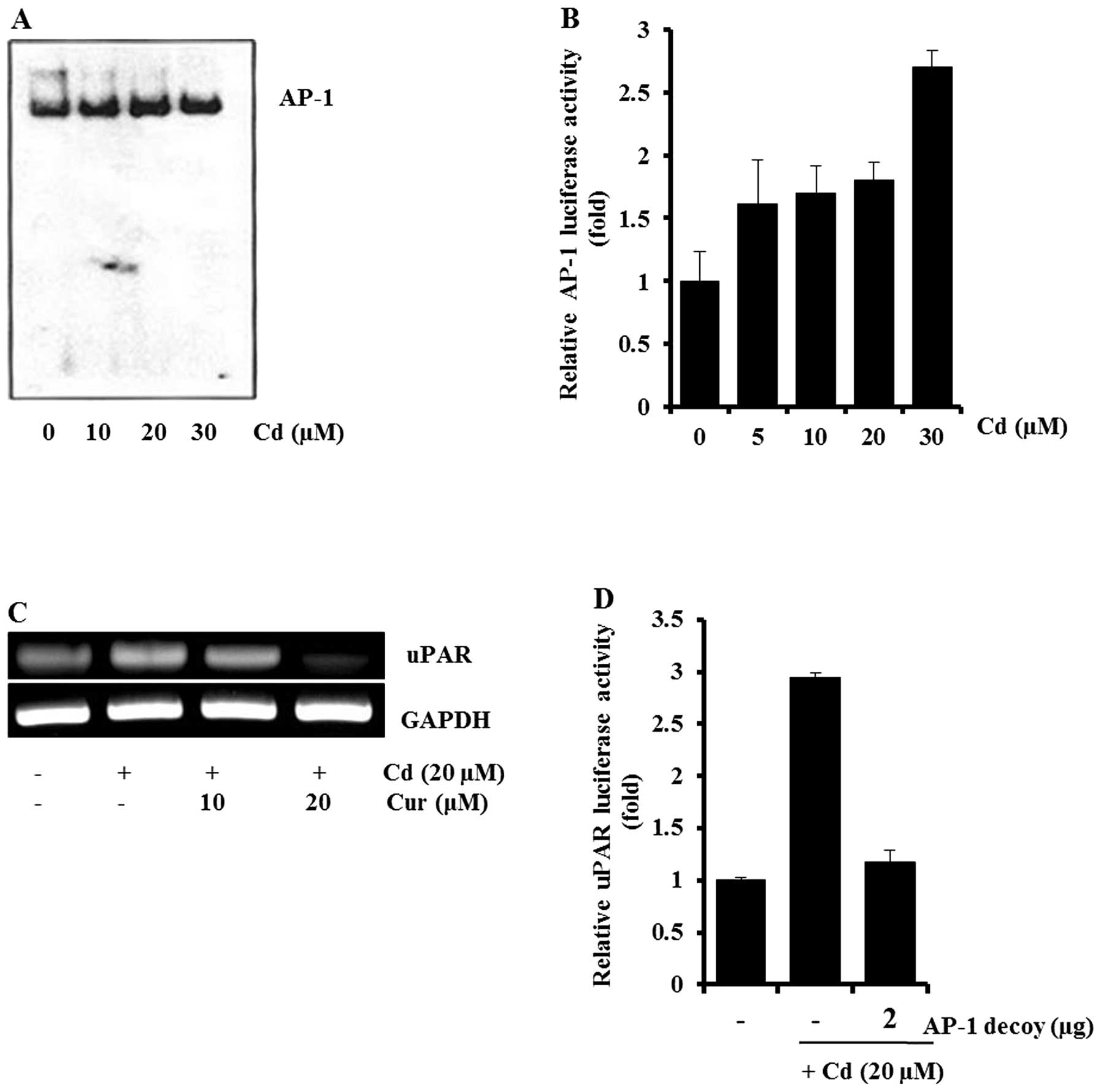
Additionally, involvement of AP-1 in cadmium-induced uPAR
expression was examined. As shown in Fig. 5A and B, cadmium treatment caused an
increase in the amount of AP-1-DNA complex and AP-1-dependent
transcriptional activity. AP-1 inhibitors (curcumin and the AP-1
decoy) blocked cadmium-induced uPAR expression and uPAR promoter
activity, respectively (Fig. 5C and
D). These results indicated that NF-κB and AP-1 may be key
molecules in cadmium-induced uPAR expression.
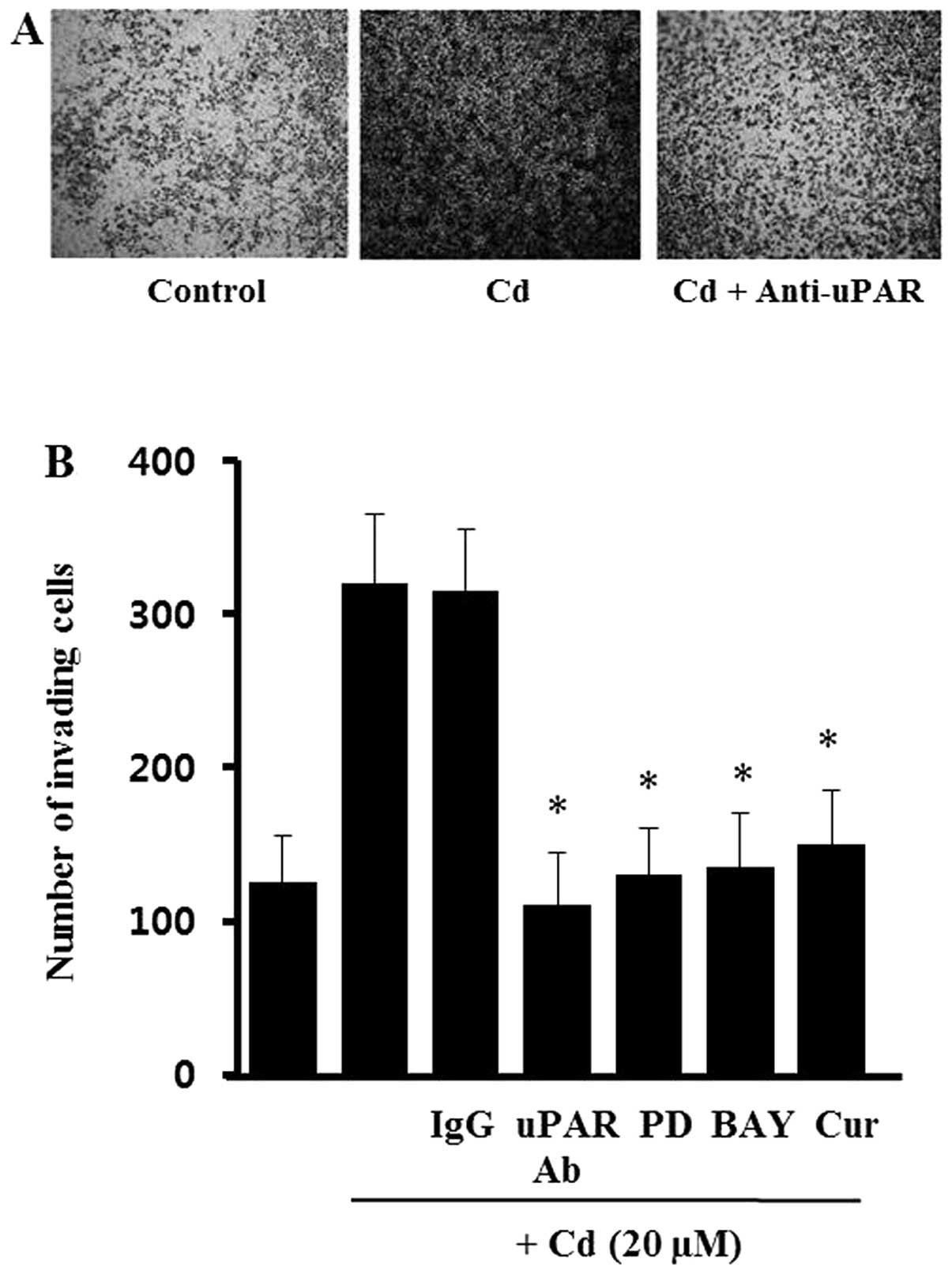
Effects of cadmium on the invasion of AGS
cells
It has been suggested that expression of uPAR is
required for the invasive phenotype of cancer cells. To evaluate
the role of cadmium-induced uPAR during AGS cell invasion, cells
were incubated with specific antibodies against uPAR in a modified
Boyden invasion chamber. As shown in Fig. 6, cell invasiveness was increased
markedly by incubation with cadmium. However, the cadmium-treated
cells partially lost the increased Matrigel invasiveness after
incubation with uPAR-neutralizing antibodies, whereas no such
effect was seen after incubation with non-specific IgG. These
results suggest that uPAR induced by cadmium has an import role in
gastric cancer cell invasiveness. To confirm that ERK-1/2, NF-κB,
and AP-1 are involved in the cadmium-induced invasiveness, the AGS
cells were treated with PD98059 (MEK inhibitor), BAY11-7082 (NF-κB
inhibitor) and curcumin (AP-1 inhibitor) before cadmium treatment.
As shown in Fig. 6B, all of the
inhibitors (PD98059, BAY11-7082, curcumin) blocked the Matrigel
invasiveness induced by cadmium. However, the inhibitors alone did
not significantly change the level of cell invasiveness (data not
shown). This suggests that the ERK-1/2, NF-κB, and AP-1 signals
activated by cadmium upregulate uPAR, leading to an increase in
gastric cancer cell invasiveness.
Discussion
A recent study reported that long-term exposure to
cadmium increased the risk of mortality from stomach cancer
(2). Because of the widespread use
of cadmium, gastrointestinal absorption of cadmium without being
aware is unavoidable. Several studies about carcinogenic effects of
cadmium have been published and the cytotoxic action and
carcinogenesis of cadmium in many organs, especially the lung,
liver, breast, and kidney, are well known (16–18).
However, the mechanisms underlying cadmium-induced carcinogenesis
have remained largely unknown. Much effort has been directed at
defining the role of cadmium in cancer development and progression,
stimulated by the following observations: i) cadmium emissions have
increased dramatically during the 20th century, one reason being
that cadmium-containing products are rarely recycled, but are often
dumped together with household waste, ii) long-term studies suggest
an association between cadmium and cancer development [the
biological half-life of cadmium in the human body is ~25–30 years,
so overexposure to cadmium can lead to its accumulation in the
human body (19)], iii) the
adverse health effects of cadmium exposure may occur at lower
exposure levels than previously anticipated, primarily in the form
of kidney damage and bone fracture (20), and iv) cadmium induces various
genes related to carcinogenesis, including IL-8 and COX-2, which
are important for cancer development and angiogenesis (21,22).
In this study, we found that cadmium could induce
uPAR expression and stimulate cell invasiveness in human gastric
cancer AGS cells, suggesting that the overexpression of uPAR by
cadmium may be involved in the increased cell invasiveness. Choi
et al (23) demonstrated
that overexpression of uPAR in human gastric carcinomas correlated
with their invasiveness and tumorigenicity. Previously, we reported
that uPAR is upregulated by various stimuli such as H.
pylori, ROS, and EGF (12) in
gastric cancer cells. The serine protease uPA and its receptor uPAR
system have the ability to convert inactive plasminogen to active
plasmin, which, in turn, activate certain matrix metalloproteinases
(MMPs), which break down the collagen components of the
extracellular matrix and accelerate tumor invasion and metastasis
(24). Cellular responses to
cadmium stimulation trigger a cascade of protein kinases that
transmit signals from the cell surface to the nucleus and these
signals ultimately regulate gene expression. These signaling
molecules including epidermal growth factor receptor (EGFR),
phosphatidyl inositol 3-kinase (PI3K), AKT, and mammalian target of
rapamycin (mTOR) have been reported to be involved in
carcinogenesis and cancer progression (25).
Several studies have documented that the MAPKs have
roles in cadmium-induced signal transduction, but the profiles of
cadmium-induced kinase activation appear to vary in a cell
type-dependent manner. Three major MAPKs have been identified in
mammalian cells: ERK-1/2, JNK, and P38 MAPK. Our results show that
cadmium promoted the activation of ERK-1/2 and induced uPAR
expression in human gastric AGS cells. Activation of ERK-1/2, JNK,
and p38 MAPK preceded the induction of uPAR mRNA expression, and
this upregulation was attenuated by a selective inhibitor of
ERK-1/2, suggesting that the ERK-1/2 signaling pathway is
implicated in the activation of the uPAR gene by cadmium. This
suggestion was further supported by observations that expression of
a vector encoding a mutated-type MEK-1 (K97M) resulted in a marked
reduction in uPAR promoter activity. It remains to be determined
how cadmium mediates ERK-1/2 activation in human gastric AGS cells.
Park et al (22) reported
that crosstalk between oxidative stress and activation of the
family of MAPK in cadmium-treated C6 cells, in which cadmium
treatment primarily lowered cellular GSH, subsequently leading to
activation of ERK-1/2. Multiple pathways have been proposed in
different cell types leading to ERK-1/2 activation by ROS. Mukhin
et al (26) suggested a
simple model for ERK-1/2 activation by ROS (NAD(P)H oxidase → Giβγ
→ Src → ERK-1/2 pathway) using CHO cells. Concurrent inhibition of
tyrosine phosphatase (PTPase) by ROS has been suggested to be
another mechanism of ERK-1/2 activation (18). Because all PTPases have a conserved
cysteine residue in their catalytic domain, the inhibition of
PTPase activity by ROS may account for activation of ERK-1/2 by
ROS.
Our results are consistent with those of earlier
studies implicating the involvement of transcription factors such
as NF-κB and AP-1 in uPAR expression in gastric cancer cells
(27). Related to this, Valko
et al (28) confirmed that
metals activate signaling pathways and the carcinogenic effect of
metals has been related to activation of mainly redox-sensitive
transcription factors, including NF-κB, AP-1, and p53. The roles of
NF-κB with cadmium in cells vary depending on the cell type. NF-κB
is important in cadmium-induced TNF-α, IL-1β, IL-6, and IL-8 in
THP-1 monocytic cells. (29).
Furthermore, NF-κB activation is apparently involved in
cadmium-induced apoptosis in lung epithelia and kidney proximal
tubule cells (30,31). However, cadmium induced the
proinflammatory cytokine IL-8 in lung epithelial cells in a
NF-κB-independent manner. ROS production by cadmium has been
suggested to activate NF-κB in cells. Li and Engelhardt (32) reported that the recruitment of NIK
to TRAF6 is mechanistically important for the ROS induction of
NF-κB activation. Storz and Toker (33) showed that protein kinase D (PKD) is
essential for ROS-induced NF-κB activation by inducing tyrosine
phosphorylation of IKK. Src and Abl mediate PKD activation in
response to H2O2 stimulation, via the
phosphorylation of Tyr463 in the PKD pleckstrin homology domain.
Similarly, Fan et al (34)
reported that ROS controlled NF-κB activation through
c-Src-dependent tyrosine phosphorylation of I-κB.
In subsequent experiments, the role of AP-1 in
cadmium-induced uPAR expression was also investigated. The
transcription factor AP-1 is composed of Fos and Jun homodimers and
heterodimers. Cascades of MAPKs are involved in the activation of
AP-1. In our system ERK-1/2 may be supposed to induce c-Fos and
AP-1 activity. Escobar Mdel et al (35) reported the role of MAPK induced by
cadmium in AP-1 activation in HepG2 cells. AP-1 activation
decreased by 74% with ERK inhibition, by 83% with p38 inhibition,
and by 70% with JNK inhibition. In mouse epidermal JB6 cells, the
induction of AP-1 activity by cadmium appeared to involve
activation of ERK-1/2, because the induction of AP-1 activity by
cadmium was blocked by pretreatment of the cells with PD98058
(36).
We also found in this study that cadmium can
stimulate the cell invasion in AGS cells via overexpression of
uPAR. Several laboratories have provided convincing evidence that
cadmium enhances cell invasion and tumorigenesis. Juang et
al (37) reported that cadmium
upregulated metallothionein 3, an androgen-upregulated gene, and
enhanced cell invasion in prostate carcinoma cells. Cadmium also
induced cell invasion via AKT/GSK-3β/β-catenin signaling in mouse
BEAS-2B cells (10). In
conclusion, the present report describes for the first time the
role of cadmium in the regulation of uPAR in gastric cancer cells.
Further studies are needed to determine the remaining details of
the regulatory mechanism.
Acknowledgements
This study was supported by a research grant
(0720570) from the National Cancer Center, by a Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science, and Technology
(2010-0009910), and by a Medical Research Center (2012-000-9442)
grant from the Korean Science and Engineering Foundation.
References
|
1
|
García-Esquinas E, Pollan M, Tellez-Plaza
M, Francesconi KA, Goessler W, Guallar E, Umans JG, Yeh J, Best LG
and Navas-Acien A: Cadmium exposure and cancer mortality in a
prospective cohort: the strong heart study. Environ Health
Perspect. 122:363–370. 2014.PubMed/NCBI
|
|
2
|
Wang M, Song H, Chen WQ, Lu C, Hu Q, Ren
Z, Yang Y, Xu Y, Zhong A and Ling W: Cancer mortality in a Chinese
population surrounding a multi-metal sulphide mine in Guangdong
province: an ecologic study. BMC Public Health. 11:3192011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Engström KS, Vahter M, Johansson G, Lindh
CH, Teichert F, Singh R, Kippler M, Nermell B, Raqib R, Strömberg U
and Broberg K: Chronic exposure to cadmium and arsenic strongly
influences concentrations of 8-oxo-7,8-dihydro-2′-deoxyguanosine in
urine. Free Radic Biol Med. 48:1211–1217. 2010.PubMed/NCBI
|
|
4
|
Ochi T and Ohsawa M: Participation of
active oxygen species in the induction of chromosomal aberrations
by cadmium chloride in cultured Chinese hamster cells. Mutat Res.
143:137–142. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fang MZ, Mar W and Cho MH: Cadmium affects
genes involved in growth regulation during two-stage transformation
of Balb/3T3 cells. Toxicology. 177:253–265. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jin P and Ringertz NR: Cadmium induces
transcription of proto-oncogenes c-jun and c-myc in rat L6
myoblasts. J Biol Chem. 265:14061–14064. 1990.PubMed/NCBI
|
|
7
|
Jing Y, Liu LZ, Jiang Y, Zhu Y, Guo NL,
Barnett J, Rojanasakul Y, Agani F and Jiang BH: Cadmium increases
HIF-1 and VEGF expression through ROS, ERK, and AKT signaling
pathways and induces malignant transformation of human bronchial
epithelial cells. Toxicol Sci. 125:10–19. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qu W, Fuquay R, Sakurai T and Waalkes MP:
Acquisition of apoptotic resistance in cadmium-induced malignant
transformation: specific perturbation of JNK signal transduction
pathway and associated metallothionein overexpression. Mol
Carcinog. 45:561–571. 2006. View
Article : Google Scholar
|
|
9
|
Goldfarb RH and Liotta LA: Proteolytic
enzymes in cancer invasion and metastasis. Semin Thromb Hemost.
12:294–307. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Son YO, Wang L, Poyil P, Budhraja A,
Hitron JA, Zhang Z, Lee JC and Shi X: Cadmium induces
carcinogenesis in BEAS-2B cells through ROS-dependent activation of
PI3K/AKT/GSK-3β/β-catenin signaling. Toxicol Appl Pharmacol.
264:153–160. 2012.PubMed/NCBI
|
|
11
|
Waltz DA, Fujita RM, Yang X, Natkin L,
Zhuo S, Gerard CJ, Rosenberg S and Chapman HA: Nonproteolytic role
for the urokinase receptor in cellular migration in vivo. Am J
Respir Cell Mol Biol. 22:316–322. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baek MK, Kim MH, Jang HJ, Park JS, Chung
IJ, Shin BA, Ahn BW and Jung YD: EGF stimulates uPAR expression and
cell invasiveness through ERK, AP-1, and NF-kappaB signaling in
human gastric carcinoma cells. Oncol Rep. 20:1569–1575.
2008.PubMed/NCBI
|
|
13
|
Shariat SF, Semjonow A, Lilja H, Savage C,
Vickers AJ and Bjartell A: Tumor markers in prostate cancer I:
blood-based markers. Acta Oncol. 50(Suppl 1): 61–75. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Raghu H, Sodadasu PK, Malla RR, Gondi CS,
Estes N and Rao JS: Localization of uPAR and MMP-9 in lipid rafts
is critical for migration, invasion and angiogenesis in human
breast cancer cells. BMC Cancer. 10:6472010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y1, Dang J, Johnson LK, Selhamer JJ
and Doe WF: Structure of the human urokinase receptor gene and its
similarity to CD59 and the Ly-6 family. Eur J Biochem. 227:116–122.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Person RJ, Tokar EJ, Xu Y, Orihuela R,
Ngalame NN and Waalkes MP: Chronic cadmium exposure in vitro
induces cancer cell characteristics in human lung cells. Toxicol
Appl Pharmacol. 273:281–288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hyder O, Chung M, Cosgrove D, Herman JM,
Li Z, Firoozmand A, Gurakar A, Koteish A and Pawlik TM: Cadmium
exposure and liver disease among US adults. J Gastrointest Surg.
17:1265–1273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lubovac-Pilav Z, Borràs DM, Ponce E and
Louie MC: Using expression profiling to understand the effects of
chronic cadmium exposure on MCF-7 breast cancer cells. PLoS One.
8:e846462013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hartwig A: Mechanisms in cadmium-induced
carcinogenicity: recent insights. Biometals. 23:951–960. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Järup L: Hazards of heavy metal
contamination. Br Med Bull. 68:167–182. 2003.
|
|
21
|
Cormet-Boyaka E, Jolivette K,
Bonnegarde-Bernard A, Rennolds J, Hassan F, Mehta P, Tridandapani
S, Webster-Marketon J and Boyaka PN: An NF-κB-independent and
Erk1/2-dependent mechanism controls CXCL8/IL-8 responses of airway
epithelial cells to cadmium. Toxicol Sci. 125:418–429. 2012.
|
|
22
|
Park YK, Hong H and Jang BC:
Transcriptional and translational regulation of COX-2 expression by
cadmium in C6 glioma cells. Int J Mol Med. 30:960–966.
2012.PubMed/NCBI
|
|
23
|
Choi YK, Yoon BI, Kook YH, Won YS, Kim JH,
Lee CH, Hyun BH, Oh GT, Sipley J and Kim DY: Overexpression of
urokinase-type plasminogen activator in human gastric cancer cell
line (AGS) induces tumorigenicity in severe combined
immunodeficient mice. Jpn J Cancer Res. 93:151–156. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Festuccia C, Dolo V, Guerra F, Violini S,
Muzi P, Pavan A and Bologna M: Plasminogen activator system
modulates invasive capacity and proliferation in prostatic tumor
cells. Clin Exp Metastasis. 16:513–528. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Carpenter RL and Jiang BH: Roles of EGFR,
PI3K, AKT, and mTOR in heavy metal-induced cancer (Review). Curr
Cancer Drug Targets. 13:252–266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mukhin YV, Garnovskaya MN, Collinsworth G,
Grewal JS, Pendergrass D, Nagai T, Pinckney S, Greene EL and
Raymond JR: 5-Hydroxytryptamine 1A receptor/Gibetagamma stimulates
mitogen-activated protein kinase via NAD(P)H oxidase and reactive
oxygen species upstream of src in chinese hamster ovary
fibroblasts. Biochem J. 347:61–67. 2000. View Article : Google Scholar
|
|
27
|
Lee K and Esselman WJ: Inhibition of PTPs
by H(2)O(2) regulates the activation of distinct MAPK pathways.
Free Radic Biol Med. 33:1121–1132. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Valko M, Morris H and Cronin MT: Metals,
toxicity and oxidative stress (Review). Curr Med Chem. 12:1161–208.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Freitas M and Fernandes E: Zinc, cadmium
and nickel increase the activation of NF-κB and the release of
cytokines from THP-1 monocytic cells. Metallomics. 3:1238–1243.
2011.PubMed/NCBI
|
|
30
|
Napolitano JR, Liu MJ, Bao S, Crawford M,
Nana-Sinkam P, Cormet-Boyaka E and Knoell DL: Cadmium-mediated
toxicity of lung epithelia is enhanced through NF-κB-mediated
transcriptional activation of the human zinc transporter ZIP8. Am J
Physiol Lung Cell Mol Physiol. 302:L909–L918. 2012.PubMed/NCBI
|
|
31
|
Thévenod F, Friedmann JM, Katsen AD and
Hauser IA: Up-regulation of multidrug resistance P-glycoprotein via
nuclear factor-kappaB activation protects kidney proximal tubule
cells from cadmium- and reactive oxygen species-induced apoptosis.
J Biol Chem. 275:1887–1896. 2000.
|
|
32
|
Li Q and Engelhardt JF: Interleukin-1beta
induction of NFkappaB is partially regulated by
H2O2-mediated activation of NFkappaB-inducing
kinase. J Biol Chem. 281:1495–1505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Storz P and Toker A: NF-kappaB signaling -
an alternate pathway for oxidative stress responses. Cell Cycle.
2:9–10. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fan C, Li Q, Ross D and Engelhardt JF:
Tyrosine phosphorylation of I kappa B alpha activates NF kappa B
through a redox-regulated and c-Src-dependent mechanism following
hypoxia/reoxygenation. J Biol Chem. 278:2072–2080. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
del Escobar MC, Souza V, Bucio L,
Hernández E, Gómez-Quiroz LE and Gutiérrez Ruiz MC: MAPK activation
is involved in cadmium-induced Hsp70 expression in HepG2 cells.
Toxicol Mech Methods. 19:503–509. 2009.PubMed/NCBI
|
|
36
|
Huang C, Zhang Q, Li J, Shi X, Castranova
V, Ju G, Costa M and Dong Z: Involvement of Erks activation in
cadmium-induced AP-1 transactivation in vitro and in vivo. Mol Cell
Biochem. 222:141–147. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Juang HH, Chung LC, Sung HC, Feng TH, Lee
YH, Chang PL and Tsui KH: Metallothionein 3: an
androgen-upregulated gene enhances cell invasion and tumorigenesis
of prostate carcinoma cells. Prostate. 73:1495–1506. 2013.
View Article : Google Scholar : PubMed/NCBI
|