Introduction
Amino acids are essential for cellular homeostasis,
growth and proliferation via their contribution to a diverse range
of cellular processes. Cells use a number of mechanisms to sense
and maintain their homeostatic levels. Intracellular levels of
amino acids are maintained by the balance between their influx,
utilization and recycling (1). The
transport of serum amino acids into cells is an active process that
is facilitated by plasma membrane-localized amino acid
transporters. The members of the L-type amino acid transporter
(LAT) family are Na+-independent transporters that
deliver neutral amino acids into cells (2). Although the LAT family plays
important roles in the development and function of normal tissues,
they are frequently increased in cancer cells (3). LAT-1 is most commonly upregulated in
multiple types of cancer and has also been used as a biomarker for
malignant cancer (4,5). In contrast to the influx of amino
acids, the degradation of proteins is also the important
intracellular mechanism for releasing free amino acids both under
steady-state conditions and during cellular stresses.
Macroautophagy (thereafter designated as autophagy) is a
self-digestive system conserved in all eukaryotic cells. Cellular
proteins and organelles are engulfed into a double-membrane vesicle
to form an autophagosome. The cargos are transported to the
lysosome and are degraded by lysosomal hydrolytic enzymes via
membrane fusion between the autophagosome and the lysosome, which
is designated as the autolysosome (6). As the recycling of amino acids by
this self-digestive mechanism is essential for supplying the
intracellular amino acid pool for cellular metabolism, autophagy is
markedly upregulated when the cells are exposed to a
nutrient-starving condition (7).
On the other hand, angiogenesis is indispensable for supporting
progressive tumor cell growth. Due to the insufficient
vascularization, autophagy is believed to be accelerated as a form
of adaptation of tumor cells particularly in the central region of
the tumor mass in a hyponutrient and hypoxic microenvironment
(6,8).
Autophagy can be potentiated by treatment with
chemical inducers or chemotherapeutic agents, as well as in
response to amino acid deprivation. We and others have reported
that epidermal growth factor receptor (EGFR)-tyrosine kinase
inhibitors (TKIs), such as gefitinib (GEF) and erlotinib potently
induce autophagy in several cancer cell lines, as well as in murine
embryonic fibroblasts (MEFs) (9–13).
EGFR is overexpressed or activated in its tyrosine kinase activity
by somatic mutation in a broad range of human cancers, including
non-small cell lung cancer (NSCLC) (14,15).
It has been reported that inactivated endosomal, but not cell
surface EGFR interacts with lysosomal-associated transmembrane
protein B4 (LAPTM-4B), resulting in a complex formation for
recruiting Rubicon from Beclin-1 (Atg6) (16). Since Rubicon is a potent inhibitory
protein for autophagy via the molecular interaction with Beclin-1,
the dissociation of Rubicon from Beclin-1 results in the initiation
of autophagy. Thus, the survival of cancer cells with a higher EGFR
expression can be supported by the efficient induction of autophagy
under various metabolic stresses (16).
We have also reported that macrolide antibiotics,
such as azithromycin (AZM) and clarithromycin (CAM) exert an
inhibitory effect on autophagy flux in myeloma and squamous cell
carcinoma cell lines (17,18). Of note, AZM and CAM exert cytotoxic
effects under amino acid-depleted culture conditions in these cell
lines along with the upregulation of the pro-apoptotic
transcription factor, CHOP/GADD153, although they exhibit no
cytotoxicity in complete culture medium (18). Mammalian target of rapamycin
(mTOR), is a master regulator that combines amino acid availability
to cell growth and autophagy (19–21).
Therefore, the shortage of the intracellular amino acid pool
appears to determine the sensitivity to various cellular stresses.
In this context, it is important to examine whether the effect of
GEF is further enhanced when cancer cells are exposed to an amino
acid starvation (AAS) culture condition.
Historically, three types of cell death have been
identified on the basis of morphological criteria, which include
type I (apoptosis), type II (autophagic cell death) and type III
(necrosis) (22). Although
physiological levels of autophagy are essential for the maintenance
of cellular homeostasis during various stress conditions, excessive
or uncontrolled levels of autophagy can induce autophagic cell
death (22,23). Autophagic cell death was originally
defined as a type of cell death accompanied by the large-scale
autophagic vacuolization of the cytoplasm and was described during
animal development, under tissue homeostasis and in diseased
tissues, as well as in cultured cells treated with chemotherapeutic
agents or other toxic compounds (22). On the other hand, necrosis has been
stereotypically considered as an accidental and passive cell death,
as opposed to apoptosis. Necroptosis, in which necrosis is
regulated, was originally described in a FADD-deficient variant of
human Jurkat T cells treated with TNF-α, which is characterized as
receptor-interacting serine/threonine-protein kinase
(RIPK)1-dependent in association with the morphological features of
cell swelling and plasma membrane integrity, but without chromatin
condensation and nuclear fragmentation (24,25).
Thereafter, non-apoptotic cell death with morphologically necrotic
features associated with the cell death inhibition induced by
necrostatin 1 (NEC-1), a specific inhibitor of RIPK1, has been
'passively' described as necroptosis.
In this study, we found that the cell killing effect
of GEF was apparently pronounced along with the upregulation of
autophagy when the cells were cultured under AAS culture conditions
plus GEF. Of note, this pronounced effect was not mediated either
by the induction of apoptosis or autophagic cell death, but
apparently by an atypical type of cell death. The molecular
mechanisms and phenotype regarding this atypical cell death are
precisely discussed.
Materials and methods
Reagents
GEF, which was purchased from Cayman Chemical Co.
(Ann Arbor, MI, USA), was dissolved in dimethyl sulfoxide (DMSO) to
yield 10 mM stock solutions. Z-VAD-fmk, which is a pan-caspase
inhibitor, was purchased from Peptide Institute (Osaka, Japan) and
3-methyladenine (3-MA), SP600125, doxycycline hyclate (DOX) and
puromycin dihydrochloride were obtained from Sigma-Aldrich (St.
Louis, MO, USA). NEC-1, a specific inhibitor of RIPK1, was
purchased from Enzo Life Sciences (Farmingdale, NY, USA).
Cycloheximide was obtained from Calbiochem (La Jolla, CA, USA).
Recombinant human TNF-α, staurosporine, amino acid-free Dulbecco's
modified Eagle's medium (DMEM) (048-33575), MEM essential amino
acids solution (X100; 132-15641) and MEM non-essential amino acids
solution (X50; 139-15651) were obtained from Wako Pure Chemical
Industries (Osaka, Japan).
Cell lines and culture conditions
The human oral squamous cell carcinoma cell line,
CAL 27, the human pharyngeal carcinoma cell line, Detroit 562, the
human NSCLC cell line, A549, the human pancreatic cancer cell line,
PANC-1, the human colorectal adenocarcinoma cell line, HT-29 and
the breast cancer cell line, MDA-MB-231 were obtained from the
American Type Culture Collection (ATCC, Manassas, VA, USA). The
human NSCLC cell line, PC-9, which carries delE746-A750 in exon 19
of the EGFR gene and exhibits exquisite sensitivity to EGFR TKIs
(26), was obtained from the RIKEN
BioResource Center (Tsukuba, Japan). The murine embryonic
fibroblast (MEF) cell line (DR-wild-type) established by SV-40
immortalization was also obtained from the ATCC. All cell lines
were cultured in DMEM plus 10% fetal bovine serum (FBS; Biosera,
Ringmer, UK) and 1% penicillin/streptomycin solution (Wako Pure
Chemical Industries). The 'AAS' culture conditions described in
this study indicate amino acid-free DMEM (048-33575; Wako Pure
Chemical Industries) plus 10% FBS and 1%
penicillin/streptomycin.
The m5–7 cell line, an Atg5 tet-off MEF
system, was a kind gift from Professor Noboru Mizushima (University
of Tokyo, Tokyo, Japan). The m5–7 cells were maintained in DMEM
containing 10% FBS. For the knockdown of the Atg5 gene for
the full inhibition of autophagy, the cells were further cultured
in the presence of 10 ng/ml DOX for 4 days (27). All cell lines were cultured in a
humidified incubator containing 5% CO2 and 95% air at
37°C. All cell lines were used for the experiments within 10
passages after thawing.
For the typical induction of necroptosis, the HT-29
cells were pre-treated with Z-VAD-fmk (20 µM) for 30 min
followed by an additional treatment with cycloheximide (CHX, 10
mg/ml) and human TNF-α (20 ng/ml) for 8 h as previously described
in the literature (28).
Assessment of cell growth inhibition
Cell growth inhibition was measured by the
CellTiter-Blue cell viability assay (Promega, Madison, WI, USA).
The cells were treated with or without GEF (5 to 50 µM) for
24 and 48 h in 96-well plates (Thermo Fisher Scientific, San Jose,
CA, USA) in pentaplicate. During the last 4 h, CellTiter-Blue
reagent was added to each well, and fluorescence was measured at
560 nm excitation and 590 nm emission. The percentage of the mean
fluorescence measured to that in untreated cells and was expressed
as % cell growth inhibition.
Morphological assessment
The cells were spread on glass slides using a
Cytospin 4 Centrifuge (Thermo Fisher Scientific) to make glass
slide preparations, then stained with May-Grünwald-Giemsa, and
examined under a digital microscope (BZ-8000; Keyence Co., Osaka,
Japan).
Flow cytometry
For the assessment of apoptosis, the cells were
stained with Annexin V and propidium iodide (PI) using the Annexin
V-FITC Apoptosis Detection kit (Nacalai Tesque, Kyoto, Japan)
according to the manufacturer's instructions and subjected to flow
cytometry using the Attune® Acoustic Focusing Cytometer
(Life Technologies, Carlsbad, CA, USA).
Immunoblotting
Immunoblotting was performed as previously described
(29). Briefly, the cells were
lysed with RIPA lysis buffer (Nacalai Tesque) supplemented with a
protease and phosphatase inhibitor cocktail (Nacalai Tesque).
Cellular proteins were quantified by Bradford assay (Thermo Fisher
Scientific). Equal amounts of proteins were loaded onto the gels,
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred onto an Immobilon-P
membrane (Millipore, Bedford, MA, USA). The membranes were probed
with primary antibodies (Abs), such as anti-microtubule-associated
protein 1 light chain 3 (LC3) B antibody (Ab) (NB600-1384; Novus
Biologicals, Inc., Littleton, CO, USA, at 1/4,000 dilution),
anti-ATG5 Ab (#12994S, at 1/1,000 dilution), anti-caspase-3 Ab,
(#9662S, 1/1,000), anti-poly(ADP-ribose) polymerase (PARP) Ab
(#9542S, 1/1,000), anti-LAT1 (#9166S, 1/1,000), anti-RIPK1 (#4926S,
1/1,000), anti-phospho-RIPK1 (Ser166) (#65746S, 1/1,000),
anti-RIPK3 Abs (#13526S, 1/1,000) (Cell Signaling Technology,
Danvers, MA, USA), anti-mixed lineage kinase domain like
pseudokinase (MLKL), anti-phospho-MLKL (Ser358) (ab184718, 1/1,000)
Abs (Abcam, Cambridge, MA, USA) and anti-EGFR Ab (sc-03, 1/1,000),
anti-phospho-EGFR (Tyr1173) Ab (sc-101668, 1/1,000), anti-p62
(sequestosome-1) mAb (sc-28359, 1/1,000), anti-GAPDH mAb (sc-32233,
1/1,000) and anti-β-actin mAb (sc-47778, 1/1,000) (Santa Cruz
Biotechnology, Santa Cruz, CA, USA). Immunoreactive proteins were
detected with horseradish peroxidase-conjugated secondary Abs (anti
mouse Ab: 115-035-003, at 1/5,000 dilution, anti rabbit Ab:
711-035-152, at 1:5,000 dilution; Jackson ImmunoResearch, West
Grove, PA, USA) and an enhanced chemiluminescence reagent (ECL)
(Millipore). Densitometry was performed using a Molecular Imager,
ChemiDoc XRS system (Bio-Rad Laboratories, Richmond, CA, USA).
Immunoprecipitation
The harvested cells were washed with
physiological-buffered saline (PBS) and lysed with lysis buffer (10
mM Tris-HCl, 150 mM NaCl, 1% NP-40, pH 8.0) supplemented with a
protease and phosphatase inhibitor cocktail (Nacalai Tesque). The
cell lysates were spun down at 16,000 × g for 15 min. The soluble
fraction was collected, and the protein concentration was
determined by Bradford assay. Subsequently, 800 µg of the
extracted protein solution in lysis buffer were immunoprecipitated
overnight with either anti-RIPK1 Ab or anti-RIPK3 Ab at 4°C.
Antibodies were collected with Protein A SureBeads (Bio-Rad
Laboratories) for 1-h rotation at 4°C, then washed 3 times with the
lysis buffer. The binding proteins on the beads were eluted with 2X
SDS loading buffer by 5-min boiling in a heat block.
Electron microscopy
Following treatment with/without GEF (25 µM)
under either complete culture condition or AAS culture condition
for 24 h, CAL 27 cells were fixed with 2.5% glutaraldehyde in 0.1 M
phosphate buffer (pH 7.3) for 1 h. The samples were further fixed
in 1% osmium tetroxide for 1 h, dehydrated in graded ethanol
(30–100%), and embedded in Quetol 812 Epoxy Resin (Nisshin EM Co.,
Ltd., Tokyo, Japan). Ultrathin sections were cut using an Ultracut
J microtome (Reichert Jung, Vienna, Austria). These sections were
stained with lead nitrate and uranium acetate (Merck, Darmstadt,
FRG) and subjected to electron microscopic analysis using a
scanning electron microscope (JEM-1200EX II; JEOL Ltd., Tokyo,
Japan).
RNA interference
For the gene silencing of LAT1 and RIPK1 in the CAL
27 cells, the LAT1 siRNA and control siRNA sequences are described
as follows: LAT1 sense, GGAAC AUUGUGCUGGCAUUdTdT and antisense,
AAUGCCAGCACAAUGUUCCdTdT; control sense, GUUAAAGGUUUGACUCGCGdTdT and
antisense, CGCGAGUCAAACCUUUAACdTdT. RIPK1 siRNA (HSS112847) and
control siRNA (12935-300) were purchased from Life Technologies
(Grand Island, NY, USA). siRNAs were diluted to a final
concentration of 10 nM in Opti-MEM I (Life Technologies).
Transfection was performed with the cells at 40% confluency using
Lipofectamine RNAiMAX transfection reagent (Life Technologies)
according to the manufacturer's instructions. The knockdown
efficiency was assessed by immunoblotting.
Stable transfection of mCherry-EGFP-LC3B
plasmid into A549 and CAL 27 cells
In the present study, a pBABE-puro-mCherry-EGFP-LC3B
plasmid vector (no. 22418) was purchased from Addgene (Cambridge,
MA, USA). The A549 and CAL 27 cells were transfected with this
plasmid DNA using Lipofectamine 2000 (Life Technologies) according
to the manufacturer's instructions and as previously described
(30). Briefly, 4 µg of
pBABE-puro-mCherry-EGFP-LC3B plasmid solution and 10 µl of
Lipofectamine 2000 were incubated in 500 µl of serum-free
Opti-MEM (Life Technologies) for 20 min and mixed into
2×106 cells cultured in 1.5 ml of antibiotic-free DMEM
with 10% FBS in a 60-mm dish. At 48 h after transfection, the cells
were seeded into a 150-mm dish with puromycin dihydrochloride (at 2
µg/ml for A549 cells and at 0.5 µg/ml for CAL 27
cells). The individual puromycin-resistant clones were isolated
using a cloning ring. After cloning,
A549/pBABE-puro-mCherry-EGFP-LC3B and
CAL27/pBABE-puro-mCherry-EGFP-LC3B were used for the following
experiments.
Time-lapse imaging
On the first day, the
A549/pBABE-puro-mCherry-EGFP-LC3B cells (clone #3) and CAL
27/pBABE-puro-mCherry-EGFP-LC3B cells (clone #7) were seeded into
the CELLview™ Cell Culture dish, 35 mm glass bottom (627860;
Greiner Bio-One, Frickenhausen, Germany), and cultured for 24 h to
make them adhere to the glass bottom. On the second day, the cells
were washed twice with PBS and incubated in either the AAS culture
medium containing 10% FBS or the complete culture medium containing
10% FBS with/without GEF (25 µM). Thereafter, observation
began at 5-min intervals using a confocal laser scanning microscope
(LSM 700; Carl Zeiss, Oberkochen, Germany) for the detection of
EGFP at 488 nm laser wavelength and mCherry at 555 nm laser
wavelength. In this time-series scanning, a 63X oil immersion lens
was used, and bright field images were obtained simultaneously. ZEN
2012 Black Edition software (Carl Zeiss) was used for the
analysis.
Statistical analysis
All quantitative data are expressed as the means ±
standard deviation (SD). Statistical analysis was performed using a
two-tailed non-paired Student's t-test. To assess the synergism of
the combined GEF and AAS treatment, multivariate linear regression
analysis using both GEF and AAS as independent variables was first
performed to determine whether an additive effect was observed
between GEF and AAS. Subsequently, interaction terms were added in
the model to clarify the existence of a synergistic effect, as
previously described (31). The
criterion for statistical significance was taken as P<0.05.
Results
Amino acid starvation enhances
GEF-induced cytotoxicity in EGFR-expressing cell lines
It is now well known that the depletion of amino
acids in the cell culture medium promotes the induction of
autophagy for the adaptation to the shortage of the intracellular
amino acid pool (18–21). It has also been reported that
EGFR-TKIs, such as GEF induce cytoprotective autophagy in various
EGFR-expressing cell lines (9–13).
Therefore, in this study, we first examined whether the
cytotoxicity of GEF is pronounced under AAS culture conditions
using the head and neck cancer cell lines, CAL 27 and Detroit 562,
the NSCLC cell lines, A549 and PC-9, and the pancreatic cancer cell
line, PANC-1, all of which express EGFR at a higher level, as well
as immortalized MEFs (Fig. 1A).
All the cell lines were cultured in the presence of GEF at various
concentrations in either amino acid-depleted DMEM containing 10%
FBS or complete culture medium for up to 48 h (Fig. 1B). Normal DMEM contains a
sufficient amount of 20 amino acids including 75 mg/l L-glutamic
acid and 30 mg/l L-methionine, whereas AAS (amino acid-free DMEM
plus 10% FBS) only contains amino acids less than 1/10 of the
normal DMEM with 10% FBS. Although AAS itself exerted some cell
growth inhibitory effects, the apparent enhanced cytotoxic effect
of GEF was observed even after 24 h of exposure in the CAL 27, A549
and PANC-1 cells compared to treatment with either AAS or GEF
alone. After 48 h of exposure to GEF under AAS culture conditions,
the effect became more apparent in all cell lines except for the
PC-9 cells. As previously reported, the PC-9 cells have EGFR
mutation with a higher sensitivity to GEF, resulting in a
sufficiently strong cell killing effect by GEF alone, which may
produce no further enhancement even by combining GEF and AAS
(26,32). The PANC-1 cells were resistant to
GEF, but exhibited a significant cytotoxicity under AAS culture
conditions. After 48 h of treatment, multivariate linear regression
analysis revealed that these pronounced effects were synergistic in
the CAL 27, Detroit 562, A549 and PANC-1 cells.
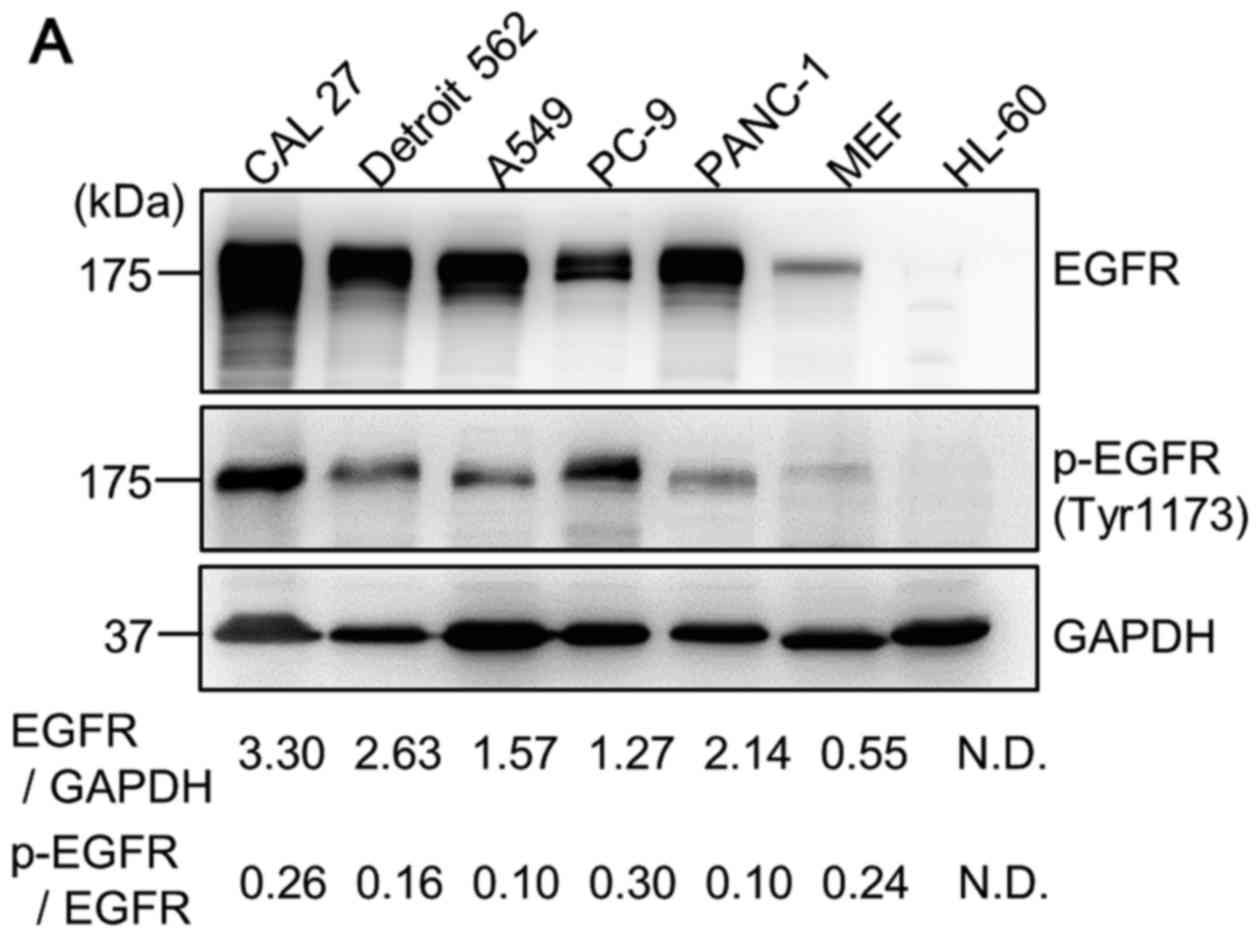 | Figure 1Cell growth inhibition following
treatment with gefitinib (GEF) under amino acid starvation (AAS)
culture conditions in EGFR-expressing cancer cell lines. (A)
Cellular proteins in the CAL 27, Detroit 562, A549, PC-9, PANC-1
and MEF cells in the exponential growth phase in the complete
culture condition containing 10% FBS were lysed as described in
Materials and methods. The cellular proteins extracted from the
1×105 cells were loaded on each lane and separated by
11.25% SDS-PAGE and immunoblotted with anti-EGFR antibody (Ab) and
anti-phospho-EGFR (Tyr1173) Ab. Immunoblotting with anti-GAPHD mAb
was performed as an internal control. The cell lysate of HL-60
leukemia cells was used as a negative control, as previously
described (11). Numbers indicate
the ratios of EGFR to GAPDH and the ratios of the phospho-EGFR to
EGFR in each lane. N.D., not detectable. (B) Cells were cultured
under the complete culture condition and AAS culture condition in
the presence of GEF (0–25 µM) for 24 and 48 h. Viable cell
number was assessed by CellTiter-Blue cell viability assay as
described in Materials and methods (*P<0.05 and
**P<0.01, complete culture condition vs. AAS by a
two-tailed non-paired Student's t-test). By multivariate linear
regression analysis using both GEF and AAS as independent
variables, the synergistic effect was observed in the 24-h
treatment of CAL-27 cells (P<0.001) and PANC-1 cells (P=0.008),
as well as in the 48-h treatment of CAL-27 cells (P<0.001),
Detroit 562 cells (P=0.023), A549 cells (P<0.001) and PANC-1
cells (P<0.001). In the PC-9 cells, the additive cytotoxic
effect was observed in the 48-h exposure (P<0.001), but not in
the 24-h exposure (P=0.335). |
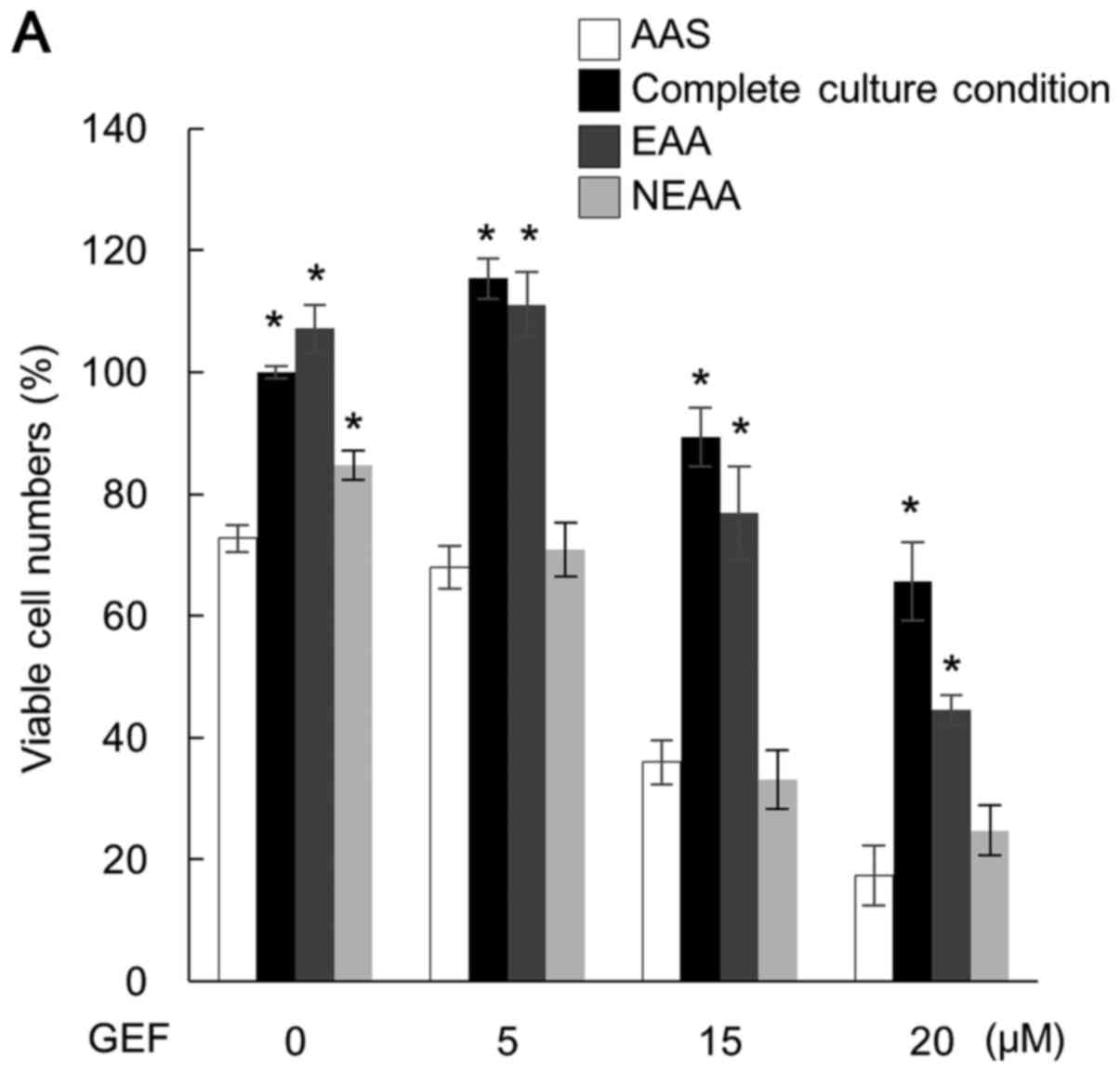
The supplementation of essential amino acids, but
not non-essential amino acids, into the AAS culture medium almost
cancelled the pronounced cytotoxicity in CAL 27 cells (Fig. 2A). This strongly suggests that the
enhanced cytotoxicity by the combined treatment with GEF plus AAS
is due to the shortage of the intracellular amino acid pool. To
confirm this hypothesis, we subsequently knock down LAT-1 in the
CAL 27 cells. It has been reported that among the amino acid
transporters, LAT-1 is specifically expressed at a higher level in
cancer cells, by which the essential amino acids are imported into
cancer cells for supporting cellular metabolism (3,33).
As shown in Fig. 2B, the high
LAT-1 expression was considerably suppressed by transfection with
LAT-1 siRNA in the CAL 27 cells. LAT-1 knockdown resulted in a
significant decrease in the viable cell number in the presence of
GEF compared with the cells transfected with control siRNA and
treated with GEF. Notably, LAT-1 knockdown itself exerted no effect
in the absence of GEF, suggesting some metabolic compensation or
adaptation for cell survival when the cells were cultured in the
complete culture medium.
Sensitization of GEF under AAS conditions
is not mediated by the induction of apoptosis
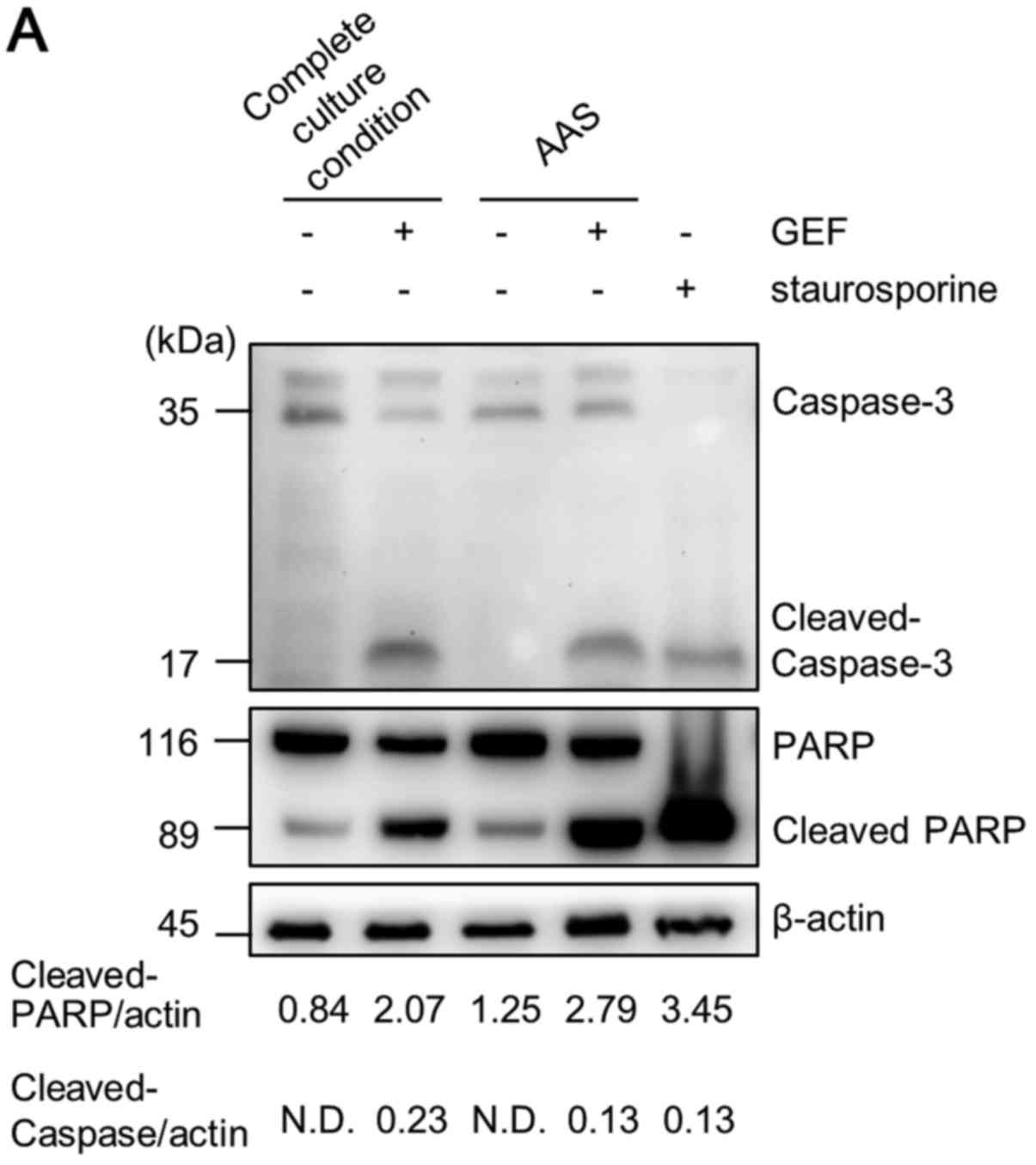
We subsequently examined whether apoptosis is
involved in the enhanced GEF-induced cell death under AAS
conditions. First, immunoblotting was performed to confirm the
cleavage of caspase-3 and PARP as indicators of apoptosis. As shown
in Fig. 3A, GEF treatment under
complete culture conditions led to some cleavage of caspase-3 and
PARP. However, GEF treatment under AAS culture conditions did not
lead to any apparent increase in the cleavage of caspase-3 as
compared with GEF treatment in complete culture medium, although
the cytotoxic effect was pronounced. As shown by flow cytometry,
GEF treatment under AAS culture conditions increased the number of
Annexin V+/PI+ double-stained cells compared
with either GEF treatment under complete culture medium conditions
or culture of the cells in AAS medium without GEF. Unlike
staurosporine treatment for the typical induction of apoptosis, the
number of Annexin V+/PI− cells indicating the
cells undergoing an early phase of apoptosis was not increased
(Fig. 3B). It was recently
reported that phosphatidylserine labeled with Annexin V is exposed
to the cell surface prior to loss of cell integrity during
necroptosis induction (34).
Therefore, Annexin V positive staining does not necessarily
indicate cells undergoing apoptosis. As regards the morphological
findings, we could not observe any chromatin condensation and
nuclear fragmentation under AAS culture conditions plus GEF
treatment, whereas cells treated with staurosporine exhibited the
typical apoptotic karyorrhexis and blebbing of the plasma membrane.
However, we observed the translucent cytoplasm and rupture of the
plasma membrane of CAL 27 cells (Fig.
3C). These results indicate that non-apoptotic cell death
appeared to be involved in the pronounced cytotoxicity induced by
AAS culture conditions and GEF treatment.
Induction of autophagy in response to GEF
and AAS conditions
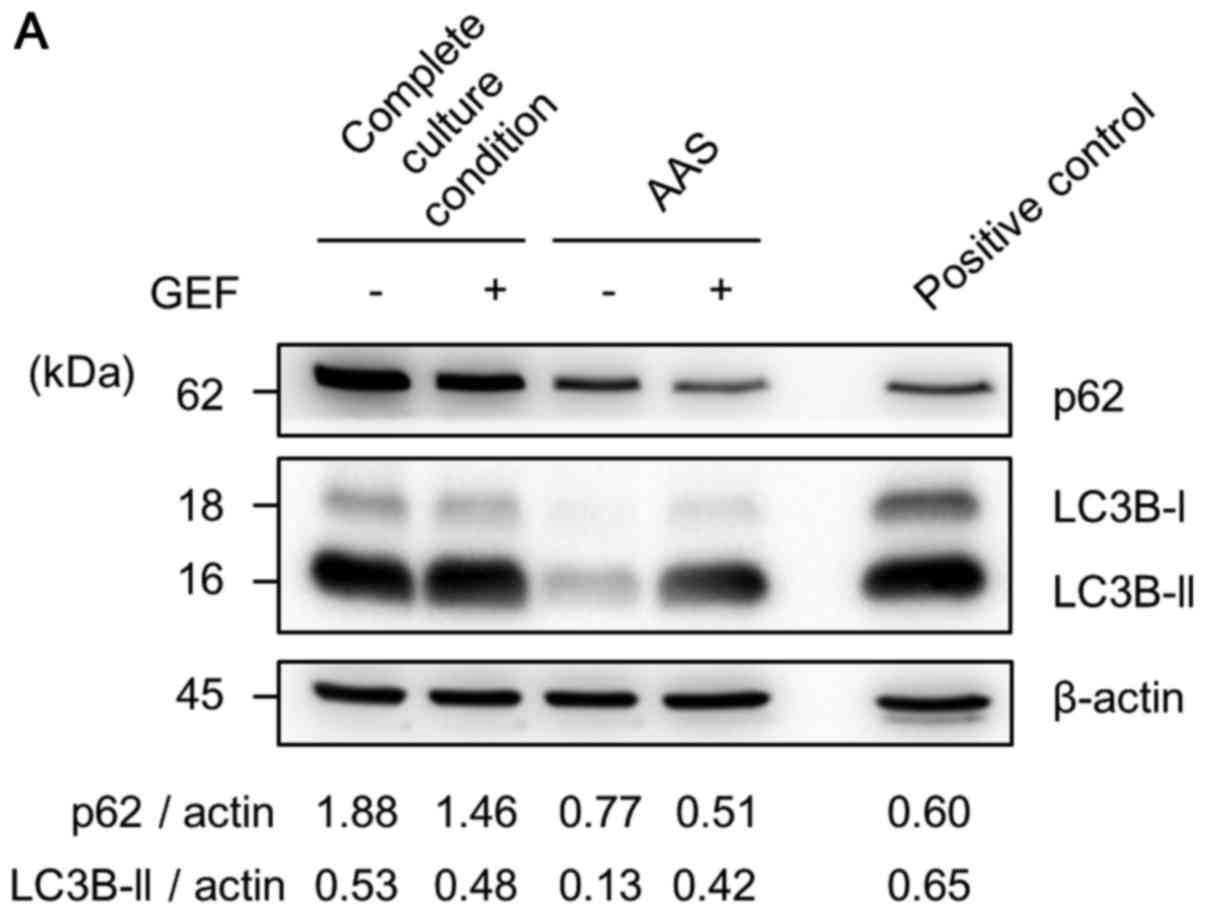
We then examined whether autophagy is induced in
response to GEF under either complete culture conditions or AAS
culture conditions by immunoblotting with anti-LC3B Ab and anti-p62
Ab. As shown in Fig. 4A, the
expression of p62, a substrate of autophagy, was decreased in
response to 24 h of treatment with GEF in complete culture medium
compared with that in the cells cultured without GEF in complete
culture medium as a control (lane 1 vs. 2). p62 expression (lane 3)
decreased in response to AAS without GEF and further decreased in
response to 24 h of exposure to GEF plus AAS (lane 4). These
results indicate that autophagy was accelerated by GEF under AAS
culture conditions (Fig. 4A). In
the CAL 27 cells, the expression ratio of LC3B-II to β-actin, a
hallmark of autophagosome formation (35), was increased even under the
complete culture condition (lane 1). This suggests the upregulation
of autophagy under the normal culture condition in this cell line,
which was consistent with the electron microscopic findings with an
increased number of autophagosomes even in the control culture
condition (data not shown). This was possibly due to the fact that
the CAL 27 cells expressed a higher level of EGFR (Fig. 1A), implying a sufficient number of
endosomal EGFR for autophagy induction (16). However, as regards the expression
ratio of LC3B-II to β-actin, the CAL 27 cells under AAS culture
conditions exhibited a decrease in this ratio compared with the
control cells (lane 1 vs. 3). In addition, both the LC3B-I and
LC3B-II bands became faint under AAS culture conditions, although
AAS should induce more autophagy. We previously observed the same
phenomenon when the cells had markedly upregulated autophagy along
with leading LC3B-II degradation (18). As the expression level of LC3B-II
is determined by the balance of synthesis and breakdown, the
degradation of LC3B-II appeared to occur more rapidly than the
induction of LC3B-II under AAS culture conditions. To confirm the
accelerated induction of autophagy under AAS culture conditions
plus GEF treatment, we carefully monitored autophagosome formation
during a shorter exposure to GEF in either complete culture
condition or AAS culture conditions using the A549 and CAL 27
cells, which stably express the tandem fluorescent-tagged LC3B
(mCherry-EGFP-LC3B) by confocal microscopic time-lapse imaging
(30). The number of fluorescent
puncta indicating autophagosomes increased when the cells were
cultured in the AAS medium in the absence of GEF. Both cell lines
treated with GEF in the complete culture medium exhibited a greater
number of mCherry-EGFP-puncta than the cells cultured in the AAS
medium (Fig. 4B). The turnover of
puncta appeared to be rapid, and most dots disappeared within 20
min after formation (data not shown). When the cells began to be
cultured in the presence of GEF under AAS culture conditions, we
observed a marked enhancement of autophagosome formation even
within 30 min (Fig. 4B).
Therefore, the induction of autophagy is accelerated in response to
GEF plus AAS.
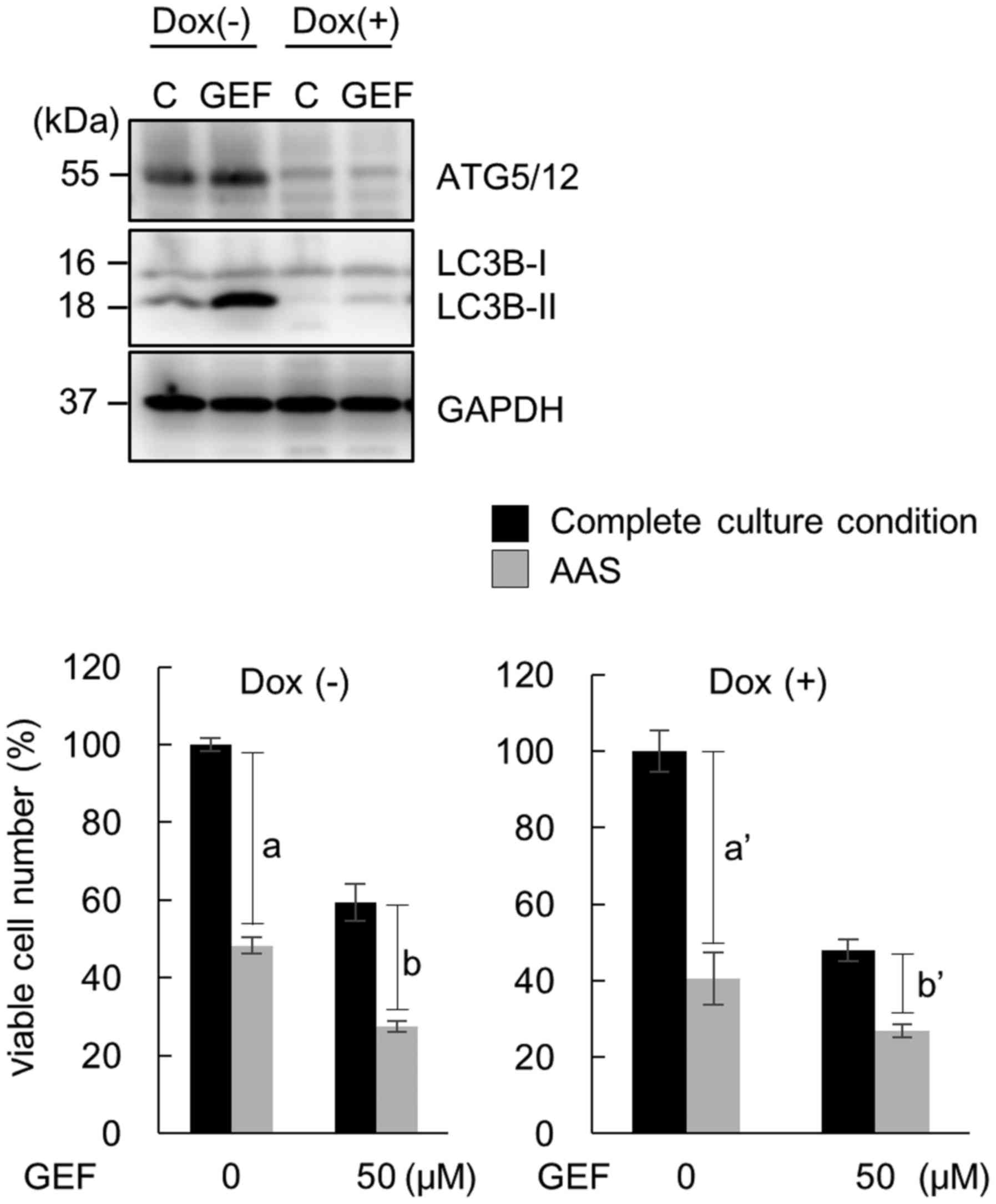
Induction of atypical necroptosis after
treatment with GEF under AAS culture conditions
To examine whether the enhanced cytotoxicity is due
to induction of 'autophagic cell death' (22,23),
we used the Atg5 tet-off MEF cell line, named m5–7 (27). This cell line can be conditionally
transformed into knockout the Atg5 gene, as a useful system
for investigating the effects of autophagy in our study.
Additionally, as MEF cells express EGFR, we intended to investigate
whether our findings in cancer cell lines can be extended to
immortalized fibroblasts.
Pre-treatment of the m5–7 cells with Dox, which
leads to Atg5 knockout, results in the inhibition of
autophagy (27). As shown in
Fig. 5, the pronounced
cytotoxicity by GEF (50 µM) plus AAS was observed regardless
of the deletion of the autophagic process in the m5–7 cells.
To investigate the molecular mechanisms responsible
for this pronounced cell killing effect, we further examined the
type of cell death involved in the GEF plus AAS treatment using CAL
27 cells. The cells were cultured with GEF either in the control
medium or the AAS culture medium in the presence or absence of
various inhibitors: 3-Methyl adenine (3-MA) for blocking
autophagosome formation, SP600125 for blocking autophagic cell
death (36), Z-VAD-fmk for
inhibiting pan-caspases for blocking apoptosis, and NEC-1, a RIPK-1
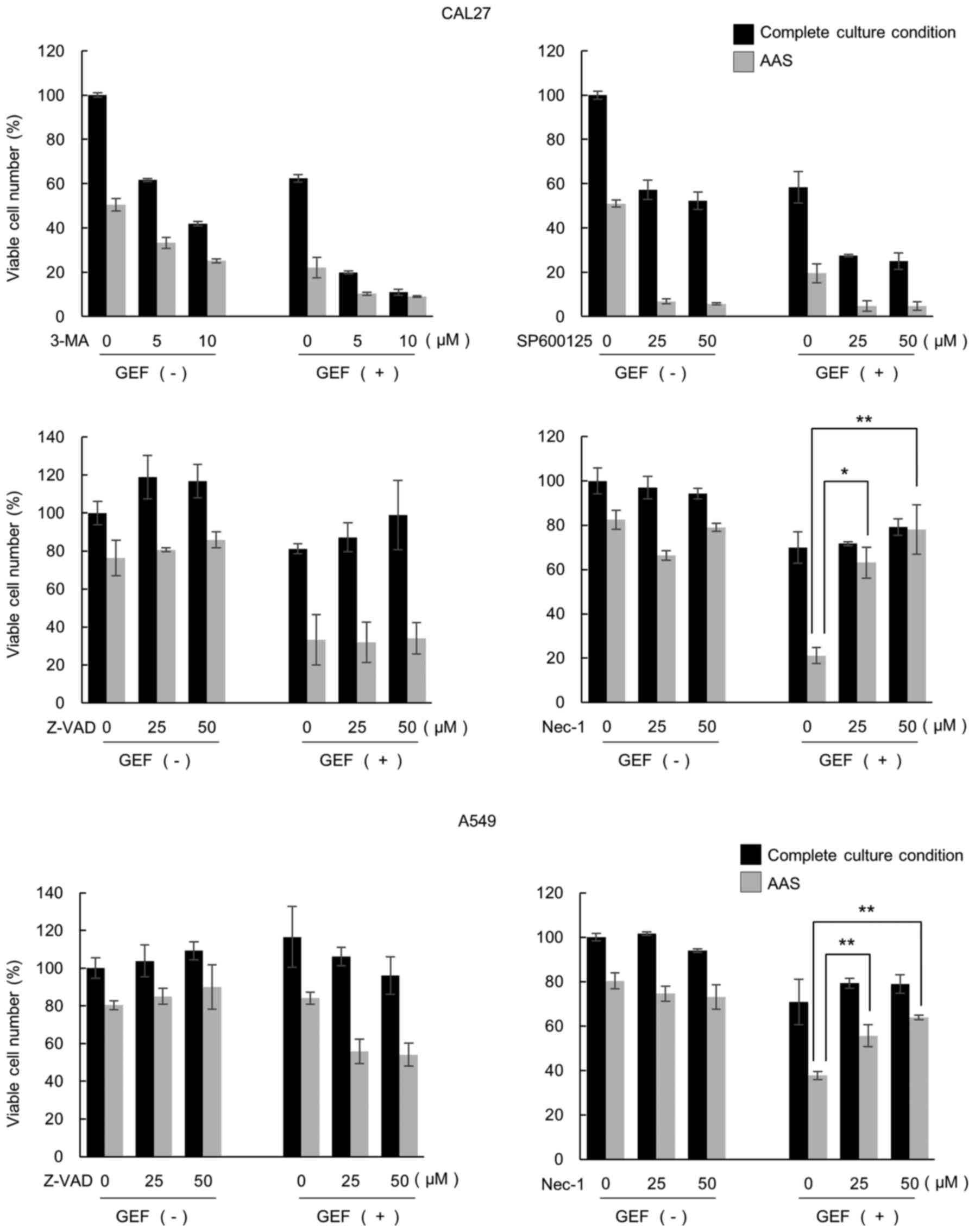
inhibitor for blocking necroptosis (24). As shown in Fig. 6, treatment with 3-MA and SP600125
enhanced the cytotoxic effects of GEF and AAS possibly due to the
cytotoxicity of the inhibitors. Taking the results of these
inhibitors and m5–7 cells shown in Fig. 5 together, autophagic cell death
appeared to be excluded. Z-VAD-fmk attenuated the GEF-induced
cytotoxic effect, but did not exert any effects on the pronounced
cytotoxicity of treatment with GEF plus AAS. Notably, in the
presence of NEC-1, the pronounced cytotoxicity was almost
completely cancelled. This cytotoxicity cancellation was also
observed in the A549 cells treated with GEF and AAS. Therefore, the
induction of necroptosis in response to culture under AAS
conditions plus GEF treatment was strongly suggested.
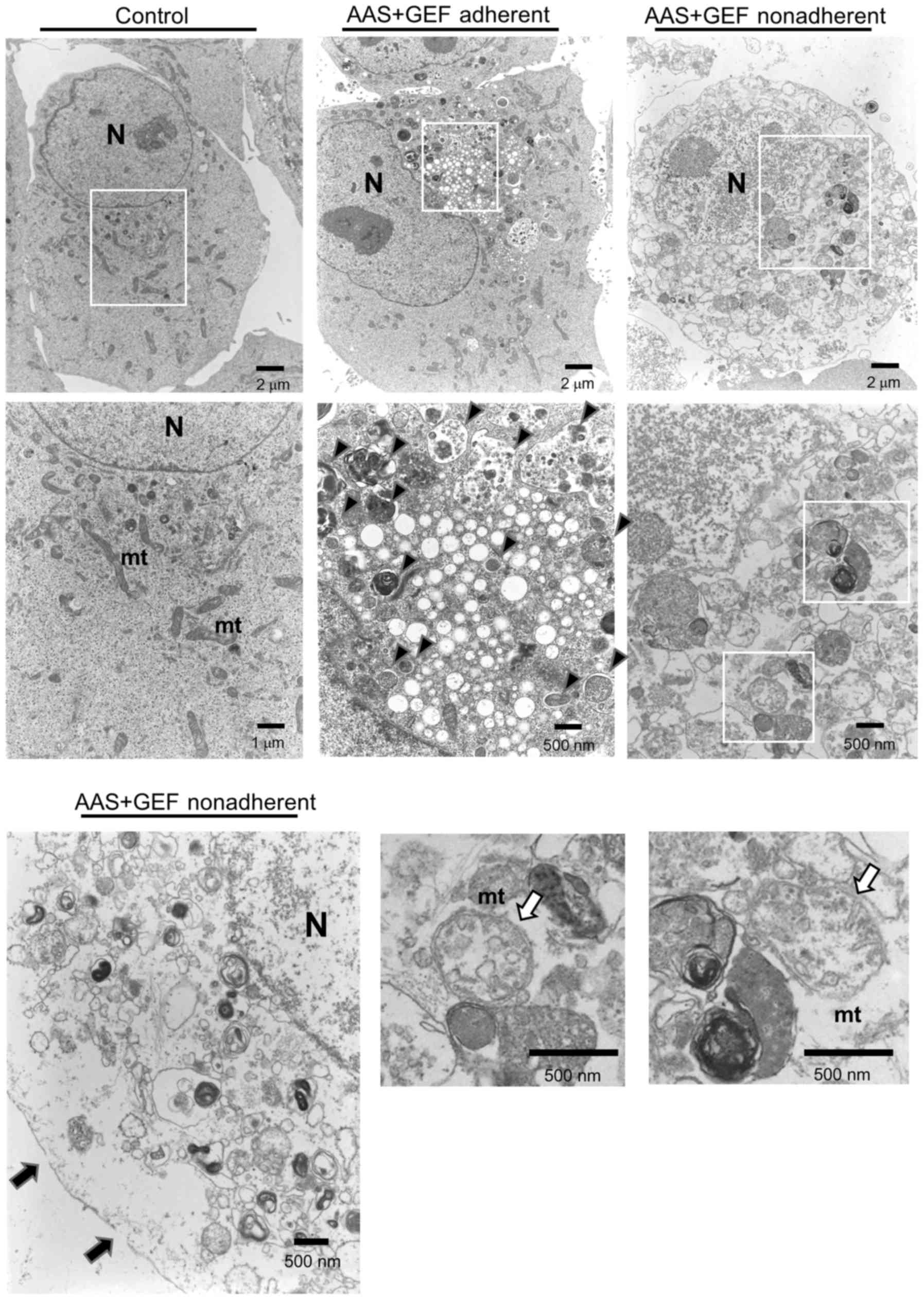
The CAL 27 cells adhere to the culture flask bottom
in the normal culture condition, and they become non-adherent while
undergoing cell death. As regards electron microscopy, the
non-adherent CAL 27 cells treated with GEF plus AAS exhibited a
translucent cytoplasm, cytoplasmic vacuoles, mitochondrial
swelling, and plasma membrane rupture with an increased number of
autophagosomes (Fig. 7). As
similarly shown in Fig. 3C, no
nuclear fragmentation, chromatin condensation, or apoptotic bodies
were observed. Therefore, all these morphological findings also
support the induction of necroptosis.
It is now well known that necroptosis is a
RIPK-1/3-dependent form of cell death (38–40).
In TNF-α-induced necroptosis, RIPK-1 is activated in response to
TNF-α stimuli and forms a cytosolic complex, known as the
necrosome, which is formed by RIPK-1 in complex with RIPK3 and MLKL
(39,40). MLKL is recruited to the necrosome
and phosphorylated by RIPK3. This allows MLKL to form the MLKL
oligomer for insertion into the plasma membrane to induce
necroptosis (41,42). Therefore, in this study, we knocked
down RIPK-1 in CAL 27 cells using siRNA. The knockdown of RIPK-1
led to a significant attenuation of GEF plus AAS-induced
cytotoxicity compared with the cells transfected with control siRNA
(Fig. 8A). No differences were
observed between the control siRNA- and RIPK-1 siRNA-treated cells
under the GEF-containing culture conditions and AAS conditions.
However, we could not detect the phosphorylation of RIPK-1 and MLKL
by immunoblotting using specific Abs, which are known to be
involved in the signaling pathways of TNFα-induced necroptosis
(40,41) (Fig.
8B). In the immunoprecipitation assay using anti-RIPK-3 Ab,
HT-29 cells pre-treated with Z-VAD-fmk followed by an additional
treatment with cycloheximide and human TNF-α for the induction of
necroptosis showed the co-precipitated phosphorylated (p)-MLKL, as
well as p-RIPK-1; however, we could not detect the co-precipitated
p-MLKL and p-RIPK-1 in the CAL 27 cells treated with GEF plus AAS
(Fig. 8C). This suggested that
culture under AAS conditions plus GEF treatment did not induce
necrosome formation in the CAL 27 cells.
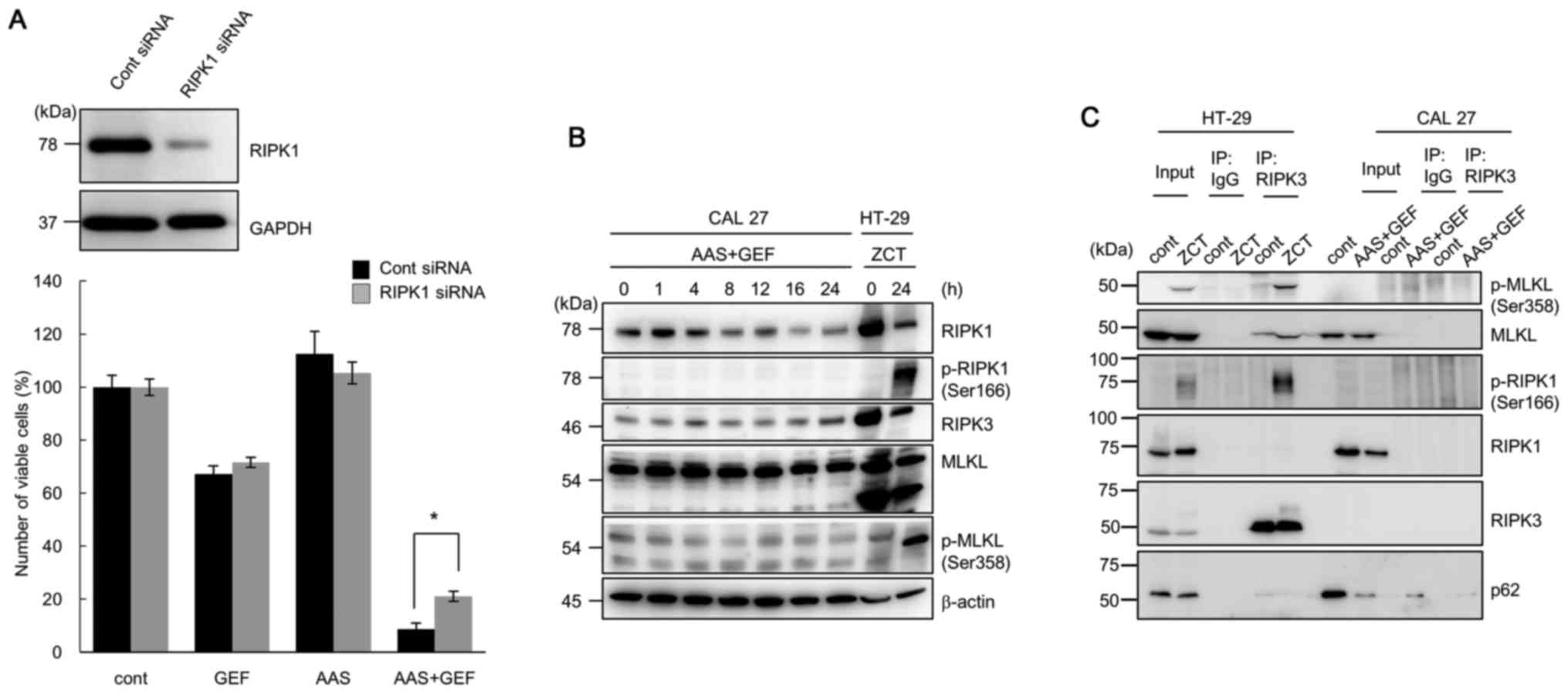 | Figure 8Assessment of the induction of
necroptosis in CAL 27 cells following treatment with gefitinib
(GEF) under amino acid starvation (AAS) conditions. (A) Following
pre-treatment with RIPK1 siRNA or control siRNA for 48 h, the CAL
27 cells were further cultured with/without GEF (25 µM)
under either AAS conditions with 10% FBS or DMEM containing 10%
FBS, and viable cell number was assessed. Immunoblotting with
anti-RIPK1 antibody was performed after 24 h of treatment with
siRNAs. Immunoblotting with anti-GAPDH monoclonal antibody was
performed as an internal loading control (*P<0.05,
control siRNA vs. RIPK1 siRNA). (B) Following treatment of the CAL
27 cells with GEF (25 µM) under AAS culture conditions for
the indicated time periods, the cells were lysed and the cellular
proteins were separated by SDS-PAGE, and then immunoblotted with
anti-RIPK1 antibody (Ab), anti-RIPK3 Ab, anti-MLKL Ab, and phosphor
(p)-specific Abs for RIPK1 MLKL Abs. As for the positive control
for the typical necroptosis induction, HT-29 cells were pre-treated
with Z-VAD-fmk (20 µM) for 30 min followed by an additional
treatment with cycloheximide (CHX, 10 µg/ml) and human TNF-α
(20 ng/ml) for 8 h as previously reported (28). This treatment is indicated as ZCT.
(C) The CAL 27 cells treated with GEF (25 µM) under AAS
conditions for 24 h were used for co-immunoprecipitation assay. The
CAL 27 cells treated under the complete culture condition without
GEF were used as control. Cell lysates were immunoprecipitated with
anti-RIPK3 Ab or the isotype matched IgG. Immunoprecipitates were
separated by SDS-PAGE and immunoblotted with either anti-p-MLKL,
anti-MLKL, anti-p-RIPK1, anti-RIPK1, anti-RIPK3, or anti-p62 Ab.
HT-29 cells treated with ZCT as described above were used as
positive control for necrosome formation. |
Taking all the data together, although the
morphological findings and the effect of NEC-1 fit well with the
induction of necroptosis, it would be more appropriate to describe
the phenomenon as follows: The enhanced killing effect by the
combined treatment with GEF plus AAS is mediated by 'atypical
necroptosis' or alternatively, 'necroptosis-like cell death' in the
CAL 27 cells.
Discussion
To the best of our knowledge, this is the first
report showing that AAS sensitizes the killing effect of GEF in
EGFR-expressing cancer cell lines (Fig. 1B). As a solid tumor appears to
proliferate by adapting to nutrient insufficiency along with
angiogenesis and autophagy, this has important clinical
implications, such as the application of combination therapy of
EGFR-TKI and LAT-1 inhibitors or anti-angiogenesis therapy for
cancer patients. Indeed, it was previously reported that the phase
II clinical trial of the anti-vascular endothelial growth factor
monoclonal antibody bevacizumab in combination with erlotinib for
advanced EGFR mutation-positive NSCLC resulted in an encouraging
antitumor activity, and this could be a new first-line regimen for
EGFR mutation-positive NSCLC (43,44).
In addition, we previously reported that macrolide antibiotics,
which have an inhibitory effect on autophagy, exerted a
CHOP/GADD153-mediated cell killing effect under amino acid
starvation conditions in head and neck squamous cell carcinoma cell
lines (18). As shown in Fig. 2A, the addition of amino acids to
the culture medium resulted in the almost complete cancellation of
cytotoxicity, implying that the cell killing effect appeared to be
mediated by the shortage of intracellular amino acids owing to the
blockage of their recycling process. Therefore, 'tumor-starving
therapy' may be a novel concept for cancer therapy.
However, as shown in Fig. 1B, the response to AAS culture in
terms of cell growth inhibition differed significantly in each cell
line (e.g., the pancreatic cancer cell line, PANC-1, exhibited
potent cytotoxicity in response to 48 h of AAS culture compared
with other cancer cell lines). In addition, there was the
significant difference in cell death under AAS conditions in the
MEF cell line used in Fig. 1B and
in m5–7, an Atg5 tet-off MEF cell line used in Fig. 5. The m5–7 cell line was generated
by Hosokawa et al (27),
and has been cloned for the complete inhibition of the autophagy
machinery. During this cloning process, the m5–7 cell line appeared
to have acquired a different phenotype including its response to
AAS treatment compared with those in the immortalized MEF cell
line. Therefore, the demand for intercellular amino acid pool
appears to be varied among the cell lines, which is possibly due to
the difference of cellular metabolism.
We deduced that the enhanced cell killing effect by
GEF plus AAS was mediated by the induction of apoptosis. However,
we could not detect any signs of enhanced apoptosis in the CAL 27
cells during the pronounced cytotoxicity (Figs. 3 and 7). Treatment with GEF alone induced
caspase-3 and PARP cleavage to a certain extent, but much less than
that induced by staurosporine (Fig.
3A). As the CAL 27 cells treated with staurosporine exhibited
typical apoptotic features, such as PARP/caspase-3 cleavage, an
increased number of the Annexin+/PI− cell
population as shown by flow cytometry, and morphological changes
showing nuclear fragmentation and chromatin condensation (Fig. 3), the canonical machinery for
apoptosis execution should be conserved in this cell line. The
question that remains to be answered is which type of cell death
phenotype was observed in this study and what cellular signals
determine this phenotype.
According to the results shown in Figs. 3 and 6, autophagic cell death can be excluded
in this case. The enhanced cytotoxicity shown in this study fits
well with the necroptosis definition, that is, the morphological
features of cell swelling and plasma membrane integrity, without
chromatin condensation and nuclear fragmentation, and cell death
inhibition by NEC-1 (Figs. 3,
6 and 7). The knockdown of RIPK-1 by siRNA,
which significantly attenuated the enhanced cytotoxicity, also
supports the induction of necroptosis (Fig. 8A). However, recent reports have
revealed the molecular mechanism of necroptosis (40,41)
as follows: Various stresses that induce necroptosis appear to be
mediated by the interaction of activated RIPK3 and MLKL. MLKL is a
substrate for RIPK3 kinase activity, and MLKL phosphorylation at
Thr357 and Ser358 by RIPK3 results in tetramer formation via the
four-helical bundle domain (4HBD) in the N-terminal region. This
oligomerization subsequently leads to MLKL migration to the plasma
membrane to bind phosphatidyl inositol lipids (42). MLKL tetramer accumulation in the
membrane site finally forms an octamer and acts as the cation
channel for ion influx, which increases the intracellular osmotic
pressure leading to cellular destruction (45). As shown in Fig. 4B, autophagy was considerably
upregulated in response to GEF and AAS. It has recently been
reported that, in association with the autophagic machinery for
TRAIL-induced necroptosis, p62/SQSTM1 recruits RIPK1 and mediates
the formation of a multimolecular complex designated as necrosome
consisting of RIPK1, RIPK3 and MLKL for the upregulation of RIPK3
kinase activity (46). Therefore,
the rapid upregulation of autophagosomes in response to GEF plus
AAS appears to function as a scaffold of necrosome formation,
switching toward necroptosis rather than the induction of
apoptosis. However, there were no signs of the phosphorylation of
RIPK1 and MLKL, or necrosome formation (Fig. 8B and C). Although other unclarified
machineries for necroptosis may still exist, these data indicate a
non-standard execution of necroptosis. As the enhanced cytotoxicity
by GEF plus AAS was significantly attenuated in the presence of
NEC-1, as well as the knockdown of RIPK1, this cell death phenotype
appears to be dependent on RIPK1, at least in part (Figs. 6 and 8A). The RIPK3 expression level was much
lower in the CAL 27 cells than in the HT-29 cells (Fig. 8B). Thus, the lower RIPK3 expression
level in CAL 27 cells may affect the execution of necroptosis in
our case.
In contrast to necroptosis acceleration, autophagy
has been reported to rather suppress necroptosis in RCC4 cells, a
human renal cell carcinoma cell line (47). The inhibition of mTOR with CCL-779,
a specific mTOR inhibitor, has been shown to stimulate autophagy
and lead to the elimination of RIPKs via autophagy-mediated
degradation (47). The
simultaneous inhibition of mTOR with CCL-779 and autophagy with
chloroquine resulted in necroptosis induction. Autophagy promotes
the apoptosis induced by Fas ligand/CD95 due to its ability to
degrade Fap-1, a negative regulator of CD95 signaling (48). On the other hand, autophagy
protects the apoptosis induced by TNF-α by controlling the levels
of PUMA, a pro-apoptotic BCL-2 family protein (49). As shown in Fig. 8B, RIPK1 expression was
downregulated during the 24-h exposure to AAS plus GEF in the CAL
27 cells. Therefore, during the upregulated autophagic degradation,
the delicate balance of the molecular components for cell death
execution may also determine the cell fate and cell death
phenotype.
It has recently been reported that when the MLKL
activation is limited or reversed, the ESCRT-III machinery controls
the duration of plasma membrane integrity, which may exert an
efficient in vivo immune response (34). In contrast to apoptosis, necrosis
and necroptosis induce a series of immunological responses
resulting in inflammation in vivo. Therefore, a more
fine-tuned system consisting of many regulatory molecules might
exist. Modulation of the necroptotic process by degradation of
regulators and effectors may have some unknown biological roles
including tumor immunity. Although the precise molecular mechanism
for executing the pronounced cell killing effect in our case
remains to be clarified, the varying amounts of degradation of
molecular components for cell death execution might interfere with
the cell death phonotype, but 'not in a stereotypical manner'.
Acknowledgments
We would like to thank Professor Noboru Mizushima
(University of Tokyo, Tokyo, Japan) for kindly providing us with
the tet-off Atg5 m5–7 cell line. We would also like to thank
Dr Edward Barroga (http://orcid.org/0000-0002-8920-2607), Associate
Professor and Senior Medical Editor of Tokyo Medical University for
editing the manuscript. This study was supported by funds provided
through a MEXT-supported program of the Strategic Research
Foundation at Private Universities (S1411011, 2014–2018) from the
Ministry of Education, Culture, Sports, Science and Technology of
Japan, and partially supported by JSPS KAKENHI Grant Numbers
JP26460478, JP16K19187, JP16K21388, and JP16K21387, and a
Grant-in-Aid from Tokyo Medical University Cancer Research to
N.T.
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Kuma A and Mizushima N: Physiological role
of autophagy as an intracellular recycling system: With an emphasis
on nutrient metabolism. Semin Cell Dev Biol. 21:683–690. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Segawa H, Fukasawa Y, Miyamoto K, Takeda
E, Endou H and Kanai Y: Identification and functional
characterization of a Na+-independent neutral amino acid
transporter with broad substrate selectivity. J Biol Chem.
274:19745–19751. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yanagida O, Kanai Y, Chairoungdua A, Kim
DK, Segawa H, Nii T, Cha SH, Matsuo H, Fukushima J, Fukasawa Y, et
al: Human L-type amino acid transporter 1 (LAT1): Characterization
of function and expression in tumor cell lines. Biochim Biophys
Acta. 1514:291–302. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oda K, Hosoda N, Endo H, Saito K,
Tsujihara K, Yamamura M, Sakata T, Anzai N, Wempe MF, Kanai Y, et
al: L-type amino acid transporter 1 inhibitors inhibit tumor cell
growth. Cancer Sci. 101:173–179. 2010. View Article : Google Scholar
|
|
5
|
Sakata T, Ferdous G, Tsuruta T, Satoh T,
Baba S, Muto T, Ueno A, Kanai Y, Endou H and Okayasu I: L-type
amino-acid transporter 1 as a novel biomarker for high-grade
malignancy in prostate cancer. Pathol Int. 59:7–18. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Galluzzi L, Pietrocola F, Levine B and
Kroemer G: Metabolic control of autophagy. Cell. 159:1263–1276.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J,
Ge W, Feng L, Lin X, Wang X, et al: EGFR tyrosine kinase inhibitors
activate autophagy as a cytoprotective response in human lung
cancer cells. PLoS One. 6:e186912011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fung C, Chen X, Grandis JR and Duvvuri U:
EGFR tyrosine kinase inhibition induces autophagy in cancer cells.
Cancer Biol Ther. 13:1417–1424. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sugita S, Ito K, Yamashiro Y, Moriya S,
Che XF, Yokoyama T, Hiramoto M and Miyazawa K: EGFR-independent
autophagy induction with gefitinib and enhancement of its cytotoxic
effect by targeting autophagy with clarithromycin in non-small cell
lung cancer cells. Biochem Biophys Res Commun. 461:28–34. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mukai S, Moriya S, Hiramoto M, Kazama H,
Kokuba H, Che XF, Yokoyama T, Sakamoto S, Sugawara A, Sunazuka T,
et al: Macrolides sensitize EGFR-TKI-induced non-apoptotic cell
death via blocking autophagy flux in pancreatic cancer cell lines.
Int J Oncol. 48:45–54. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wei Y, Zou Z, Becker N, Anderson M,
Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et
al: EGFR-mediated Beclin 1 phosphorylation in autophagy
suppression, tumor progression, and tumor chemoresistance. Cell.
154:1269–1284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sordella R, Bell DW, Haber DA and
Settleman J: Gefitinib-sensitizing EGFR mutations in lung cancer
activate anti-apoptotic pathways. Science. 305:1163–1167. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ciardiello F and Tortora G: EGFR
antagonists in cancer treatment. N Engl J Med. 358:1160–1174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tan X, Thapa N, Sun Y and Anderson RA: A
kinase-independent role for EGF receptor in autophagy initiation.
Cell. 160:145–160. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moriya S, Che XF, Komatsu S, Abe A,
Kawaguchi T, Gotoh A, Inazu M, Tomoda A and Miyazawa K: Macrolide
antibiotics block autophagy flux and sensitize to bortezomib via
endoplasmic reticulum stress-mediated CHOP induction in myeloma
cells. Int J Oncol. 42:1541–1550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hirasawa K, Moriya S, Miyahara K, Kazama
H, Hirota A, Takemura J, Abe A, Inazu M, Hiramoto M, Tsukahara K,
et al: Macrolide antibiotics exhibit cytotoxic effect under amino
acid-depleted culture condition by blocking autophagy flux in head
and neck squamous cell carcinoma cell lines. PLoS One.
11:e01645292016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dalle Pezze P, Ruf S, Sonntag AG,
Langelaar-Makkinje M, Hall P, Heberle AM, Razquin Navas P, van
Eunen K, Tölle RC, Schwarz JJ, et al: A systems study reveals
concurrent activation of AMPK and mTOR by amino acids. Nat Commun.
7:132542016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptotic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Teng X, Degterev A, Jagtap P, Xing X, Choi
S, Denu R, Yuan J and Cuny GD: Structure-activity relationship
study of novel necroptosis inhibitors. Bioorg Med Chem Lett.
15:5039–5044. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar
|
|
26
|
Kawahara A, Yamamoto C, Nakashima K, Azuma
K, Hattori S, Kashihara M, Aizawa H, Basaki Y, Kuwano M, Kage M, et
al: Molecular diagnosis of activating EGFR mutations in non-small
cell lung cancer using mutation-specific antibodies for
immunohistochemical analysis. Clin Cancer Res. 16:3163–3170. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hosokawa N, Hara Y and Mizushima N:
Generation of cell lines with tetracycline-regulated autophagy and
a role for autophagy in controlling cell size. FEBS Lett.
581:2623–2629. 2007.PubMed/NCBI
|
|
28
|
Sun XM, MacFarlane M, Zhuang J, Wolf BB,
Green DR and Cohen GM: Distinct caspase cascades are initiated in
receptor-mediated and chemical-induced apoptosis. J Biol Chem.
274:5053–5060. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miyahara K, Kazama H, Kokuba H, Komatsu S,
Hirota A, Takemura J, Hirasawa K, Moriya S, Abe A, Hiramoto M, et
al: Targeting bortezomib-induced aggresome formation using
vinorelbine enhances the cytotoxic effect along with ER stress
loading in breast cancer cell lines. Int J Oncol. 49:1848–1858.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hansen TE and Johansen T: Following
autophagy step by step. BMC Biol. 9:392011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Golden SH, Folsom AR, Coresh J, Sharrett
AR, Szklo M and Brancati F: Risk factor groupings related to
insulin resistance and their synergistic effects on subclinical
atherosclerosis: The atherosclerosis risk in communities study.
Diabetes. 51:3069–3076. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang J, Chen X, Su L, Li P, Liu B and Zhu
Z: LAT-1 functions as a promotor in gastric cancer associated with
clinicopathologic features. Biomed Pharmacother. 67:693–699. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gong YN, Guy C, Olauson H, Becker JU, Yang
M, Fitzgerald P, Linkermann A and Green DR: ESCRT-III acts
downstream of MLKL to regulate necroptotic cell death and its
consequences. Cell. 169:286–300.e16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen L, Meng Y, Guo X, Sheng X, Tai G,
Zhang F, Cheng H and Zhou Y: Gefitinib enhances human colon cancer
cells to TRAIL-induced apoptosis of via autophagy- and JNK-mediated
death receptors upregulation. Apoptosis. 21:1291–1301. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Degterev A, Hitomi J, Germscheid M, Ch'en
IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Weinlich R, Oberst A, Beere HM and Green
DR: Necroptosis in development, inflammation and disease. Nat Rev
Mol Cell Biol. 18:127–136. 2017. View Article : Google Scholar
|
|
39
|
Li J, McQuade T, Siemer AB, Napetschnig J,
Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et
al: The RIP1/RIP3 necrosome forms a functional amyloid signaling
complex required for programmed necrosis. Cell. 150:339–350. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ,
Chen X, Cai Q, Yang ZH, Huang D, Wu R, et al: RIP1
autophosphorylation is promoted by mitochondrial ROS and is
essential for RIP3 recruitment into necrosome. Nat Commun.
8:143292017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ros U, Peña-Blanco A, Hänggi K, Kunzendorf
U, Krautwald S, Wong WW and García-Sáez AJ: Necroptosis execution
is mediated by plasma membrane nanopores independent of calcium.
Cell Reports. 19:175–187. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X, et al: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Seto T, Kato T, Nishio M, Goto K, Atagi S,
Hosomi Y, Yamamoto N, Hida T, Maemondo M, Nakagawa K, et al:
Erlotinib alone or with bevacizumab as first-line therapy in
patients with advanced non-squamous non-small-cell lung cancer
harbouring EGFR mutations (JO25567): An open-label, randomised,
multi-centre, phase 2 study. Lancet Oncol. 15:1236–1244. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ichihara E, Hotta K, Nogami N, Kuyama S,
Kishino D, Fujii M, Kozuki T, Tabata M, Harada D, Chikamori K, et
al: Phase II trial of gefitinib in combination with bevacizumab as
first-line therapy for advanced non-small cell lung cancer with
activating EGFR gene mutations: The Okayama Lung Cancer Study Group
Trial 1001. J Thorac Oncol. 10:486–491. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Huang D, Zheng X, Wang ZA, Chen X, He WT,
Zhang Y, Xu JG, Zhao H, Shi W, Wang X, et al: The MLKL channel in
necroptosis is an octamer formed by tetramers in a dyadic process.
Mol Cell Biol. 37:e00497-e162017. View Article : Google Scholar
|
|
46
|
Goodall ML, Fitzwalter BE, Zahedi S, Wu M,
Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD and
Thorburn A: The autophagy machinery controls cell death switching
between apoptosis and necroptosis. Dev Cell. 37:337–349. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bray K, Mathew R, Lau A, Kamphorst JJ, Fan
J, Chen J, Chen HY, Ghavami A, Stein M, DiPaola RS, et al:
Autophagy suppresses RIP kinase-dependent necrosis enabling
survival to mTOR inhibition. PLoS One. 7:e418312012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gump JM, Staskiewicz L, Morgan MJ, Bamberg
A, Riches DW and Thorburn A: Autophagy variation within a cell
population determines cell fate through selective degradation of
Fap-1. Nat Cell Biol. 16:47–54. 2014. View Article : Google Scholar
|
|
49
|
Thorburn J, Andrysik Z, Staskiewicz L,
Gump J, Maycotte P, Oberst A, Green DR, Espinosa JM and Thorburn A:
Autophagy controls the kinetics and extent of mitochondrial
apoptosis by regulating PUMA levels. Cell Rep. 7:45–52. 2014.
View Article : Google Scholar : PubMed/NCBI
|