Introduction
Colorectal cancer (CRC) is the second leading cause
of cancer-related death, and 5-fluorouracil (5-FU) is still the
main chemotherapeutic agent used in the first-line treatment of
this disease. The response rate of 5-FU monotherapy is 15% and thus
modulators are added for higher response rates: combination therapy
of 5-FU with irinotecan or oxaliplatin (40%), and the newly
developed combination therapy of 5-FU with bevacizumab and
cetuximab (60–70%) (1). However,
resistance to chemotherapy is a major cause of mortality in CRC
patients. Therefore, other compounds are needed in order to
increase treatment efficacy and overcome the 5-FU resistance.
Furthermore, the current therapy options with 5-FU cause severe
side-effects. Thus, novel agents are urgently required. Natural
products are a rich source of anticancer agents and provide novel
and more effective anticancer agents for therapeutic use. Gambogic
acid (GA) is a small molecule extracted from the traditional
Chinese medicine herb Garcinia hanburyi Hook. f. which has
been used for a long time in China. GA has a strong cytotoxic
effect on a variety of cancers but has very weak effect on the
hematologic system (2–5). Importantly, GA has been approved by
the China Food and Drug Administration (CFDA) for phase II clinical
trial in solid tumor therapy (6).
There have been many research studies published demonstrating the
anticancer activity of GA (3,7–10).
However, the mechanisms of action for the GA anticancer effects are
not fully understood. Therefore, further molecular studies need to
be conducted in order to further elucidate the mechanism of GA
activity. In the present study, we have established an acquired
5-FU resistant cell line to explore the anticancer effect of GA. We
demonstrated that GA directly inhibited proliferation and induced
apoptosis in both drug sensitive and drug resistant colorectal
cancer cells and induced apoptosis via activating the JNK signaling
pathway. Data presented here demonstrate that GA activates the JNK
signaling pathway and overcomes drug resistance in CRC cells. Thus,
it could be a promising medicinal compound for colorectal cancer
therapy.
Materials and methods
Cell culture
Human epithelial colorectal adenocarcinoma HCT-15
cells were purchased from the Culture Collection of Chinese Academy
of Science (Shanghai, China). Cells were cultured in Dulbecco's
modified Eagle's medium (Gibco Life Technologies, Carlsbad, CA,
USA) supplemented with 10% inactivated fetal bovine serum (Gibco
Life Technologies), 100 units/ml penicillin and 10 μg/ml
streptomycin (Gibco Life Technologies) in a humidified atmosphere
of 5% CO2 at 37°C. The 5-FU resistant cell line
(HCT-15R) was established from its parental cell line HCT-15 by
stepwise exposure to increasing the concentrations of 5-FU,
starting at 1 μM and ending at 100 μM. 5-FU (1 μM) was included in
the culture medium for HCT-15R to maintain the drug resistance. The
cells were maintained in 5-FU free medium at least 2 weeks before
the experiments.
Reagents
5-Fluorouracil (Sigma-Aldrich, St. Louis, MO, USA)
was dissolved in dimethyl sulphoxide (DMSO) to a 200 mM solution
and stored at −20°C. SP600125 (Sigma-Aldrich) was dissolved in DMSO
to a 50 mM solution and stored at −20°C. Gambogic acid
(Sigma-Aldrich) was dissolved in DMSO to a 10 mM stock solution and
stored at −20°C. PARP, caspase-3, cleaved-caspase-3, caspase-8,
Mcl-1, Bcl-xl, Bcl-2, XIAP, survivin, cytochrome c, AIF,
cyclin D1, p53, JNK and phospho-JNK at Thr183/Tyr185 were from Cell
Signaling Technology (Beverly, MA, USA). Cleaved-caspase-9 was
purchased from Abcam (Cambridge, MA, USA). Antibodies against
caspase-9, β-actin and anti-mouse immunoglobulin G and anti-rabbit
immunoglobulin G horseradish peroxidase-conjugated secondary
antibodies were from Proteintech Group (Chicago, IL, USA).
Cell proliferation assay by real-time
cell impedance analysis
For real-time cell analysis (RTCA), xCELLigence
system (Roche Applied Science, Mannheim, Germany) was used to
dynamically monitor cell proliferation rates. The experiments were
performed following the standard protocol developed by Roche
Applied Science. Briefly, cells were seeded into 100 μl of media in
an E-Plate. Cell proliferation was monitored at set intervals via
measuring electrical impedance across microelectrodes on the bottom
of E-Plate. The impedance was expressed as cell index (CI), an
arbitrary unit. RTCA software, supplied by the manufacturer, was
used to analyze the measurements.
Cell viability assay
The effect of GA on cell viability assay was
analyzed by MTS assay (Promega, Madison, WI, USA). Briefly, a total
of 5×104/ml cells in 100 μl were treated with GA for 72
h. Four hours before culture termination, 20 μl MTS labeling
mixture (MTS/PMS) was added to each well. The absorbance density
was read on a 96-well plate reader at wavelength 490 nm.
Cell cycle analysis by flow
cytometry
Cells incubated with 1 μM GA for the indicated
times, were collected, washed and fixed with 75% cold ethanol at
4°C overnight. After 75% ethanol was moved, cells were washed twice
in phosphate-buffered saline (PBS) and labeled with propidium
iodide (BD Biosciences, Franklin Lakes, NJ, USA). The cell cycle
distribution was analyzed by BD FACSCanto II flow cytometry.
Analysis of cell apoptosis by flow
cytometry
Apoptosis was determined by flow cytometry using an
Annexin V-FITC/PI dual staining kit (Nanjing KeyGen Biotech Co.,
Ltd., Nanjing, China). Cells incubated with 2 μM GA for the
indicated times, were collected, washed and stained in working
solution (500 μl binding buffer with 5 μl Annexin V-FITC and 5 μl
propidium iodide) for 15 min at room temperature in the dark. Cells
were then washed and resuspended with binding buffer. Apoptotic
cells were determined by BD FACSCanto II flow cytometry. Annexin
V-FITC-positive and Annexin V-FITC-plus-PI positive cells were
determined as apoptotic cells.
Mitochondrial membrane potential
measurement
Mitochondrial membrane potential was detected using
a JC-1 mitochondrial membrane potential assay kit (Nanjing KeyGen
Biotech Co., Ltd.), following the manufacturer's protocol. Briefly,
after treatment, cells were incubated with JC-1 staining solution
at 37°C for 20 min and rinsed twice with incubation buffer provided
by the kit. Fluorescence intensity of both mitochondrial JC-1
monomers (Green) and aggregates (Red) were detected using a BD
FACSCanto II flow cytometry. JC-1 stains the mitochondria of
healthy cells red, and apoptotic cells green.
Western blot analysis
Western blot analysis was performed as previously
described (11). Briefly, cells
were lysed in lysis buffer containing protease and phosphatase
inhibitors (Nanjing KeyGen Biotech Co., Ltd.). Protein
concentrations were measured using a Bio-Rad assay kit (Bio-Rad
Laboratiries, Hercules, CA, USA). Total cellular proteins were
separated by SDS-PAGE and transferred to PVDF membranes followed by
probed with a primary antibody overnight at 4°C. The next day, the
membrane was washed and incubated with HRP-conjugated secondary
antibody at room temperature for 2 h followed by ECL detection.
After detection of protein bands, the membrane was stripped and
re-probed with anti-β-actin antibody to confirm equal loading of
samples.
Preparation of cytoplasmic fractions
GA-treated cells were pelleted by centrifugation and
rinsed with PBS. Whole cell lysates were prepared in ice-cold lysis
buffer (10 mM Hepes, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.4% NP-40
with 1 mM DTT, 0.5 mM PMSF, 1 mM NaF and 1 mM Complete protease
inhibitor mix) by pipetting up and down (without bubbling) ~10
times. After incubation on ice for 10 min, the lysates were
centrifuged at 15,000 × g for 1 min. The supernatants were
transferred to fresh tubes and referred to as cytoplasmic
extracts.
Colony formation assay
HCT-15P and HCT-15R cells were seeded in a 6-well
plate, with 500 cells/well. The cells were treated with either
different concentration of GA or 0.1% DMSO (vehicle control) and
cultured in an atmosphere of 5% CO2 at 37°C for the
indicated times. Medium was changed every 3 days. The cells were
washed with PBS and fixed in ice-cold methanol for 5 min, and
stained with crystal violet. Images of the colonies were obtained
using an Epson scanner. Each treatment was evaluated in
triplicates, and representative images are shown.
Statistical analysis
All experiments were performed at least 3 times, and
results are expressed as mean ± SD where applicable. Statistical
analysis was performed by one-way analysis of variance followed by
Tukey's test by GraphPad Prism software (San Diego, CA, USA).
P<0.05 was considered statistically significant.
Results
Establishment and characterization of the
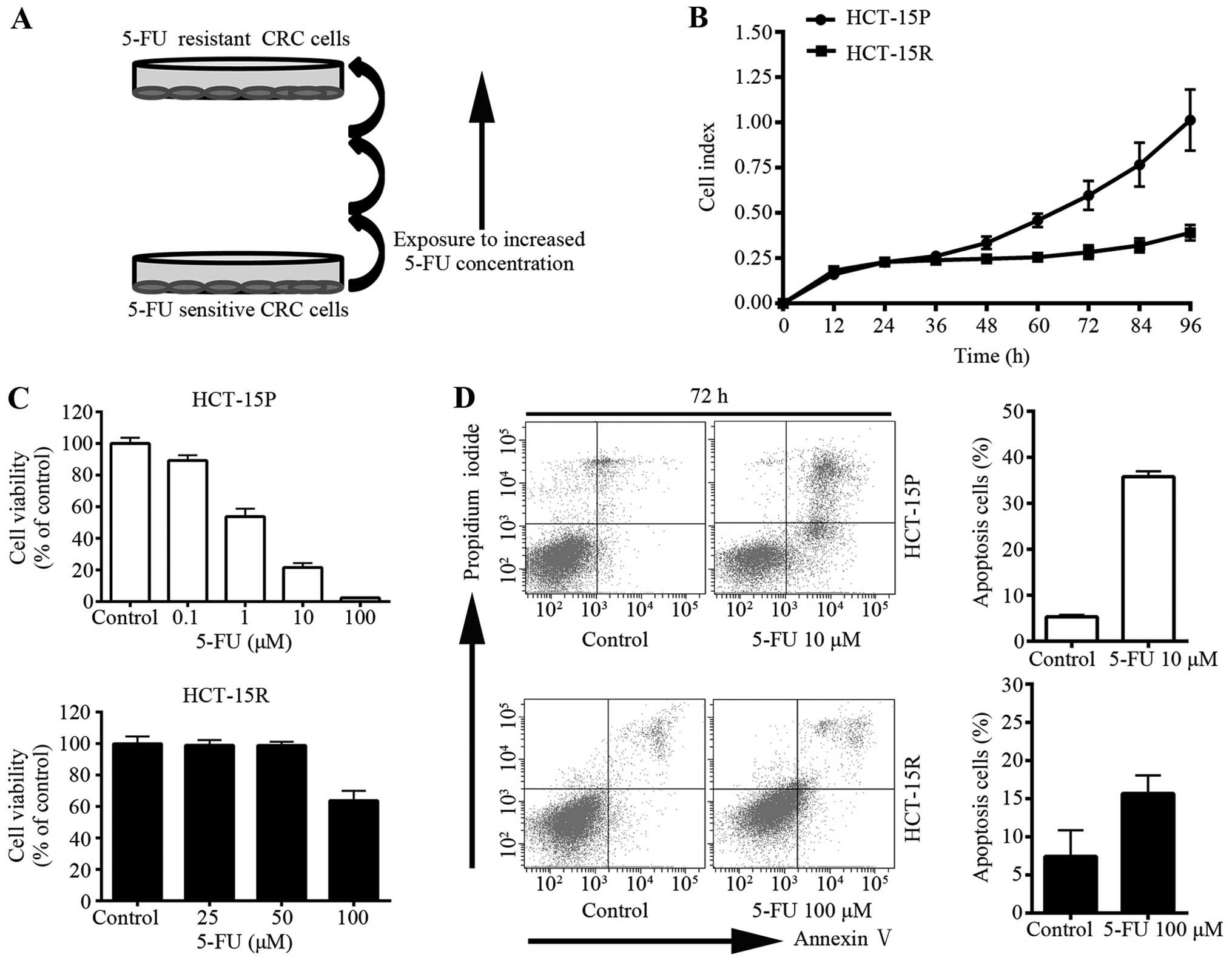
5-FU resistant CRC cells
To explore the anticancer effect of GA on 5-FU
resistant cells, the CRC cell line HCT-15 was subjected to
successively increasing concentrations of 5-FU (Fig. 1A) to establish 5-FU resistant cells
(HCT-15R). HCT-15R cells in culture grew more slowly than
drug-sensitive parental cells (HCT-15P) as shown in Fig. 1B. In order to identify the
establishment of resistance to 5-FU, the sensitivities of the
HCT-15P cells and the HCT-15R cells were compared. Cell viability
was measured using the MTS assay. HCT-15R cells were not as
sensitive as HCT-15P to 5-FU (Fig.
1C). Additionally, the anti-apoptotic activity was measured
using flow cytometry with Annexin V-FITC/PI dual staining (Fig. 1D). After being treated with
different concentrations of 5-FU (HCT-15P cells were treated with
10 μM 5-FU while HCT-15R cells were treated with 100 μM), there was
increased apoptosis in both cell lines; however, HCT-15R cells were
treated with 10 times the concentration of 5-FU and the apoptotic
rate of HCT-15R was much lower than that of HCT-15P, indicating
that HCT-15R showed more resistance to 5-FU.
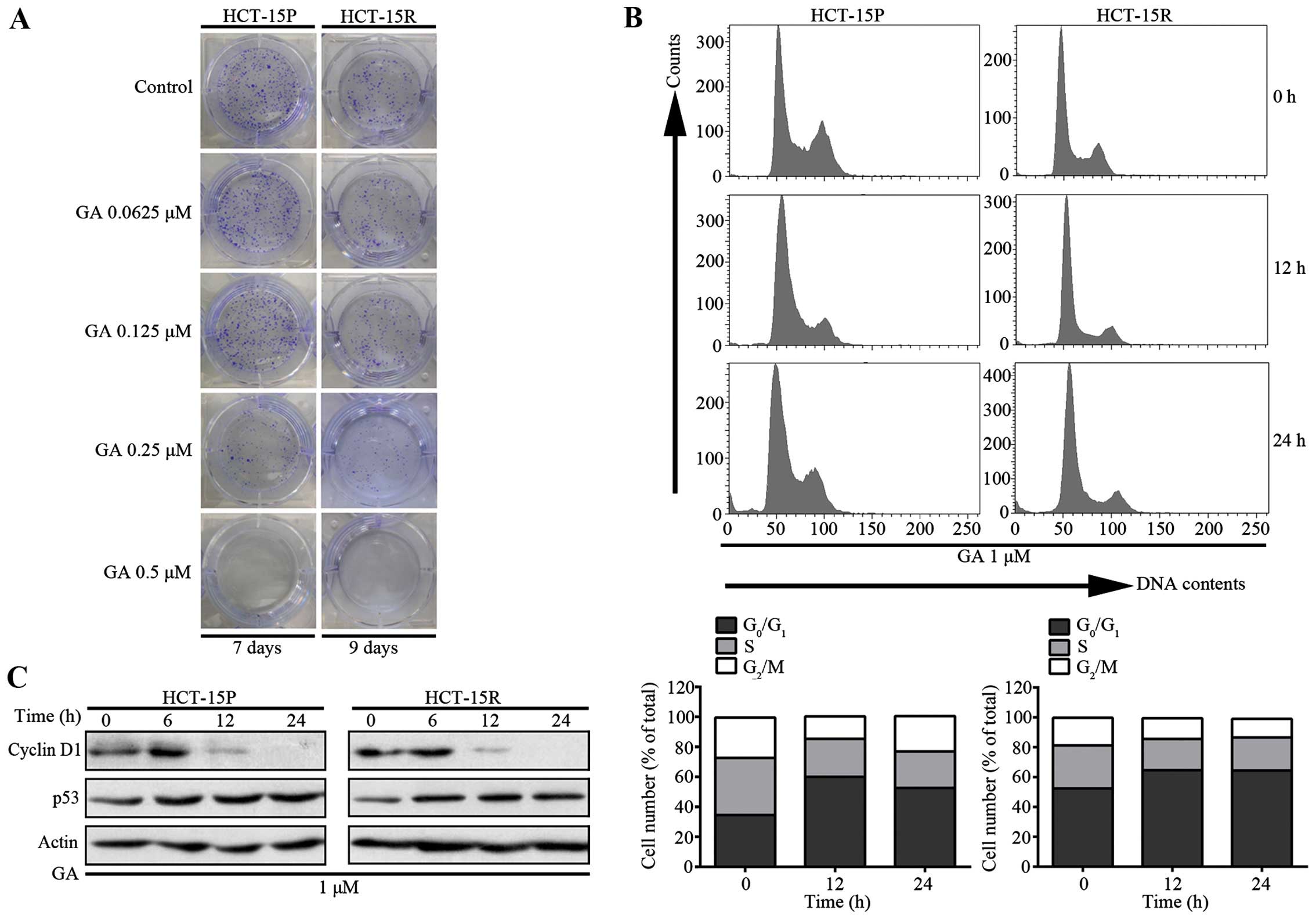
GA effects on the proliferation and cell
cycle arrest of both 5-FU sensitive and resistant cells
To investigate the anti-cancer effect of GA on both
5-FU sensitive and resistant CRC cells, we performed a colony
formation assay. As shown in Fig.
2A, treatment of HCT-15P and HCT-15R cells with GA at
concentrations from 0.0625 to 0.5 μM for 7 days and 9 days,
respectively, resulted in fewer and smaller colonies than those of
the control group. In addition, flow cytometry analysis was used to
determine the effect of GA on cell cycle distribution. After
exposing CRC cells to 2 μM GA, cells were fixed, stained and
analyzed by flow cytometry. As shown in Fig. 2B, there was accumulation of cells
at the G1 phase, which meant GA caused G1
arrest in CRC cells. To further explore the mechanism of GA-induced
cell cycle arrest, we investigated the expression of cyclin D1 and
p53 proteins, which control the G1 checkpoint (12,13).
The results presented in Fig. 2C
demonstrate that the addition of GA decreased the level of cyclin
D1 proteins and increased the level of p53, which also confirmed
that GA caused G1 arrest in CRC cells.
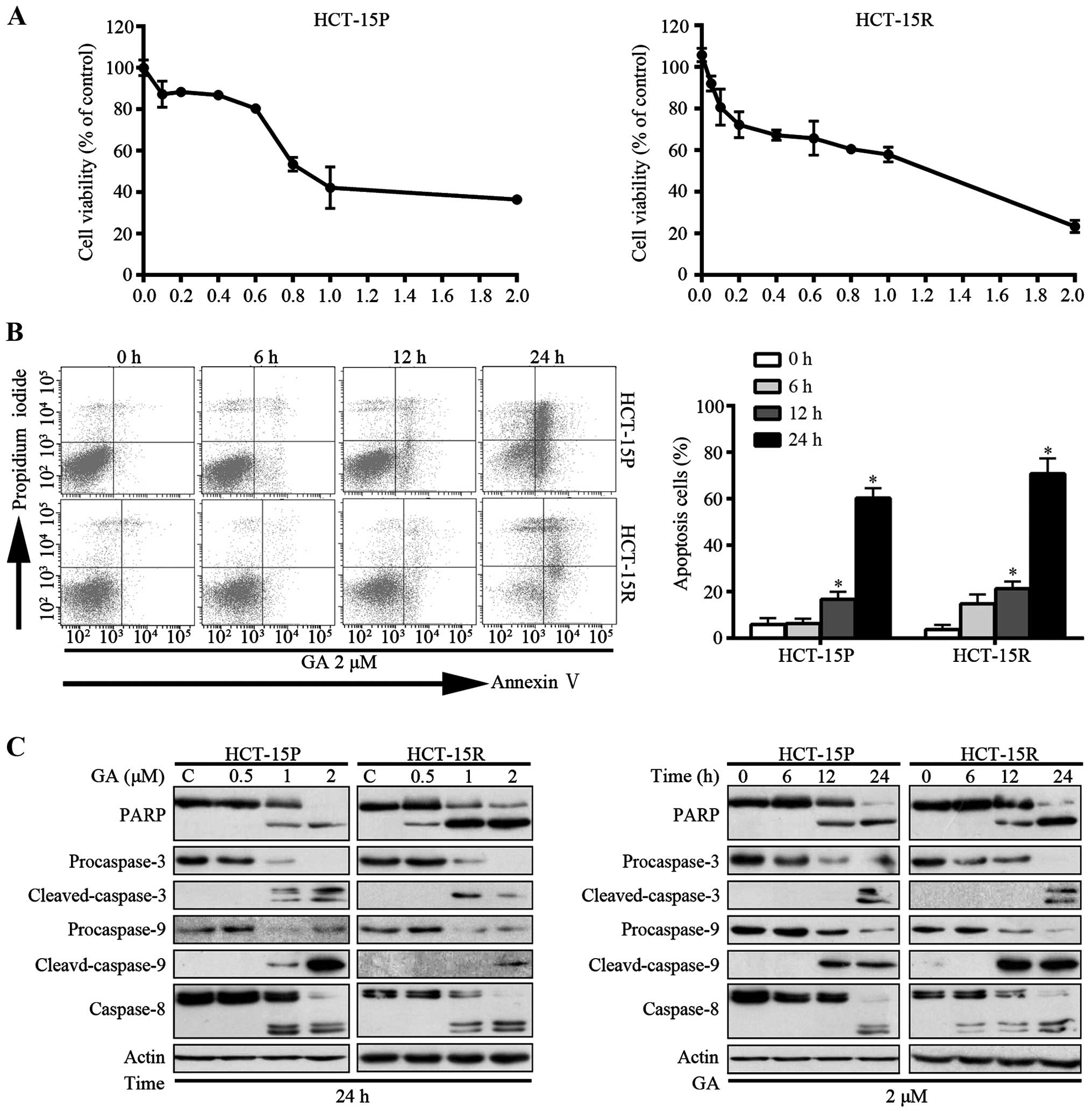
GA-induced apoptosis is associated with
caspase activation in both HCT-15P and HCT-15R cells
Since deregulated proliferation and inhibition of
apoptosis lie at the heart of all tumor development, they are the
targets for therapeutic intervention in all cancers (14). We examined the ability of GA to
induce cell death in HCT-15P and HCT-15R cell lines. Initially, the
effect of GA on cell viability was measured by MTS assay. As shown
in Fig. 3A, GA decreased the cell
viability in a dose-dependent manner, with IC50 values
of 1.08 and 0.87 μM, respectively. Additionally, the capacity of GA
to induce cell apoptosis was measured by flow cytometry with
Annexin V-FITC/PI dual staining. The proportion of apoptotic cells,
in a time-dependent manner, increased in both HCT-15P and HCT-15R
cells (Fig. 3B). To further verify
the induction of apoptosis, apoptosis-associated proteins were
measured by western blot assay. As shown in Fig. 3C, GA induced the cleavage of PARP
(an indicator of apoptosis) in both dose- and time-dependent manner
in both cell lines. Consistently, the levels of the precursor forms
of caspases-3, -8 and -9 were decreased and that of the active
forms of caspases-3, -8 and -9 were increased after GA treatment,
matching the pattern of PARP cleavage, demonstrating that GA
triggers CRC cell apoptosis via caspase activation.
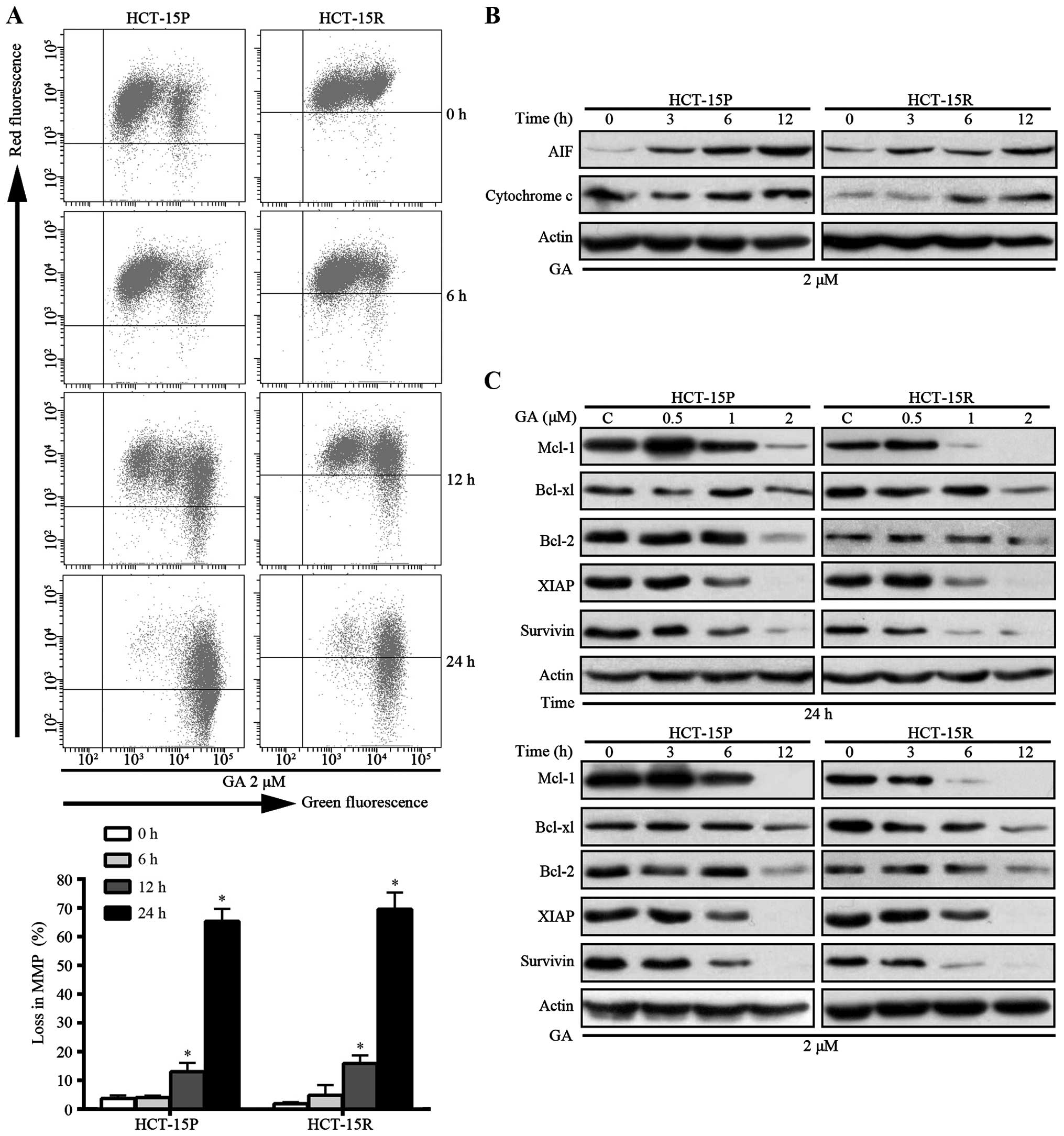
GA-induced apoptosis is associated with
the loss of mitochondrial membrane potential (MMP) and decreased
expression of anti-apoptotic proteins in HCT-15P and HCT-15R
cells
Mitochondria are well known to have a central role
in apoptosis, which is involved in a variety of key invents,
including loss of MMP, mitochondrial swelling and release of
mitochondria proteins such as cytochrome c and AIF from
mitochondria to cytosol and/or the nucleus, which are recognized as
indicators of the early stage of apoptosis (15). Since loss of MMP is a crucial step
and subsequently triggers the release of mitochondria proteins.
First, we measured the loss of MMP in GA treatment CRC cells. As
shown in Fig. 4A, Both HCT-15P and
HCT-15R cells treated with 2 μM GA exhibited an increased green
fluorescence signal and a decreased red fluorescence signal in a
time-dependent manner. The percentage for loss of MMP increased to
65.37 and 69.57% in HCT-15P and HCT-15R cells, respectively, with
GA in 24 h (Fig. 4A).
Subsequently, the levels of cytosolic cytochrome c and AIF
were detected by western blot assay. As shown in Fig. 4B, after GA treatment, the levels of
mitochondrial cytochrome c and AIF increased in a
time-dependent manner in both cell lines. The release of cytochrome
c and other apoptotic proteins from mitochondria are known
to be regulated by the Bcl-2 family of proteins (16). Therefore, the expression of Bcl-2,
Bcl-xl and other anti-apoptotic proteins were measured. As shown in
Fig. 4C, GA decreased the level of
anti-apoptotic proteins Bcl-2, Bcl-xl, Mcl-1, XIAP and survivin in
both HCT-15P and HCT-15R cells in a dose- and time-dependent
manner. These results demonstrated that GA-induced apoptosis is
associated with loss of MMP and decreasing of anti-apoptotic
proteins in both HCT-15P and HCT-15R cells.
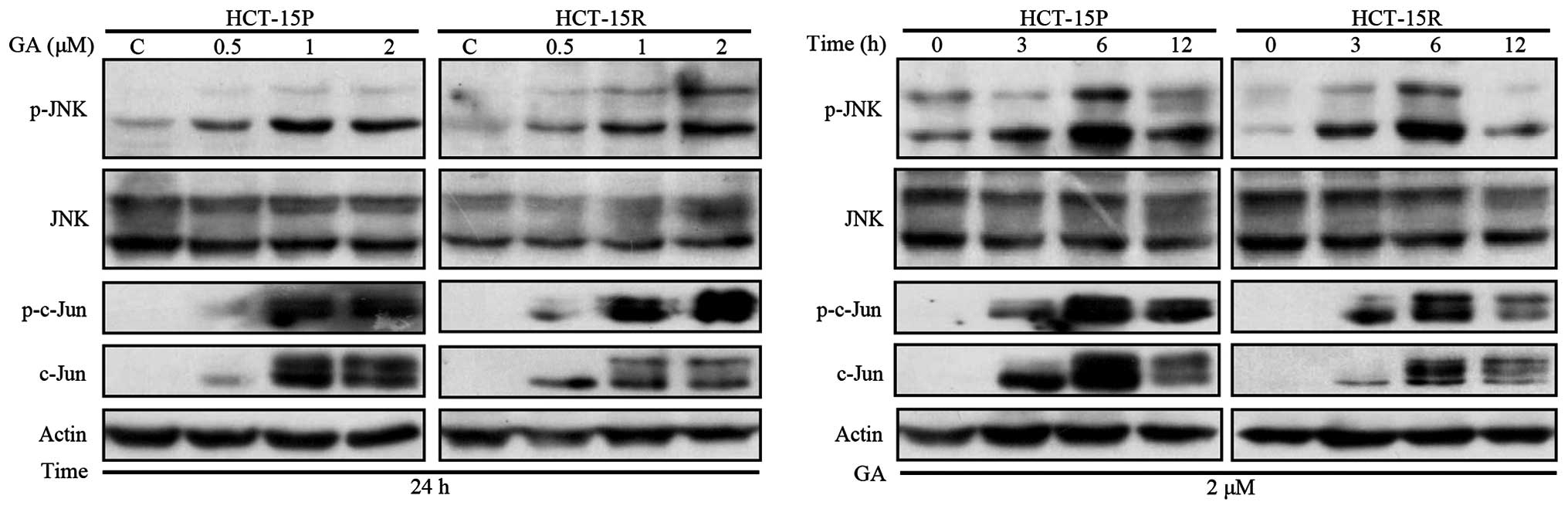
GA-induced apoptosis is associated with
activation of JNK signaling pathway in HCT-15P and HCT-15R
cells
JNK activation can lead to cytotoxic effect in
cancer cells. Therefore, we examined the effect of GA on the
expression of this signaling pathway. The level of phosphorylated
JNK (p-JNK) and total JNK was examined by western blot assay. As
shown in Fig. 5, GA treatment of
HCT-15P and HCT-15R cells resulted in activation of JNK, in a dose-
and time-dependent manner. In addition, c-Jun expression level was
elevated in parallel with increased c-Jun phosphorylation in GA
treated HCT-15P and HCT-15R cells. To identify whether the JNK
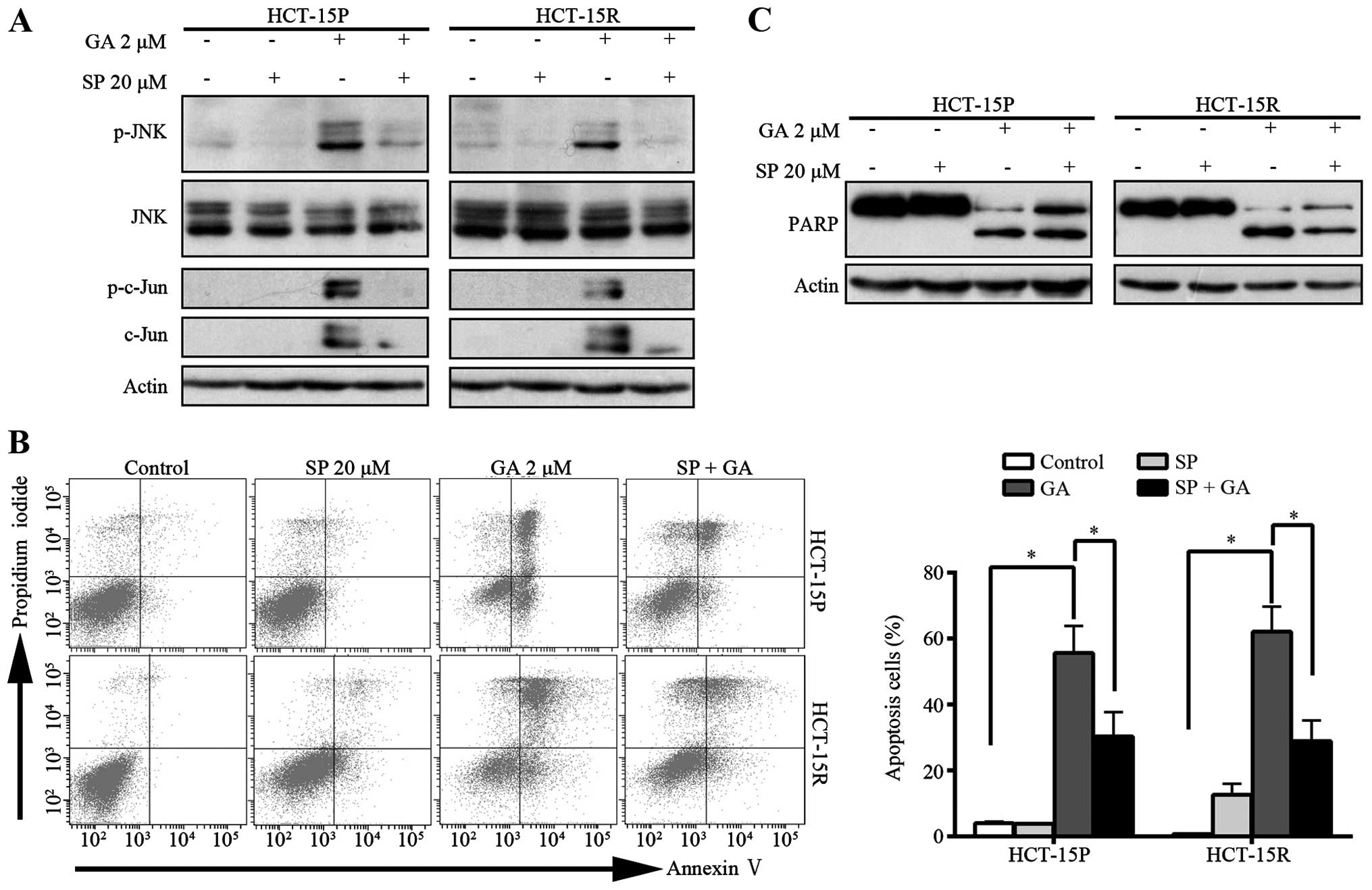
signaling pathway was involved in GA-mediated apoptosis, SP600125,
a general inhibitor of JNK, was pre-treated with GA in HCT-15P and
HCT-15R cells and cell apoptosis was then determined using flow
cytometry with Annexin V-FITC/PI dual staining. The cleavage of
PARP also was detected by western blot assay. Pre-treatment with
JNK inhibitor SP600125 completely blocked GA-induced
phosphorylation of JNK and c-Jun (Fig.
6A). As shown in Fig. 6B,
SP600125 pretreatment rescued the GA-induced apoptosis. The
percentage of apoptotic cells decreased from 55.63 and 62.13 to
30.27 and 28.86% in HCT-15P and HCT-15R cells, respectively.
Western blot assay showed that SP600125 inhibited the cleavage of
PARP induced by GA (Fig. 6C),
further confirming SP600125 can rescue the GA-induced apoptosis.
Together, these results demonstrated that GA induced both HCT-15P
and HCT-15R cell apoptosis via JNK activation.
Discussion
CRC is one of the most prevalent malignancies with
high morbidity and mortality worldwide. Metastatic colorectal
cancer (mCRC) at diagnosis is found in ~25% of patients with
colorectal cancer and almost 50% of patients will develop
metastasis (17). For decades, the
only treatment option available for mCRC patients was 5-FU. In
recent years, due to new approaches in the treatment of mCRC,
involving chemotherapy regimens including bevacizumab, cetuximab or
panitumumab, response rates and overall survival has significantly
improved (18). However, 5-FU is
still widely used in the treatment of various types of cancer,
including colorectal (19), breast
(20) and pancreatic cancer
(21). The mechanism of 5-FU
resistance still remains largely unknown despite extensive
investigations. Great efforts have been made to develop novel
chemotherapeutic strategies for mCRC patients and to overcome
chemotherapy resistance. Multiple factors could be causing
chemotherapy resistance such as the alteration of drug transport,
enhancement of the drug detoxification system, modification of DNA
damage tolerance mechanisms and disruption of apoptotic cell death
pathways. Among them, inhibition of apoptosis is considered as the
most important factor of 5-FU resistance (22).
GA, a candidate drug, has been approved by CFDA for
phase II clinical trial in solid tumor therapy (6). Previous studies showed that GA could
induce apoptosis in a broad range of human cancer cells (3,8,23–25).
Thus, it is possible that GA could overcome 5-FU resistant in CRC
by promoting apoptosis. To explore the potential capacity of GA to
overcome 5-FU resistance in CRC cells, we established the 5-FU
resistant colorectal cancer cell line HCT-15R, displaying stable
biological characteristics and resistance to 5-FU. We reasoned that
GA has an antitumor effect on 5-FU sensitive and 5-FU resistant
cells. To test this hypothesis, we performed a series of
cytotoxicity experiments on both 5-FU sensitive and 5-FU resistant
cells. Our results demonstrated that GA, inhibited cell
proliferation and caused G1 arrest in both 5-FU
sensitive and 5-FU resistant cells. Additionally, it induced
apoptosis in both 5-FU sensitive and resistant CRC cells. Western
blot analyses showed that GA induced caspase-dependent apoptosis.
The above data demonstrate that GA might become an ideal agent for
overcoming 5-FU resistance.
GA was reported to induce apoptosis through both
death receptor apoptotic pathway and mitochondrial apoptotic
pathway in hepatocellular carcinoma cells (26). On the other hand, GA was reported
to induce apoptosis via a mitochondrial pathway in breast cancer
cells (8) and mantle cell lymphoma
cells (27). Conversely, other
reports have shown the viability of an inhibition effect of GA via
mitochondria-independent apoptotic pathway on osteosarcoma cells
(28). In the present study, we
found that GA triggered the loss of MMP and subsequently released
cytochrome c and AIF to the cytosol. Cytochrome c
releasing from mitochondria can activate caspase-9, which in turn
activates executioner caspase-3 via cleavage induction, and PARP,
an important substrate of caspase-3 was cleaved. Thus our results
suggest that GA could, at least partially, cause apoptosis through
a mitochondrial pathway in both 5-FU sensitive and 5-FU resistant
cells.
MAPKs families play an important role in complex
cellular programs such as proliferation, differentiation,
development, transformation and apoptosis (28). In mammalian cells, the MAPK family
includes ERKs (classical MAPK), JNKs and p38 MAPKs. Usually, ERKs
are activated by mitogens (29).
In addition, JNKs and p38 MAPKs are most potently activated by
cytokines, a variety of chemical and radiant stress and in response
to cellular stress (30–32). GA was reported to induce cancer
cell apoptosis through p38 MAPK activation in human breast cancer
MCF-7 cells (33),
Doxorubicin-resistant MCF-7/ADR cells (34) and in hepatocellular carcinoma
SMMC-7721 cells (25). Evidence
that JNK signaling to c-Jun contributes to stress-induced cell
apoptosis have come from studies in neuron cells (32). The activation of JNK pathway
impairs survival of colorectal cancer cells by promoting cell
apoptosis (35). However, whether
or not the JNK signaling pathway plays similar roles in GA-mediated
apoptosis in colorectal cancer cells, especially in drug resistant
colorectal cancer cells, is still unclear. Therefore, we
investigated JNK signaling pathway after GA treatment. As the
activation of JNK requires phosphorylation (p-JNK), it was observed
that treatment with GA markedly promoted the expression of p-JNK.
Several studies indicated that c-Jun is a specific target of JNK
(32). On one hand, a variety of
cellular stresses such as oxidative stress can strongly activate
the JNKs, which inhibit c-Jun ubiquitination and promote the c-Jun
activation through c-Jun phosphorylation. On the other hand,
because the transcription factor c-Jun can regulate itself,
expression via binding to its own promoter as a c-Jun/ATF-2 dimer,
specific phosphorylation of c-Jun by JNK could lead to increased
expression of c-Jun (36).
Consistently, the active form of c-Jun (p-c-Jun) and total c-Jun
protein were remarkably increased dose- and time-dependently.
Furthermore, our results also showed that the inhibition with
SP600125, a JNK inhibitor, partially inhibited apoptosis induced by
GA, suggesting the involvement of JNK on the GA effect.
Downregulation of JNK activation by SP600125, a JNK inhibitor,
decreased GA-induced apoptosis, indicating that GA required the
activation of JNK signaling pathways in both 5-FU sensitive and
resistant cells in order to trigger the apoptotic pathway. Several
studies in other cancers, such as cervical (23), breast cancer (33) and hepatic carcinoma (25), reported that inhibition of either
JNK or p38 pathway led to inhibition of GA-induced apoptosis. Our
results, together with these reports, highlight the important role
of JNK signaling pathway in GA-induced apoptosis.
In conclusion, our results demonstrate that GA can
directly inhibit proliferation and induce apoptosis in both 5-FU
sensitive and 5-FU-resistant colorectal cancer cells and induce
apoptosis via activating JNK signaling pathway. Therefore, data
presented here suggest an alternative strategy to overcome 5-FU
resistance in CRC, highlighting the importance of targeting JNK
signaling pathways in anticancer strategies and their modulation by
GA in CRC cells. Furthermore, we show the potential of GA to be
used in CRC chemotherapy.
Acknowledgements
This study was supported by the Guangdong Provincial
Department of Science and Technology (2012B050500004;
2011B090400559); the Guangdong Innovative Research Team
Program(2009010058); the Science and Information Technology Bureau
of Guangzhou, Guangdong (2011J5200009); the Overseas Excellent
Professor Project, Ministry of Education, China; and the Japan
Ministry of Education, Culture, Sports, Science and Technology
(MEXT) for Program of Japan Initiative for Global Research Network
on Infectious Diseases (J-GRID).
References
|
1
|
Akasaka T, Tsujii M, Kondo J, Hayashi Y,
Ying J, Lu Y, Kato M, Yamada T, Yamamoto S, Inoue T, et al: 5-FU
resistance abrogates the amplified cytotoxic effects induced by
inhibiting checkpoint kinase 1 in p53-mutated colon cancer cells.
Int J Oncol. 46:63–70. 2015.
|
|
2
|
Qi Q, You Q, Gu H, Zhao L, Liu W, Lu N and
Guo Q: Studies on the toxicity of gambogic acid in rats. J
Ethnopharmacol. 117:433–438. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi X, Chen X, Li X, Lan X, Zhao C, Liu S,
Huang H, Liu N, Liao S, Song W, et al: Gambogic acid induces
apoptosis in imatinib-resistant chronic myeloid leukemia cells via
inducing proteasome inhibition and caspase-dependent Bcr-Abl
down-regulation. Clin Cancer Res. 20:151–163. 2014. View Article : Google Scholar :
|
|
4
|
Chuah LO, Yeap SK, Ho WY, Beh BK and
Alitheen NB: In vitro and in vivo toxicity of garcinia or
hydroxycitric Acid: A review. Evid Based Complement Alternat Med.
2012:1979.202012.
|
|
5
|
Márquez F, Babio N, Bulló M and
Salas-Salvadó J: Evaluation of the safety and efficacy of
hydroxycitric acid or Garcinia cambogia extracts in humans. Crit
Rev Food Sci Nutr. 52:585–594. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chi Y, Zhan XK, Yu H, Xie GR, Wang ZZ,
Xiao W, Wang YG, Xiong FX, Hu JF, Yang L, et al: An open-labeled,
randomized, multicenter phase IIa study of gambogic acid injection
for advanced malignant tumors. Chin Med J (Engl). 126:1642–1646.
2013.
|
|
7
|
Wang LH, Yang JY, Yang SN, Li Y, Ping GF,
Hou Y, Cui W, Wang ZZ, Xiao W and Wu CF: Suppression of NF-κB
signaling and P-glycoprotein function by gambogic acid
synergistically potentiates adriamycin-induced apoptosis in lung
cancer. Curr Cancer Drug Targets. 14:91–103. 2014. View Article : Google Scholar
|
|
8
|
Li C, Qi Q, Lu N, Dai Q, Li F, Wang X, You
Q and Guo Q: Gambogic acid promotes apoptosis and resistance to
metastatic potential in MDA-MB-231 human breast carcinoma cells.
Biochem Cell Biol. 90:718–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li X, Liu S, Huang H, Liu N, Zhao C, Liao
S, Yang C, Liu Y, Zhao C, Li S, et al: Gambogic acid is a
tissue-specific proteasome inhibitor in vitro and in vivo. Cell
Rep. 3:211–222. 2013. View Article : Google Scholar
|
|
10
|
Yi T, Yi Z, Cho SG, Luo J, Pandey MK,
Aggarwal BB and Liu M: Gambogic acid inhibits angiogenesis and
prostate tumor growth by suppressing vascular endothelial growth
factor receptor 2 signaling. Cancer Res. 68:1843–1850. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song M, Chen D, Lu B, Wang C, Zhang J,
Huang L, Wang X, Timmons CL, Hu J, Liu B, et al: PTEN loss
increases PD-L1 protein expression and affects the correlation
between PD-L1 expression and clinical parameters in colorectal
cancer. PLoS One. 8:e658212013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shapiro GI and Harper JW: Anticancer drug
targets: Cell cycle and checkpoint control. J Clin Invest.
104:1645–1653. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pines J: Four-dimensional control of the
cell cycle. Nat Cell Biol. 1:E73–E79. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang X: The expanding role of mitochondria
in apoptosis. Genes Dev. 15:2922–2933. 2001.PubMed/NCBI
|
|
16
|
Tsujimoto Y: Role of Bcl-2 family proteins
in apoptosis: Apoptosomes or mitochondria? Genes Cells. 3:697–707.
1998. View Article : Google Scholar
|
|
17
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar
|
|
18
|
Temraz S, Mukherji D, Alameddine R and
Shamseddine A: Methods of overcoming treatment resistance in
colorectal cancer. Crit Rev Oncol Hematol. 89:217–230. 2014.
View Article : Google Scholar
|
|
19
|
Gustavsson B, Carlsson G, Machover D,
Petrelli N, Roth A, Schmoll HJ, Tveit KM and Gibson F: A review of
the evolution of systemic chemotherapy in the management of
colorectal cancer. Clin Colorectal Cancer. 14:1–10. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gandhi S, Fletcher GG, Eisen A, Mates M,
Freedman OC, Dent SF and Trudeau ME: Adjuvant chemotherapy for
early female breast cancer: A systematic review of the evidence for
the 2014 Cancer Care Ontario systemic therapy guideline. Curr
Oncol. 22(Suppl 1): S82–S94. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thota R, Pauff JM and Berlin JD: Treatment
of metastatic pancreatic adenocarcinoma: A review. Oncology
(Williston Park). 28:70–74. 2014.
|
|
22
|
Johnstone RW, Ruefli AA and Lowe SW:
Apoptosis: A link between cancer genetics and chemotherapy. Cell.
108:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Krajarng A, Imoto M, Tashiro E, Fujimaki
T, Shinjo S and Watanapokasin R: Apoptosis induction associated
with the ER stress response through up-regulation of JNK in HeLa
cells by gambogic acid. BMC Complement Altern Med. 15:262015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao W, Zhou SF, Zhang ZP, Xu GP, Li XB
and Yan JL: Gambogic acid inhibits the growth of osteosarcoma cells
in vitro by inducing apoptosis and cell cycle arrest. Oncol Rep.
25:1289–1295. 2011.PubMed/NCBI
|
|
25
|
Nie F, Zhang X, Qi Q, Yang L, Yang Y, Liu
W, Lu N, Wu Z, You Q and Guo Q: Reactive oxygen species
accumulation contributes to gambogic acid-induced apoptosis in
human hepatoma SMMC-7721 cells. Toxicology. 260:60–67. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee PN and Ho WS: Antiproliferative
activity of gambogic acid isolated from Garcinia hanburyi in Hep3B
and Huh7 cancer cells. Oncol Rep. 29:1744–1750. 2013.PubMed/NCBI
|
|
27
|
Xu J, Zhou M, Ouyang J, Wang J, Zhang Q,
Xu Y, Xu Y, Zhang Q, Xu X and Zeng H: Gambogic acid induces
mitochondria-dependent apoptosis by modulation of Bcl-2 and Bax in
mantle cell lymphoma JeKo-1 cells. Chin J Cancer Res. 25:183–191.
2013.PubMed/NCBI
|
|
28
|
Zhao W, You CC, Zhuang JP, Zu JN, Chi ZY,
Xu GP and Yan JL: Viability inhibition effect of gambogic acid
combined with cisplatin on osteosarcoma cells via
mitochondria-independent apoptotic pathway. Mol Cell Biochem.
382:243–252. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fang JY and Richardson BC: The MAPK
signalling pathways and colorectal cancer. Lancet Oncol. 6:322–327.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zarubin T and Han J: Activation and
signaling of the p38 MAP kinase pathway. Cell Res. 15:11–18. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dunn C, Wiltshire C, MacLaren A and
Gillespie DA: Molecular mechanism and biological functions of c-Jun
N-terminal kinase signalling via the c-Jun transcription factor.
Cell Signal. 14:585–593. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen J, Gu HY, Lu N, Yang Y, Liu W, Qi Q,
Rong JJ, Wang XT, You QD and Guo QL: Microtubule depolymerization
and phosphorylation of c-Jun N-terminal kinase-1 and p38 were
involved in gambogic acid induced cell cycle arrest and apoptosis
in human breast carcinoma MCF-7 cells. Life Sci. 83:103–109. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang S, Wang L, Chen M and Wang Y:
Gambogic acid sensitizes resistant breast cancer cells to
doxorubicin through inhibiting P-glycoprotein and suppressing
survivin expression. Chem Biol Interact. 235:76–84. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Favata MF, Horiuchi KY, Manos EJ, Daulerio
AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F,
et al: Identification of a novel inhibitor of mitogen-activated
protein kinase kinase. J Biol Chem. 273:18623–18632. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Raivich G, Bohatschek M, Da Costa C, Iwata
O, Galiano M, Hristova M, Nateri AS, Makwana M, Riera-Sans L,
Wolfer DP, et al: The AP-1 transcription factor c-Jun is required
for efficient axonal regeneration. Neuron. 43:57–67. 2004.
View Article : Google Scholar : PubMed/NCBI
|