Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common cancer worldwide and among the leading causes of
cancer-related deaths. In the majority of cases, HCC develops as a
result of chronic inflammation and cirrhosis caused by viral
hepatitis B and C, ethanol or aflatoxins (1). HCC is associated with a very poor
prognosis (2), because only a
limited number of patients are eligible for potentially curative
treatment options such as surgical resection followed by orthotopic
liver transplantation. Therefore, there is an urgent need to
develop effective therapeutic strategies for the large number of
HCC patients with advanced disease.
For a long time, hepatocarcinogenesis has been
considered to be the result of different genetic alterations that
ultimately lead to malignant transformation (3). Nowadays, cancer development is no
longer thought to be induced only by genetic and genomic
alterations, but also closely associated with lipid metabolism
(4). The studies of Karagozian
et al indicate an adipogenic lifestyle alone is sufficient
for the development of nonalcoholic steatohepatitis, hepatic stem
cell activation and hepatocarcinogenesis in wild-type mice
(5). In recent years, metabolism
is talked highly important in cancer research, especially the lipid
metabolism. Researchers believe that the de novo fatty acid
synthesis plays an important role in tumor development (6). Sphingolipids, a family of membrane
lipids, are bioactive molecules that participate in diverse
functions controlling fundamental cellular processes such as cell
division, differentiation and cell death (7). Given that most of these cellular
processes form the basis for several pathologies, it is not
surprising that sphingolipids are key players in several
pathological processes, such as diabetes, Alzheimer’s disease and
hepatocellular carcinoma (7).
miRNA regulation of gene expression plays a role in
development, differentiation, proliferation and apoptosis (8–13).
Expressions of miRNAs are abnormal in a variety of cancers and
miRNA may play a role in tumorigenesis (14–21).
Several miRNA have been identified that may act as tumor suppressor
genes (22–27). The microRNAs 449 are potent
inducers of cell death, cell cycle arrest and/or cell
differentiation. They belong to the same family as the
p53-responsive microRNAs miR-34. Instead of p53, however, the cell
cycle regulatory transcription factor E2F1 induces miR-449. All
members of this microRNA family are capable of mediating cell cycle
arrest and apoptosis and might thereby contribute to tumor
suppression. Underlying mechanisms include the downregulation of
histone acetyl transferases and consecutive activation of p53, but
also the targeting of cyclin dependent kinases and their
association partners. Thus, miR-34 and miR-449 provide an
asymmetric feedback loop to balance E2F and p53 activities. More
recently, it was discovered that miR-449 helps to ensure proper
cell function but also to avoid cancer, marking a close link
between cell differentiation and tumor suppression (28,29).
We predicted SIRT1 as miR-449 target gene. Recent
data suggest that SIRT1 may function as an oncogene and play a role
in tumorigenesis (30–33). Because sterol regulatory element
binding protein (SREBP)-1c is a transcription factor that controls
the expression of genes related to fatty acid and triglyceride
synthesis in tissues with high lipid synthesis rates such as
adipose tissue and liver. Previous studies indicate that SIRT1 can
regulate the expression and function of SREBP-1c in liver. Recent
in vivo studies have suggested a crucial role for SIRT1 in
regulating SREBP-1c expression and activity in liver. In
vivo, SIRT1 activation decreased expression of SREBP-1c and
SREBP-1c target genes (34).
Conversely, SIRT1 knockdown in liver induced the expression of
SREBP-1c and its target genes encoding lipid-synthesizing enzymes
(35). Furthermore, SREBP-1c
deacetylation by SIRT1 on Lys-289 and Lys-309 inhibits SREBP-1c
activity by decreasing its stability and its association with
lipogenic target genes (34).
Targeting the aberrant SREBP-lipogenesis-cholesterogenesis pathway
related to SIRT1 may lead to new approaches to the treatment of
HCC. MiR-449 might regulate cellular proliferation through a
SIRT1-SREBP-1c pathway implicating energy metabolism; it will be
revelatory for further study on the possible applications in
clinical treatment for HCC.
Materials and methods
Culture cells
Human hepatoma cell lines HepG2 and Huh7 from ATCC
(Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 10% fetal bovine serum (FBS)
(Gibco-BRL Co. Ltd., USA) and 1% penicillin-streptomycin-neomycin
(PSN) Antibiotic Mixture (Invitrogen, Carlsbad, CA, USA) at 37°C in
a humidified 5% CO2/95% air environment.
Reagents
Human miRNA precursors, miRNA inhibitors, antisense
oligonucleotides or small interfering RNA (siRNA), TaqMan miRNA
assay, mirVana™ miRNA isolation kit and Lipofectamine 2000 were
purchased from Life Technologies (Invitrogen). CellTiter 96H
AQueous One Solution Cell Proliferation Assay (mitochondrial MTS
assay).
Bioinformatics analysis of human
SIRT1
The 3′ untranslated region (3′-UTR) sequences were
retrieved from the Entrez Nucleotide database (http://www.ncbi.nlm.nih.gov/nuccore). The
potential miRNA targets within the conserved regions in 3′-UTR of
SIRT1 were predicted by miRBase (www.mirbase.org/)
(36).
Treatments
The expression plasmid was pcDNA 4-SIRT1 (gifts from
Leonard Guarente, USA). For transfection, cells in 12-well plates
were transfected with the indicated plasmids (1 μg/well), miRNA
mimics, antisense oligonucleotides or small interfering RNA (siRNA)
at the indicated final concentrations in the culture medium using
Lipofectamine 2000. After transfection for 72 h, cells were
subsequently harvested for immunoblot analysis.
Quantitative real-time reverse
transcription-polymerase chain reaction (qRT-PCR)
Total RNA was prepared from cells using the RNeasy
Mini kit (Qiagen, Valencia, CA) and subjected to reverse
transcription by SuperScriptH III reverse transcriptase (Life
Technologies) according to the manufacturer’s instructions. A hot
start at 95°C for 5 min was followed by 40 cycles at denaturation
at 95°C for 15 sec, annealing of the primers at 58°C for 30 sec and
elongation at 72°C for 30 sec using ABI 7500 Fast Real-Time PCR
System (Invitrogen). Data were normalized to 18S rRNA or GAPDH and
represented as the average fold of three independent duplicates. To
determine intrinsic miR-449 expression, miRNA was prepared from
cells using the mirVana™ miRNA isolation kit (Invitrogen). Mature
miRNA was quantified by the TaqMan miRNA assay (Invitrogen) in
accordance with the manufacturer’s instructions. The data were
normalized by RNU6B.
Western blot analysis
Cell lysates were prepared from miR-449, NC
(miR-negative control)-transfected or non-transfected cells using a
lysis buffer [50 mM Tris (pH 8), 150 mM NaCl, 0.02%
NaN3, 0.1% SDS, 1% NP-40 and 0.5% sodium deoxycholate]
containing 1 mM phenylmethylsulfonyl fluoride and protease
inhibitor cocktail (Roche Applied Science, Indianapolis, IN).
Protein concentration was determined by Bradford assay using
Coomassie Plus Protein Reagent (Thermo Scientific, Rockford, IL,
USA). Western blot was performed using the Novex system
(Invitrogen).
Primary antibodies anti-SIRT1, SREBP-1c, FASN, HMGCR
and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA), and
secondary antibodies which were conjugated with horseradish
peroxidase (GE Healthcare, Piscataway, NJ) were used. Detection of
protein bands was done using Enhanced Chemiluminescence Western
Blotting Detection Reagents.
BrdU labeling and mitotic index
BrdU labeling was used to evaluate DNA synthesis.
After released from the second thymidine arrest at indicated time
points, cells grown in 12-well plate were pulse labeled with BrdU
(50 μM) for 30 min. After three washes of PBS, cells were fixed
with 1 ml of Carnoy’s fixative (3 parts methano 1:1 part glacial
acetic acid) at −20°C for 20 min, and followed by three washes of
PBS. Subsequently, DNA was denatured by incubation of 2M HCL at
37°C for 60 min, followed by three washes in borate buffer (0.1 M
borate buffer, pH 8.5). After incubation with the blocking buffer,
cells were stained with anti-BrdU antibody (BD Biosciences, 1:100)
overnight at 4°C. After washes in PBS, cells were incubated with
Texas Red-conjugated anti-mouse goat IgG for 30 min at RT. After
washes, cells were mounted and BrdU positive cells were manually
scored under an immunofluorescence microscope.
Mitotic events were scored by time-lapse
video-microscopy and DNA staining. Cells were synchronized as
described above. Real-time images were captured every 10 min with
Openlab software. Mitotic events of control, cells were scored by
their morphological change (from flat to round-up). For each
experiment, at least 800 cells were videotaped, tracked and
analyzed. Alternatively, nocodazole (100 ng/ml) was added into the
medium after release, cells were collected, fixed and stained with
DNA dye (Hoechst 33258). Mitotic cells were scored by nuclear
morphology and DNA condensation.
Cell growth and proliferation assay
Cell growth was determined by the colorimetric
tetrazolium derived XTT (sodium 3′-(1-(phenylaminocarbonyl)-3,
4-tetrazolium)-bis (4-methoxy-6-nitro) benzene sulfonic acid
hydrate) assay (Roche Applied Science, Mannheim, Germany), and DNA
synthesis of cells was assessed by the BrdU (bromodeoxyuridine)
incorporation assay (Roche Applied Science). For the cell growth
and proliferation assay, at 48 h after transfection of treatment,
the cells of each group were re-seeded in 96-well plates at a
density of 0.3-1×104 cells per well. After 48 h, XTT and
incorporated BrdU were measured colorimetrically using a microtiter
plate reader at a wavelength of 450 nm (37).
Fatty acid and cholesterol
quantification
The amounts of long chain fatty acids and
cholesterols were determined using the Free Fatty Acid
Quantification Kit and Cholesterol/Cholesteryl Ester Detection Kit
(Abcam, Cambridge, MA) in cells transfected with miR-449 or NC. The
amounts of fatty acids and cholesterols were normalized by total
cell numbers. The relative fatty acid or cholesterol (%) was
assigned as 100% in NC cells.
Statistical analysis
Continuous normally distributed variables were
represented graphically as mean ± standard deviation (SD). For
statistical comparison of quantitative data between groups,
analysis of variance (ANOVA) or t-test was performed. To determine
differences between groups not normally distributed, medians were
compared using Kruskal-Wallis ANOVA. χ2 test was used
when necessary for qualitative data. The degree of association
between variables was assessed using Spearman’s non-parametric
correlation. All statistical analyses were carried out using SPSS
software version 13.0 (SPSS Inc., Chicago, IL, USA). Probabilities
of 0.05 or less were considered to be statistically
significant.
Results
miR-449 inhibits mRNA expression of
SIRT1, SREBP1-c and their downstream genes in hepatoma cells
To investigate whether miRNAs regulate the SIRT1 and
its downstream SREBP-lipogenesis-cholesterogenesis metabolic
pathway in hepatoma cells, we first used DIANA microT v4.0 online
software (http://diana.cslab.ece.ntua.gr/) to predict if one or
more miRNAs target SIRT1, the key factor that regulate fatty acid,
lipid and cholesterol biosynthesis and homeostasis. The miR-449 was
retrieved that potentially co-targeted 3′-UTRs of SIRT1 mRNAs. To
further verify if miR-449 directly binds with 3′-UTRs of SIRT1, we
performed 3′-UTR luciferase reporter assay and found that the
relative 3′-UTR luciferase activities of SIRT1 were significantly
decreased in miR-449 transfected hepatoma cells HepG2 and Huh7
(Fig. 1A and B). The results
confirm that SIRT1 mRNAs are direct targets of miR-449. To validate
whether the miRNAs control the SREBP-lipogenesis-cholesterogenesis
pathway in hepatoma cells, we performed qRT-PCR quantification
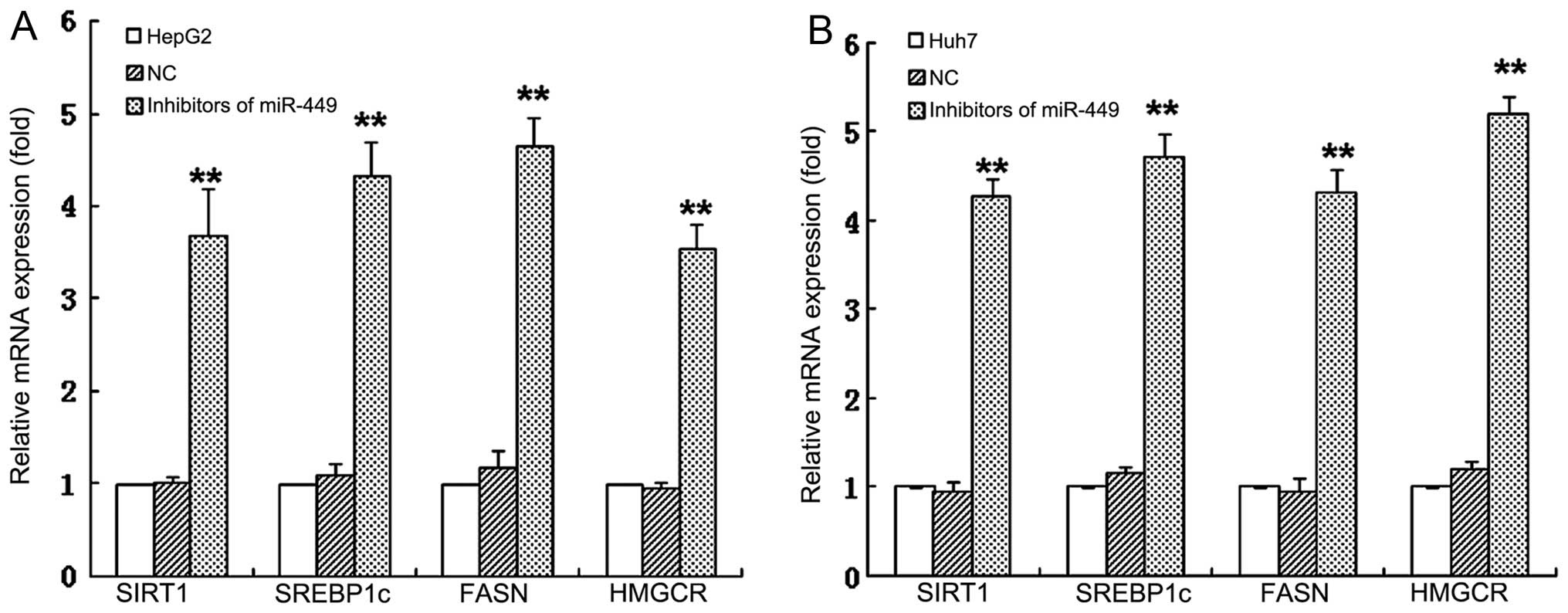
analyses. The miR-449 inhibited the mRNA expression of SIRT1 in
HepG2 and Huh7 hepatoma cell lines, respectively. Expression of
SREBP, FASN and HMGCR, which are downstream regulated genes of
SIRT1, was decreased in mRNA levels of HepG2 and Huh7 cells
(Fig. 1C and D) by miR-449, and
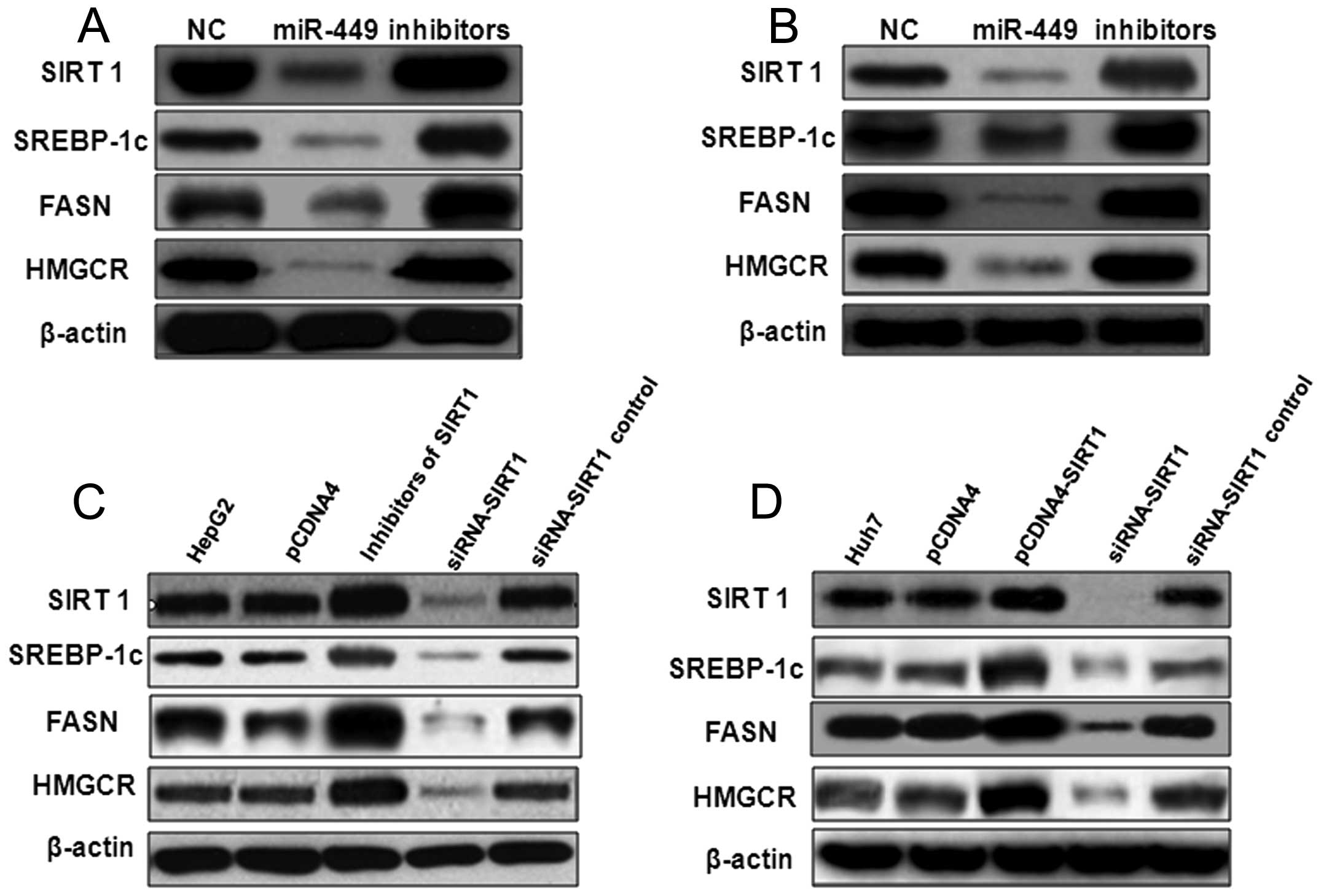
could be upregulated by inhibitors of miR-449 (Fig. 2). The protein level of SIRT1,
SREBP-1c, FASN and HMGCR was inhibited also by miR-449 in HepG2
(Fig. 3A) and Huh7 (Fig. 3B), similarly to treatment with
SIRT1 siRNA (Fig. 3C and D), but
were upregulated by inhibitors of miR-449 or overexpression of
SIRT1 (Fig. 3).
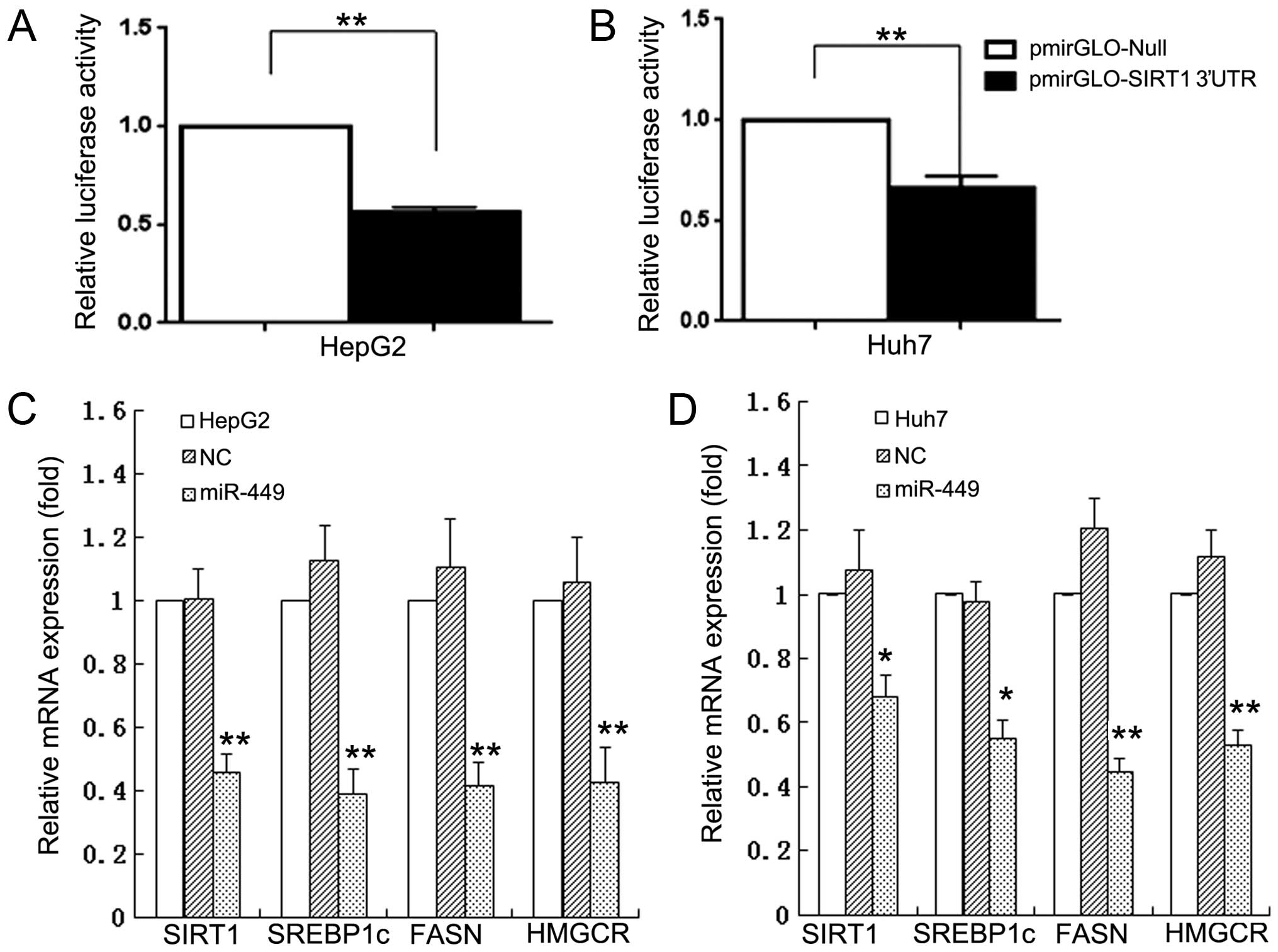 | Figure 1MiR-449 directly binds with 3′-UTRs
of SIRT1, and inhibits mRNA expression of SIRT1, SREBP1-c and their
downstream genes in hepatoma cell lines. To investigate whether
miRNAs regulate the SIRT1 and its downstream
SREBP-lipogenesis-cholesterogenesis metabolic pathway in hepatoma
cells, we first used DIANA microT v4.0 online software (http://diana.cslab.ece.ntua.gr/) to predict if
one or more miRNAs target SIRT1, the key factor that regulate fatty
acid, lipid and cholesterol biosynthesis and homeostasis. The
miRNAs, miR-449, were retrieved that potentially co-targeted
3′-UTRs of SIRT1 mRNAs. To further verify if miR-449 directly binds
with 3′-UTRs of SIRT1, we performed 3′-UTR luciferase reporter
assay and found that the relative 3′-UTR luciferase activities of
SIRT1 were significantly decreased in miR-449 transfected hepatoma
cells HepG2 and Huh7 (A and B). The results confirm that SIRT1
mRNAs are direct targets of miR-449. To validate whether the miRNAs
control the SREBP-lipogenesis-cholesterogenesis pathway in hepatoma
cells, we performed qRT-PCR quantification analyses. The miR-449
inhibited the mRNA expression of SIRT1 in HepG2 and Huh7 hepatoma
cell lines, respectively. The mRNA expression of SREBP, FASN and
HMGCR, which are downstream regulated genes of SIRT1, was decreased
in mRNA levels of HepG2 and Huh7 cells (C and D) by miR-449. |
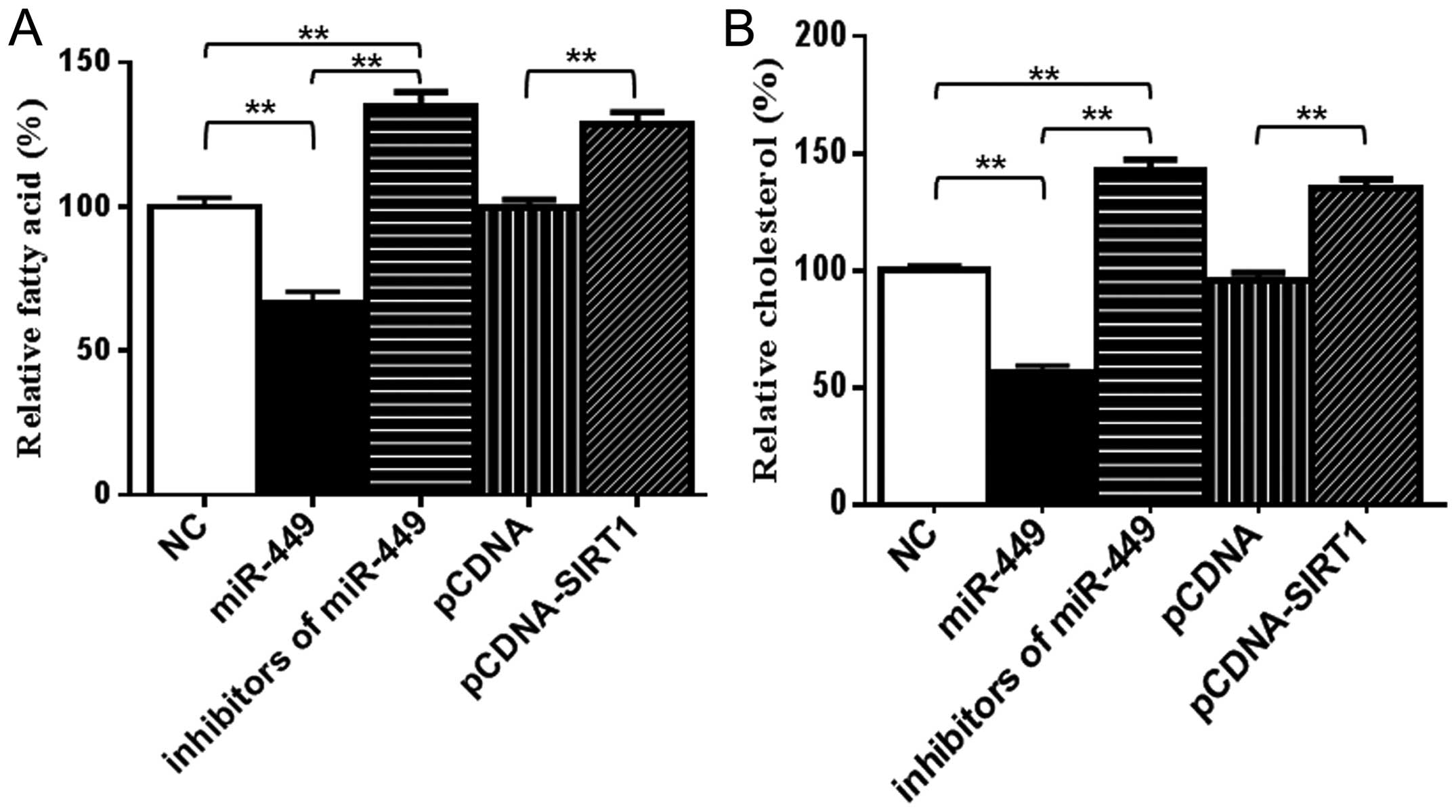
Because SREBP1c, FASN and HMGCR are important
enzymes for de novo synthesis of fatty acid and cholesterol
individually (34), we
subsequently examined the levels of fatty acid and cholesterol in
cells. As shown in Fig. 4, the
amounts of intracellular fatty acid and cholesterol were
significantly decreased in miR-449-transfected HepG2 (Fig. 4A) and Huh7 (Fig. 4B) cells compared with the control
groups. To further test the specificity of miR-449 for
SREBP-lipogenesis-cholesterogenesis, antisense oligonucleotides
against miR-449 were used as miR-449 inhibitors. By blocking
endogenous miR-449 in hepatoma cells, the amounts of intracellular
fatty acid and cholesterol were upregulated by inhibitors of
miR-449 similarly to cells treated with overexpression of SIRT1
(Fig. 4A and B). These results
suggested that miR-449 inhibitors increased SIRT1, SREBP-1c and
their downstream gene expression, and miR-449 could inhibit
SIRT1-SREBP signaling through reduction expression of SIRT1 and
decreased the levels of fatty acid and cholesterol in hepatoma
cells.
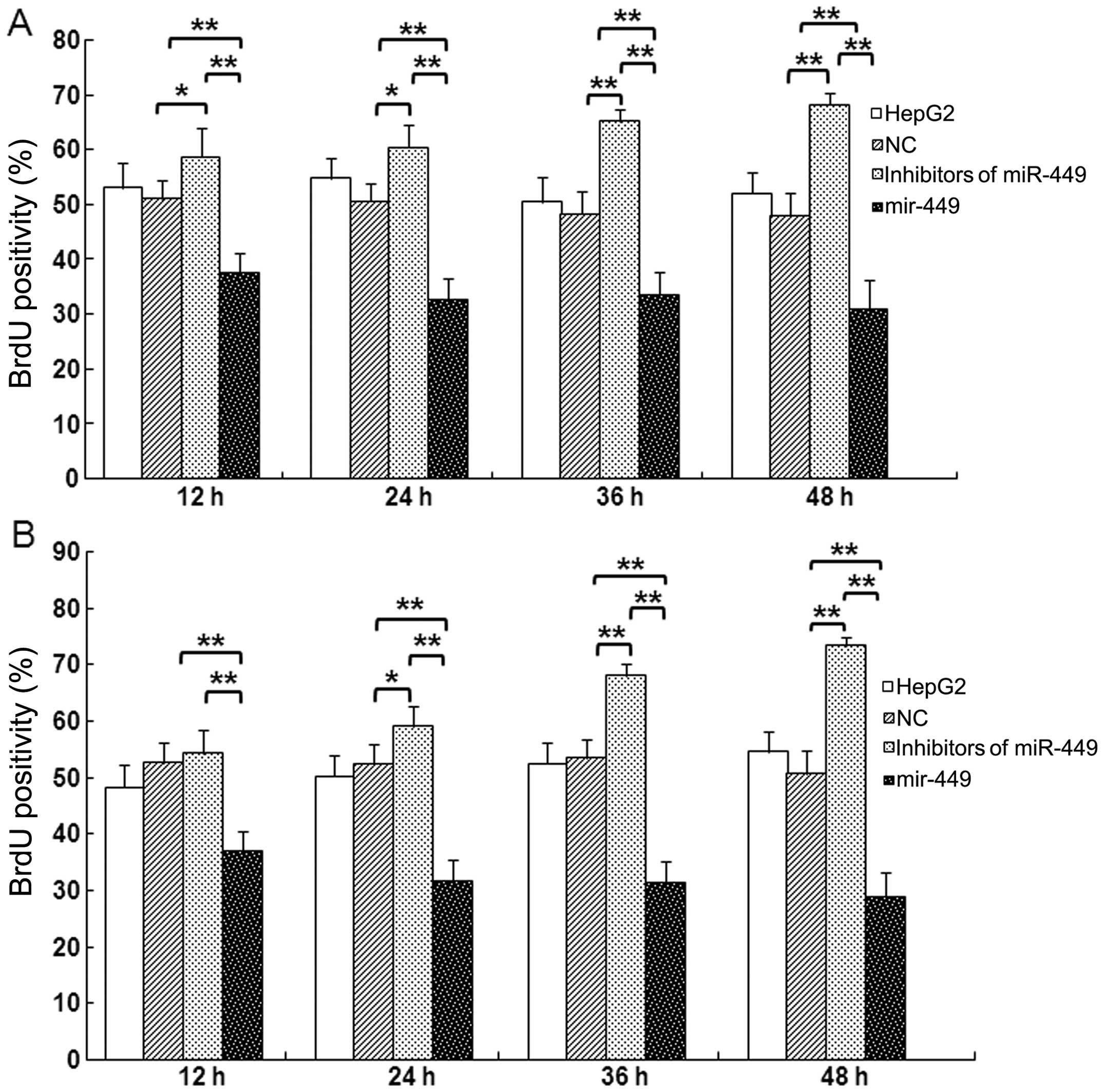
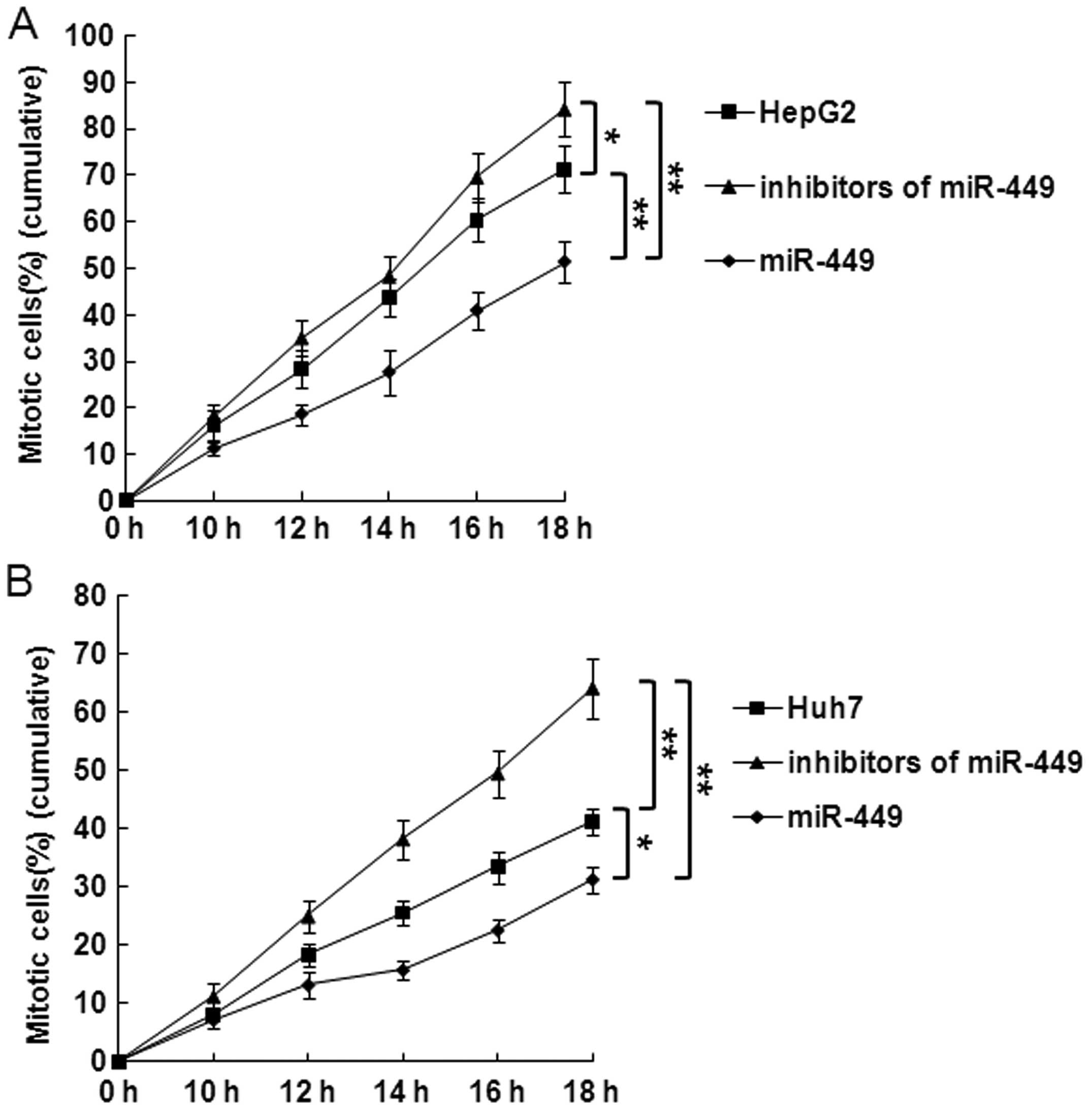
miR-449 suppresses DNA synthesis and
mitotic entry in hepatoma cells
Cells were synchronized at the G1/S boundary by
double thymidine block, and then released into mitosis. After 24 h,
BrdU was added into the medium at indicated time points to evaluate
DNA synthesis. As shown in Fig. 5A and
B, incorporation of BrdU into the control, accumulation of
mitotic HepG2 and Huh7 cells was significantly delayed in
miR-449-treated cells in each point. In contrast, accumulation of
mitotic HepG2 or Huh7 cells was significantly promoted at every
time point by inhibitors of miR-449.
To further examine the specific effect of the
overexpression of miR-449 on mitotic entry, we repeated this
experiment and evaluated the mitotic entry. Overexpression miR-449
significantly delayed mitotic entry of HepG2 or Huh7 cells
(Fig. 6A and B). In contrast,
mitotic entry of HepG2 or Huh7 cells was significantly promoted in
miR-449-treated cells at each point (Fig. 6A and B).
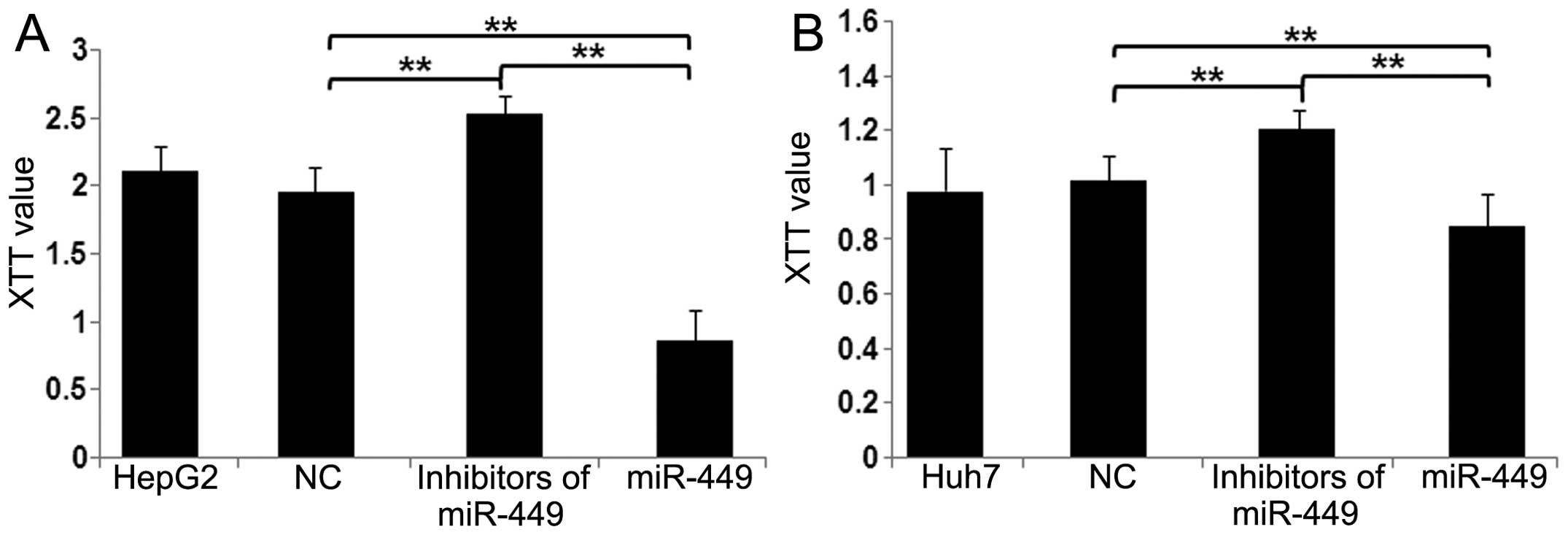
miR-449 restrains cell proliferation in
hepatoma cells
To assess the potential for biological consequences
elicited by miR-449 by XTT assays, we used mimics of miR-449 in
HepG2 and Huh7 cells and performed a series of functional assays
relevant to tumorigenicity and cancer progression. When HepG2
(Fig. 7A) and Huh7 (Fig. 7B) cells were transfected with
miR-449, proliferation of both cell types was inhibited in
comparison with miR-NC cells. Conversely, by blocking miR-449 in
hepatoma cells, miR-449 inhibitors increased cell proliferation
(Fig. 7A and B). These data
suggest that miR-449 suppresses the pathways relevant to
tumorigenicity and cancer progression.
Discussion
Sterol regulatory element-binding protein (SREBP) is
a basic helix-loop-helix leucine zipper transcription factor with
important metabolic roles in lipogenesis and cholesterogenesis
(38–40). Three major SREBP isoforms have been
identified, SREBP-1a, SREBP-1c and SREBP-2 (41). SREBP-1 controls genes involved in
fatty acid, lipid and cholesterol biosynthesis (42), whereas SREBP-2 more specifically
regulates cholesterol metabolism and homeostasis (45). Dysregulation of SREBPs and their
downstream targeted genes associated with lipogenesis and
cholesterogenesis has been implicated in cancer. Examples include
fatty acid synthase (FASN), a metabolic oncogene (44,45),
and 3-hydroxy-3-methylglutaryl CoA reductase (HMGCR), the
rate-limiting step in cholesterol biosynthesis; both proteins have
been reported to be involved in the development and progression of
cancer (44,46–49).
Targeting the aberrant SREBP-lipogenesis-cholesterogenesis pathway
may lead to new approaches to the treatment of cancer. Altered
lipid metabolism has been recently linked to HCC pathogenesis.
Upregulation of lipogenesis and cholesterogenesis in
cancer cells is associated with increased need for cell membrane
components and activation of lipid raft-related intercellular
signaling transduction during uncontrolled cell proliferation and
division as well as cancer development and progression (50–53).
Blockade of abnormal lipogenesis and cholesterogenesis provides a
promising therapeutic approach for prevention or treatment of
prostatic malignancy. MiRNA has been reported to regulate multiple
important biological processes including metabolism (54,55)
and is of potential use in cancer therapy (56–58).
However, how miRNA mediates aberrant fat metabolism and homeostasis
in hepatoma cells remains unclear. In this study, we identified one
miRNAs that play an important role in the regulation of lipogenesis
and cholesterogenesis in hepatoma cells. MiR-449 inhibited fatty
acid and cholesterol biosynthesis through downregulation of SIRT1
and key lipogenic or cholesterogenic transcription factors,
SREBP-1c, and their downstream regulated genes including FASN and
HMGCR.
SIRT1 also regulates metabolism and modulates
metabolic diseases such as diabetes. Cellular studies showed that
SIRT1 modulates fat accumulation, regulates mitochondrial
biogenesis and activates fatty acid oxidation (59). Despite extensive studies for
several decades, the role of SIRT1 in human cancer remains
controversial. Increased SIRT1 expression has been found to reduce
tumor formation in a mouse model of colon tumor (60,61),
whereas SIRT1 mutant mice have exhibited increased DNA instability
and are more susceptible to tumor development (62). Reduced levels of SIRT1 mRNA and
proteins are observed in breast tumors when compared with normal
tissue (62). Intriguingly,
however, SIRT1 overexpression is found in prostate (63), ovarian (64), gastric (65) and colorectal cancers (66), suggesting a role in tumorigenesis.
Therefore, the function of SIRT1 may be tumor-type specific and
also depend on the stage of tumorigenesis being assessed. In liver
cancer, especially in HCC, Wang et al (62) used pooled microarray data from HCC
samples to suggest that SIRT1 mRNA is expressed at comparable
levels in both malignant and benign tissues. They further concluded
that SIRT1 protein expression is reduced in HCC on the basis of one
paired HCC specimen (61). In
contrast, Chen et al (67)
have demonstrated that SIRT1 protein was overexpressed by a
post-transcriptional mechanism in a subset of HCC. They have also
shown that SIRT1 expression is low in normal and premalignant
livers, and its overexpression is closely associated with poorly
differentiated histology (67). In
agreement with the results of Bae et al, their study
confirmed that SIRT1 protein was overexpressed in a subset of
patients with HCC, whereas SIRT1 mRNA levels did not differ between
HCC and non-tumoral tissues in a large cohort of HCC patients.
Furthermore, they demonstrated that SIRT1 inactivation suppressed
in vitro cell growth and proliferation of liver cancer
cells. Many previous reports have suggested that histone
deacetylase (HDAC)-mediated gene suppression can cause uncontrolled
cell growth because HDACs repress the transcription of
cyclin-dependent kinase inhibitors, allowing continued cellular
proliferation (68,69). These results clearly support the
suggestion of oncogenic SIRT1 in hepatocellular malignant
proliferation and transformation. However, regulation of SIRT1
activity or expression leading to oncogenic SIRT1 overexpression
has not been reported yet.
Recent studies have suggested that SIRT1 expression
is regulated by miRNAs in various tissues, and miRNAs regulating
SIRT1 are involved in many cellular pathways, including embryonic
stem cell differentiation, apoptosis, senescence and hypoxia. These
findings led us to hypothesize that SIRT1 overexpression at the
post-transcriptional level may be induced by the loss or
suppression of a miRNA that selectively targets SIRT1. The
induction of SIRT1 protein by Dicer knockdown helped us to identify
endogenous miRNAs that blocked SIRT1 mRNA translation in HCC cells.
In HCC cells, upregulation of HDAC1–3 reduces expression of
miR-449. MiR-449 binds c-MET mRNA to reduce its levels, promoting
apoptosis and reducing proliferation of liver cells. Expression of
miR-449 slows growth of HCC xenograft tumors in mice; this miR
might function as a tumor suppressor (70).
We have predicted SIRT1 as miR-449 target gene.
SIRT1 could regulate SREBP-1c expression and activity in liver.
SREBP-1 and FASN have been shown to be a potentially oncogenic
transcription factor (71) and a
metabolic oncogene (72,73), respectively. By integrative
analyses, Li et al not only identified the miRNA predictors
of survival, but also analysed the functional roles of miRNAs in
the context of hepatoma progression. In conclusion, systematic
analysis of the miRNA-mRNA regulatory network (FMRN) provides new
insights into post-transcriptional gene regulation in the
progression of hepatoma (74). The
authors focused on the identification of the miRNA signatures
associated with the hepatoma progression by considering their
topological central roles in the FMRN. Similarly, the ‘in-degree’
for genes measured their central roles in the FMRN. By computing
the correlation between in-degree of gene in the FMRN and the
negative log-transformed P-value of survival analysis, in-degree is
significantly positively associated with the power for survival
analysis. These results suggest that the in-degree may be a good
indicator for identifying the genes involved in the progression of
hepatomas, extending the application of the integrative method.
We confirmed that SIRT1 mRNAs are direct targets of
miR-449. The miR-449 inhibited the expression of SIRT1, SREBP-1c
and their downstream gene FASN, HMGCR in HepG2 and Huh7 hepatoma
cell lines, respectively. Our study suggested that miR-449 could
inhibit SIRT1-SREBP signaling and DNA synthesis, mitotic entry,
cell proliferation through reduction expression of SIRT1 and
decreased the levels of fatty acid and cholesterol in hepatoma
cells.
In summary, our study demonstrates for the first
time that MiR-449 inhibits the expression of SIRT1 and SREBP-1c as
well as their downstream regulated genes, and re-programs
lipogenesis and cholesterogenesis in hepatoma cells. MiR-449
suppresses tumorigenicity and cell proliferation, which suggests
that MiR-449 play a tumor-suppressive role in hepatoma.
Collectively, miR-449 directly or indirectly regulates a cohort of
genes with significant biological roles in lipid and cholesterol
anabolism and homeostasis, cell proliferation and progression in
hepatoma cells. The small non-coding RNA therefore provides
potential therapeutic agents for treatment of hepatoma
malignancy.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (nos. 30600524, 81341067,
81071990 and 81201758), Science and Technology Planning Project of
Guangdong Province (nos. 2012A030400055, 2010B080701088,
2011B080701096 and 2011B031800184), Science and Technology projects
of Guangzhou (nos. 2011J410010 and 2011J4300066), Special Project
of Beijing Health Development and Scientific Research, China (no.
2011-5041-02), Key Projects in the PLA’s logistics Program during
the Twelfth Five-year Plan Period (no. BWS11J029). The study
sponsors had no involvement in the study.
References
|
1
|
Herold C, Reck T, Fischler P, et al:
Prognosis of a large cohort of patients with hepatocellular
carcinoma in a single European centre. Liver. 22:23–28. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Okuda K: Hepatocellular carcinoma. J
Hepatol. 32:225–237. 2000. View Article : Google Scholar
|
|
3
|
Bhalla KN: Epigenetic and chromatin
modifiers as targeted therapy of hematologic malignancies. J Clin
Oncol. 23:3971–3993. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dowman JK, Hopkins LJ, Reynolds GM, et al:
Development of hepatocellular carcinoma in a murine model of
nonalcoholic steatohepatitis induced by use of a high-fat/fructose
diet and sedentary lifestyle. Am J Pathol. 184:1550–1561. 2014.
View Article : Google Scholar
|
|
5
|
Karagozian R, Derdák Z and Baffy G:
Obesity-associated mechanisms of hepatocarcinogenesis. Metabolism.
63:607–617. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Qin H and Ruan ZH: The role of
monoacylglycerol lipase (MAGL) in the cancer progress. Cell Biochem
Biophys. Mar 16–2014.(Epub ahead of print).
|
|
7
|
Pralhada Rao R, Vaidyanathan N, Rengasamy
M, Mammen Oommen A, Somaiya N and Jagannath MR: Sphingolipid
metabolic pathway: an overview of major roles played in human
diseases. J Lipids. 2013:1789102013.PubMed/NCBI
|
|
8
|
Lee RC, Feinbaum RL and Ambros V: The
C. elegans heterochronic gene lin-4 encodes small RNAs with
antisense complementarity to lin-14. Cell. 75:843–854. 1993.
|
|
9
|
Xu P, Vernooy SY, Guo M and Hay BA: The
Drosophila microRNA Mir-14 suppresses cell death and is
required for normal fat metabolism. Curr Biol. 13:790–795.
2003.
|
|
10
|
Wightman B, Ha I and Ruvkun G:
Post-transcriptional regulation of the heterochronic gene lin-14 by
lin-4 mediates temporal pattern formation in C. elegans.
Cell. 75:855–862. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reinhart BJ, Slack FJ, Basson M, et al:
The 21-nucleotide let-7 RNA regulates developmental timing in
Caenorhabditis elegans. Nature. 403:901–906. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brennecke J, Hipfner DR, Stark A, Russell
RB and Cohen SM: Bantam encodes a developmentally regulated
microRNA that controls cell proliferation and regulates the
proapoptotic gene hid in Drosophila. Cell. 113:25–36. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hatfield SD, Shcherbata HR, Fischer KA,
Nakahara K, Carthew RW and Ruohola-Baker H: Stem cell division is
regulated by the microRNA pathway. Nature. 435:974–978. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Michael MZ, O’ Connor SM, van Holst
Pellekaan NG, Young GP and James RJ: Reduced accumulation of
specific microRNAs in colorectal neoplasia. Mol Cancer Res.
1:882–891. 2003.PubMed/NCBI
|
|
15
|
Calin GA, Sevignani C, Dumitru CD, et al:
Human microRNA genes are frequently located at fragile sites and
genomic regions involved in cancers. Proc Natl Acad Sci USA.
101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Iorio MV, Ferracin M, Liu CG, et al:
MicroRNA gene expression deregulation inhumanbreast cancer. Cancer
Res. 65:7065–7070. 2005. View Article : Google Scholar
|
|
17
|
Lu J, Getz G, Miska EA, et al: MicroRNA
expression profiles classify human cancers. Nature. 435:834–838.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He L, Thomson JM, Hemann MT, et al: A
microRNA polycistron as a potential human oncogene. Nature.
435:828–833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Volinia S, Calin GA, Liu CG, et al: A
microRNA expression signature of human solid tumors defines cancer
gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pekarsky Y, Santanam U, Cimmino A, et al:
Tcl1 expression in chronic lymphocytic leukemia is regulated by
miR-29 and miR-181. Cancer Res. 66:11590–11593. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Slack FJ and Weidhaas JB: MicroRNAs as a
potential magic bullet in cancer. Future Oncol. 2:73–82. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cimmino A, Calin GA, Fabbri M, et al:
miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl
Acad Sci USA. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Johnson SM, Grosshans H, Shingara J, et
al: RAS is regulated by the let-7 microRNA family. Cell.
120:635–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takamizawa J, Konishi H, Yanagisawa K, et
al: Reduced expression of the let-7 microRNAs in human lung cancers
in association with shortened postoperative survival. Cancer Res.
64:3753–3756. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mayr C, Hemann MT and Bartel DP:
Disrupting the pairing between let-7 and Hmga2 enhances oncogenic
transformation. Science. 315:1576–1579. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Morris JP and McManus MT: Slowing down the
Ras lane: miRNAs as tumor suppressors? Sci STKE.
16:412005.PubMed/NCBI
|
|
27
|
He L, He X, Lowe SW and Hannon GJ:
MicroRNAs join the p53 network - another piece in the
tumour-suppression puzzle. Nat Rev Cancer. 7:819–822. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lizé M, Klimke A and Dobbelstein M:
MicroRNA-449 in cell fate determination. Cell Cycle. 10:2874–2882.
2011.PubMed/NCBI
|
|
29
|
Lizé M, Pilarski S and Dobbelstein M:
E2F1-inducible microRNA 449a/b suppresses cell proliferation and
promotes apoptosis. Cell Death Differ. 17:452–458. 2010.PubMed/NCBI
|
|
30
|
Hida Y, Kubo Y, Murao K and Arase S:
Strong expression of a longevity-related protein, SIRT1, in Bowen’s
disease. Arch Dermatol Res. 299:103–106. 2007.PubMed/NCBI
|
|
31
|
Yeung F, Hoberg JE, Ramsey CS, et al:
Modulation of NF-kappaB-dependent transcription and cell survival
by the SIRT1 deacetylase. EMBO J. 23:2369–2380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Walker AK, Yang F, Jiang K, et al:
Conserved role of SIRT1 orthologs in fasting-dependent inhibition
of the lipid/cholesterol regulator SREBP. Genes Dev. 24:1403–1417.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rodgers JT and Puigserver P:
Fasting-dependent glucose and lipid metabolic response through
hepatic sirtuin 1. Proc Natl Acad Sci USA. 104:12861–12866. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ponugoti B, Kim DH, Xiao Z, et al: SIRT1
deacetylates and inhibits SREBP-1C activity in regulation of
hepatic lipid metabolism. J Biol Chem. 285:33959–33970. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Truman JP, García-Barros M, Obeid LM and
Hannun YA: Evolving concepts in cancer therapy through targeting
sphingolipid metabolism. Biochim Biophys Acta. Dec 30–2013.(Epub
ahead of print).
|
|
36
|
Zhou B, Li C, Qi W, et al: Downregulation
of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic
insulin sensitivity. Diabetologia. 55:2032–2043. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ford J, Jiang M and Milner J:
Cancer-specific functions of SIRT1 enable human epithelial cancer
cell growth and survival. Cancer Res. 65:10457–10463. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hamamoto R, Furukawa Y, Morita M, et al:
SMYD3 encodes a histone methyltransferase involved in the
proliferation of cancer cells. Nat Cell Biol. 6:731–740. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang H, Liu H, Chen K, et al: SIRT1
promotes tumorigenesis of hepatocellular carcinoma through
PI3K/PTEN/AKT signaling. Oncol Rep. 28:311–318. 2012.PubMed/NCBI
|
|
40
|
Eberle D, Hegarty B, Bossard P, Ferre P
and Foufelle F: SREBP transcription factors: master regulators of
lipid homeostasis. Biochimie. 86:839–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dif N, Euthine V, Gonnet E, et al: Insulin
activates human sterol-regulatory-element-binding protein-1c
(SREBP-1c) promoter through SRE motifs. Biochem J. 400:179–188.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guillet-Deniau I, Mieulet V, Le Lay S, et
al: Sterol regulatory element binding protein-1c expression and
action in rat muscles: insulin-like effects on the control of
glycolytic and lipogenic enzymes and UCP3 gene expression.
Diabetes. 51:1722–1728. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rome S, Lecomte V, Meugnier E, et al:
Microarray analyses of SREBP-1a and SREBP-1c target genes identify
new regulatory pathways in muscle. Physiol Genomics. 34:327–337.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Giandomenico V, Simonsson M, Gronroos E
and Ericsson J: Coactivator dependent acetylation stabilizes
members of the SREBP family of transcription factors. Mol Cell
Biol. 23:2587–2599. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Calkin AC and Tontonoz P: Transcriptional
integration of metabolism by the nuclear sterol-activated receptors
LXR and FXR. Nat Rev Mol Cell Biol. 13:213–224. 2012.PubMed/NCBI
|
|
46
|
Yoshikawa T, Shimano H, Amemiya-Kudo M, et
al: Identification of liver X receptor-retinoid X receptor as an
activator of the sterol regulatory element-binding protein 1c gene
promoter. Mol Cell Biol. 21:2991–3000. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Repa JJ, Liang G, Ou J, et al: Regulation
of mouse sterol regulatory element-binding protein-1c gene
(SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev.
14:2819–2830. 2000. View Article : Google Scholar
|
|
48
|
Cozzone D, Debard C, Dif N, et al:
Activation of liver X receptors promotes lipid accumulation but
does not alter insulin action in human skeletal muscle cells.
Diabetologia. 49:990–999. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Swinnen JV, Heemers H, van de Sande T, et
al: Androgens, lipogenesis and prostate cancer. J Steroid Biochem
Mol Biol. 92:273–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yamashita T, Honda M, Takatori H, et al:
Activation of lipogenic pathway correlates with cell proliferation
and poor prognosis in hepatocellular carcinoma. J Hepatol.
50:100–110. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Di Vizio D, Solomon KR and Freeman MR:
Cholesterol and cholesterol-rich membranes in prostate cancer: an
update. Tumori. 94:633–639. 2008.PubMed/NCBI
|
|
52
|
Swinnen JV, Brusselmans K and Verhoeven G:
Increased lipogenesis in cancer cells: new players, novel targets.
Curr Opin Clin Nutr Metab Care. 9:358–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Freeman MR, Cinar B and Lu ML: Membrane
rafts as potential sites of nongenomic hormonal signaling in
prostate cancer. Trends Endocrinol Metab. 16:273–279. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fernandez-Hernando C, Suarez Y, Rayner KJ
and Moore KJ: MicroRNAs in lipid metabolism. Curr Opin Lipidol.
22:86–92. 2011. View Article : Google Scholar
|
|
55
|
Krutzfeldt J and Stoffel M: MicroRNAs: a
new class of regulatory genes affecting metabolism. Cell Metab.
4:9–12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Henry JC, Azevedo-Pouly AC and Schmittgen
TD: MicroRNA replacement therapy for cancer. Pharm Res.
28:3030–3042. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Garzon R, Marcucci G and Croce CM:
Targeting microRNAs in cancer: rationale, strategies and
challenges. Nat Rev Drug Discov. 9:775–789. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bader AG, Brown D and Winkler M: The
promise of microRNA replacement therapy. Cancer Res. 70:7027–7030.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lee J and Kemper JK: Controlling SIRT1
expression by microRNAs in health and metabolic disease. Aging
(Albany, NY). 2:527–534. 2010.PubMed/NCBI
|
|
60
|
Firestein R, Blander G, Michan S, et al:
The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon
cancer growth. PLoS One. 3:e20202008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bae HJ, Noh JH, Kim JK, et al:
MicroRNA-29c functions as a tumor suppressor by direct targeting
oncogenic SIRT1 in hepatocellular carcinoma. Oncogene.
33:2557–2567. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang RH, Sengupta K, Li C, et al: Impaired
DNA damage response, genome instability, and tumorigenesis in SIRT1
mutant mice. Cancer Cell. 14:312–323. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Huffman DM, Grizzle WE, Bamman MM, et al:
SIRT1 is significantly elevated in mouse and human prostate cancer.
Cancer Res. 67:6612–6618. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jang KY, Kim KS, Hwang SH, et al:
Expression and prognostic significance of SIRT1 in ovarian
epithelial tumours. Pathology. 41:366–371. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cha EJ, Noh SJ, Kwon KS, et al: Expression
of DBC1 and SIRT1 is associated with poor prognosis of gastric
carcinoma. Clin Cancer Res. 15:4453–4459. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Stunkel W, Peh BK, Tan YC, et al: Function
of the SIRT1 protein deacetylase in cancer. Biotechnol J.
2:1360–1368. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chen J, Zhang B, Wong N, et al: Sirtuin 1
is upregulated in a subset of hepatocellular carcinomas where it is
essential for telomere maintenance and tumor cell growth. Cancer
Res. 71:4138–4149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Noh JH, Jung KH, Kim JK, et al: Aberrant
regulation of HDAC2 mediates proliferation of hepatocellular
carcinoma cells by deregulating expression of G1/S cell cycle
proteins. PLoS One. 6:e281032011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Xie HJ, Noh JH, Kim JK, et al: HDAC1
inactivation induces mitotic defect and caspase-independent
autophagic cell death in liver cancer. PLoS One. 7:e342652012.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Buurman R, Gürlevik E, Schäffer V, et al:
Histone deacetylases activate hepatocyte growth factor signaling by
repressing microRNA-449 in hepatocellular carcinoma cells.
Gastroenterology. 143:811–820. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Huang WC, Li X, Liu J, Lin JT and Chung
LW: Activation of androgen receptor, lipogenesis and oxidative
stress converged by SREBP-1 is responsible for regulating growth
and progression of prostate cancer cells. Mol Cancer Res.
10:133–142. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Menendez JA, Decker JP and Lupu R: In
support of fatty acid synthase (FAS) as a metabolic oncogene:
extracellular acidosis acts in an epigenetic fashion activating FAS
gene expression in cancer cells. J Cell Biochem. 94:1–4. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Baron A, Migita T, Tang D and Loda M:
Fatty acid synthase: a metabolic oncogene in prostate cancer? J
Cell Biochem. 91:47–53. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Li Y, Xu J, Chen H, et al: Comprehensive
analysis of the functional microRNA-mRNA regulatory network
identifies miRNA signatures associated with hepatoma malignant
progression. Nucleic Acids Res. 41:e2032013. View Article : Google Scholar : PubMed/NCBI
|