Introduction
Osteosarcoma derives from primitive bone-forming
mesenchymal cells and is the most common primary bone malignancy.
The incidence (and 95% confidence intervals) of osteosarcoma for
all races and both sexes is 4.0% (3.5–4.6) among those aged 0–14
years and 5.0% (4.6–5.6) among those aged 0–19 years per year
(1). Therapy for osteosarcoma has
made good progress with the development of surgery and screening
technologies and with the combination of neoadjuvant chemotherapy.
However, the problems of metastasis, recurrence and chemoresistance
have yet to be solved. In recent years, drugs that target specific
molecules have been developed as treatments for human malignancies
(2). These drugs often selectively
inhibit specific molecules, such as growth factor receptors or
intracellular signaling proteins that are related to tumor
proliferation, migration and/or metastasis (3).
Mammalian target of rapamycin (mTOR) is an essential
serine/threonine kinase that belongs to the phosphoinositide-3-OH
kinase (PI3K)-related kinase family (4). It regulates multiple cellular
processes, including survival, cell growth, proliferation,
migration and angiogenesis, in many kinds of cancers (5,6). The
inhibition of mTOR affects mTOR pathway-mediated cellular
transcription and translation, leading to cell cycle arrest and
anti-angiogenesis. Regarding the antitumor mechanisms of mTOR
inhibitors, several studies have indicated that inhibitors of Akt
and its downstream target, mTOR signaling, have anticancer effects
as the inhibition of the mTOR pathway contributes to the initiation
of autophagy (7–9).
mTOR nucleates two complexes: mTOR complex 1
(mTORC1) and mTOR complex 2 (mTORC2). These complexes are key and
essential regulators of cellular pathways that control the
initiation of mRNA translation and ribosome biogenesis and exhibit
important monitoring effects on cell metabolism, lipolysis and
autophagy (10–12). Autophagy is activated during
starvation to provide an alternative energy source through
self-digestion. Thus, autophagy serves as a temporary survival
mechanism. Autophagy is also important in the induction of tumor
cell death (13) and excessive
autophagy triggers autophagic cell death in tumors (14,15).
However, it remains under debate whether chemotherapy-induced
autophagy in tumor cells is a protective response or is invoked to
promote cell death (16). Not
surprisingly, there is an intricate relationship between autophagy
and apoptosis. Recent studies indicate that autophagy can function
as a self-defense mechanism in cells that are exposed to antitumor
agents, and that blocking autophagy can trigger the activation of
apoptosis (17–19).
Recently, a potent small molecule inhibitor of
autophagy named Spautin-1 (specific and potent autophagy
inhibitor-1) was discovered. This molecule promotes the degradation
of Vps34-PI3 kinase complexes by inhibiting two
ubiquitin-specific peptidases, USP10 and USP13, that target the
Beclin-1 subunit of Vps34 complexes. USP10 mediates the
deubiquitination of p53, and as Beclin-1 regulates the
deubiquitination activity of USP10 and USP13, Beclin-1 can control
the levels of p53. Through this mechanism, Spautin-1 increases
cancer cell death under the condition of nutrient deprivation when
autophagy would normally act as a survival mechanism in the
metabolically stressed cells (20).
The aim of this study was to examine the effects of
the mTOR inhibitor rapamycin on osteosarcoma cells. We investigated
whether rapamycin modulated the phosphorylation of proteins in the
Akt/mTOR signaling pathway and/or induced autophagy in osteosarcoma
cells. In addition, we hypothesized that the combination of
rapamycin and Spautin-1 would induce strong antitumor effects in
osteosarcoma cells by blocking self-defensive autophagy.
Materials and methods
Chemical reagents
Rapamycin (CCI-779) and the autophagy inhibitor,
Spautin-1, were purchased from Calbiochem (San Diego, CA, USA).
Both reagents were dissolved in dimethyl sulfoxide (DMSO) and
rapamycin was stored at −20°C and Spautin-1 was stored at 8°C.
Chloroquine diphosphate salt (CQ) was purchased from Sigma-Aldrich
(St. Louis, MO, USA) and dissolved in distilled water to a
concentration of 100 mM, and was stored at room temperature.
Cell lines and cell culture
The MG63 cell line, derived from an osteogenic
sarcoma, was used in this study. The cells were grown in Dulbecco's
modified Eagle's medium (DMEM; Sigma-Aldrich) containing 10% fetal
bovine serum (FBS; Sigma-Aldrich) and 100 U/ml penicillin, and were
routinely maintained at 37°C in a humidified 5% CO2
atmosphere. Cultures at the mid-log phase were used for the
experiments.
For the detection of autophagy and the apoptosis
assay, cells were divided into four groups: control (no inhibitor),
Rap (rapamycin), Spa (Spautin-1), and Rap-plus-Spa (rapamycin +
Spautin-1) groups.
In vitro proliferation assay
Cell proliferation was determined by the CellTiter
96® AQueous One Solution Cell Proliferation assay
(Promega Corp., Madison, WI, USA). Briefly, cells were trypsinized
and seeded at a density of ~1×104 cells/well in 96-well
cell culture plates containing 200 μl/well of culture medium with
10% FBS. After 48 h, the medium was replaced with medium containing
10% FBS and supplemented with rapamycin at a concentration of 0,
0.4, 2, 10, or 50 μM. After 24 or 48 h, the medium was replaced
with fresh medium containing
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) reagent (100 μl of medium plus 20 μl of MTS regent/well). In
the experiments testing the combined effect of rapamycin and
Spautin-1, cells were treated with 20 μM rapamycin and 100 μM
Spautin-1 for 24 h. In the experiments testing the effect of
rapamycin or Spautin-1 alone, cells were treated with either 20 μM
rapamycin or 100 μM Spautin-1 for 24 h. The optical density of the
samples was measured at 490 nm using an automatic microplate reader
(SpectraMax® Plus384 microplate reader; Molecular
Devices, Sunnyvale, CA, USA) after 2 h of further incubation with
MTS reagent. Absorbance was directly proportional to the number of
living cells. The percent viability of the cells in each well was
calculated. At least three independent experiments were performed
for each condition.
Western blot analyses
Cells were trypsinized and seeded at a density of
~6×105 cells/well in 6-well cell culture plates with 2
ml/well of culture medium containing 10% FBS. After 48 h, cells
were treated with medium containing 10% FBS and rapamycin at a
concentration of 0, 0.4, 2, 10, or 50 μM for 24 h. In the
experiments testing the combined effect of rapamycin and Spautin-1,
the cells were treated with 20 μM rapamycin and 100 μM Spautin-1
for 24 h. In the experiments testing the effect of rapamycin or
Spautin-1 alone, the cells were treated with either 20 μM rapamycin
or 100 μM Spautin-1 for 24 h. Following treatment, the culture
medium was replaced with lysis buffer (Cell Signaling Technology,
Beverly, MA, USA) and the cells were lysed on ice for 20 min. The
cell lysates were spun at 15,000 × g using a Tabletop Micro
Refrigerated Centrifuge 3500 (Kubota Shoji Co., Ltd., Tokyo, Japan)
at 4°C for 30 min. The supernatant was then collected as the total
cellular protein extract. The protein concentrations were
determined using the Protein Assay Bicinchoninate kit (Nacalai
Tesque, Inc., Kyoto, Japan) and standardized using bovine serum
albumin. The total cellular protein samples were loaded onto an SDS
polyacrylamide gel (10 or 12.5% commercial precast gels; Wako,
Tokyo, Japan), and the proteins were separated by SDS-PAGE under
reducing conditions. The separated proteins were
electrophoretically transferred onto nitrocellulose membranes (GE
Healthcare Bio-Sciences, Piscataway, NJ, USA). The membranes were
blocked for 90 min in blocking buffer that contained Tris-buffered
saline (TBS-T) and EzBlock Chemi (Atto Co., Tokyo, Japan). They
were then incubated overnight at 4°C with the appropriate primary
antibody (Table I) diluted in
blocking buffer. The specific horseradish peroxidase-conjugated
secondary antibody incubation was performed overnight at 4°C with
gentle agitation. Bound secondary antibodies were detected using
the ECL Plus Western Blotting Detection system (GE Healthcare
Bio-Sciences) and an LAS-1000 Plus Image Analyzer (Fujifilm Co.,
Tokyo, Japan). Specific signals were quantified by densitometric
analysis using NIH ImageJ software.
 | Table IPrimary antibodies used in western
blot analysis. |
Table I
Primary antibodies used in western
blot analysis.
| Target | Source | Host | Dilution | Second antibody |
|---|
| LC-3 | MBL | Rabbit | 1:1,000 | Anti-rabbit |
| p62/SQSTM1 | MBL | Rabbit | 1:1,000 | Anti-rabbit |
| 4E-BP1 | Cell Signaling | Rabbit | 1:1,000 | Anti-rabbit |
| phospho-4E-BP1 | Cell Signaling | Rabbit | 1:1,000 | Anti-rabbit |
| cleaved PARP | Cell Signaling | Rabbit | 1:1,000 | Anti-rabbit |
| α-tubulin | Sigma | Mouse | 1:1,000 | Anti-mouse |
Immunocytochemical staining for LC3
Cells were trypsinized and seeded at a density of
~1×106 cells/well on 25-mm circular coverslips
(Matsunami Glass Industries, Ltd., Osaka, Japan) in 2 ml of culture
medium containing 10% FBS overnight. In the experiments testing the
effect of rapamycin, cells were treated with 20 μM rapamycin for 24
h. Cells were then fixed in 4% paraformaldehyde in phosphate buffer
for 30 min at room temperature and washed in phosphate-buffered
saline (PBS).
For the detection of autophagy, cells were incubated
with anti-LC3 antibody (code no. PM036, MBL, Nagoya, Japan) for 1 h
at room temperature. Cells were then washed two times with PBS,
incubated with anti-IgG secondary antibody (Alexa Fluor®
488, code no. A11008; Invitrogen, Carlsbad, CA, USA) for 30 min at
room temperature and examined under an epifluorescence microscope
(FSX100, Olympus Optical Co., Ltd., Tokyo, Japan) with a 50×
objective lens.
Quantification of GFP-LC3 puncta
The GFP-LC3 puncta was quantified for the detection
of autophagy. MG63 cells were transfected with GFP-LC3 (P36235,
Invitrogen) for 24 h and a CQ assay was used to determine the
dynamic turnover of GFP-LC3 in autolysosomes. Because CQ is a
lysosome inhibitor, GFP-LC3 puncta will increase even CQ alone.
However, if we could prove that GFP-LC3 puncta is increased in the
Rap group treated with CQ compared to CQ alone, it would mean
autophagy induced by rapamycin is not caused by the lysosome
inhibitor, therefore we used CQ assay. The transfected cells were
pre-treated with 50 μM CQ for 12 h. They were then washed with PBS
two times and treated with or without 20 μM rapamycin for 24 h.
Next, cells were fixed with 4% paraformaldehyde for 30 min. Images
of individual GFP-LC3-expressing cells were taken under an
epifluorescence microscope (FSX100, Olympus Optical Co.) with a 50×
objective lens.
Fluorescence microscopy imaging of the
Annexin V/propidium iodide (PI) and Hoechst 33342 triple-staining
assay
Cells were trypsinized and seeded at a density of
~1×106 cells/well on 25-mm circular coverslips in 2 ml
of culture medium containing 10% FBS for 24 h. Next, cells were
washed with PBS and treated with 20 μM rapamycin and/or 100 μM
Spautin-1 for 24 h. After treatment, cells were incubated for 15
min in a dark room with Annexin V-FITC, PI and Hoechst 33342 using
the Promokine Apoptotic/Necrotic/Healthy Cells Detection kit
(PromoCell GmbH, Heidelberg, Germany). Cells were then examined
under an epifluorescence microscope (FSX100, Olympus Optical Co.)
with a 50× objective lens.
In vivo xenograft studies
Four-week-old female athymic BALB/c nude mice (Clea
Japan, Inc., Tokyo, Japan) were maintained in pathogen-free
conditions and in accordance with institutional principals. MG63
cells (3.0×106 cells in 0.5 ml of medium) were injected
subcutaneously into the dorsal area of mice. For this study on
antitumor activity, rapamycin and Spautin-1 were purchased and used
to determine whether rapamycin and/or Spautin-1 affect tumor
volume. Twenty-four mice were randomly divided into four groups: a
rapamycin group (Rap; n=6), a Spautin-1 group (Spa; n=6), a
combination of rapamycin and Spautin-1 group (Rap-plus-Spa; n=6)
and a control group (control; n=6). After allowing 14 days for
implantation, intraperitoneal injections of the drugs were started.
Throughout the experimental period, the mice were slowly injected
intraperitoneally with 0.2 ml of the following five times a week
for 4 weeks: 8 mg/kg of rapamycin for the Rap group; 40 mg/kg of
Spautin-1 for the Spa group; 8 mg/kg of rapamycin plus 40 mg/kg of
Spautin-1 for the Rap-plus-Spa group; and PBS only for the control
group. After implantation, the tumor dimensions were measured once
a week. Tumor volume was calculated according to the formula V =
π/6 × a2 × b, where a and b represent the shortest and
the longest dimensions of the tumor. All in vivo studies
were performed in accordance with The Guide for the Care and Use of
Laboratory Animals (Washington, DC: National Academy Press, 1996)
and approved by the Institutional Animal Care and Use Committee of
our institution.
Statistical analysis
Statistical analyses for the cell proliferation
assay were performed using GraphPad Prism 5 software (GraphPad, San
Diego, CA, USA) with one- or two-way ANOVA followed by post hoc
analysis. A value of p<0.05 was considered to indicate a
statistically significant difference.
Results
Rapamycin inhibits the proliferation of
MG63 cells
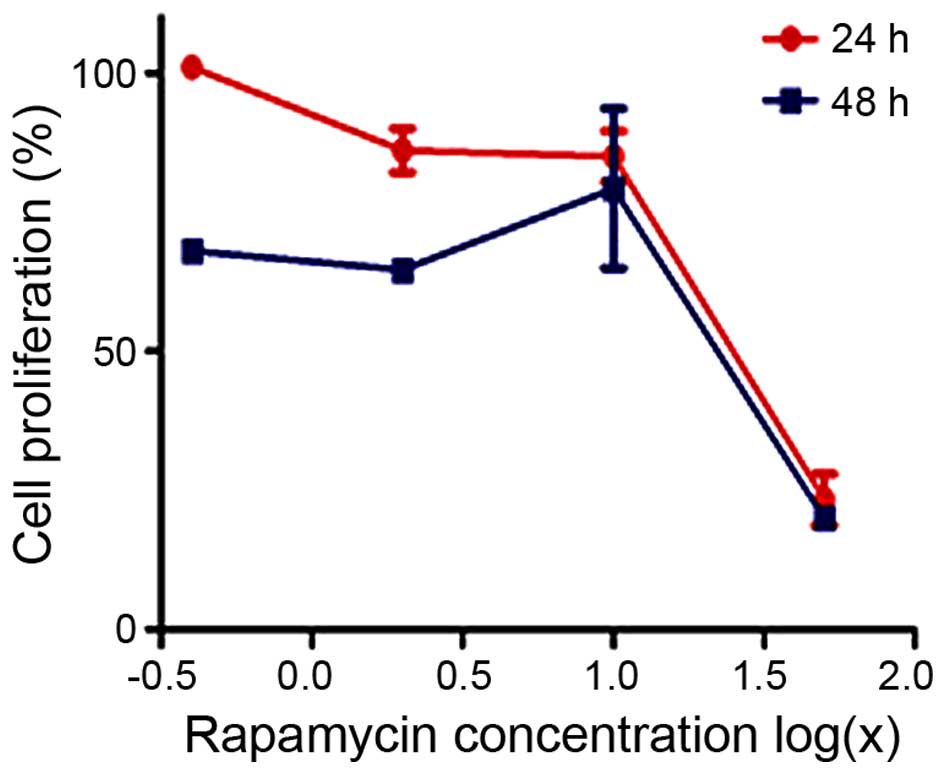
First, we assessed the effects of rapamycin on
cellular proliferation using the CellTiter 96R AQueous One Solution
Cell Proliferation assay. MG63 cells were cultured in the presence
of increasing doses of rapamycin for 24 or 48 h. As shown in
Fig. 1, rapamycin inhibited MG63
proliferation in a dose- and time-dependent manner. The
IC50 value of rapamycin at 24 h was 19.36 μM.
Rapamycin-induced MG63 cell death is
enhanced by Spautin-1
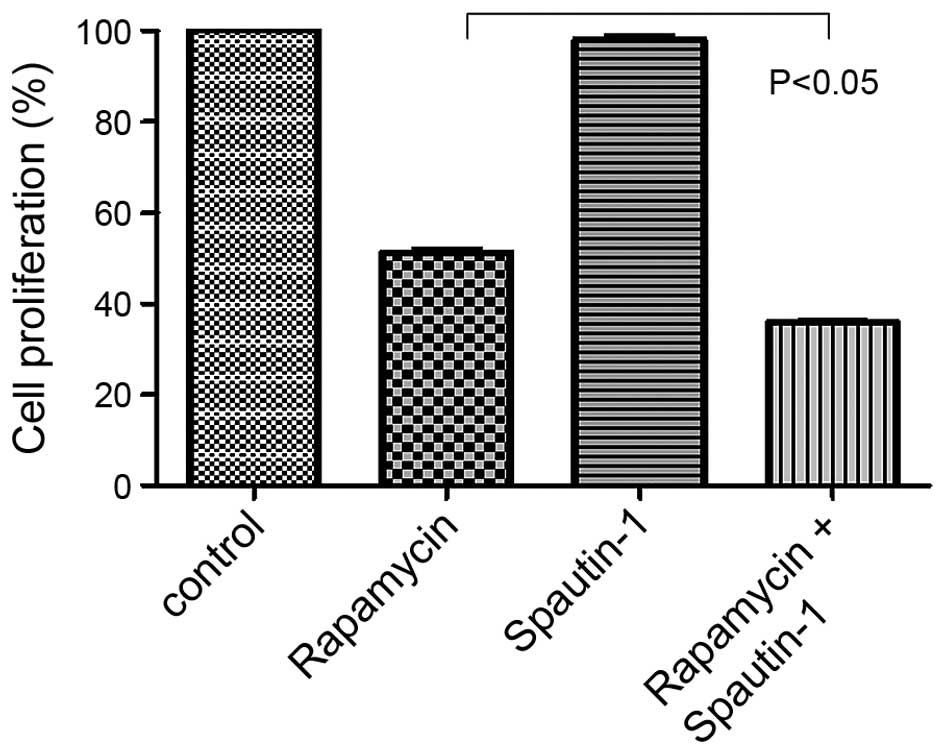
We then examined the effects of rapamycin and/or
Spautin-1 on MG63 cell proliferation. Based on the 24-h
IC50 of rapamycin, we examined the proliferation of MG63
cells treated for 24 h with 20 μM rapamycin, 100 μM Spautin-1, or
20 μM rapamycin and 100 μM Spautin-1. Cell proliferation was
significantly lower in the Rap-plus-Spa group than in the Rap group
(p<0.05) (Fig. 2).
Western blot analysis
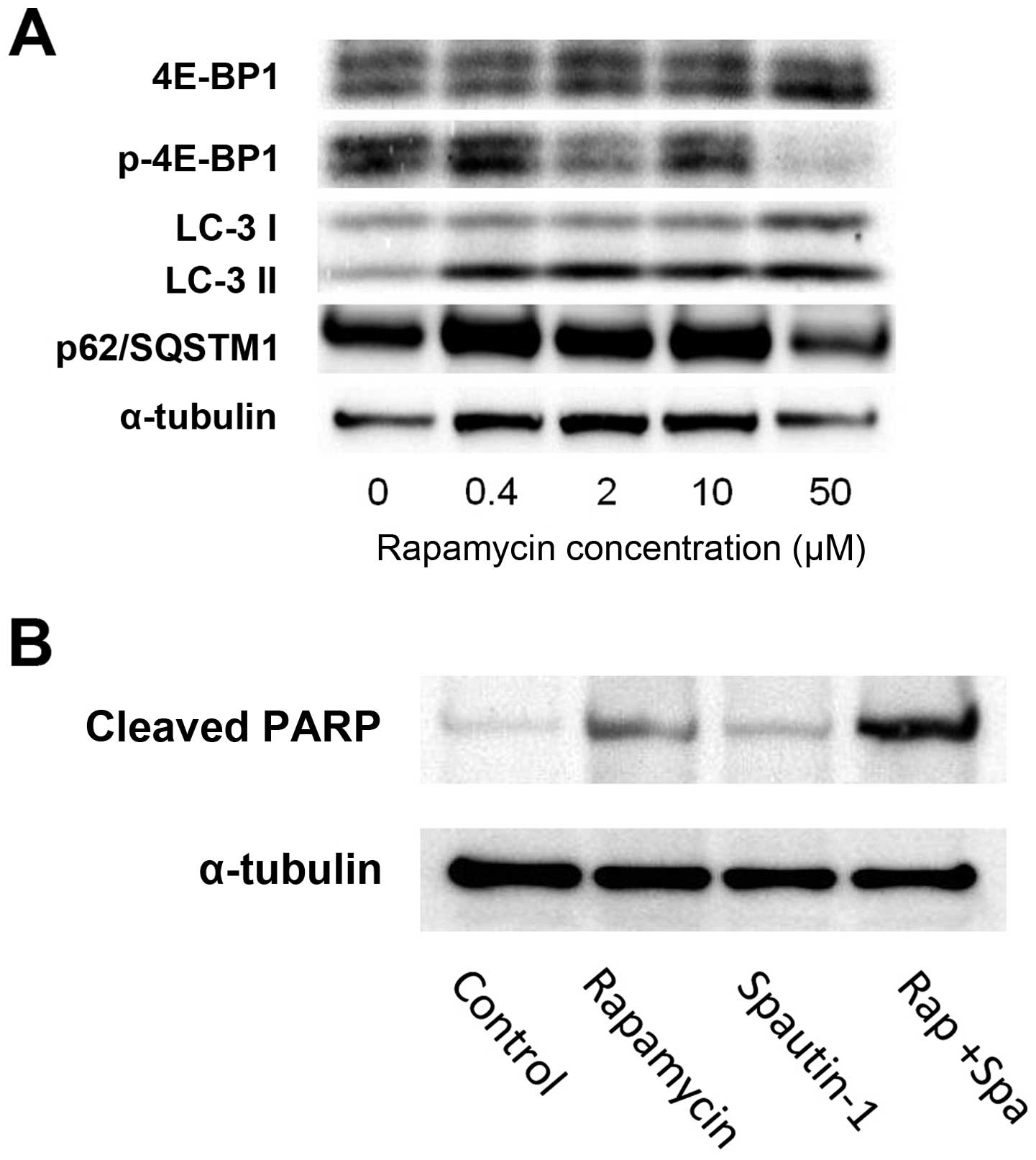
Western blot analysis demonstrated that treatment
with rapamycin induced the phosphorylation of 4E-binding protein
(4E-BP1), one of the key components in the mTOR pathway.
Additionally, we examined the expression of the autophagy-related
gene complex, p62/SQSTM1, and LC-3 in MG63 cells exposed to various
concentrations of rapamycin (ranging from 0.4 to 50 μM) for 24 h
(Fig. 3A). Treatment with
rapamycin resulted in a dose-dependent decrease in the levels of
phospho-4E-BP1, which is a downstream effector of mTOR. These
findings indicate that rapamycin affected the mTOR pathway by
inhibiting the phosphorylation of downstream effectors of mTOR.
LC-3II expression was used as an autophagic marker. The p62
protein, also called sequestosome 1 (SQSTM1), is commonly found in
inclusion bodies containing polyubiquitinated protein aggregates,
which are degraded by autophagy (21). Treatment with rapamycin resulted in
a dose-dependent increase in the expression of LC-3II in the MG63
cells. In contrast, p62/SQSTM1 expression decreased in a
dose-dependent manner (Fig. 3A).
In cells treated with rapamycin, the production of cleaved PARP
slightly increased. On the other hand, in cells treated with
rapamycin plus Spautin-1, the production of cleaved PARP strongly
increased (Fig. 3B).
Immunocytochemistry of LC3 for the
detection of autophagy
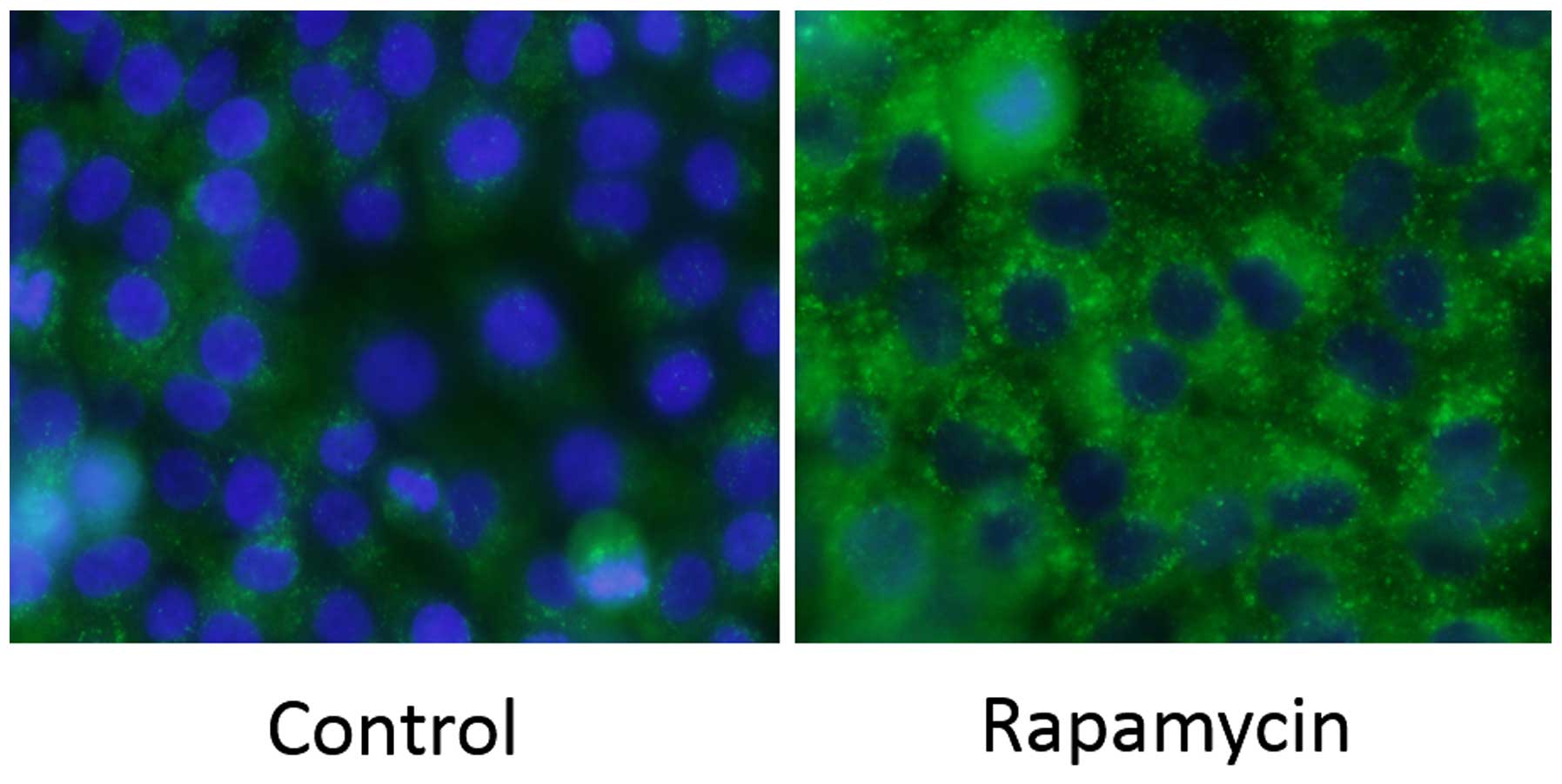
Immunochemical staining for LC3 was performed on
MG63 cells. There was a strong increase in LC3-positive puncta
(autophagosomes) in the Rap group (Fig. 4).
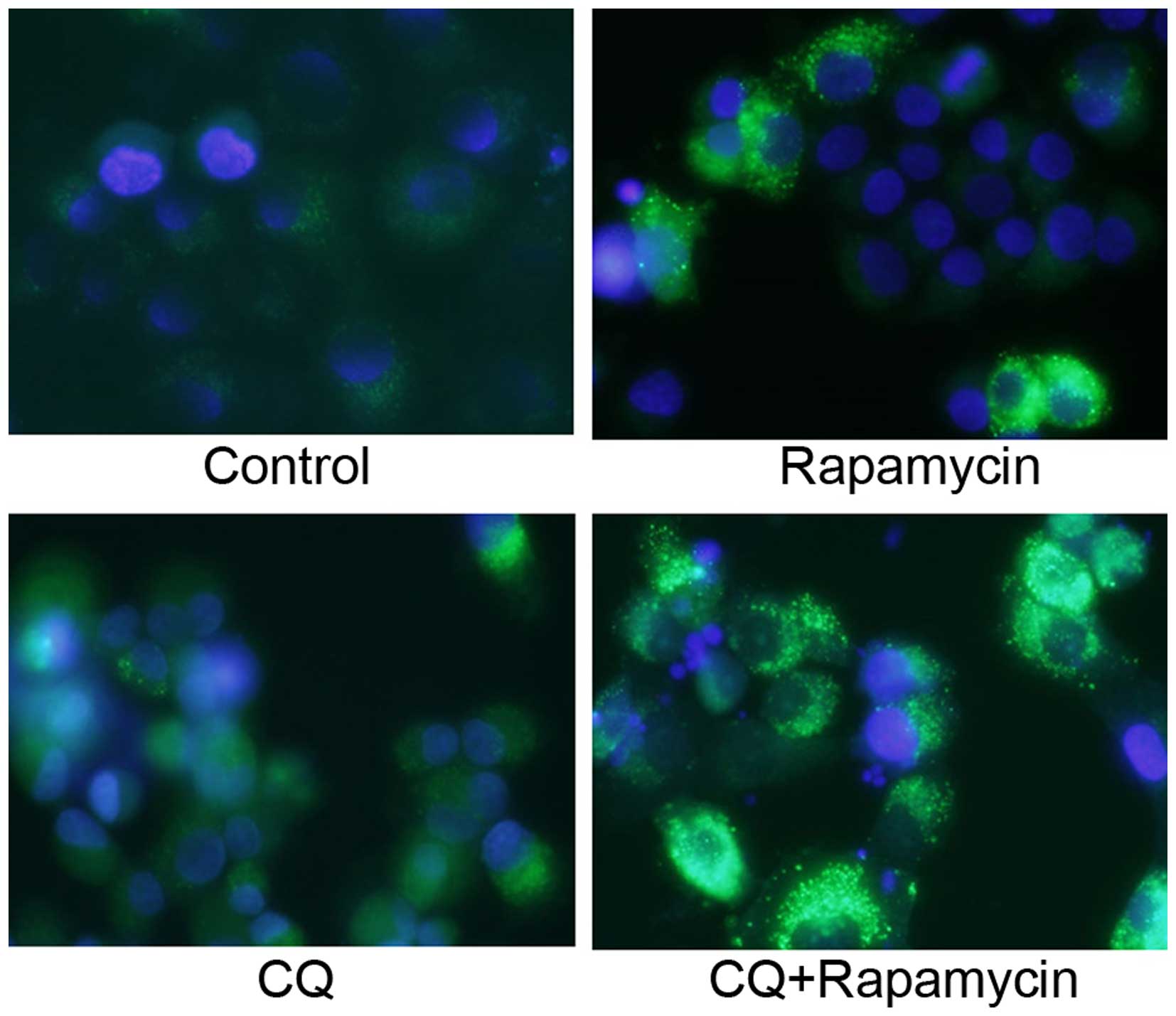
Quantification of GFP-LC3 puncta
Similar to the immunocytochemical staining assay of
LC3, GFP-LC3-positive puncta were slightly increased in the Rap
group and were significantly increased in the Rap group treated
with CQ (Fig. 5).
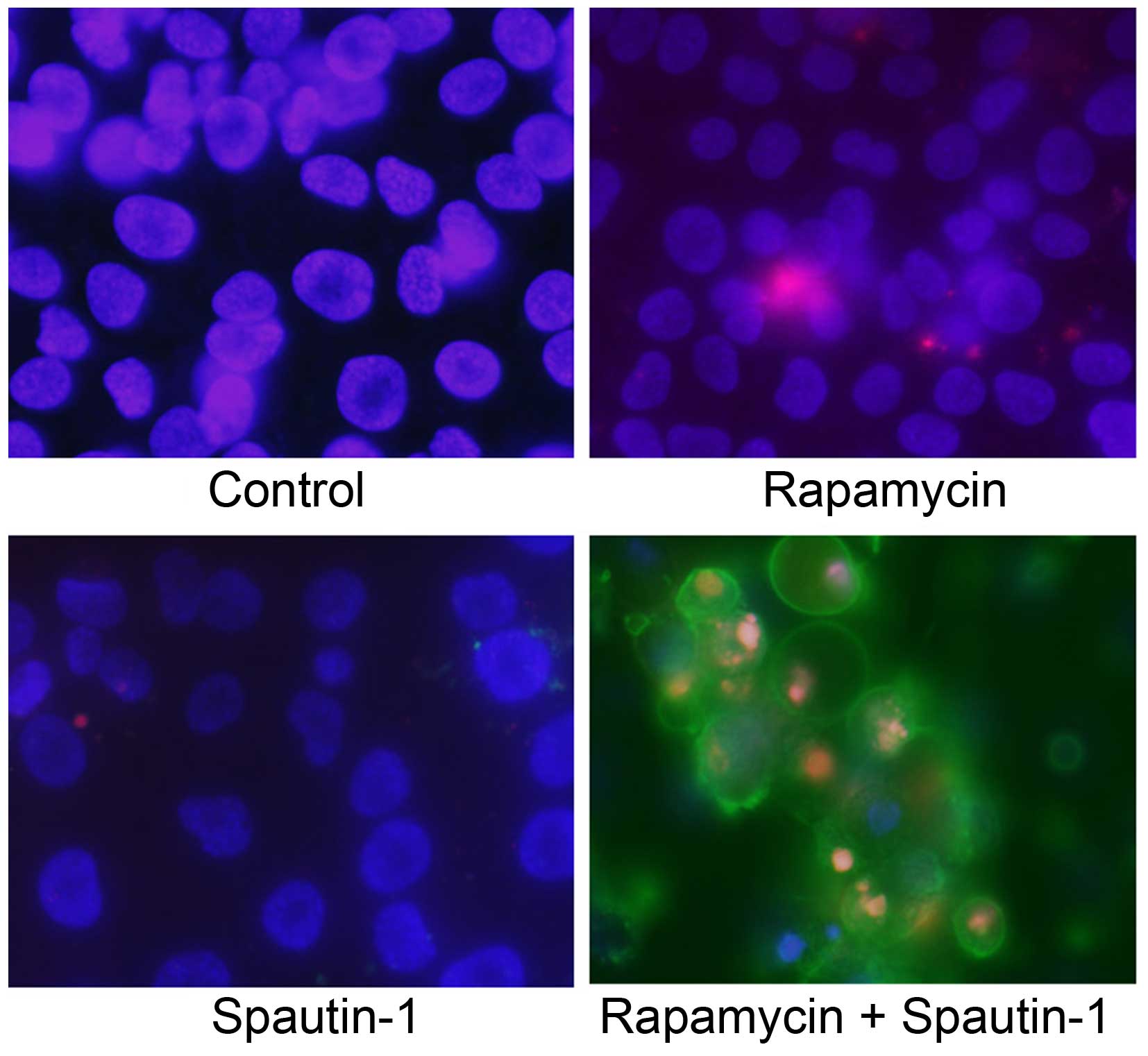
Fluorescence microscopy images of the
Annexin V/PI and Hoechst 33342 triple-staining assay for the
detection of apoptosis
We next used an Annexin V/PI and Hoechst 33342
triple-staining assay to detect apoptotic cells. Hoechst 33342
(blue) is a marker for live cells, Annexin V-FITC (green) is a
marker for early apoptosis, and PI (red) is a marker for late
apoptosis and necrosis. We observed several Annexin V-FITC-positive
cells (early stage of apoptosis) and a high number of Annexin
V-FITC plus PI-positive cells (late stage of apoptosis) in the
Rap-plus-Spa group (Fig. 6).
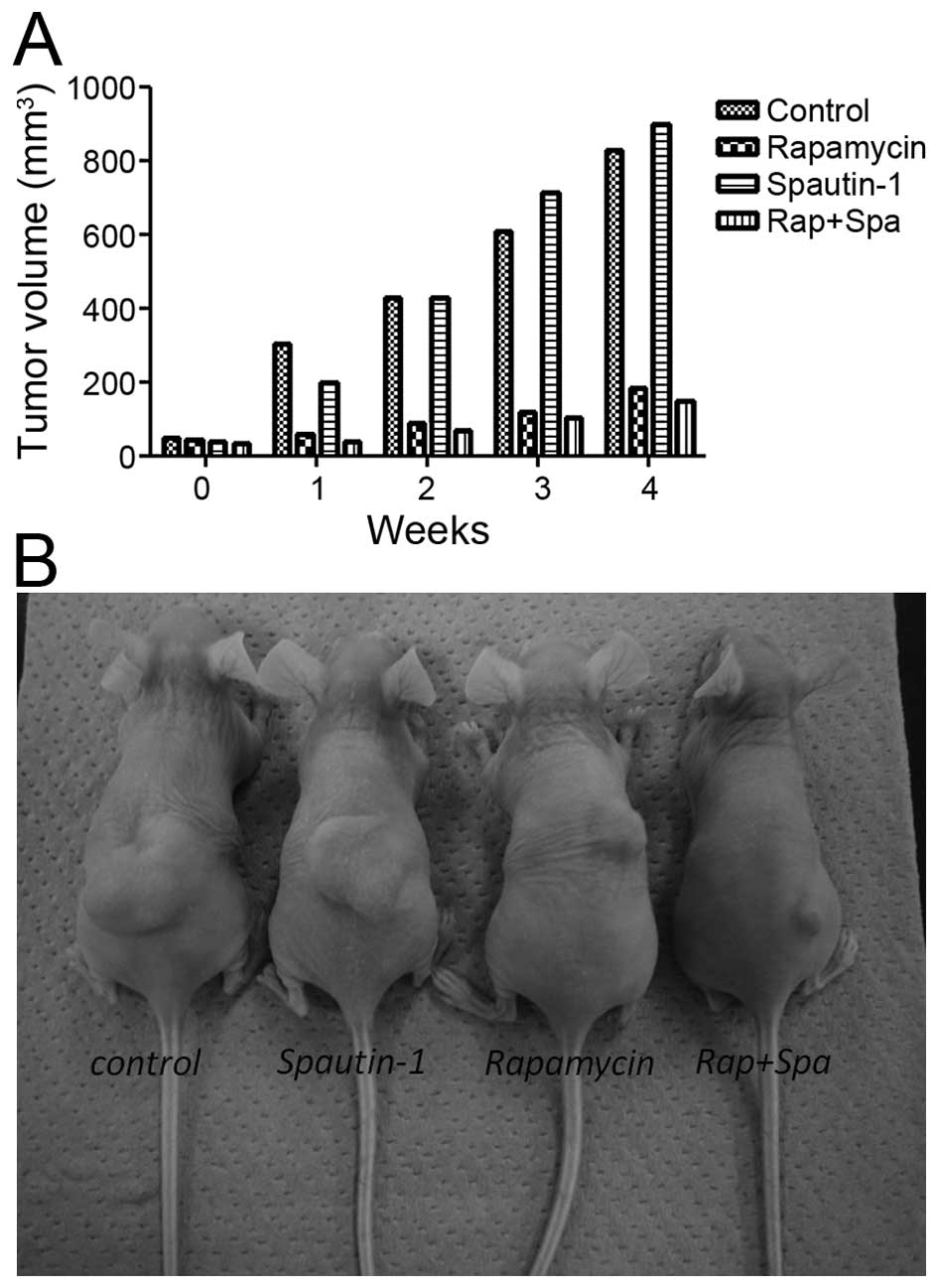
Effect of rapamycin and Spautin-1 on
tumor growth in xenograft models
The antitumor activities of rapamycin and Spautin-1
in nude mice bearing MG63 xenografts was investigated. Implantation
of 3.0×106 cells into the dorsal area of nude mice
resulted in the development of tumors in 100% of the animals. After
1 week, tumor growth in the Rap group and the Rap-plus-Spa group
was significantly inhibited when compared with that in the control
group and the Spa group. At the end of the experimental period, the
mean tumor volume of the Rap group and the Rap-plus-Spa group was
181 and 146 mm3, respectively (Fig. 7B). Although there was no
significant difference between the Rap group and the Rap-plus-Spa
group, tumor growth in the Rap-plus-Spa group was less than that
seen in the Rap group (Fig. 7A).
Throughout the experimental period, no side effects, such as body
weight loss, were observed in the treatment groups.
Discussion
mTOR signaling pathway
mTOR is a serine/threonine kinase with a catalytic
subunit of two biochemically distinct complexes called mTORC1 and
mTORC2. mTORC1 contains Raptor, mLST8 and PRAS40, while mTORC2
contains mTOR, Rictor, mSIN1 and Protor-1. mTORC1 senses several
cellular/environmental signals, including protein misfolding,
nutrient availability and growth signals. mTORC1 controls cell
growth through the phosphorylation of S6K and 4E-BP1, and regulates
autophagy; in contrast, mTORC2 is not a direct regulator of
autophagy. Rapamycin, which is a specific inhibitor of mTORC1, has
been shown to selectively and completely block mTORC1-dependent
p70S6K phosphorylation and partially block 4E-BP1 phosphorylation
(22). In this study, we observed
that rapamycin suppressed the phosphorylation of 4E-BP1 in a
dose-dependent manner. This result demonstrated that the mTOR
pathway is a target of rapamycin in MG63 cells, as previously shown
in several reports.
Apoptosis induced by rapamycin is
enhanced by an autophagy inhibitor
Autophagy is a process in which subcellular
membranes undergo dynamic morphological changes (autophagosomes
form and fuse with lysosomes) leading to the degradation of
cellular proteins and cytoplasmic organelles. Autophagy plays a
protective role when cells encounter environmental stress such as
starvation or pathogen infection (23,24).
Autophagy also occurs under pathological conditions, such as
neurodegenerative diseases or hereditary myopathies (25). Recent accumulating evidence
indicates that autophagy often plays a role in malignant diseases.
Specifically, autophagy is believed to play an important role in
tumor development. During the early stages of tumor formation,
autophagy functions as a tumor suppressor, and autophagic activity
is often impaired in cancer cells. However, autophagy has also been
shown to play a role as a self-defense mechanism in promoting tumor
cell resistance to chemotherapy (26). Some anticancer drugs that lead to
apoptosis can also induce autophagy-related cell death in cancer
cell lines (27,28). In this study, we demonstrated that
apoptosis induced by an mTOR inhibitor was enhanced by the addition
of an autophagy inhibitor. As such, apoptosis is considered to be
closely related to autophagy-related cell death.
Autophagy is induced by an mTOR
inhibitor
In this study, our western blot results revealed
that rapamycin treatment suppressed the phosphorylation of 4E-BP1
and induced a dose-dependent upregulation of the expression of the
autophagy marker LC3-II and downregulation of p62/SQSTM1 in MG63
cells. In addition, immunocytochemical analysis by the staining of
LC3 was performed. In rapamycin-treated cells, the number of
LC3-positive puncta was markedly increased. Similar to the results
of the immunocytochemical staining assay for LC3, the number of
GFP-LC3 puncta was slightly increased in the Rap group and was
significantly increased in the Rap group treated with CQ. These
results indicate that suppression of the mTOR pathway is involved
in the induction of autophagy. Furthermore, GFP-LC3-transfected
cells treated with a combination of CQ and rapamycin demonstrated
activation of the autophagy pathway. Our previous study also
demonstrated that rapamycin can induce cytoprotective autophagy in
human malignant fibrous histiocytoma (MFH) cell lines by activating
the MEK/ERK signaling pathway, and that the rapamycin-induced
apoptosis can be enhanced by a MEK inhibitor that was used as an
autophagy inhibitor (29).
Autophagy inhibitor Spautin-1
Spautin-1 is an inhibitor of autophagy. It promotes
the degradation of Vps34 complexes by inhibiting USP10 and USP13,
two ubiquitin-specific peptidases that target the deubiquitination
of Beclin-1. Several reports have reported the use of Spautin-1 as
an autophagy inhibitor for the treatment of malignancy. For
example, Correa et al reported that a combination of AKT
inhibition and autophagy blockage would prove efficacious to reduce
residual epithelial ovarian cancer cells that cause ovarian cancer
recurrence (30). However, there
have been no reports on the use of Spautin-1 for the treatment of
osteosarcoma. Thus, to the best of our knowledge, this is the first
report of the use of Spautin-1 for the treatment of
osteosarcoma.
Combination therapy with rapamycin and
Spautin-1
Recent reports indicate that when an autophagy
inhibitor, such as 3-methyladenine (3-MA), is combined with
chemotherapeutic drugs, it triggers apoptosis in some cancer cells
(31). Our previous study
demonstrated that the combination of temsirolimus (CCI-779, an
analog of rapamycin) and 3-MA suppressed autophagy and induced
apoptosis in MFH cell lines (32).
In this study, cell proliferation was significantly lower in the
Rap-plus-Spa group than in the Rap group, indicating that Spautin-1
enhanced the rapamycin-mediated suppression of osteosarcoma cell
proliferation. In addition, we demonstrated by western blot
analysis that the production of cleaved PARP strongly increased in
the Rap-plus-Spa group, and by immunocytochemical analysis, that
the number of Annexin V-positive cells markedly increased in the
Rap-plus-Spa group. Furthermore, in vivo experiments
demonstrated that tumor growth was inhibited in the Rap-plus-Spa
group when compared with that in the Rap group despite no
significant difference in body weight between the two groups.
Autophagy appears to function as a protective mechanism in
rapamycin-treated MG63 cells and blocking autophagy using Spautin-1
can promote the activation of apoptosis. These results suggest that
the combination of rapamycin and Spautin-1 can potently induce
apoptotic cell death in MG63 cells with the inhibition of
autophagy.
In this study, only MG63 cell line was examined.
Furthermore, in vivo experiment, the autophagic and
apoptotic activities were not proved in treated mouse xenografts.
Of course, further experiments are needed for these limitations.
For example, other osteosarcoma cell lines should be used, and
immunostaining assays for detection of autophagy and apoptosis
should be analyzed in excised tumor models in vivo.
In conclusion, this study demonstrated that
rapamycin induced autophagy in MG63 cells by inhibiting
phosphorylation of mTOR pathway components, and that
rapamycin-induced apoptosis was enhanced by Spautin-1. These
results suggest that self-protective mechanisms involving mTOR
inhibitors are hindered by the inhibition of autophagy in MG63
cells. Therefore, the combination of an mTOR inhibitor (e.g.,
rapamycin) and an autophagy inhibitor (e.g., Spautin-1) may offer
an effective treatment for osteosarcoma as this combination
effectively activates apoptotic pathways.
Acknowledgements
The authors thank Mr. Toshitaka Nakagawa (Division
of Research Instrument and Equipment, Kagawa University School of
Medicine, Kagawa, Japan) for providing technical assistance with
epifluorescence microscopy.
References
|
1
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar
|
|
2
|
Wardelmann E, Chemnitz JM and Wendtner CM:
Targeted therapy of soft tissue sarcomas. Onkologie. 35(Suppl 1):
21–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arslan MA, Kutuk O and Basaga H: Protein
kinases as drug targets in cancer. Curr Cancer Drug Targets.
6:623–634. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gridelli C, Maione P and Rossi A: The
potential role of mTOR inhibitors in non-small cell lung cancer.
Oncologist. 13:139–147. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mita MM and Tolcher AW: The role of mTOR
inhibitors for treatment of sarcomas. Curr Oncol Rep. 9:316–322.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim KW, Mutter RW, Cao C, Albert JM,
Freeman M, Hallahan DE and Lu B: Autophagy for cancer therapy
through inhibition of pro-apoptotic proteins and mammalian target
of rapamycin signaling. J Biol Chem. 281:36883–36890. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hung JY, Hsu YL, Li CT, Ko YC, Ni WC,
Huang MS and Kuo PL: 6-Shogaol, an active constituent of dietary
ginger, induces autophagy by inhibiting the AKT/mTOR pathway in
human non-small cell lung cancer A549 cells. J Agric Food Chem.
57:9809–9816. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tan ML, Ooi JP, Ismail N, Moad AI and
Muhammad TS: Programmed cell death pathways and current antitumor
targets. Pharm Res. 26:1547–1560. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zoncu R, Efeyan A and Sabatini DM: mTOR:
From growth signal integration to cancer, diabetes and ageing. Nat
Rev Mol Cell Biol. 12:21–35. 2011. View
Article : Google Scholar
|
|
12
|
Beauchamp EM and Platanias LC: The
evolution of the TOR pathway and its role in cancer. Oncogene.
32:3923–3932. 2013. View Article : Google Scholar
|
|
13
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Roca H, Varsos Z and Pienta KJ: CCL2
protects prostate cancer PC3 cells from autophagic death via
phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation.
J Biol Chem. 283:25057–25073. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rami A and Kögel D: Apoptosis meets
autophagy-like cell death in the ischemic penumbra: Two sides of
the same coin? Autophagy. 4:422–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu D, Yang Y, Liu Q and Wang J:
Inhibition of autophagy by 3-MA potentiates cisplatin-induced
apoptosis in esophageal squamous cell carcinoma cells. Med Oncol.
28:105–111. 2011. View Article : Google Scholar
|
|
18
|
Kanematsu S, Uehara N, Miki H, Yoshizawa
K, Kawanaka A, Yuri T and Tsubura A: Autophagy inhibition enhances
sulforaphane-induced apoptosis in human breast cancer cells.
Anticancer Res. 30:3381–3390. 2010.PubMed/NCBI
|
|
19
|
Ren Y, Huang F, Liu Y, Yang Y, Jiang Q and
Xu C: Autophagy inhibition through PI3K/Akt increases apoptosis by
sodium selenite in NB4 cells. BMB Rep. 42:599–604. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L,
Cai Y, Norberg HV, Zhang T, Furuya T, et al: Beclin1 controls the
levels of p53 by regulating the deubiquitination activity of USP10
and USP13. Cell. 147:223–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim MS, Kuehn HS, Metcalfe DD and
Gilfillan AM: Activation and function of the mTORC1 pathway in mast
cells. J Immunol. 180:4586–4595. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meijer AJ and Codogno P: Regulation and
role of autophagy in mammalian cells. Int J Biochem Cell Biol.
36:2445–2462. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mori M, Hitora T, Nakamura O, Yamagami Y,
Horie R, Nishimura H and Yamamoto T: Hsp90 inhibitor induces
autophagy and apoptosis in osteosarcoma cells. Int J Oncol.
46:47–54. 2015.
|
|
26
|
Liang X, Tang J, Liang Y, Jin R and Cai X:
Suppression of autophagy by chloroquine sensitizes
5-fluorouracil-mediated cell death in gallbladder carcinoma cells.
Cell Biosci. 4:102014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu B, Cheng Y, Zhang B, Bian HJ and Bao
JK: Polygonatum cyrtonema lectin induces apoptosis and autophagy in
human melanoma A375 cells through a mitochondria-mediated
ROS-p38-p53 pathway. Cancer Lett. 275:54–60. 2009. View Article : Google Scholar
|
|
28
|
Wang Q, Chen Z, Diao X and Huang S:
Induction of autophagy-dependent apoptosis by the survivin
suppressant YM155 in prostate cancer cells. Cancer Lett. 302:29–36.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakamura O, Hitora T, Yamagami Y, Mori M,
Nishimura H, Horie R, Yamaguchi K and Yamamoto T: The combination
of rapamycin and MAPK inhibitors enhances the growth inhibitory
effect on Nara-H cells. Int J Mol Med. 33:1491–1497.
2014.PubMed/NCBI
|
|
30
|
Correa RJ, Valdes YR, Peart TM, Fazio EN,
Bertrand M, McGee J, Préfontaine M, Sugimoto A, DiMattia GE and
Shepherd TG: Combination of AKT inhibition with autophagy blockade
effectively reduces ascites-derived ovarian cancer cell viability.
Carcinogenesis. 35:1951–1961. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nishikawa T, Tsuno NH, Okaji Y, Shuno Y,
Sasaki K, Hongo K, Sunami E, Kitayama J, Takahashi K and Nagawa H:
Inhibition of autophagy potentiates sulforaphane-induced apoptosis
in human colon cancer cells. Ann Surg Oncol. 17:592–602. 2010.
View Article : Google Scholar
|
|
32
|
Nakamura O, Hitora T, Akisue T, Kawamoto
T, Yamagami Y and Yamamoto T: Inhibition of induced autophagy
increases apoptosis of Nara-H cells. Int J Oncol. 39:1545–1552.
2011.PubMed/NCBI
|