Cancer cells display a number of abnormal properties
in order to maintain their unrestrained growth and proliferation
(1). Ribosome biogenesis and
protein synthesis are in this context critical cellular processes
necessary for sustained cancer cell growth. Historically, ribosomes
were considered to be relatively stable entities. However, with the
discoveries of mutations affecting ribosomal protein (RP) genes in
the Diamond-Blackfan anemia (DBA) syndrome it became evident that
mutant RPs may cause complex, variable, and viable phenotypes
(2). Of note, DBA and other
syndromes involving mutant ribosomal or nucleolar proteins are
often associated with an increased life time risk of cancer
(3). Recently, a number of studies
using next generation sequencing technologies describe RP gene
mutations also in cancers without a previous known history of bone
marrow failure disorder. By applying whole-exome sequencing, RNA
seq, or whole-genome sequencing, RP gene mutations have been
detected in the genome of cancer cells, including from endometrial
cancer, T-cell acute lymphoblastic leukemia (T-ALL), chronic
lymphocytic leukemia (CLL), colorectal carcinomas, and high grade
gliomas (4–7). The mechanisms underlying cancer
development in the setting of a ribosome biogenesis defect remain
poorly understood. In this review, the most recent studies are
summarized and possible mechanisms by which mutant ribosomal
proteins are linked to cancer development are discussed.
The basis of protein synthesis is the translation of
messenger RNA (mRNA) to an amino acid sequence. Translation of mRNA
is carried out by the ribosome, transfer RNA (tRNA), with the
assistance of an army of different helper proteins. The intrinsic
catalytic activity of ribosomes is thought to be dependent on
ribosomal RNA (rRNA), that is involved in mRNA decoding and
formation of peptide bonds. Certain chemical modifications on rRNA,
including pseudo-uridylation and ribose methylation, are critical
for maintaining proper rRNA structure and modulate the interactions
between rRNA and proteins (8). The
ribosome consists of two subunits, each of which is made up of
ribosomal RNA (rRNA) and many RPs. Eukaryotes have 80S ribosomes,
consisting of the small (40S) and the large (60S) subunit. The
large 60S subunit is composed of a 5S rRNA, a 28S rRNA, a 5.8S
subunit, and ~46 RPs. The small 40S subunit has an 18S rRNA and ~33
RPs. Note that the 5S rRNA is transcribed by RNA polymerase III,
while 28S, 5.8S, and 18S rRNAs are processed from a long precursor
(pre-) rRNA transcribed by RNA polymerase I (9). The maturation of pre-rRNA occurs in
the nucleolus involving both endo- and exonucleases that remove
external and internal transcribed sequences. In the nucleolus the
45S pre-RNA associates with RPs, ribonucleases, RNA helicases,
small nucleolar RNPs and other accessory factors, to form 90S
pre-ribosomes. During the maturation process, the 90S pre-ribosome
is separated into pre-40S and pre-60S subunits that are exported to
the cytoplasm where maturation is completed (9). It should be noted that RPs are
synthesized by pre-existing ribosomes in the cytoplasm and imported
into the nucleus where majority of the RPs home into the nucleolus
to assemble with rRNA, and majority of RPs are essential in
ribosome biogenesis (10–12). Strikingly, RPs have high
isoelectric points allowing them to interact with rRNAs, mRNAs, and
tRNAs (13). The names of the RPs
belonging to the large subunit include the prefix L and the names
of the RPs of the smaller subunit include the prefix S. A new
universal nomenclature has been launched and we will provide both
names at their first mentioning in the text (14).
Some cells have the potential to produce ribosomes
with a different composition of RPs, and post-translational
modifications, in response to changing extracellular demands. These
adaptations have mostly been studied in bacteria, plants, and yeast
but recently also in mammalian cells (15). There are a number of potential
mechanisms leading to ribosome heterogeneity (16), although the nature of the
heterogeneity is variable, from subtle changes in
post-translational modification patterns to the loss of an RP.
Duplicated RP genes exist in the genomes of some species such as
plants. These extra RP genes are sometimes encoding for a variant
protein (paralog) that may differ in amino acid sequence (17). Paralogs might have specific
functional roles. For example, Rpl22−/− mice have
only subtle phenotypes with no significant translation defects
because in these mice there is a compensatory increase in
Rpl22-like1 (Rpl22l1) expression and incorporation into
ribosomes (17). Importantly,
knockdown of Rpl22l1 impairs growth of cells lacking Rpl22
(17). Post-translational
modifications of RPs (e.g. ubiquitination and phosphorylation) have
been described and these may alter the functional properties of
ribosomes (18). Another layer of
ribosome heterogeneity may stem from differences in modification of
the rRNA itself (8). RP genes also
generate a large number of processed pseudogenes that are dispersed
throughout the genome (13,19).
While the pseudogenes have been considered to be inactive there are
studies indicating that they have the potential to produce
functional coding RNA and protein (20). Finally, it should be added that
long non-coding RNAs are involved in regulating mRNA translation, a
number of long non-coding RNAs associates with cytoplasmic
ribosomes, and if we also include these regulatory levels, the
complexity becomes even higher (21,22).
Taken together, there are a number of potential different
mechanisms contributing to ribosome heterogeneity, and these are
probably functionally relevant to both normal and cancer cells. One
may suspect that certain mechanisms are dominant in cancer cells
when compared to normal cells. It will be important to identify
these differences as it might open up novel avenues for anti-cancer
treatment.
A critical issue to keep in mind concerns the fate
of pre-ribosomes in the context of an RP mutation or deletion
(23). It is known that the
synthesis of ribosomes is a process regulated and balanced at
multiple levels (24), and that
RPs produced in excess are rapidly degraded in the nucleus
(25,26). Depletion of an individual RP in
normal cultured cells often, but not always, results in a decrease
in the total level of the other RPs belonging to the same ribosomal
subunit, thus creating an unbalanced ribosome assembly pathway
(27,28). In the setting of an RP loss by
deletion or an early truncating mutation one may therefore expect
reduced numbers of ribosomes to be a common outcome. Normal and
cancer cells may try to compensate a ribosome deficit by activation
of pathways that boost ribosome production, e.g. the mTOR
pathway (29). This situation may
create a pressure to mutate components in the cell that normally
restrains the pathway activity in question.
Congenital diseases found in humans that are linked
to genetic defects in RPs or ribosome biogenesis factors are
collectively known as the ribosomopathies (40–42).
These include Dyskeratosis congenita (DKC), Diamond-Blackfan anemia
(DBA), and Shwachman-Diamond syndrome (SDS) that constitute major
inherited bone marrow failure syndromes (41). The ribosomopathies are
characterized by a number of abnormalities including birth defects
and anemia (41). DBA is a
dominant autosomal bone marrow failure syndrome associated with
mutations in RP genes including RPS19(eS19), RPS17(eS17),
RPS24(eS24), RPL35A(eL33), RPS7(eS7), RPL5(uL18), RPL11(uL5),
RPL26(uL24), RPL27(eL27), RPS10(eS10), RPS26(eS26), RPS27(eS27),
RPL15(eL15), RPS28(eS28), RPL31(eL31) and RPS29(uS14) (2,3,43–45).
Patients with DBA experience a block in erythroid progenitor cell
division in the bone marrow coupled to an increased apoptosis
(46). DBA patients have a 5-fold
higher lifetime risk of cancer than the general population,
specifically a 28- to 36-fold higher risk of developing AML,
osteosarcoma, or colon cancer (3).
Although a somatic mosaic disorder, and not congenital,
RPS14(uS11) heterozygous loss is associated with 5q-
syndrome and the development of anemia (47). Patients with 5q- syndrome or SDS
are at higher risk of developing AML (48–50).
DKC is a syndrome characterized by premature aging and increase in
cancer susceptibility. X-linked DKC, which has a more severe
phenotype compared with the autosomal dominant form of DKC, is
caused by a mutation in DKC1, which encodes dyskerin
(51). Dyskerin is in part a
nucleolus located protein associated with the snoRNPs involved in
rRNA modification (52,53). Patients with X-linked DKC are
predisposed to AML, lymphoma, and a variety of solid tumors
including squamous carcinoma (54). Note that both DKC and SDS have a
higher risk of cancer development than DBA, especially the risk of
leukemia, although some cohorts are rather small thus causing
estimates with greater differences among the studies (3,48,49,54,55).
It should be emphasized that the main problem in DBA patients is
related to acute effects from bone marrow failure or complications
due to chronic blood transfusions and not cancer per se
(56).
Genome-wide sequencing indicates that RP gene
mutations are relatively frequent in some cancer types.
RPS15(uS19) mutations have been found in CLL and even more
frequently in relapsed CLL (up to 19.5% of cases) (57, 58). Moreover, ~10% of children with
T-ALL have mutations in RP genes including RPL10(uL16), RPL5,
RPL11, and RPL22 (4,59,60).
In fact, 6.5% of T-ALL patients presented with an identical RPL10
Arg98Ser missense mutation (4)
(Table I). A separate study in
T-ALL patients identified a 10% incidence of heterozygous deletions
in the region of chromosome 1p that harbors RPL22 (60), and a number of T-ALL cell lines and
relapse cases had point mutations in RPL22 (60). In line with its potential role as
tumor suppressor, RPL22 is also mutated or have decreased
expression in other cancers as well, including endometrial cancers,
colorectal cancer, gastric cancer, breast carcinoma, and non-small
cell lung carcinoma (7,61–63).
Internal deletions and insertions resulting in early truncating
frame shifts are most commonly seen, examples include RPL22
Lys15Arg and Lys16Glu (Table I).
Truncating frame shift mutations in RPL5 have been detected
in glioblastoma (5) and
RPL5 (as well as RPL22) is identified as being
mutated at a significant frequency in cancer (5). A closer look at TCGA data using the
cBioportal website suggests that RPL10 and RPL22 are
deleted in cases of diffuse large B cell lymphoma, adrenocortical
carcinoma, and sarcoma, and that RPL5 is mutated in a few
cases of human MPNSTs (64), and
potentially in other cancer types including endometrial carcinoma
and lung adenocarcinoma. Genetic linkage analysis and exome
sequencing led to the identification of a truncating germline
mutation in RPS20(uS10) predisposing to colorectal cancer,
which is interesting given the association of DBA with colon
cancer, but a previous history of DBA appeared unlikely (6). Deep sequencing uncovered the
existence of RPL39(eL39) mutations in cells from breast
cancer lung metastatic lesions (65). A more complete picture of the
relevant RPs in cancer will emerge from additional sequencing
projects and from functional studies. One must also recall that
possibly not all relevant RP mutations are detected since the
mutations may be present in a small subpopulation of cells
(66). Especially solid cancers
often exhibit cell heterogeneity that may prevent the
identification of specific mutations. Cell sorting in combination
with single cell genome sequencing and single cell RNA-seq may
provide more detailed information in the future.
Are the DBA associated RP gene mutations different
from the mutations that have been found in cancer? A
cross-comparison of TCGA data with associated recent publications
and information available in the DBA database (67), indicates that the mutations
described to date usually are different but a few are actually in
common. As with regard to RPL5, mutations Lys5fs, Val6fs, Arg35fs,
Asn57fs and Asp59fs have been found in cancer and also the RPL5
point mutants Glu82Lys and Arg54Cys (Table I). There are a large number of DBA
associated RPL5 mutants including Met1Arg and Arg58Lys. Most
interestingly, two of the RPL5 mutations seen in DBA were also
found in T-ALL namely Arg179X and Arg58LysfsX55. The region in RPL5
between Arg54 to Asp59 appears to be a ‘hot spot’ in both DBA and
cancer. RPS15 frequently mutated in CLL is rarely so in DBA and the
Met70Val DBA mutant has to date not been found in CLL. Also, the
few RPL11 mutations in T-ALL described have so far not been
observed in DBA. It will be important to investigate whether cancer
associated mutations in RPs occur in the setting of an underlying
ribosome biogenesis disorder.
The mechanism(s) by which RP mutations increase the
risk of developing cancer remains an important unanswered question
and several hypotheses have been proposed (79,80).
RP deficiency often causes complex phenotypes during development.
These different phenotypes may arise from altered translation
and/or from the effects of activation of cell stress responses
including cell cycle arrest and apoptosis (81). This complexity is seen in a number
of different mouse models. Rpl24(eL24)+/− mice
display a size decrease of approximately 20%, white ventral midline
spots, white hind feet, and kinked tails (82). Rpl29 (eL2)+/−
mice suffer from a global growth deficiency and shortened lifespan.
Rpl38(eL38)+/− mice present with tissue-specific
patterning defects due to the perturbation of a subset of Homeobox
mRNAs (83). Given these
pleiotropic phenotypes several mechanisms could also be involved in
cancer development.
The best known response to ribosome biogenetic
defects involves the tumor suppressor p53 that induces cell cycle
arrest, senescence, apoptosis, or differentiation (84,85).
A number of mouse models confirm the involvement of p53 in
mediating certain phenotypes. For example, deletion of only one
allele of Rps6 is enough to impair ribosome biogenesis, but
the early embryonic lethality is due to activation of p53-dependent
cell cycle arrest and apoptosis rather than to a general
downregulation of protein synthesis (86). Furthermore, mutations in
Rps19(eS19) and Rps20(uS10) in mice result in
p53-dependent pigmentation defects (abnormal melanocyte
proliferation), reduced body size, and anemia (87). Rpl22 deficient mice develop T
lymphopenia by blocking αβ-T cell development in a p53-dependent
manner (37,88). Supporting observations also came
from studies on the 5q-syndrome. The haplo-insufficiency of RPS14
has a critical role in the development of the anemia that
characterizes 5q- syndrome (47).
Bone marrow cells from a mouse model of 5q- syndrome shows elevated
level of p53 and intercross with Trp53−/− mice
rescued the macrocytic anemia and dysplasia phenotypes of the 5q-
mouse (89). For a more exhaustive
list of the different mouse models having mutations in ribosomal
protein genes we refer the reader to an informative overview by
Terzian and Box (90).
Activation of checkpoints for quality control of
ribosome biogenesis is contributing to the disease manifestations
among the ribosomopathies (91,92).
The hematopoietic phenotype in DBA patients is for example at least
partially linked to the activation of p53 (93). What is the mechanism sensing
ribosome dysfunction leading to p53 activation? It is now
established that two RPs, namely RPL11 (uL5) and RPL5 (uL18),
control p53-dependent cell cycle arrest, senescence or apoptosis in
response to impaired ribosome biogenesis (91,94,95).
Loss of RPL5 or RPL11 also impairs ribosome biogenesis and stalls
cell proliferation similar to other essential RPs (27,95),
but in the case of RPL11 or RPL5 there is no distinct cell cycle
arrest (95). RPL11 and RPL5
regulate p53 as key components of the 5S ribonucleo-protein
particle (5S RNP), in which the 5S rRNA is essential as well
(96–98). When ribosome biogenesis is blocked,
the 5S RNP pre-ribosomal complex is re-directed from assembly into
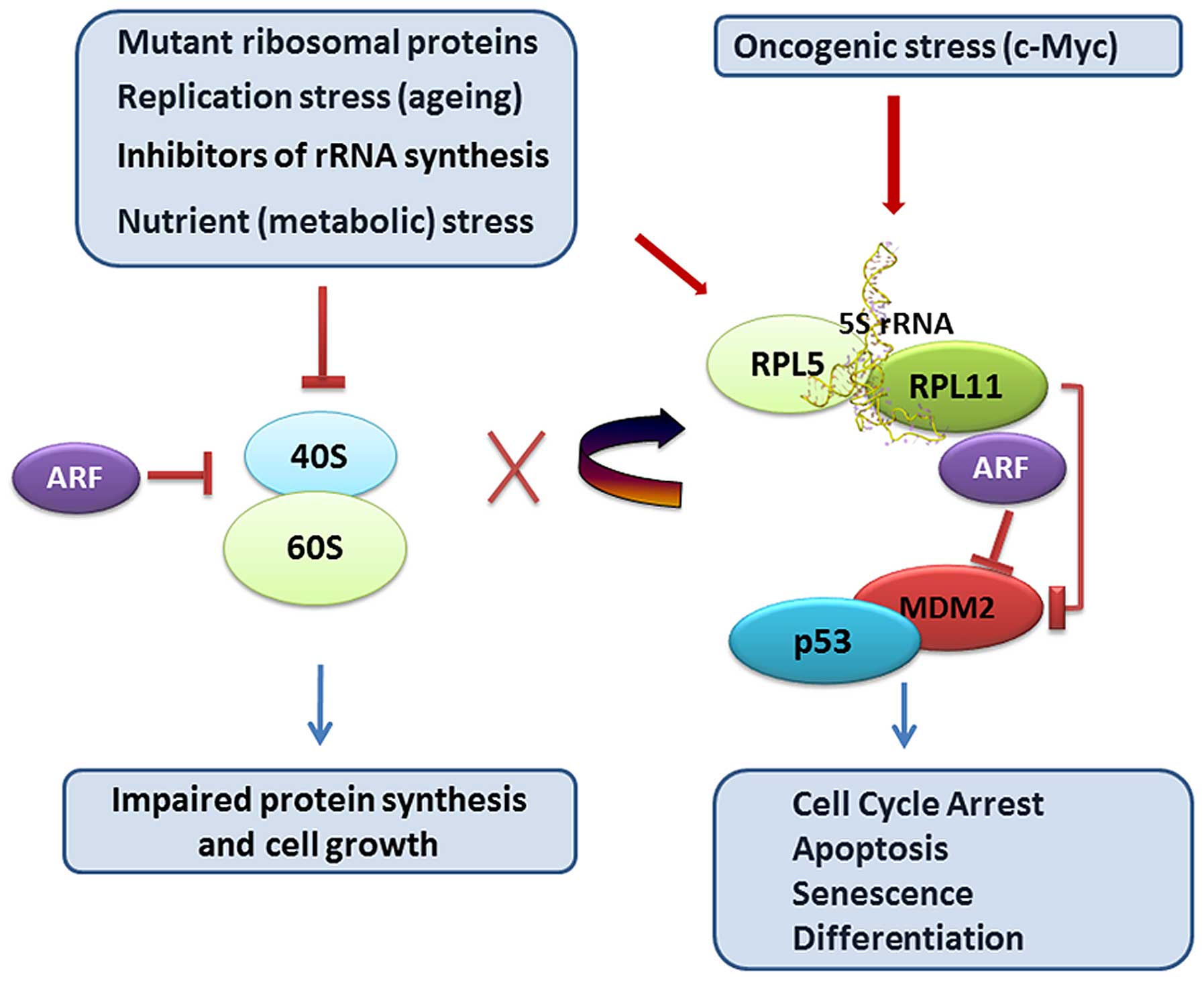
60S ribosomes to MDM2 E3 ligase inhibition (99–101) (Fig.
1). 5S RNP promotes cellular senescence in response to
oncogenic or replicative stress, given that oncogenic stress
accelerates rRNA transcription while replication stress delays rRNA
processing both causing imbalances in ribosome production (102) (Fig.
1). The 5S RNP complex also act as a sensor responsible for
stimulating fatty acid oxidation in response to nutrient depletion
(103), and sets the level of p53
activation by ARF (p14ARF, p19Arf), a protein induced by oncogenes
(97). The ARF and RP-MDM2
interactions are distinct regulatory pathways and function in
non-redundant manner to boost the p53 response to oncogenic c-Myc
yet to some extent they rely on each other (104). ARF is a joker in the game and
there are now a number of unresolved issues regarding the
functional interplay between ARF and 5S RNP. 5S RNP (RPL11/RPL5/5S
rRNA and MDM2) has now with these findings emerged as a critical
coordinator of signaling pathways at the interface of cell growth
and proliferation control. Intuitively, p53 would then be
influenced by a number of other factors regulating 5S RNP (96,97).
What is the functional relevance of the 5S
RNP-Mdm2-p53 pathway in DBA? Mice with reduced levels of Rps19,
that display hallmarks of DBA and p53 activation, were crossed with
Mdm2C305F knock-in mice (105). The Mdm2C305F mice have
a disrupted 5S RNP-Mdm2 interaction (98) since the MDM2C305F
mutation causes a collapse of the MDM2 zinc finger, with subsequent
loss of RPL5 and RPL11 binding (106–108). Upon induction of Rps19
deficiency, a disrupted 5S RNP-Mdm2 interaction by
Mdm2C305F was able to partially reverse the p53 response
and improve the expansion of hematopoietic progenitors in
vitro, and the anemia became less severe (105). One may then conclude that p53
activation through 5S RNP plays a role in DBA pathogenesis although
it is not the only mechanism involved. The role of p53 is debated,
and the anemia seen in zebrafish models of DBA is sometimes not
ameliorated by the concomitant inactivation of p53 (109–113). Discrepancies among the studies
may in part be explained by the fact that a more complete knockdown
of an RP often results in severe p53-independent phenotypes,
whereas a milder reduction generates a milder p53-dependent
phenotype. We must also keep in mind that p63 and p73 in some
settings may serve as a back-up for p53 functions (109).
Deregulation of the 5S RNP-MDM2-p53 pathway may have
a functional role in the evolution of 5q- syndrome and DBA into
malignancy such as leukemia. It is not far-fetched to suggest that
the chronic growth inhibition caused by p53 in turn could select
for mutations that promote unrestricted growth and overcomes p53
function (for example in RPL11, RPL5, MDM2 or TP53).
Mutation in TP53 is considered a critical step in the
progression of the 5q- syndrome to AML (114,115). An unresolved issue at the moment
relates to the involvement of RPL11 and RPL5 since
they are frequently mutated in DBA to begin with, and thus raising
questions about the role of 5S RNP checkpoint in these patients.
Indeed, heterozygous conditional loss of Rpl11 in adult mice
triggered anemia similar to DBA patients (38), but the mice were more prone to
radiation-induced lymphomagenesis, and also failed to induce p53
when treated with agents triggering ribosomal stress for example
Actinomycin D (38). This is
similar to MDM2C305F knock-in mice that fail to mount a
p53 response upon treatment with Actinomycin D (98). Most studies that describe an
increased association of RPL11/RPL5 with MDM2 rely on Actinomycin D
treatment or a severe reduction of an RP. DBA, however, develops on
a heterozygous (RP+/−) background as a consequence of RP
gene haploinsufficiency in hematopoiesis. Whether in the
Rpl11+/− cells there is sufficient residual RPL11
and/or RPL5 for the checkpoint to operate is not clear. There is a
need to better understand the dynamics of RPL5/L11 binding to MDM2
in the context of reduced levels of one component of the 5S RNP
complex and explore 5S RNP-independent mechanisms. For example, one
such mechanism potentially relevant to cancer development is
related to the AKT pathway. RP mutations in zebrafish suppress
activity of the AKT pathway resulting in proteasomal degradation of
p53 and by re-activating the AKT pathway stabilization of p53 was
restored (116,117). In support, RP-deficient zebrafish
embryos (similar to RP haploinsufficient zebrafish tumor cells)
exhibited normal p53 transcription, but reduced levels of p53
protein, and an impaired p53 response to DNA damage (36,116,117). The role of AKT pathway in RPL11
deficient cells should therefore be explored. In summary,
accumulating evidence from cell culture, mouse models and DBA
patients indicate the importance of maintaining a normal 5S RNP
regulation of p53, although a number of unresolved issues remains
(38). Encouraging for the future
is that the molecular anatomy of the MDM2-RPL11 complex have been
resolved in greater detail and this allows for efforts to design
tailor-made drugs (118). Such
compounds may then either enhance or block the p53 response with
potential benefits to cancer and DBA patients, respectively.
In addition to the 5S RNP complex, other possible
signaling molecules are thought to be activated in ribosome
deficient cells and that may converge on p53 to increase its
activity. ATR and ATM kinases are key components of the replication
stress and DNA-damage checkpoints contributing to p53 activation.
The ATR-Chk1 pathway was implicated in cell cycle arrest induced by
inhibition of rRNA synthesis using Actinomycin D although in the
absence of DNA damage (119), and
was also found activated in RPS19-deficient human cells (120). Increased levels of DNA damage
response markers including γH2AX were detected in U2OS cancer cells
depleted of RPS9 (uS4) (27).
Another potential mechanism could be related to maintenance of
proper nucleolar structure and genome stability. The nucleolus
plays an important role in the spatial organization of certain
heterochromatin enriched chromosome domains (121). Disruption of the heterochromatin
architecture surrounding nucleoli has been described in cells
depleted of RPs indicating there is a fine balance between ribosome
biogenesis and chromatin organization (122). Altered organization of
heterochromatin including silent rDNA may predispose cells to
genome instability and DNA damage (123).
Autophagy is probably a relatively common cellular
response to loss of an RP. Autophagy could be dependent or
independent of mTOR and p53 in a cell type-specific manner
(117,124). There are other p53-independent
effects seen in cells with defects in ribosome biogenesis for
example directed degradation of the E2F-1 transcription factor.
p53-independent ribosome biogenesis effects have been reviewed
(84,125–127). In essence it is clear that
activation of specific cell protective mechanisms appears as a
common response to a shortage in ribosomes.
Other potential mechanisms that may play a role in
cancers with RP mutations and in the ribosomopathies are related to
the hypothesis that defective maturation of ribosomal subunits
could delay translation of certain mRNAs or that malfunction of
accumulated ribosomal precursors may cause aberrant translation
(reduced fidelity). It may involve differential translation of
specific mRNA transcripts or the use of alternative translation
initiation sites. Both quantitative variations in actual ribosome
numbers and qualitative alterations such as lack of rRNA
modifications of the ribosomes have been reported. A first example
is X-linked DKC, caused by a mutation in DKC1, which encodes
dyskerin (51). Nucleolar dyskerin
associates with a specific group of snoRNPs known as H/ACA, which
function in the pseudo-uridylation of rRNAs, but mutant DKC1 alters
the rRNA pseudo-uridylation pattern of ribosomes reducing
translation of some mRNAs (53). A
second example is fibrillarin, a nucleolar rRNA methyl-transferase
(52). p53 represses fibrillarin
by direct protein-protein interaction and high levels of
fibrillarin are accompanied by abnormal rRNA methylation patterns
and impaired translational fidelity (128). In this setting, p53 acts as a
surveyor of protein synthesis by its ability to regulate ribosome
activity (128). The translation
fidelity model has gathered additional experimental evidence. The
RPL10 Arg98Ser mutant, the most commonly identified ribosomal
mutation in acute T-ALL, was functionally evaluated in yeast
(129). The mutation leads to a
failure to produce 60S followed by degradation of the defective
ribosomes (129). The 60S subunit
shortage puts pressure on cells to select for suppressors of the
ribosome biogenesis defect, allowing the yeast cells to boost
ribosome production to sustain cell proliferation (129). However, the consequence of this
bypass is synthesis of defective ribosomes that wreak havoc in the
mRNA translation process (129).
Whether similar mechanisms exist in humans and how they function
remains to be investigated. It is interesting to note that some of
the RPs mutated in cancer including RPL5, RPL10 and RPS20 are known
to bind directly to mRNAs, moreover, two of them RPL5 and RPL10,
have a preferential association with monosomes reflecting ribosome
heterogeneity (15).
Another possibility to explain how defects in the
synthesis or function of the ribosomes could affect the pattern of
translated mRNAs and possibly lead to cell transformation involves
changes in the mRNA translation patterns. A study in mice revealed
a selective reduction in the translation of Hox mRNAs following
deletion of Rpl38 (83),
and as another example serves the transcription factor GATA1 being
critical for normal erythropoiesis. Its mRNA is inefficiently
translated in DBA patients (130), while mutated in other DBA cases
(131). In an interesting twist,
GATA1 binding to RP gene promoters is important to sustain high
levels of RPs in erythroid cells (132). A more specific hypothesis that
has been discussed is that a ribosome deficit may impact on the
translation patterns favoring the synthesis of oncogenic proteins
by altering the ratio between translation initiation and elongation
(133). Related to this is the
hypothesis that a reduced number of ribosomes may cause a selective
reduced translation of mRNAs that are difficult to translate while
other mRNA could become increasingly translated. Indeed, a decrease
in p53 mRNA translation has been suspected to be of relevance
during tumor development (36).
Reduced mRNA translation may also result in a shortage of DNA
replication and repair factors as well as histones that in turn may
result in genome instability. Ribosome profiling will in the
contexts of pre-existing ribosome biogenesis or mature ribosome
defects become an essential tool to study changes in translation
patterns and finding novel targets for intervention (134).
RPs are often regulated in surprisingly
sophisticated manner and several RPs possess extra-ribosomal
functions. In addition to their roles in ribosome biogenesis and
mature ribosome function, some RPs are involved in DNA repair,
transcription, RNA processing and apoptosis (82,135–137). A few of these extra ribosomal
functions are relevant to discuss in the context of cancer
development. To begin with, a number of RPs may affect cell growth
to promote cancer cell proliferation. For example, overexpression
of RPS3A leads to the transformation of mouse NIH3T3 cells and the
formation of tumors in nude mice (138). Another example is RPS13 (uS15)
that promotes gastric cancer growth by decreasing levels of p27Kip1
(139). Upregulation of RPS13
accelerated the growth, enhanced in vitro colony formation
and soft agar growth, and promoted in vivo tumor formation
whereas downregulation of RPS13 in gastric cancer cells led to
G1 arrest (139).
RPS13 as well as RPL23 (uL14) may also suppress drug-induced
apoptosis of gastric cancer cells (140). Growth inhibitory functions of RPs
have been described as well. The most obvious examples are perhaps
RPL5 and RPL11 that when overexpressed inhibit MDM2 (141). Many other RPs including RPS15
also bind MDM2 and may impact on the p53 response (57,142). Decreased levels of RPL41 (eL41)
led to anchorage-independent growth of NIH3T3 cells in soft agar
and increased tumor growth in mice (143), while in contrast the enforced
expression of RPL41 triggered cell cycle arrest and sensitized
cancer cells to cisplatin (143).
One must emphasize that cells are sensitive to enforced
disturbances in the balance of RPs, and even that certain tags when
fused to RPs including GFP, HA or FLAG may prevent or interfere
with an RPs assembly into ribosomes (144). Therefore, anti-proliferative
effects stemming from the ectopic overexpression of RPs may be
indirect.
There are more elaborate mechanisms relevant to
bring up in the context of the cancer-associated mutations
occurring in RPL5, RPL22 and RPL10. For example, RPL10 has been
linked to regulation of the oncogenic transcription factor JUN and
other non-ribosome related proteins (145), and these functions could
potentially be altered by the RPL10-Arg98Ser mutant with
implications for cancer development. Another intriguing example is
the inactivation of RPL22 that enhances transformation potential
through induction of the Lin28B stemness factor (60). The mechanism whereby a deficiency
in RPL22 induces Lin28B is not known. RPL22 is an RNA binding
protein (146) but it also
associates with chromatin and is involved in gene repression
through complex formation with linker histone H1 (147). The possibility that RPL22 has
specific functions in gene regulation on a transcriptional level
must therefore be taken into consideration. This finding, together
with the unusual mode of Rpl22 regulation in mice (17) and a number of links to p53
regulation (37,88,148,149) suggest that RPL22 is a very
interesting candidate for use in diagnostic, prognostic and
therapeutic applications related to cancer.
The rate of cell growth is often in proportion to
the numbers of new ribosomes made (150,151). It may therefore not come as a
surprise to learn that many anticancer drugs interfere with RNA pol
I transcription or ribosomal RNA (rRNA) metabolism leading to
preferential targeting of dividing cancer cells. Inhibition of
ribosome biogenesis by chemotherapeutic drugs may contribute
significantly to the efficacy of therapeutic regimens. Ribosome
biogenesis has the potential to be more effectively exploited as a
target in anticancer therapy given that it is one of the major
biosynthetic activities in a cancer cell. RNA Pol I, the
multiprotein complex that synthesizes rRNA, is very active in most
cancer cells (152). Selective
inhibitors of RNA Pol I may therefore offer a general therapeutic
strategy to block cancer cell proliferation and small molecule
compounds that specifically inhibit rDNA transcription have been
developed by academic teams and biotech companies (153). One compound CX-3543, target rRNA
synthesis by disrupting G-quadruplex DNA structures in the G-rich
region of the rRNA repeat, thereby altering the binding of proteins
required for rRNA transcription (154). A second compound, CX-5461 is an
inhibitor of RNA pol I transcription that works by specifically
impairing the binding of SL1/TIF-1B to the rDNA promoter thereby
inhibiting the initiation of rRNA synthesis (155). This latter compound selectively
inhibits Pol I-driven transcription relative to Pol II-driven
transcription, DNA replication, and translation. CX-5461
selectively kills B-lymphoma cells in vivo by induction of
p53-dependent apoptotic signaling (156). The small molecule and acridine
derivative, BMH-21 was found to have potent antitumorigenic
activity (157). BMH-21
intercalates into GC-rich sequences in rDNA genes, and represses
RNA Pol I transcription (158). A
related compound, the acridine derivative CID-765471, inhibits rDNA
transcription and activates p53 through 5S RNP also in the absence
of detectable DNA damage (159).
The mechanism involved in the case of CID-765471 is similar to
BMH-21 in that there is a selective degradation of the RPA194
subunit of RNA polymerase I. Degradation of RPA194 could be a
common event in the case of nucleolar disruption by non-genotoxic
acridines, however it is not a general feature of all rDNA
intercalating compounds (159).
The type of anticancer activity and non-genotoxic activation of p53
represented by these different compounds mentioned holds great
promise in future anticancer therapy, but whether selective
targeting of ribosome biogenesis will be of broad clinical value in
anti-cancer treatment remain to be seen.
One may of course also consider other targets in
the ribosome biogenesis machinery including ribosomal proteins
themselves. RPS2 (uS5) was reported to be a novel therapeutic
target in prostate cancer whereas knock down of RPS2 expression had
little effect on normal cells (160), in similar ways knock down of
RPL19 (eL19) abrogated the aggressive phenotype of human prostate
cancer (161). Depletion of the
primary rRNA binding RPS9 (uS4) induced p53-dependent cell cycle
arrest and differentiation in glioma cells (27). As an interesting example, RPL39 was
found to be a protein that affects breast cancer stem cell
self-renewal through a non-biased screening approach (65). Depletion of RPL39 reduced tumor
growth and metastasis associated with fewer cancer stem cells with
a potential link to the nitric oxide synthase pathway (65). Clearly, additional studies
targeting ribosomal components in various in vivo cancer
models are warranted. Finally, one may envisage that acquired
ribosome defects, or ‘cancer-specific’ ribosomes, may become novel
targets in anticancer therapy (162).
From studies on the ribosomopathies it is clear
that impaired ribosome biogenesis is to be considered a risk factor
for cancer initiation. Remarkably, distinct and recurrent mutations
in genes encoding for ribosomal proteins (RPs) have recently been
implicated in cancer development in patients without a previous
known history of a ribosomopathy. This has been a wake-up call in
the tumor biology field and one may compare this with the parallel
and equally remarkable discovery of histone H3.3 mutations in
pediatric gliomas (163). The
role of RPs in cancer is a complex issue and while some exert a
direct effect on proto-oncogenes and tumor suppressor genes,
e.g. p53, it is possible that mutations in other RPs may
have general effects on mRNA translation. The trend evident from
the assembled sequencing data suggests that RP mutations or changes
in the expression patterns of RPs could be functionally relevant in
a large number of cancer types and cases. A more complete picture
of the relevant RPs in cancer is due to emerge from additional
cancer genome analysis projects and functional studies.
This work was supported by Karolinska Institutet
and the Swedish Research Council (VR-MH project
K2012-99X-21969-01-3) to M.L.
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Draptchinskaia N, Gustavsson P, Andersson
B, Pettersson M, Willig TN, Dianzani I, Ball S, Tchernia G, Klar J,
Matsson H, et al: The gene encoding ribosomal protein S19 is
mutated in Diamond-Blackfan anaemia. Nat Genet. 21:169–175. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vlachos A, Rosenberg PS, Atsidaftos E,
Alter BP and Lipton JM: Incidence of neoplasia in Diamond Blackfan
anemia: A report from the Diamond Blackfan Anemia Registry. Blood.
119:3815–3819. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
De Keersmaecker K, Atak ZK, Li N, Vicente
C, Patchett S, Girardi T, Gianfelici V, Geerdens E, Clappier E,
Porcu M, et al: Exome sequencing identifies mutation in CNOT3 and
ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic
leukemia. Nat Genet. 45:186–190. 2013. View Article : Google Scholar
|
|
5
|
Lawrence MS, Stojanov P, Mermel CH,
Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander
ES and Getz G: Discovery and saturation analysis of cancer genes
across 21 tumour types. Nature. 505:495–501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nieminen TT, O'Donohue MF, Wu Y, Lohi H,
Scherer SW, Paterson AD, Ellonen P, Abdel-Rahman WM, Valo S,
Mecklin JP, et al: Germline mutation of RPS20, encoding a ribosomal
protein, causes predisposition to hereditary nonpolyposis
colorectal carcinoma without DNA mismatch repair deficiency.
Gastroenterology. 147:595–598.e5. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Novetsky AP, Zighelboim I, Thompson DM Jr,
Powell MA, Mutch DG and Goodfellow PJ: Frequent mutations in the
RPL22 gene and its clinical and functional implications. Gynecol
Oncol. 128:470–474. 2013. View Article : Google Scholar
|
|
8
|
Sharma S and Lafontaine DL: ‘View From A
Bridge’: A new perspective on eukaryotic rRNA base modification.
Trends Biochem Sci. 40:560–575. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boisvert FM, van Koningsbruggen S,
Navascués J and Lamond AI: The multifunctional nucleolus. Nat Rev
Mol Cell Biol. 8:574–585. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ferreira-Cerca S, Pöll G, Gleizes PE,
Tschochner H and Milkereit P: Roles of eukaryotic ribosomal
proteins in maturation and transport of pre-18S rRNA and ribosome
function. Mol Cell. 20:263–275. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ferreira-Cerca S, Pöll G, Kühn H, Neueder
A, Jakob S, Tschochner H and Milkereit P: Analysis of the in vivo
assembly pathway of eukaryotic 40S ribosomal proteins. Mol Cell.
28:446–457. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Robledo S, Idol RA, Crimmins DL, Ladenson
JH, Mason PJ and Bessler M: The role of human ribosomal proteins in
the maturation of rRNA and ribosome production. RNA. 14:1918–1929.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kenmochi N, Kawaguchi T, Rozen S, Davis E,
Goodman N, Hudson TJ, Tanaka T and Page DC: A map of 75 human
ribosomal protein genes. Genome Res. 8:509–523. 1998.PubMed/NCBI
|
|
14
|
Ban N, Beckmann R, Cate JH, Dinman JD,
Dragon F, Ellis SR, Lafontaine DL, Lindahl L, Liljas A, Lipton JM,
et al: A new system for naming ribosomal proteins. Curr Opin Struct
Biol. 24:165–169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Slavov N, Semrau S, Airoldi E, Budnik B
and van Oudenaarden A: Differential stoichiometry among core
ribosomal proteins. Cell Rep. 13:865–873. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gilbert WV: Functional specialization of
ribosomes? Trends Biochem Sci. 36:127–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
O'Leary MN, Schreiber KH, Zhang Y, Duc AC,
Rao S, Hale JS, Academia EC, Shah SR, Morton JF, Holstein CA, et
al: The ribosomal protein Rpl22 controls ribosome composition by
directly repressing expression of its own paralog, Rpl22l1. PLoS
Genet. 9. pp. e10037082013, View Article : Google Scholar
|
|
18
|
Xirodimas DP, Sundqvist A, Nakamura A,
Shen L, Botting C and Hay RT: Ribosomal proteins are targets for
the NEDD8 pathway. EMBO Rep. 9:280–286. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ishii K, Washio T, Uechi T, Yoshihama M,
Kenmochi N and Tomita M: Characteristics and clustering of human
ribosomal protein genes. BMC Genomics. 7:372006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Branca RM, Orre LM, Johansson HJ, Granholm
V, Huss M, Pérez-Bercoff Å, Forshed J, Käll L and Lehtiö J: HiRIEF
LC-MS enables deep proteome coverage and unbiased proteogenomics.
Nat Meth. 11:59–62. 2014. View Article : Google Scholar
|
|
21
|
Lafontaine DL: Noncoding RNAs in
eukaryotic ribosome biogenesis and function. Nat Struct Mol Biol.
22:11–19. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van Heesch S, van Iterson M, Jacobi J,
Boymans S, Essers PB, de Bruijn E, Hao W, MacInnes AW, Cuppen E and
Simonis M: Extensive localization of long noncoding RNAs to the
cytosol and mono- and polyribosomal complexes. Genome Biol.
15:R62014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lafontaine DL: A ‘garbage can’ for
ribosomes: How eukaryotes degrade their ribosomes. Trends Biochem
Sci. 35:267–277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Perry RP: Balanced production of ribosomal
proteins. Gene. 401:1–3. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lam YW, Lamond AI, Mann M and Andersen JS:
Analysis of nucleolar protein dynamics reveals the nuclear
degradation of ribosomal proteins. Curr Biol. 17:749–760. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Warner JR: In the absence of ribosomal RNA
synthesis, the ribosomal proteins of HeLa cells are synthesized
normally and degraded rapidly. J Mol Biol. 115:315–333. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lindström MS and Nistér M: Silencing of
ribosomal protein S9 elicits a multitude of cellular responses
inhibiting the growth of cancer cells subsequent to p53 activation.
PLoS One. 5:e95782010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Badhai J, Fröjmark AS, Razzaghian HR,
Davey E, Schuster J and Dahl N: Posttranscriptional down-regulation
of small ribosomal subunit proteins correlates with reduction of
18S rRNA in RPS19 deficiency. FEBS Lett. 583:2049–2053. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Payne EM, Virgilio M, Narla A, Sun H,
Levine M, Paw BH, Berliner N, Look AT, Ebert BL and Khanna-Gupta A:
L-Leucine improves the anemia and developmental defects associated
with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR
pathway. Blood. 120:2214–2224. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lambertsson A: The minute genes in
Drosophila and their molecular functions. Adv Genet. 38:69–134.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stewart MJ and Denell R: Mutations in the
Drosophila gene encoding ribosomal protein S6 cause tissue
overgrowth. Mol Cell Biol. 13:2524–2535. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Watson KL, Konrad KD, Woods DF and Bryant
PJ: Drosophila homolog of the human S6 ribosomal protein is
required for tumor suppression in the hematopoietic system. Proc
Natl Acad Sci USA. 89:11302–11306. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin JI, Mitchell NC, Kalcina M,
Tchoubrieva E, Stewart MJ, Marygold SJ, Walker CD, Thomas G,
Leevers SJ, Pearson RB, et al: Drosophila ribosomal protein mutants
control tissue growth non-autonomously via effects on the
prothoracic gland and ecdysone. PLoS Genet. 7:e10024082011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Amsterdam A, Sadler KC, Lai K, Farrington
S, Bronson RT, Lees JA and Hopkins N: Many ribosomal protein genes
are cancer genes in zebrafish. PLoS Biol. 2:E1392004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lai K, Amsterdam A, Farrington S, Bronson
RT, Hopkins N and Lees JA: Many ribosomal protein mutations are
associated with growth impairment and tumor predisposition in
zebrafish. Dev Dyn. 238:76–85. 2009. View Article : Google Scholar :
|
|
36
|
MacInnes AW, Amsterdam A, Whittaker CA,
Hopkins N and Lees JA: Loss of p53 synthesis in zebrafish tumors
with ribosomal protein gene mutations. Proc Natl Acad Sci USA.
105:10408–10413. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stadanlick JE, Zhang Z, Lee SY, Hemann M,
Biery M, Carleton MO, Zambetti GP, Anderson SJ, Oravecz T and Wiest
DL: Developmental arrest of T cells in Rpl22-deficient mice is
dependent upon multiple p53 effectors. J Immunol. 187:664–675.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Morgado-Palacin L, Varetti G, Llanos S,
Gómez-López G, Martinez D and Serrano M: Partial Loss of Rpl11 in
Adult mice recapitulates diamond-blackfan anemia and promotes
lymphomagenesis. Cell Rep. 13:712–722. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kazerounian S, Ciarlini PD, Yuan D,
Ghazvinian R, Alberich-Jorda M, Joshi M, Zhang H, Beggs AH and
Gazda HT: Development of soft tissue sarcomas in ribosomal proteins
L5 and S24 heterozygous mice. J Cancer. 7:32–36. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
McCann KL and Baserga SJ: Genetics.
Mysterious ribosomopathies. Science. 341:849–850. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Narla A and Ebert BL: Ribosomopathies:
Human disorders of ribosome dysfunction. Blood. 115:3196–3205.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Freed EF, Bleichert F, Dutca LM and
Baserga SJ: When ribosomes go bad: Diseases of ribosome biogenesis.
Mol Biosyst. 6:481–493. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Choesmel V, Fribourg S, Aguissa-Touré AH,
Pinaud N, Legrand P, Gazda HT and Gleizes PE: Mutation of ribosomal
protein RPS24 in Diamond-Blackfan anemia results in a ribosome
biogenesis disorder. Hum Mol Genet. 17:1253–1263. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cmejla R, Cmejlova J, Handrkova H, Petrak
J, Petrtylova K, Mihal V, Stary J, Cerna Z, Jabali Y and
Pospisilova D: Identification of mutations in the ribosomal protein
L5 (RPL5) and ribosomal protein L11 (RPL11) genes in Czech patients
with Diamond-Blackfan anemia. Hum Mutat. 30:321–327. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Farrar JE, Nater M, Caywood E, McDevitt
MA, Kowalski J, Takemoto CM, Talbot CC Jr, Meltzer P, Esposito D,
Beggs AH, et al: Abnormalities of the large ribosomal subunit
protein, Rpl35a, in Diamond-Blackfan anemia. Blood. 112:1582–1592.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Flygare J and Karlsson S: Diamond-Blackfan
anemia: Erythropoiesis lost in translation. Blood. 109:3152–3154.
2007. View Article : Google Scholar
|
|
47
|
Ebert BL, Pretz J, Bosco J, Chang CY,
Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR, et al:
Identification of RPS14 as a 5q- syndrome gene by RNA interference
screen. Nature. 451:335–339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Alter BP, Giri N, Savage SA, Peters JA,
Loud JT, Leathwood L, Carr AG, Greene MH and Rosenberg PS:
Malignancies and survival patterns in the National Cancer Institute
inherited bone marrow failure syndromes cohort study. Br J
Haematol. 150:179–188. 2010.PubMed/NCBI
|
|
49
|
Majeed F, Jadko S, Freedman MH and Dror Y:
Mutation analysis of SBDS in pediatric acute myeloblastic leukemia.
Pediatr Blood Cancer. 45:920–924. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Maserati E, Pressato B, Valli R, Minelli
A, Sainati L, Patitucci F, Marletta C, Mastronuzzi A, Poli F, Lo
Curto F, et al: The route to development of myelodysplastic
syndrome/acute myeloid leukaemia in Shwachman-Diamond syndrome: The
role of ageing, karyotype instability, and acquired chromosome
anomalies. Br J Haematol. 145:190–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Heiss NS, Knight SW, Vulliamy TJ, Klauck
SM, Wiemann S, Mason PJ, Poustka A and Dokal I: X-linked
dyskeratosis congenita is caused by mutations in a highly conserved
gene with putative nucleolar functions. Nat Genet. 19:32–38. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ge J, Rudnick DA, He J, Crimmins DL,
Ladenson JH, Bessler M and Mason PJ: Dyskerin ablation in mouse
liver inhibits rRNA processing and cell division. Mol Cell Biol.
30:413–422. 2010. View Article : Google Scholar :
|
|
53
|
Jack K, Bellodi C, Landry DM, Niederer RO,
Meskauskas A, Musalgaonkar S, Kopmar N, Krasnykh O, Dean AM,
Thompson SR, et al: rRNA pseudouridylation defects affect ribosomal
ligand binding and translational fidelity from yeast to human
cells. Mol Cell. 44:660–666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Alter BP, Giri N, Savage SA and Rosenberg
PS: Cancer in dyskeratosis congenita. Blood. 113:6549–6557. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Donadieu J, Leblanc T, Bader Meunier B,
Barkaoui M, Fenneteau O, Bertrand Y, Maier-Redelsperger M, Micheau
M, Stephan JL, Phillipe N, et al; French Severe Chronic Neutropenia
Study Group; Experience of the French Severe Chronic Neutropenia
Study Group. Analysis of risk factors for myelodysplasias,
leukemias and death from infection among patients with congenital
neutropenia. Haematologica. 90:45–53. 2005.PubMed/NCBI
|
|
56
|
Danilova N and Gazda HT: Ribosomopathies:
How a common root can cause a tree of pathologies. Dis Model Mech.
8:1013–1026. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ljungström V, Cortese D, Young E, Pandzic
T, Mansouri L, Plevova K, Ntoufa S, Baliakas P, Clifford R, Sutton
LA, et al: Whole-exome sequencing in relapsing chronic lymphocytic
leukemia: Clinical impact of recurrent RPS15 mutations. Blood. Dec
16–2015.(Epub ahead of print).
|
|
58
|
Landau DA, Tausch E, Taylor-Weiner AN,
Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M,
Böttcher S, et al: Mutations driving CLL and their evolution in
progression and relapse. Nature. 526:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tzoneva G, Perez-Garcia A, Carpenter Z,
Khiabanian H, Tosello V, Allegretta M, Paietta E, Racevskis J, Rowe
JM, Tallman MS, et al: Activating mutations in the NT5C2
nucleotidase gene drive chemotherapy resistance in relapsed ALL.
Nat Med. 19:368–371. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rao S, Lee SY, Gutierrez A, Perrigoue J,
Thapa RJ, Tu Z, Jeffers JR, Rhodes M, Anderson S, Oravecz T, et al:
Inactivation of ribosomal protein L22 promotes transformation by
induction of the stemness factor, Lin28B. Blood. 120:3764–3773.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ferreira AM, Tuominen I, van Dijk-Bos K,
Sanjabi B, van der Sluis T, van der Zee AG, Hollema H, Zazula M,
Sijmons RH, Aaltonen LA, et al: High frequency of RPL22 mutations
in microsatellite-unstable colorectal and endometrial tumours. Hum
Mutat. 35:1442–1445. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Nagarajan N, Bertrand D, Hillmer AM, Zang
ZJ, Yao F, Jacques PÉ, Teo AS, Cutcutache I, Zhang Z, Lee WH, et
al: Whole-genome reconstruction and mutational signatures in
gastric cancer. Genome Biol. 13:R1152012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yang M, Sun H, Wang H, Zhang S, Yu X and
Zhang L: Down-regulation of ribosomal protein L22 in non-small cell
lung cancer. Med Oncol. 30:6462013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lee W, Teckie S, Wiesner T, Ran L, Prieto
Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, et al: PRC2 is
recurrently inactivated through EED or SUZ12 loss in malignant
peripheral nerve sheath tumors. Nat Genet. 46:1227–1232. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Dave B, Granados-Principal S, Zhu R, Benz
S, Rabizadeh S, Soon-Shiong P, Yu KD, Shao Z, Li X, Gilcrease M, et
al: Targeting RPL39 and MLF2 reduces tumor initiation and
metastasis in breast cancer by inhibiting nitric oxide synthase
signaling. Proc Natl Acad Sci USA. 111:8838–8843. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Nakagawa H, Wardell CP, Furuta M,
Taniguchi H and Fujimoto A: Cancer whole-genome sequencing: Present
and future. Oncogene. 34:5943–5950. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Boria I, Quarello P, Avondo F, Garelli E,
Aspesi A, Carando A, Campagnoli MF, Dianzani I and Ramenghi U: A
new database for ribosomal protein genes which are mutated in
Diamond-Blackfan Anemia. Hum Mutat. 29:E263–E270. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wang W, Nag S, Zhang X, Wang MH, Wang H,
Zhou J and Zhang R: Ribosomal proteins and human diseases:
pathogenesis, molecular mechanisms, and therapeutic implications.
Med Res Rev. 35:225–285. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kowalczyk P, Woszczyński M and Ostrowski
J: Increased expression of ribosomal protein S2 in liver tumors,
post-hepactomized livers, and proliferating hepatocytes in vitro.
Acta Biochim Pol. 49:615–624. 2002.
|
|
70
|
Wang H, Zhao LN, Li KZ, Ling R, Li XJ and
Wang L: Overexpression of ribosomal protein L15 is associated with
cell proliferation in gastric cancer. BMC Cancer. 6:912006.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Vaarala MH, Porvari KS, Kyllönen AP,
Mustonen MV, Lukkarinen O and Vihko PT: Several genes encoding
ribosomal proteins are over-expressed in prostate-cancer cell
lines: Confirmation of L7a and L37 over-expression in
prostate-cancer tissue samples. Int J Cancer. 78:27–32. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bee A, Ke Y, Forootan S, Lin K, Beesley C,
Forrest SE and Foster CS: Ribosomal protein l19 is a prognostic
marker for human prostate cancer. Clin Cancer Res. 12:2061–2065.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Sim EU, Ang CH, Ng CC, Lee CW and
Narayanan K: Differential expression of a subset of ribosomal
protein genes in cell lines derived from human nasopharyngeal
epithelium. J Hum Genet. 55:118–120. 2010. View Article : Google Scholar
|
|
74
|
Yong WH, Shabihkhani M, Telesca D, Yang S,
Tso JL, Menjivar JC, Wei B, Lucey GM, Mareninov S, Chen Z, et al:
Ribosomal proteins RPS11 and RPS20, two stress-response markers of
glioblastoma stem cells, are novel predictors of poor prognosis in
glioblastoma patients. PLoS One. 10:e01413342015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yan TT, Fu XL, Li J, Bian YN, Liu DJ, Hua
R, Ren LL, Li CT, Sun YW, Chen HY, et al: Downregulation of RPL15
may predict poor survival and associate with tumor progression in
pancreatic ductal adenocarcinoma. Oncotarget. 6:37028–37042.
2015.PubMed/NCBI
|
|
76
|
Kobayashi T, Sasaki Y, Oshima Y, Yamamoto
H, Mita H, Suzuki H, Toyota M, Tokino T, Itoh F, Imai K, et al:
Activation of the ribosomal protein L13 gene in human
gastrointestinal cancer. Int J Mol Med. 18:161–170. 2006.PubMed/NCBI
|
|
77
|
Song MJ, Jung CK, Park CH, Hur W, Choi JE,
Bae SH, Choi JY, Choi SW, Han NI and Yoon SK: RPL36 as a prognostic
marker in hepatocellular carcinoma. Pathol Int. 61:638–644. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
de Las Heras-Rubio A, Perucho L, Paciucci
R, Vilardell J and LLeonart ME: Ribosomal proteins as novel players
in tumorigenesis. Cancer Metastasis Rev. 33:115–141.
2014.PubMed/NCBI
|
|
79
|
De Keersmaecker K, Sulima SO and Dinman
JD: Ribosomopathies and the paradox of cellular hypo- to
hyperproliferation. Blood. 125:1377–1382. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ruggero D and Pandolfi PP: Does the
ribosome translate cancer? Nat Rev Cancer. 3:179–192. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Warner JR and McIntosh KB: How common are
extraribosomal functions of ribosomal proteins? Mol Cell. 34:3–11.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Barkić M, Crnomarković S, Grabusić K,
Bogetić I, Panić L, Tamarut S, Cokarić M, Jerić I, Vidak S and
Volarević S: The p53 tumor suppressor causes congenital
malformations in Rpl24-deficient mice and promotes their survival.
Mol Cell Biol. 29:2489–2504. 2009. View Article : Google Scholar
|
|
83
|
Kondrashov N, Pusic A, Stumpf CR, Shimizu
K, Hsieh AC, Xue S, Ishijima J, Shiroishi T and Barna M:
Ribosome-mediated specificity in Hox mRNA translation and
vertebrate tissue patterning. Cell. 145:383–397. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Holmberg Olausson K, Nistér M and
Lindström MS: p53-dependent and -independent nucleolar stress
responses. Cells. 1:774–798. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
James A, Wang Y, Raje H, Rosby R and
DiMario P: Nucleolar stress with and without p53. Nucleus.
5:402–426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Panić L, Tamarut S, Sticker-Jantscheff M,
Barkić M, Solter D, Uzelac M, Grabusić K and Volarević S: Ribosomal
protein S6 gene haploinsufficiency is associated with activation of
a p53-dependent checkpoint during gastrulation. Mol Cell Biol.
26:8880–8891. 2006. View Article : Google Scholar
|
|
87
|
McGowan KA, Li JZ, Park CY, Beaudry V,
Tabor HK, Sabnis AJ, Zhang W, Fuchs H, de Angelis MH, Myers RM, et
al: Ribosomal mutations cause p53-mediated dark skin and
pleiotropic effects. Nat Genet. 40:963–970. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Anderson SJ, Lauritsen JP, Hartman MG,
Foushee AM, Lefebvre JM, Shinton SA, Gerhardt B, Hardy RR, Oravecz
T and Wiest DL: Ablation of ribosomal protein L22 selectively
impairs alphabeta T cell development by activation of a
p53-dependent checkpoint. Immunity. 26:759–772. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Barlow JL, Drynan LF, Hewett DR, Holmes
LR, Lorenzo-Abalde S, Lane AL, Jolin HE, Pannell R, Middleton AJ,
Wong SH, et al: A p53-dependent mechanism underlies macrocytic
anemia in a mouse model of human 5q- syndrome. Nat Med. 16:59–66.
2010. View Article : Google Scholar :
|
|
90
|
Terzian T and Box N: Genetics of ribosomal
proteins: ‘curiouser and curiouser’. PLoS Genet. 9:e10033002013.
View Article : Google Scholar
|
|
91
|
Fumagalli S, Di Cara A, Neb-Gulati A, Natt
F, Schwemberger S, Hall J, Babcock GF, Bernardi R, Pandolfi PP and
Thomas G: Absence of nucleolar disruption after impairment of 40S
ribosome biogenesis reveals an rpL11-translation-dependent
mechanism of p53 induction. Nat Cell Biol. 11:501–508. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Volarevic S, Stewart MJ, Ledermann B,
Zilberman F, Terracciano L, Montini E, Grompe M, Kozma SC and
Thomas G: Proliferation, but not growth, blocked by conditional
deletion of 40S ribosomal protein S6. Science. 288:2045–2047. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Jaako P, Flygare J, Olsson K, Quere R,
Ehinger M, Henson A, Ellis S, Schambach A, Baum C, Richter J, et
al: Mice with ribosomal protein S19 deficiency develop bone marrow
failure and symptoms like patients with Diamond-Blackfan anemia.
Blood. 118:6087–6096. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Fumagalli S, Ivanenkov VV, Teng T and
Thomas G: Supra-induction of p53 by disruption of 40S and 60S
ribosome biogenesis leads to the activation of a novel G2/M
checkpoint. Genes Dev. 26:1028–1040. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Teng T, Mercer CA, Hexley P, Thomas G and
Fumagalli S: Loss of tumor suppressor RPL5/RPL11 does not induce
cell cycle arrest but impedes proliferation due to reduced ribosome
content and translation capacity. Mol Cell Biol. 33:4660–4671.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Donati G, Peddigari S, Mercer CA and
Thomas G: 5S ribosomal RNA is an essential component of a nascent
ribosomal precursor complex that regulates the Hdm2-p53 checkpoint.
Cell Rep. 4:87–98. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Sloan KE, Bohnsack MT and Watkins NJ: The
5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar
stress. Cell Rep. 5:237–247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Macias E, Jin A, Deisenroth C, Bhat K, Mao
H, Lindström MS and Zhang Y: An ARF-independent c-MYC-activated
tumor suppression pathway mediated by ribosomal protein-Mdm2
Interaction. Cancer Cell. 18:231–243. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Deisenroth C and Zhang Y: Ribosome
biogenesis surveillance: Probing the ribosomal protein-Mdm2-p53
pathway. Oncogene. 29:4253–4260. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Miliani de Marval PL and Zhang Y: The
RP-Mdm2-p53 pathway and tumorigenesis. Oncotarget. 2:234–238. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zhang Y and Lu H: Signaling to p53:
Ribosomal proteins find their way. Cancer Cell. 16:369–377. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Nishimura K, Kumazawa T, Kuroda T,
Katagiri N, Tsuchiya M, Goto N, Furumai R, Murayama A, Yanagisawa J
and Kimura K: Perturbation of ribosome biogenesis drives cells into
senescence through 5S RNP-mediated p53 activation. Cell Rep.
10:1310–1323. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Liu Y, He Y, Jin A, Tikunov AP, Zhou L,
Tollini LA, Leslie P, Kim TH, Li LO, Coleman RA, et al: Ribosomal
protein-Mdm2-p53 pathway coordinates nutrient stress with lipid
metabolism by regulating MCD and promoting fatty acid oxidation.
Proc Natl Acad Sci USA. 111:E2414–E2422. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Meng X, Carlson NR, Dong J and Zhang Y:
Oncogenic c-Myc-induced lymphomagenesis is inhibited
non-redundantly by the p19Arf-Mdm2-p53 and RP-Mdm2-p53 pathways.
Oncogene. 34:5709–5717. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Jaako P, Debnath S, Olsson K, Zhang Y,
Flygare J, Lindström MS, Bryder D and Karlsson S: Disruption of the
5S RNP-Mdm2 interaction significantly improves the erythroid defect
in a mouse model for Diamond-Blackfan anemia. Leukemia.
29:2221–2229. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lindström MS, Deisenroth C and Zhang Y:
Putting a finger on growth surveillance: Insight into MDM2 zinc
finger-ribosomal protein interactions. Cell Cycle. 6:434–437. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Lindström MS, Jin A, Deisenroth C, White
Wolf G and Zhang Y: Cancer-associated mutations in the MDM2 zinc
finger domain disrupt ribosomal protein interaction and attenuate
MDM2-induced p53 degradation. Mol Cell Biol. 27:1056–1068. 2007.
View Article : Google Scholar :
|
|
108
|
Zhang Q, Xiao H, Chai SC, Hoang QQ and Lu
H: Hydrophilic residues are crucial for ribosomal protein L11
(RPL11) interaction with zinc finger domain of MDM2 and p53 protein
activation. J Biol Chem. 286:38264–38274. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Danilova N, Sakamoto KM and Lin S:
Ribosomal protein S19 deficiency in zebrafish leads to
developmental abnormalities and defective erythropoiesis through
activation of p53 protein family. Blood. 112:5228–5237. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Torihara H, Uechi T, Chakraborty A, Shinya
M, Sakai N and Kenmochi N: Erythropoiesis failure due to RPS19
deficiency is independent of an activated Tp53 response in a
zebrafish model of Diamond-Blackfan anaemia. Br J Haematol.
152:648–654. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Uechi T, Nakajima Y, Chakraborty A,
Torihara H, Higa S and Kenmochi N: Deficiency of ribosomal protein
S19 during early embryogenesis leads to reduction of erythrocytes
in a zebrafish model of Diamond-Blackfan anemia. Hum Mol Genet.
17:3204–3211. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Uechi T, Nakajima Y, Nakao A, Torihara H,
Chakraborty A, Inoue K and Kenmochi N: Ribosomal protein gene
knockdown causes developmental defects in zebrafish. PLoS One.
1:e372006. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Yadav GV, Chakraborty A, Uechi T and
Kenmochi N: Ribosomal protein deficiency causes Tp53-independent
erythropoiesis failure in zebrafish. Int J Biochem Cell Biol.
49:1–7. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Jädersten M, Saft L, Smith A,
Kulasekararaj A, Pomplun S, Göhring G, Hedlund A, Hast R,
Schlegelberger B, Porwit A, et al: TP53 mutations in low-risk
myelodysplastic syndromes with del(5q) predict disease progression.
J Clin Oncol. 29:1971–1979. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Saft L, Karimi M, Ghaderi M, Matolcsy A,
Mufti GJ, Kulasekararaj A, Göhring G, Giagounidis A, Selleslag D,
Muus P, et al: p53 protein expression independently predicts
outcome in patients with lower-risk myelodysplastic syndromes with
del(5q). Haematologica. 99:1041–1049. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Antunes AT, Goos YJ, Pereboom TC, Hermkens
D, Wlodarski MW, Da Costa L and MacInnes AW: Ribosomal Protein
mutations result in constitutive p53 protein degradation through
impairment of the AKT pathway. PLoS Genet. 11:e10053262015.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Heijnen HF, van Wijk R, Pereboom TC, Goos
YJ, Seinen CW, van Oirschot BA, van Dooren R, Gastou M, Giles RH,
van Solinge W, et al: Ribosomal protein mutations induce autophagy
through S6 kinase inhibition of the insulin pathway. PLoS Genet.
10:e10043712014. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zheng J, Lang Y, Zhang Q, Cui D, Sun H,
Jiang L, Chen Z, Zhang R, Gao Y, Tian W, et al: Structure of human
MDM2 complexed with RPL11 reveals the molecular basis of p53
activation. Genes Dev. 29:1524–1534. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Ma H and Pederson T: The nucleolus stress
response is coupled to an ATR-Chk1-mediated G2 arrest. Mol Biol
Cell. 24:1334–1342. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Danilova N, Bibikova E, Covey TM,
Nathanson D, Dimitrova E, Konto Y, Lindgren A, Glader B, Radu CG,
Sakamoto KM, et al: The role of the DNA damage response in
zebrafish and cellular models of Diamond Blackfan anemia. Dis Model
Mech. 7:895–905. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Padeken J and Heun P: Nucleolus and
nuclear periphery: Velcro for heterochromatin. Curr Opin Cell Biol.
28:54–60. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
O'Donohue MF, Choesmel V, Faubladier M,
Fichant G and Gleizes PE: Functional dichotomy of ribosomal
proteins during the synthesis of mammalian 40S ribosomal subunits.
J Cell Biol. 190:853–866. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Peng JC and Karpen GH: H3K9 methylation
and RNA interference regulate nucleolar organization and repeated
DNA stability. Nat Cell Biol. 9:25–35. 2007. View Article : Google Scholar
|
|
124
|
Boglev Y, Badrock AP, Trotter AJ, Du Q,
Richardson EJ, Parslow AC, Markmiller SJ, Hall NE, de Jong-Curtain
TA, Ng AY, et al: Autophagy induction is a Tor- and
Tp53-independent cell survival response in a zebrafish model of
disrupted ribosome biogenesis. PLoS Genet. 9:e10032792013.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Donati G, Brighenti E, Vici M, Mazzini G,
Treré D, Montanaro L and Derenzini M: Selective inhibition of rRNA
transcription downregulates E2F-1: A new p53-independent mechanism
linking cell growth to cell proliferation. J Cell Sci.
124:3017–3028. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Donati G, Montanaro L and Derenzini M:
Ribosome biogenesis and control of cell proliferation: p53 is not
alone. Cancer Res. 72:1602–1607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Orsolic I, Jurada D, Pullen N, Oren M,
Eliopoulos AG and Volarevic S: The relationship between the
nucleolus and cancer: Current evidence and emerging paradigms.
Semin Cancer Biol. Dec 23–2015.(Epub ahead of print). View Article : Google Scholar
|
|
128
|
Marcel V, Ghayad SE, Belin S, Therizols G,
Morel AP, Solano-Gonzàlez E, Vendrell JA, Hacot S, Mertani HC,
Albaret MA, et al: p53 acts as a safeguard of translational control
by regulating fibrillarin and rRNA methylation in cancer. Cancer
Cell. 24:318–330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Sulima SO, Patchett S, Advani VM, De
Keersmaecker K, Johnson AW and Dinman JD: Bypass of the pre-60S
ribosomal quality control as a pathway to oncogenesis. Proc Natl
Acad Sci USA. 111:5640–5645. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Ludwig LS, Gazda HT, Eng JC, Eichhorn SW,
Thiru P, Ghazvinian R, George TI, Gotlib JR, Beggs AH, Sieff CA, et
al: Altered translation of GATA1 in Diamond-Blackfan anemia. Nat
Med. 20:748–753. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Sankaran VG, Ghazvinian R, Do R, Thiru P,
Vergilio JA, Beggs AH, Sieff CA, Orkin SH, Nathan DG, Lander ES, et
al: Exome sequencing identifies GATA1 mutations resulting in
Diamond-Blackfan anemia. J Clin Invest. 122:2439–2443. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Amanatiadou EP, Papadopoulos GL,
Strouboulis J and Vizirianakis IS: GATA1 and PU.1 bind to ribosomal
protein genes in erythroid cells: Implications for ribosomopathies.
PLoS One. 10:e01400772015. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Loreni F, Mancino M and Biffo S:
Translation factors and ribosomal proteins control tumor onset and
progression: How? Oncogene. 33:2145–2156. 2014. View Article : Google Scholar
|
|
134
|
Ingolia NT: Ribosome profiling: New views
of translation, from single codons to genome scale. Nat Rev Genet.
15:205–213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Bhavsar RB, Makley LN and Tsonis PA: The
other lives of ribosomal proteins. Hum Genomics. 4:327–344. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Lindström MS: Emerging functions of
ribosomal proteins in gene-specific transcription and translation.
Biochem Biophys Res Commun. 379:167–170. 2009. View Article : Google Scholar
|
|
137
|
Wool IG: Extraribosomal functions of
ribosomal proteins. Trends Biochem Sci. 21:164–165. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Naora H, Takai I, Adachi M and Naora H:
Altered cellular responses by varying expression of a ribosomal
protein gene: Sequential coordination of enhancement and
suppression of ribosomal protein S3a gene expression induces
apoptosis. J Cell Biol. 141:741–753. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Guo X, Shi Y, Gou Y, Li J, Han S, Zhang Y,
Huo J, Ning X, Sun L, Chen Y, et al: Human ribosomal protein S13
promotes gastric cancer growth through down-regulating p27(Kip1). J
Cell Mol Med. 15:296–306. 2011. View Article : Google Scholar
|
|
140
|
Shi Y, Zhai H, Wang X, Han Z, Liu C, Lan
M, Du J, Guo C, Zhang Y, Wu K, et al: Ribosomal proteins S13 and
L23 promote multidrug resistance in gastric cancer cells by
suppressing drug-induced apoptosis. Exp Cell Res. 296:337–346.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Dai MS and Lu H: Inhibition of
MDM2-mediated p53 ubiquitination and degradation by ribosomal
protein L5. J Biol Chem. 279:44475–44482. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Daftuar L, Zhu Y, Jacq X and Prives C:
Ribosomal proteins RPL37, RPS15 and RPS20 regulate the
Mdm2-p53-MdmX network. PLoS One. 8:e686672013. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wang S, Huang J, He J, Wang A, Xu S, Huang
SF and Xiao S: RPL41, a small ribosomal peptide deregulated in
tumors, is essential for mitosis and centrosome integrity.
Neoplasia. 12:284–293. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Krüger T, Zentgraf H and Scheer U:
Intranucleolar sites of ribosome biogenesis defined by the
localization of early binding ribosomal proteins. J Cell Biol.
177:573–578. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Chan YL, Diaz JJ, Denoroy L, Madjar JJ and
Wool IG: The primary structure of rat ribosomal protein L10:
Relationship to a Jun-binding protein and to a putative Wilms'
tumor suppressor. Biochem Biophys Res Commun. 225:952–956. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Houmani JL, Davis CI and Ruf IK:
Growth-promoting properties of Epstein-Barr virus EBER-1 RNA
correlate with ribosomal protein L22 binding. J Virol.
83:9844–9853. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Ni JQ, Liu LP, Hess D, Rietdorf J and Sun
FL: Drosophila ribosomal proteins are associated with linker
histone H1 and suppress gene transcription. Genes Dev.
20:1959–1973. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Fahl SP, Harris B, Coffey F and Wiest DL:
Rpl22 Loss impairs the development of B lymphocytes by activating a
p53-dependent checkpoint. J Immunol. 194:200–209. 2015. View Article : Google Scholar :
|
|
149
|
Rashkovan M, Vadnais C, Ross J, Gigoux M,
Suh WK, Gu W, Kosan C and Möröy T: Miz-1 regulates translation of
Trp53 via ribosomal protein L22 in cells undergoing V(D)J
recombination. Proc Natl Acad Sci USA. 111:E5411–E5419. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Montanaro L, Treré D and Derenzini M:
Nucleolus, ribosomes, and cancer. Am J Pathol. 173:301–310. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Treré D, Ceccarelli C, Montanaro L, Tosti
E and Derenzini M: Nucleolar size and activity are related to pRb
and p53 status in human breast cancer. J Histochem Cytochem.
52:1601–1607. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Montanaro L, Treré D and Derenzini M: The
emerging role of RNA polymerase I transcription machinery in human
malignancy: A clinical perspective. Onco Targets Ther. 6:909–916.
2013.PubMed/NCBI
|
|
153
|
Drygin D, O'Brien SE, Hannan RD, McArthur
GA and Von Hoff DD: Targeting the nucleolus for cancer-specific
activation of p53. Drug Discov Today. 19:259–265. 2014. View Article : Google Scholar
|
|
154
|
Drygin D, Siddiqui-Jain A, O'Brien S,
Schwaebe M, Lin A, Bliesath J, Ho CB, Proffitt C, Trent K, Whitten
JP, et al: Anticancer activity of CX-3543: A direct inhibitor of
rRNA biogenesis. Cancer Res. 69:7653–7661. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Drygin D, Lin A, Bliesath J, Ho CB,
O'Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK,
Siddiqui-Jain A, et al: Targeting RNA polymerase I with an oral
small molecule CX-5461 inhibits ribosomal RNA synthesis and solid
tumor growth. Cancer Res. 71:1418–1430. 2011. View Article : Google Scholar
|
|
156
|
Bywater MJ, Poortinga G, Sanij E, Hein N,
Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, et al:
Inhibition of RNA polymerase I as a therapeutic strategy to promote
cancer-specific activation of p53. Cancer Cell. 22:51–65. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Peltonen K, Colis L, Liu H, Trivedi R,
Moubarek MS, Moore HM, Bai B, Rudek MA, Bieberich CJ and Laiho M: A
targeting modality for destruction of RNA polymerase I that
possesses anticancer activity. Cancer Cell. 25:77–90. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Colis L, Peltonen K, Sirajuddin P, Liu H,
Sanders S, Ernst G, Barrow JC and Laiho M: DNA intercalator BMH-21
inhibits RNA polymerase I independent of DNA damage response.
Oncotarget. 5:4361–4369. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Morgado-Palacin L, Llanos S,
Urbano-Cuadrado M, Blanco-Aparicio C, Megias D, Pastor J and
Serrano M: Non-genotoxic activation of p53 through the
RPL11-dependent ribosomal stress pathway. Carcinogenesis.
35:2822–2830. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Wang M, Hu Y and Stearns ME: RPS2: a novel
therapeutic target in prostate cancer. J Exp Clin Cancer Res.
28:62009. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Bee A, Brewer D, Beesley C, Dodson A,
Forootan S, Dickinson T, Gerard P, Lane B, Yao S, Cooper CS, et al:
siRNA knockdown of ribosomal protein gene RPL19 abrogates the
aggressive phenotype of human prostate cancer. PLoS One.
6:e226722011. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Marcel V, Catez F and Diaz JJ: Ribosomes:
The future of targeted therapies? Oncotarget. 4:1554–1555. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Wu G, Broniscer A, McEachron TA, Lu C,
Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al;
St. Jude Children's Research Hospital-Washington University
Pediatric Cancer Genome Project. Somatic histone H3 alterations in
pediatric diffuse intrinsic pontine gliomas and non-brainstem
glioblastomas. Nat Genet. 44:251–253. 2012. View Article : Google Scholar : PubMed/NCBI
|