Introduction
Gastric cancer (GC) is one of the most common
malignant tumors and the third most common cause of death from
malignancies worldwide (1).
Despite intensive investigations of anticancer treatments for GC,
the prognosis of unresectable advanced or recurrent GC remains
poor. The median overall survival of GC patients has reached only
~1 year using conventional cytotoxic chemotherapy (2–4).
Trastuzumab, a monoclonal antibody against human epidermal growth
factor receptor (EGFR) 2 (HER2), used in combination with
chemotherapy has shown a survival benefit when used as a first-line
treatment in patients with HER2-positive advanced GC (5). However, HER2 overexpression has only
been reported in 13–23% of GC cases (6–8).
Fibroblast growth factor receptor 2 amplification,
MET amplification and RhoA mutations have been
anticipated as new molecular targets in GC, but these aberrations
are relatively infrequent (9–12).
Therefore, new therapeutic modalities are still needed.
The mitogen-activated protein kinase (MAPK) signal
pathway cascade and its downstream factors promote cancer cell
proliferation, differentiation and survival. The three-tiered
kinase cascade consisting of RAF, mitogen-activated protein kinase
kinase (MEK), and extracellular signal-regulated kinase (ERK) is
frequently dysregulated in malignancies as a result of activating
mutations in the upstream RAS (KRAS, NRAS and
HRAS) (13,14). The activating BRAF mutation
has been detected in a variety of human cancers (15), and the success of RAF inhibitors in
BRAF-mutated melanoma has revealed a new modality and has
substantiated the contribution of the MAPK signal to carcinogenesis
(16,17). Somatic oncogenic MEK1
mutations have also been identified in several human malignancies
(18–21). In addition, our previous study
demonstrated that MEK1 mutations in poorly differentiated GC
cell lines that are hypersensitive to MEK inhibitors have
transformational abilities and that the growth of these cancer
cells is dependent on these mutations (22). Considering the addiction of cancer
cells to active MEK1 mutations for proliferation, GC with
such oncogenic MEK1 mutations might be suitable for targeted
therapy with MEK inhibitors.
A persistent problem is the development of
resistance to molecular-targeted therapies. A series of resistance
mechanisms for MEK inhibition that operate ERK-dependently or
ERK-independently have been reported, including MEK
mutations, elevated RAS or RAF protein levels, and the activation
of an alternative PI3K/AKT or STAT3 pathway (23–25).
Currently, the phosphorylation of EGFR after the inhibition of the
MAPK signal has been suggested as another potential mechanism
responsible for the resistance to MEK inhibition in various
cancers, and combination therapy with EGFR inhibitors yielded
synergistic effects (26,27). In this study, we observed the
phosphorylation of EGFR and HER2 after MEK inhibition in a
MEK1-mutated GC cell line. Then, the relevancy of this
mechanism to MEK inhibitor resistance was validated, and
combinations with other tyrosine kinase inhibitors were tested to
develop new therapeutic possibilities.
Materials and methods
Reagents and ligand
Trametinib (GSK1120212) and lapatinib (GW572016)
were purchased from Selleck Chemicals (Houston, TX, USA) and were
dissolved in dimethyl sulfoxide (DMSO) for the in vitro
experiments. Recombinant human epidermal growth factor (EGF) was
obtained from R&D Systems (Minneapolis, MN, USA) and was
constituted in sterile phosphate-buffered saline (PBS).
Antibodies
Rabbit-antibodies specific for ERK1/2, EGFR, HER2,
phospho-ERK1/2 (pERK1/2), phospho-EGFR (pEGFR), phospho-HER2
(pHER2), poly (ADP-ribose) polymerase (PARP), caspase-3, cleaved
PARP (cPARP), cleaved caspase-3 (cCaspase-3), and β-actin were
obtained from Cell Signaling Technology (Beverly, MA, USA).
Cell cultures
A poorly differentiated GC cell line harboring the
MEK1 Q56P mutation, OCUM-1, was propagated in RPMI-1640
(Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Gibco BRL, Grand Island,
NY, USA) and 1% Penicillin-Streptomycin Mixed Solution (Nacalai
Tesque, Kyoto, Japan) in a humidified atmosphere of 5%
CO2 at 37°C.
Human phospho-receptor tyrosine kinase
(RTK) array
The screening of 49 phosphorylated tyrosine kinases
in the OCUM-1 cell line was performed using the Human Phospho-RTK
Array kit (R&D Systems). Whole lyses of OCUM-1 cells incubated
in the absence (DMSO) or presence of 1 nM trametinib for 72 h were
compared in accordance with the manufacturer’s recommendation.
Protein detection was accomplished using an
anti-phospho-tyrosine-HRP detection antibody and an enhanced
chemiluminescence system (Image Quant LAS4000; GE Healthcare Life
Science, Buckinghamshire, UK).
Growth inhibition assay
The
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay was used to evaluate the growth inhibitory effect of the
drugs, as previously described (28). When the effect of EGF was
evaluated, 1% FBS was used.
Real-time reverse transcription
polymerase chain reaction (RT-PCR)
A total of 1 μg of RNA was isolated from the cells
using ISOGEN reagent (Nippon Gene, Tokyo, Japan) and then converted
to cDNA using GeneAmp RNA-PCR kit (Applied Biosystems, Foster City,
CA USA). Real-time PCR was performed using SYBR Premix Ex Taq and
Thermal Cycler Dice (Takara, Shiga, Japan), as described previously
(29). The glyceraldehyde
3-phosphate dehydrogenase (GAPD, NM_002046) gene was used to
normalize the expression levels in subsequent quantitative
analyses. To amplify the target genes encoding amphiregulin (AREG),
EGF, heparin-binding EGF-like growth factor (HB-EGF), neuregulin 1
(NRG1), transforming growth factor α (TGFA), EGFR, HER2, HER3, and
HER4 (AREG, EGF, HB-EGF, NRG1, TGFA, EGFR, ERBB2, ERBB3 and
ERBB4 genes, respectively), the following primers were used:
AREG-F, GTCGCTCTTGATAC TCGGCTCAG; AREG-R, TCCCAGAGTAGGTGTCATTG
AGGTC; EGF-F, CAACCAGTGGCTGGTGAGGA; EGF-R,
GAGCCCTTATCACTGGATACTGGAA; HB-EGF-F, CAA GGTGATTTCAGACTGCAGAGG;
HB-EGF-R, TTTGGCA CTTGAAGGCTCTGG; NRG1-F, GCCAGGAATCGGCTG CAGGT;
NRG1-R, AGCCAGTGATGCT TTGTTAATGCGA; TGFA-F, CTTTGGAAACCAGCAGGTCTGA;
TGFA-R, CCCAAATAAGCCAGGCTGTTCTA; EGFR-F, CATCCA GGCCCAACTGTGAG;
EGFR-R, CAGTGGAAGCCTT GAAGCAGAA; ERBB2-F, TGGGAGCCTGGCATTTCTG; E R
BB2-R, CG G CCATG C TGAGATGTATAG GTA; ERBB3-F,
GGGAGCATTTAATGGCAGCTA; ERBB3-R, GAATGGAATTGTCTGGGACTGG; ERBB4-F,
GCAGCT AACTTTGAATGCCTGTCTC; ERBB4-R, GCAGCTA ACTTTGAATGCCTGTCTC;
GAPD-F, GCACCGTCAAG GCTGAGAAC; and GAPD-R, ATGGTGGTGAAGACG
CCAGT.
Western blot analysis
The western blot analysis was performed as described
previously (28). Briefly,
subconfluent cells were washed with cold PBS and harvested with
Lysis A buffer containing 1% Triton X-100, 20 mM Tris-HCl (pH 7.0),
5 mM EDTA, 50 mM sodium chloride, 10 mM sodium pyrophosphate, 50 mM
sodium fluoride, 1 mM sodium orthovanadate, and a protease
inhibitor mix, Complete™ (Roche Diagnostics). Whole-cell lyses were
separated using SDS-PAGE and were blotted onto a polyvinylidene
fluoride membrane. After blocking with 3% bovine serum albumin in a
TBS buffer (pH 8.0) with 0.1% Tween-20, the membrane was probed
with the primary antibody. After rinsing twice with TBS buffer, the
membrane was incubated with a horseradish peroxidase-conjugated
secondary antibody and washed, followed by visualization using an
ECL detection system and LAS-4000. When the cells were stimulated
using EGF, the cells were incubated with 1% FBS medium.
Statistical analysis
The results are shown as the mean value ± standard
deviation (SD) for three replicate independent experiments. The
statistical analyses were performed using the Student’s t-test
(two-tailed). A P-value <0.05 was considered statistically
significant. The statistical analyses were two-tailed and were
performed using Microsoft Excel (Microsoft, Redmond, WA, USA).
Results
MEK inhibition led to the phosphorylation
of EGFR and HER2 in the MEK1-mutated OCUM-1 cell line
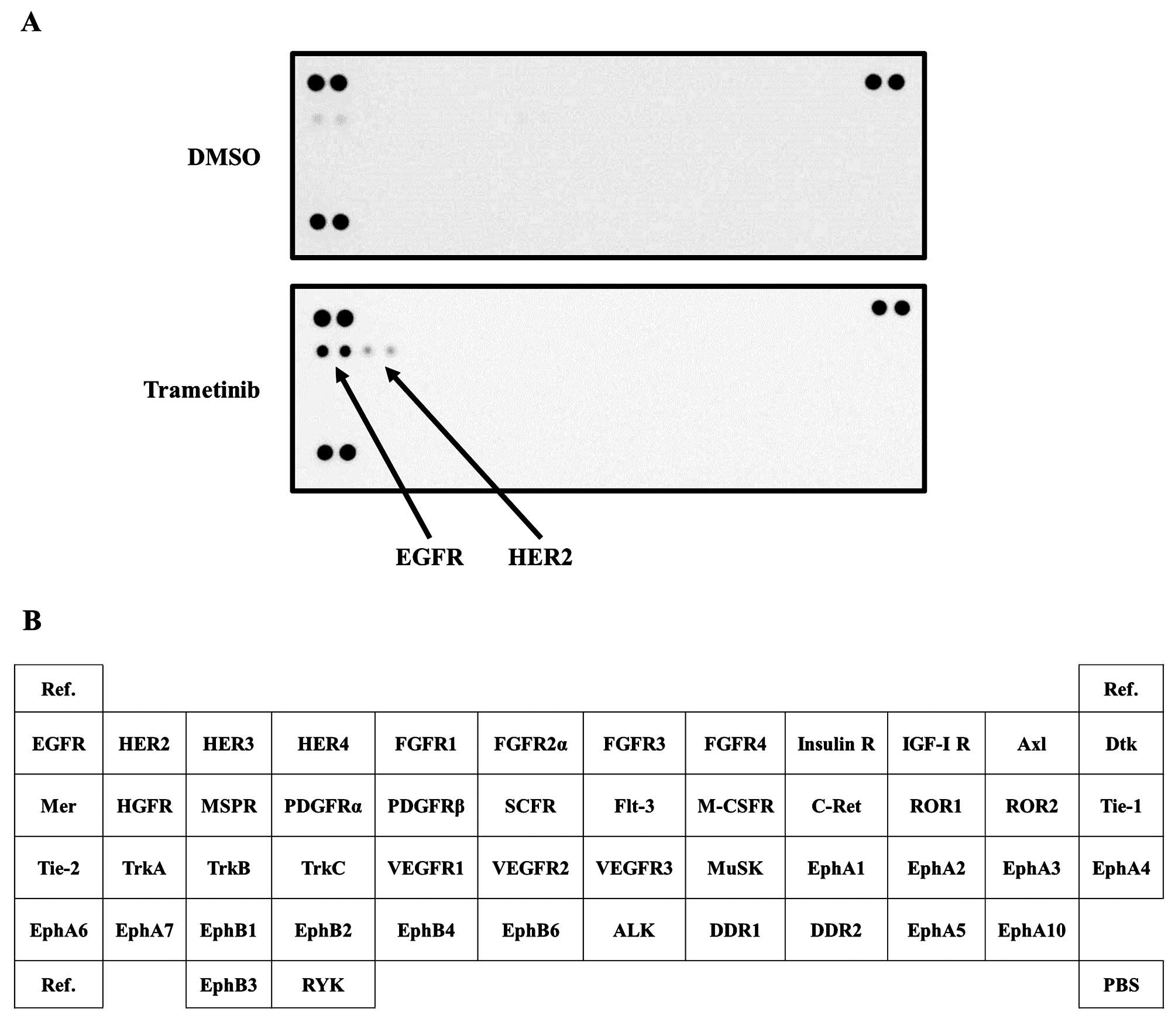
To address the phosphorylation of RTK after MEK
inhibition, we used a phospho-RTK array. The arrangement of the RTK
array is summarized in Fig. 1B.
The phosphorylation levels of EGFR and HER2 were elevated at 72 h
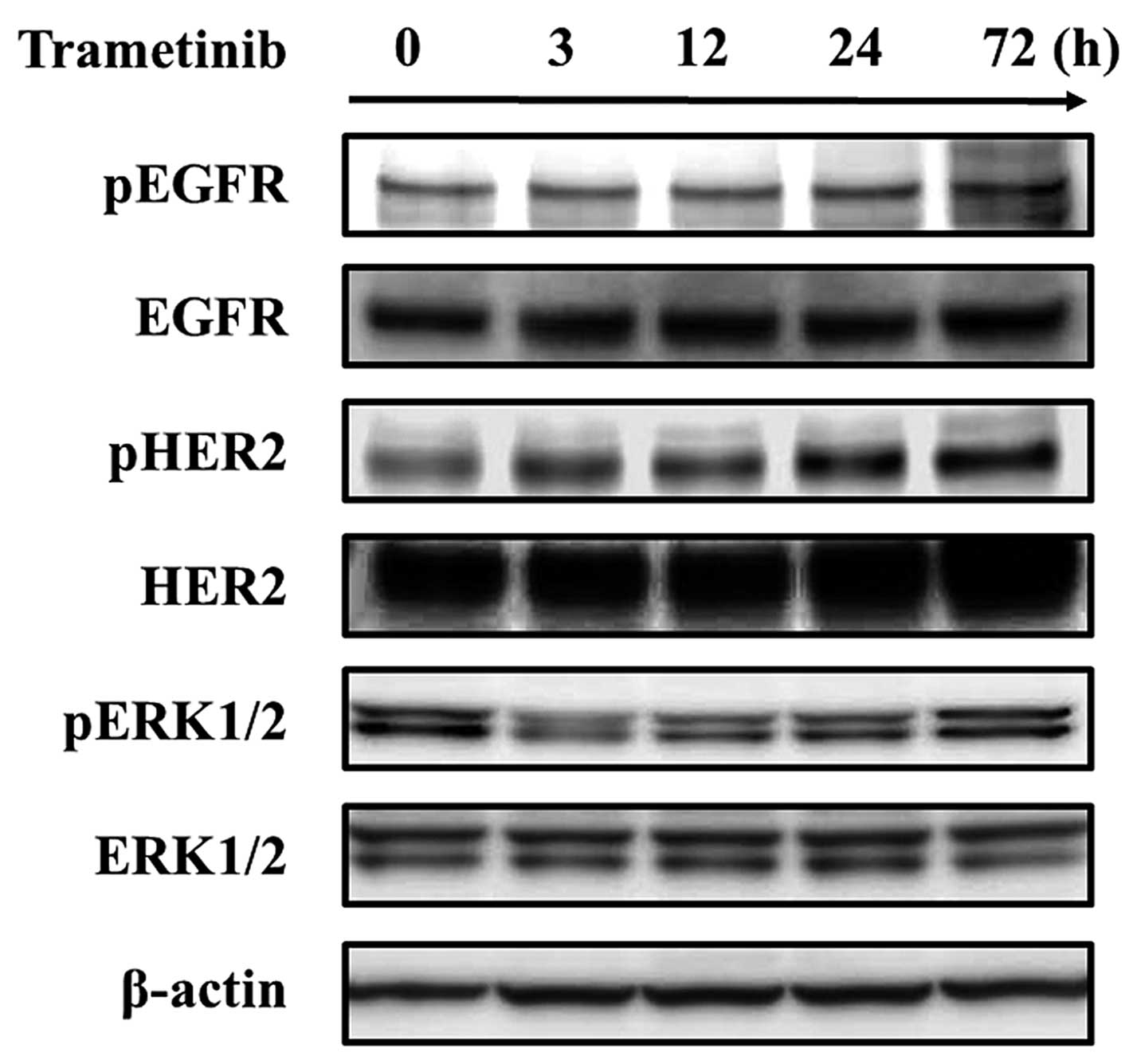
after treatment with a MEK inhibitor (trametinib) (Fig. 1A). Next, to validate the results of
the RTK array, we evaluated the phosphorylation level of EGFR and
HER2 using a western blot analysis. In addition to the inhibitory
effect on the ERK signal, trametinib led to the activation of EGFR
and HER2 signals similar to the results of the RTK array. In
addition, the phosphorylation of ERK1/2, which had been inhibited
by trametinib, was reactivated following the activation of the EGFR
and HER2 signals (Fig. 2). These
results suggest that the EGFR and HER2 signals are activated under
MEK inhibition in MEK1-mutated GC. To investigate the
mechanism responsible for the activation of the EGFR and HER2
signals, real-time RT-PCR was performed. However, no significant
changes in the mRNA expression levels of the molecules relative to
the EGFR and HER2 signals were observed (AREG, EGF, HB-EGF,
NRG1, TGFA, EGFR, HER2, HER3 and HER4; data not
shown).
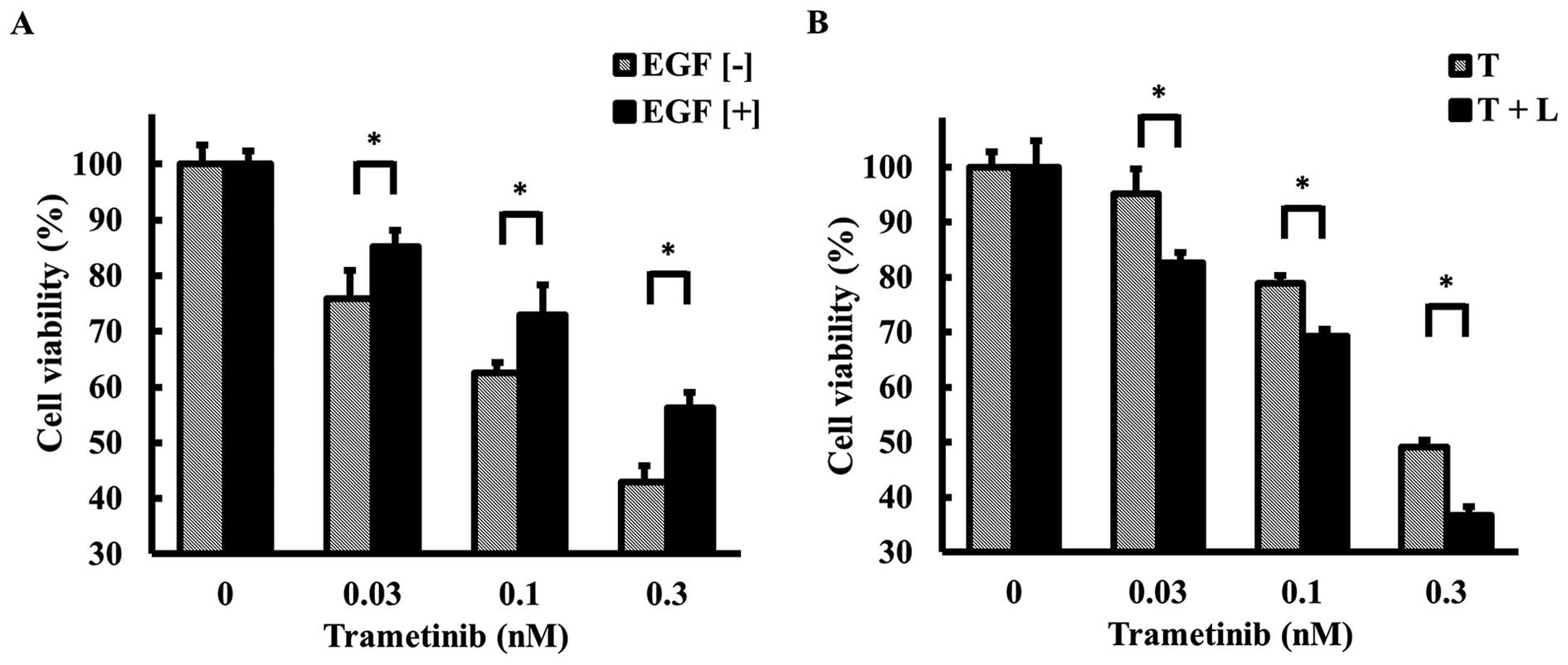
Ligand-induced EGFR and HER2 activation
led to the acquisition of resistance to a MEK inhibitor
To verify the contribution of EGFR and HER2 signals
to MEK inhibitor resistance, the experiments were performed using
recombinant human EGF (10 ng/ml) as the ligand. After the
EGF-induced activation of the EGFR and HER2 signals, the OCUM-1
cell line became resistant to trametinib (Fig. 3A). Using a western blot analysis,
EGF stimulation was shown to increase the phosphorylation levels of
EGFR and HER2, and ERK1/2, which had been suppressed by trametinib,
was re-phosphorylated (Fig.
4B).
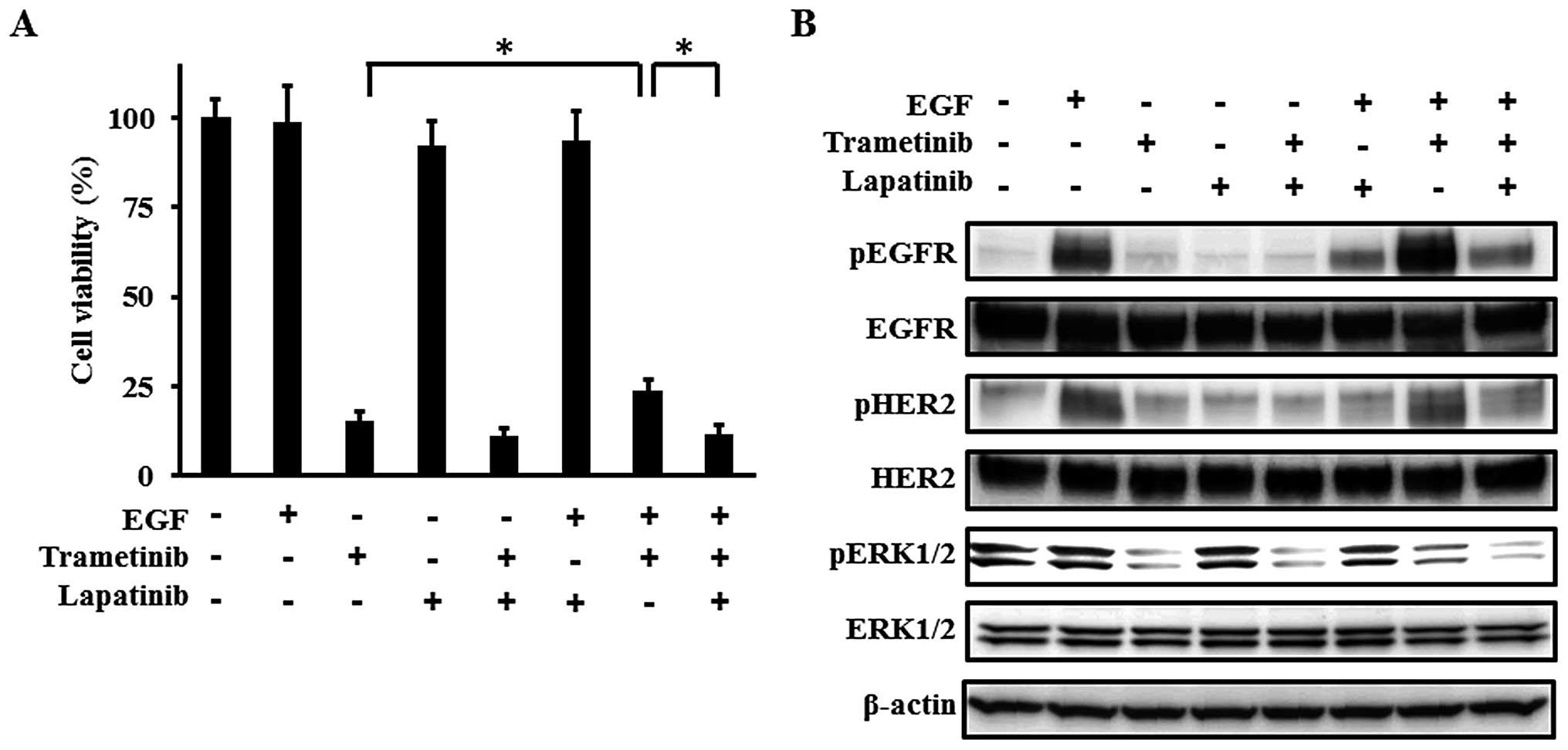 | Figure 4Effect of lapatinib on the resistance
to trametinib induced by EGF. (A) Growth inhibitory effect of
combination with trametinib and lapatinib in the presence of EGF.
The cells were exposed to 1 nM of trametinib, 10 μM of lapatinib,
and/or 10 ng/ml of EGF for 72 h and the cell growth was evaluated
using an MTT assay with 1% FBS. In the presence of EGF, the
inhibitory effect of trametinib was weakened significantly, while
the combination of trametinib and lapatinib abolished the
resistance induced by EGF. EGF or lapatinib monotherapy did not
influence the cell growth. Columns, mean of independent triplicate
experiments; error bars, SD; *P<0.05. (B)
Phosphorylation of EGFR, HER2, and ERK1/2. When the phosphorylation
levels were examined using a western blot analysis, the samples
were collected 3 h after stimulation. The following concentrations
were used: 1 nM of trametinib, 10 μM of lapatinib, and 10 ng/ml of
EGF. EGF stimulation increased the phosphorylation levels of EGFR
and HER2, resulting in the activation of ERK1/2. The decreased
phosphorylation levels of ERK1/2 induced by trametinib were
re-activated by EGF stimulation, and lapatinib abolished the
re-activation of ERK1/2 by inhibiting the phosphorylation of EGFR
and HER2. β-actin was used as an internal control. pEGFR,
phospho-EGFR; pHER2, phospho-HER2; pERK1/2, phospho-ERK1/2. |
Lapatinib abolished the resistance to
trametinib induced by EGF
Next, to investigate the effect of lapatinib (an
EGFR and HER2 dual tyrosine kinase inhibitor) on the EGF-induced
resistance, a combination therapy was tested. The EGF-induced
resistance to trametinib was abolished by lapatinib (Fig. 4A). The suppression of the
phosphorylation levels of ERK1/2 through the inhibition of EGFR and
HER2 by lapatinib can explain the restored response to trametinib
(Fig. 4B). These results suggest
that the activation of EGFR and HER2 signals potentially plays an
important role in MEK inhibitor resistance and that lapatinib may
abolish such resistance.
Combination of trametinib and lapatinib
synergistically inhibits cell growth
To investigate the role of the activated EGFR and
HER2 signals during treatment with trametinib, we examined the
synergistic anticancer effect of trametinib and lapatinib in the
OCUM-1 cell line using an MTT assay. The combination of trametinib
and lapatinib showed a synergistic effect on the inhibition of cell
proliferation (Fig. 3B).
Furthermore, lapatinib decreased the phosphorylation levels of EGFR
and HER2, which had been activated after the treatment with
trametinib, and the phosphorylation level of ERK1/2 was inhibited
to a greater degree by the combination treatment than by trametinib
monotherapy (Fig. 5A). Cleaved
PARP and cleaved caspase-3, which are hallmarks of apoptosis, had
also increased at 72 h after the trametinib and lapatinib
combination therapy, compared with after trametinib monotherapy
(Fig. 5B). These results suggest
that activated EGFR and HER2 signals are involved in salvage
pathways under MEK inhibition in MEK1-mutated GC and that
trametinib and lapatinib exert a synergistic effect by inhibiting
the salvage signals.
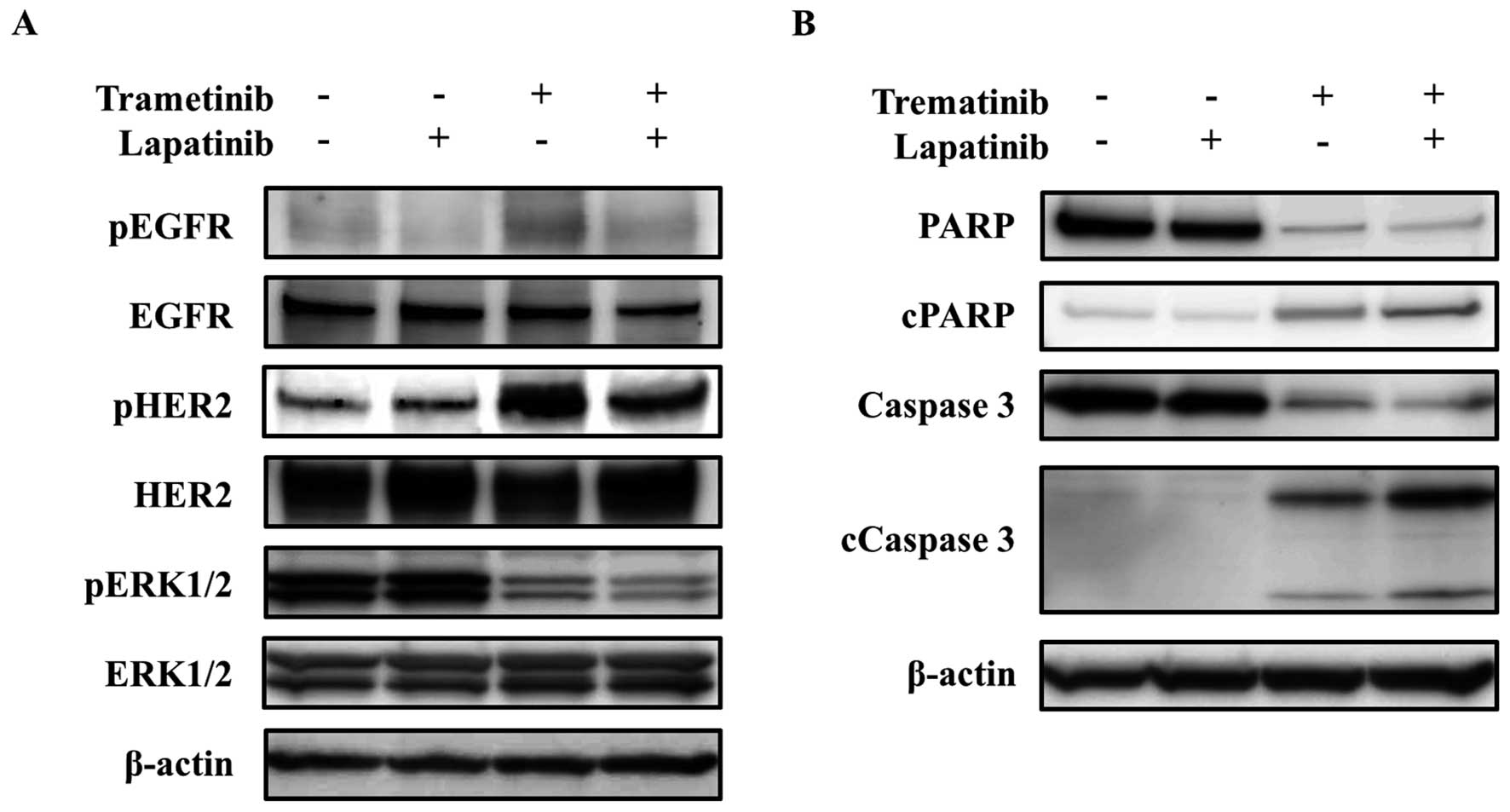 | Figure 5Synergistic effect of trametinib and
lapatinib on EGFR, HER2, and ERK signals and apoptosis-related
molecules. (A) Phosphorylation of EGFR, HER2, and ERK1/2. For the
analysis of the phosphorylation levels using a western blot
analysis, the samples were collected at 72 h after exposure. The
following concentrations were used: 1 nM of trametinib and 10 μM of
lapatinib. The phosphorylation levels of EGFR and HER2 were
elevated at 72 h after treatment with trametinib, similar to the
results obtained using the phospho-RTK array. Lapatinib reduced the
phosphorylation level of ERK1/2 by inhibiting the EGFR and HER2
signals, which were activated after trametinib treatment. β-actin
was used as an internal control. pEGFR, phospho-EGFR; pHER2,
phospho-HER2; pERK1/2, phospho-ERK1/2. (B) Expressions of
apoptosis-related molecules. The cells were exposed to the
inhibitors for 72 h, and apoptosis-related molecules were then
examined using a western blot analysis. The following
concentrations were used: 1 nM of trametinib and 10 μM of
lapatinib. Trametinib increased the expressions of cleaved PARP and
cleaved caspase-3 to a greater extent when used in combination with
lapatinib, compared with the results for trametinib monotherapy.
β-actin was used as an internal control. cPARP, cleaved PARP;
cCaspase-3, cleaved caspase-3. |
Discussion
In this study, we demonstrated the reactivation of
ERK1/2 via the activation of EGFR and HER2 signals after MEK
inhibition with trametinib, and EGF-induced resistance in the
MEK1-mutated GC cell line. Our data also identified the
receptor tyrosine kinase family members EGFR and HER2 as potent
targets for overcoming resistance to MEK inhibitors. Moreover, the
combination of trametinib and lapatinib was more effective against
a MEK1-mutated GC cell line than trametinib monotherapy. To
the best of our knowledge, this is the first report to show the
activation of EGFR and HER2 after MEK inhibition, the potential of
combination therapy as a therapeutic strategy, and the possibility
of overcoming resistance in MEK1-mutated GC.
The serine threonine kinases BRAF, MEK, and ERK are
major regulators of the MAPK signal and are frequently dysregulated
in malignancies. Given the role of these kinases in cancer, the
MAPK signal has become an attractive target for molecular targeted
drugs (30). While a number of
MAPK signal inhibitors, as well as RAF inhibitors, have been
developed and are in clinical use, the acquisition of resistance
has hindered the continuation of treatment, resulting in a limited
survival benefit. Resistance to MAPK inhibitors is mediated by many
mechanisms, including the reactivation of RAS/RAF/MEK/ERK
signals, MEK mutations, and increases in the RAS or RAF
protein levels, as well as increased signals through alternative
pathways, including the PI3K/AKT and STAT3 pathways (31). Recently, BRAF inhibition leading to
a feedback activation of EGFR and a strong synergistic effect of
BRAF and EGFR inhibition were described in BRAF-mutated
colorectal cancer cell lines (32). Furthermore, the combination of a
MEK inhibitor and a dual EGFR and HER2 inhibitor produced a
synergistic effect by inhibiting the activation of ERK signals both
in vivo and in vitro in KRAS-mutated colon and
lung cancer cell lines (27).
These results suggest that the activation of HER family members is
an important salvage signal during MAPK inhibition. Similarly, in
our data, the EGFR and HER2 signals were activated after treatment
with a MEK inhibitor, resulting in the re-activation of the ERK
signal. In addition, EGF stimulation also activated the ERK signal
via the activation of EGFR and HER2 in the MEK1-mutated GC
cell line, which led to MEK inhibitor resistance. The effects of
the EGFR and HER2 signals were abolished by the inhibitor. These
findings indicate the possibility of a novel combination therapy
comprised of a MEK inhibitor and a HER inhibitor for GC patients,
similar to results observed for other malignancies.
To address how MEK inhibition by trametinib
activates EGFR and HER2, we investigated the mRNA expression of the
corresponding genes using real-time RT-PCR. However, no significant
change in mRNA expression was observed. MEK inhibition reportedly
leads to MYC degradation and HER2 and HER3 upregulation in
KRAS-mutated colon and lung cancer (27). Another study has shown that CDC25c,
which can bind to EGFR, may be involved in the activation of EGFR
in a BRAF-mutated colorectal cancer cell line (32). However, the detailed mechanism
responsible for the activation of HER signals after MEK inhibition
remains unclear. Furthermore, the activation of HER signals has
been reported to result not only from MAPK inhibition, but also
from exposure to cytotoxic agents, including 5-FU, oxaliplatin in
colorectal cancer cells, and pemetrexed in non-small cell lung
cancer (33,34). These findings suggest that the
activation of HER signals plays a primary salvage role in many
cancers, promising that resistance to many anticancer therapies can
be overcome. Therefore, further research on approaches to
overcoming such resistance is needed.
In conclusion, the present study revealed that the
activation of EGFR and HER2 signals after MEK inhibition may serve
as a potential mechanism responsible for the resistance to a MEK
inhibitor in MEK1-mutated GC. Additionally, combined
treatment with trametinib and lapatinib exhibited a synergistic
effect by inhibiting the activation of the EGFR and HER2 signals.
These data may contribute to the development of novel therapeutic
strategies for MEK1-mutated GC.
Acknowledgements
We thank Mr. Shinji Kurashimo, Mr. Yoshihiro Mine,
Ms. Eiko Honda, Ms. Tomoko Kitayama and Ms. Ayaka Kurumatani for
their technical assistance. This study was supported in part by the
Grant-in Aid for Japan Society for Promotion of Science
Fellows.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Van Cutsem E, Moiseyenko VM, Tjulandin S,
Majlis A, Constenla M, Boni C, Rodrigues A, Fodor M, Chao Y, Voznyi
E, et al; V325 Study Group. Phase III study of docetaxel and
cisplatin plus fluorouracil compared with cisplatin and
fluorouracil as first-line therapy for advanced gastric cancer: A
report of the V325 Study Group. J Clin Oncol. 24:4991–4997. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cunningham D, Starling N, Rao S, Iveson T,
Nicolson M, Coxon F, Middleton G, Daniel F, Oates J and Norman AR;
Upper Gastrointestinal Clinical Studies Group of the National
Cancer Research Institute of the United Kingdom. Capecitabine and
oxaliplatin for advanced esophagogastric cancer. N Engl J Med.
358:36–46. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Koizumi W, Narahara H, Hara T, Takagane A,
Akiya T, Takagi M, Miyashita K, Nishizaki T, Kobayashi O, Takiyama
W, et al: S-1 plus cisplatin versus S-1 alone for first-line
treatment of advanced gastric cancer (SPIRITS trial): A phase III
trial. Lancet Oncol. 9:215–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bang YJ, Van Cutsem E, Feyereislova A,
Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T,
et al; ToGA Trial Investigators. Trastuzumab in combination with
chemotherapy versus chemotherapy alone for treatment of
HER2-positive advanced gastric or gastro-oesophageal junction
cancer (ToGA): A phase 3, open-label, randomised controlled trial.
Lancet. 376:687–697. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sakai K, Mori S, Kawamoto T, Taniguchi S,
Kobori O, Morioka Y, Kuroki T and Kano K: Expression of epidermal
growth factor receptors on normal human gastric epithelia and
gastric carcinomas. J Natl Cancer Inst. 77:1047–1052.
1986.PubMed/NCBI
|
|
7
|
Yano T, Doi T, Ohtsu A, Boku N, Hashizume
K, Nakanishi M and Ochiai A: Comparison of HER2 gene amplification
assessed by fluorescence in situ hybridization and HER2 protein
expression assessed by immunohistochemistry in gastric cancer.
Oncol Rep. 15:65–71. 2006.
|
|
8
|
Gravalos C and Jimeno A: HER2 in gastric
cancer: A new prognostic factor and a novel therapeutic target. Ann
Oncol. 19:1523–1529. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matsumoto K, Arao T, Hamaguchi T, Shimada
Y, Kato K, Oda I, Taniguchi H, Koizumi F, Yanagihara K, Sasaki H,
et al: FGFR2 gene amplification and clinicopathological features in
gastric cancer. Br J Cancer. 106:727–732. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kawakami H, Okamoto I, Arao T, Okamoto W,
Matsumoto K, Taniguchi H, Kuwata K, Yamaguchi H, Nishio K, Nakagawa
K, et al: MET amplification as a potential therapeutic target in
gastric cancer. Oncotarget. 4:9–17. 2013.PubMed/NCBI
|
|
11
|
Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi
ST, Siu HC, Deng S, Chu KM, Law S, et al: Whole-genome sequencing
and comprehensive molecular profiling identify new driver mutations
in gastric cancer. Nat Genet. 46:573–582. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kakiuchi M, Nishizawa T, Ueda H, Gotoh K,
Tanaka A, Hayashi A, Yamamoto S, Tatsuno K, Katoh H, Watanabe Y, et
al: Recurrent gain-of-function mutations of RHOA in diffuse-type
gastric carcinoma. Nat Genet. 46:583–587. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bollag G, Hirth P, Tsai J, Zhang J,
Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, et al:
Clinical efficacy of a RAF inhibitor needs broad target blockade in
BRAF-mutant melanoma. Nature. 467:596–599. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Flaherty KT, Puzanov I, Kim KB, Ribas A,
McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et
al: Inhibition of mutated, activated BRAF in metastatic melanoma. N
Engl J Med. 363:809–819. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pao W and Girard N: New driver mutations
in non-small-cell lung cancer. Lancet Oncol. 12:175–180. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Estep AL, Palmer C, McCormick F and Rauen
KA: Mutation analysis of BRAF, MEK1 and MEK2 in 15 ovarian cancer
cell lines: Implications for therapy. PLoS One. 2:e12792007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marks JL, Gong Y, Chitale D, Golas B,
McLellan MD, Kasai Y, Ding L, Mardis ER, Wilson RK, Solit D, et al:
Novel MEK1 mutation identified by mutational analysis of epidermal
growth factor receptor signaling pathway genes in lung
adenocarcinoma. Cancer Res. 68:5524–5528. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bentivegna S, Zheng J, Namsaraev E,
Carlton VE, Pavlicek A, Moorhead M, Siddiqui F, Wang Z, Lee L,
Ireland JS, et al: Rapid identification of somatic mutations in
colorectal and breast cancer tissues using mismatch repair
detection (MRD). Hum Mutat. 29:441–450. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sogabe S, Togashi Y, Kato H, Kogita A,
Mizukami T, Sakamoto Y, Banno E, Terashima M, Hayashi H, de Velasco
MA, et al: MEK inhibitor for gastric cancer with MEK1 gene
mutations. Mol Cancer Ther. 13:3098–3106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Emery CM, Vijayendran KG, Zipser MC,
Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA,
Karpova MB, et al: MEK1 mutations confer resistance to MEK and
B-RAF inhibition. Proc Natl Acad Sci USA. 106:20411–20416. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Little AS, Balmanno K, Sale MJ, Newman S,
Dry JR, Hampson M, Edwards PA, Smith PD and Cook SJ: Amplification
of the driving oncogene, KRAS or BRAF, underpins acquired
resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci
Signal. 4:ra172011.PubMed/NCBI
|
|
25
|
Corcoran RB, Dias-Santagata D, Bergethon
K, Iafrate AJ, Settleman J and Engelman JA: BRAF gene amplification
can promote acquired resistance to MEK inhibitors in cancer cells
harboring the BRAF V600E mutation. Sci Signal.
3:ra842010.PubMed/NCBI
|
|
26
|
Walters DM, Lindberg JM, Adair SJ, Newhook
TE, Cowan CR, Stokes JB, Borgman CA, Stelow EB, Lowrey BT,
Chopivsky ME, et al: Inhibition of the growth of patient-derived
pancreatic cancer xenografts with the MEK inhibitor trametinib is
augmented by combined treatment with the epidermal growth factor
receptor/ HER2 inhibitor lapatinib. Neoplasia. 15:143–155. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun C, Hobor S, Bertotti A, Zecchin D,
Huang S, Galimi F, Cottino F, Prahallad A, Grernrum W, Tzani A, et
al: Intrinsic resistance to MEK inhibition in KRAS mutant lung and
colon cancer through transcriptional induction of ERBB3. Cell Rep.
7:86–93. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Togashi Y, Sakamoto H, Hayashi H,
Terashima M, De Velasco MA, Fujita Y, Kodera Y, Sakai K, Tomida S,
Kitano M, et al: Homozygous deletion of the activin A receptor,
type IB gene is associated with an aggressive cancer phenotype in
pancreatic cancer. Mol Cancer. 13:1262014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kaneda H, Arao T, Tanaka K, Tamura D,
Aomatsu K, Kudo K, Sakai K, De Velasco MA, Matsumoto K, Fujita Y,
et al: FOXQ1 is overexpressed in colorectal cancer and enhances
tumorigenicity and tumor growth. Cancer Res. 70:2053–2063. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hingorani SR, Jacobetz MA, Robertson GP,
Herlyn M and Tuveson DA: Suppression of BRAF(V599E) in human
melanoma abrogates transformation. Cancer Res. 63:5198–5202.
2003.PubMed/NCBI
|
|
31
|
Freeman AK and Morrison DK: Mechanisms and
potential therapies for acquired resistance to inhibitors targeting
the Raf or MEK kinases in cancer. Molecular Mechanisms of Tumor
Cell Resistance to Chemotherapy: Targeted Therapies to Reverse
Resistance. Bonavita B: pp. 47–67. 2013, View Article : Google Scholar
|
|
32
|
Prahallad A, Sun C, Huang S, Di
Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A
and Bernards R: Unresponsiveness of colon cancer to BRAF(V600E)
inhibition through feedback activation of EGFR. Nature.
483:100–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Van Schaeybroeck S, Karaiskou-McCaul A,
Kelly D, Longley D, Galligan L, Van Cutsem E and Johnston P:
Epidermal growth factor receptor activity determines response of
colorectal cancer cells to gefitinib alone and in combination with
chemotherapy. Clin Cancer Res. 11:7480–7489. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li T, Ling YH, Goldman ID and Perez-Soler
R: Schedule-dependent cytotoxic synergism of pemetrexed and
erlotinib in human non-small cell lung cancer cells. Clin Cancer
Res. 13:3413–3422. 2007. View Article : Google Scholar : PubMed/NCBI
|