Introduction
Paclitaxel (PTX) is a natural-product anticancer
drug that is isolated from the bark of Taxus brevifolia. This drug
has generated much interest due to its broad-spectrum
antineoplastic activity, which is mediated by preventing
microtubule depolymerization and subsequently stimulating M-phase
cell cycle arrest and apoptosis (1,2).
Furthermore, it has been reported that the chemotherapeutic use of
PTX can induce generation of reactive oxygen species, increase
production of hydrogen peroxide and stimulate the immune system to
combat tumors (3–5). To overcome the extremely poor water
solubility of PTX, the pharmaceutical company Bristol-Myers Squibb
developed Taxol, which consists of PTX micelles in a solvent of
50:50 (v/v) Cremophor EL (CrEL) and dehydrated ethanol (6). At present, Taxol is broadly used as
in antineoplastic regimen for the treatment of numerous types of
cancer, including ovarian, breast, lung, head-and-neck, prostate
and pancreatic cancer, and acquired immune deficiency
syndrome-related Kaposi's sarcoma (1,7).
However, CrEL is known to induce acute systemic side effects within
10 min of initiation of drug infusion, including anaphylactic
hypersensitivity reactions (HSRs), hypotension, neutropenia,
cardiotoxicity, neuropathy and hyperlipidemia (8–10).
Consequently, corticosteroids and antihistamines are sometimes used
as premedications. As an additional measure, Taxol infusion rate
may be slowed to reduce the intensity and incidence of
CrEL-associated toxicity. However, ~40% of patients still suffer
from HSRs, even after premedication (11). Alongside CrEL-mediated toxicity,
Taxol-based delivery produces undesirable nonlinear pharmacokinetic
behaviors associated with the disposition and metabolism of PTX
(12,13). Therefore, a CrEL-free delivery
system for PTX is urgently required to mitigate the side effects
and improve the pharmacokinetics.
Recently, various PTX delivery systems have been
investigated and developed to reduce CrEL-induced toxicities
associated with Taxol. Emulsions, micelles, polymers, nanocrystals,
cyclodextrins, hydrogels, liposomes and water-soluble prodrugs have
all been explored (14,15). Among these novel formulations, the
only one to successfully gain Food and Drug Administration (FDA)
approval and commercialization is nanoparticle albumin-bound PTX,
Abraxane, which is also called nab-PTX (Celgene Corporation,
Summit, NJ, USA) (16,17). Compared with Taxol, Abraxane
significantly increases the maximum tolerated dosage (300 vs. 175
mg/m2), overall response rate (33 vs. 19%) and time to
progression (23.0 vs. 16.9 weeks) (18). In addition, numerous
Taxol-associated adverse effects, including HSR, grade 3/4
neutropenia, hypotension and bradycardia, are alleviated in
patients receiving Abraxane. However, electrocardiogram (ECG)
irregularities are not rectified by Abraxane, and up to 60% of
patients receiving Abraxane treatment exhibit abnormal ECG readings
(19). Furthermore, the incidence
of myocardial infarction is 44% in the first month following
Abraxane treatment, which is decreased to 33% in the first
half-year and 22% in the second half-year post-therapy (20). Although HSRs associated with
Abraxane are significantly less frequent than with Taxol,
myocardial toxicity remains a major issue (18–20).
Therefore, a novel CrEL-free delivery system for PTX with reduced
cardiac toxicity and a similar therapeutic profile to Taxol is
urgently required to satisfy the clinical need.
For several drugs, liposomal formulations offer
unsurpassed advantages, including the ability to carry a
hydrophobic payload, ease of synthesis, favorable manufacturing
control and excellent biocompatibility. Liposomal PTX has
previously been reported to reduce CrEL-associated side effects at
the expense of poor stability (21–25).
The present study introduced a novel liposomal PTX (lipo-PTX)
preparation that is composed of PTX/lipid at a molar ratio of 2%,
with a PTX concentration of 2 mg/ml in PBS. This lipo-PTX
formulation exhibited long-term stability when stored at 4°C, and
possessed a similar therapeutic efficacy to Taxol in various cancer
models. Notably, this novel formulation exhibited major reductions
in CrEL-associated systemic side effects.
Materials and methods
Materials
Soybean phosphatidylcholine (SPC; Lipoid S100) and
N-(carbonyl-methoxy-poly-ethylene glycol
2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine,
sodium salt (mPEG-DSPE; Lipoid PE 18:0/18:0-PEG2000) were purchased
from Lipoid GmbH (Ludwigshafen, Germany). Cholesterol (C8667) and
MTT (M5655) were obtained from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). PTX (SPF2039) was provided by ScinoPharm
Taiwan, Ltd. (Tainan, Taiwan). Oregon Green®
488-conjugated PTX (OG488-PTX; P22310) was purchased from Thermo
Fisher Scientific, Inc. (Waltham, MA USA). Lissamine rhoda-mine
B-conjugated
1,2-dipalmitoyl-sn-glycero-3-phosphoeth-anolamine-N-(lissamine
rhodamine B sulfonyl) (ammonium salt) (Rh-DPPE; 810158P) was
supplied by Avanti Polar Lipids (Alabaster, AL, USA). Taxol was
obtained from Bristol-Myers Squibb Company (New York, NY, USA) and
Abraxane was provided by Celgene Corporation. Roswell Park Memorial
Institute (RPMI)-1640 medium (31800-022), Dulbecco's modified
Eagle's medium (DMEM; 12100-061), DMEM/Nutrient Mixture F-12
(DMEM/F12; 12400-024), fetal bovine serum (FBS; 10437028),
penicillin-streptomycin solution (P/S; 15140-122) and 2.5% (w/v)
trypsin solution (15090-046) were purchased from Gibco (Thermo
Fisher Scientific, Inc.)
Animals
Non-obese diabetic/severe combined immunodeficiency
(NOD/SCID, NOD.CB17-Prkdcscid/J) mice (female;
age, 4–6 weeks; weight, 18–22 g) were obtained from National
Laboratory Animal Center (Taipei, Taiwan), and outbred ICR
(Bltw:CD1) mice (female; age, 5–8 weeks; weight, 25–30 g) were
supplied by BioLASCO Taiwan Co., Ltd. (Taipei, Taiwan). Mice were
maintained in a specific pathogen-free animal room, with ad
libitum access to food and water intake, at 18–23°C, 40–60%
humidity under a 12-h light/dark cycle at the Institute of Cellular
and Organismic Biology, Academia Sinica (Taipei, Taiwan). All
animal care and experimental procedures were conducted in
accordance with the principles in the Guide for the Care and Use of
Laboratory Animals (26), and the
present study was approved by the Institutional Animal Care and Use
Committee of Academia Sinica.
Preparation of PTX-loaded liposomes
PTX-loaded liposomes were prepared according to the
thin film hydration method described previously (27–30).
Briefly, SPC, cholesterol, mPEG-DSPE and PTX were dissolved in
chloroform (C2432; Sigma-Aldrich; Merck KGaA) at a molar ratio of
95:2:1:2 in a round-bottom flask. A dry lipid film was formed on
the bottom of the flask after the organic solvent was removed by
rotary evaporation at 40°C, and the film was hydrated in PBS (pH
7.4) at 45°C. The lipid suspension was downsized through 10
freeze-thaw cycles, and a LIPEX® Extruder (TRANSFERRA
Nanosciences Inc., Burnaby, BC, Canada) with 0.2 µm
polycarbonate filter membranes (WHA110406; Sigma-Aldrich; Merck
KGaA) was used to finally obtain homogeneous unilamellar liposomes.
To evaluate the concentration and encapsulation efficiency of PTX,
the encapsulated drug was extracted using 40% acetonitrile in fresh
distilled-deionized water, and 40 µl of the extraction was
assessed using a high-performance lipid chromatography (HPLC)
system, which comprised of the 2707 Autosampler (Waters
Corporation, Milford, MA, USA), 600 Controller and Pump (Waters
Corporation), reverse phase HPLC column (4.6×100 mm, 2 µm
macropores in diameter; 1.02129.0001; Merck KGaA) and 2489
UV/Visible Detector (Waters Corporation). HPLC was performed under
a linear gradient of acetonitrile:water from 40:60 to 95:5 (v/v)
within 15 min at a flow rate of 0.8 ml/min at 20°C. Signal was
detected at a single wavelength of 227 nm. The encapsulation
efficiency (EE) was calculated as follows: EE (%) = PTX
concentration in the filtered liposomes / PTX concentration in the
unfiltered liposomes × 100%. The phosphorus in the liposome
suspension was determined by Bartlett assay (31). The lipo-PTX preparations were
stored at 4°C and the stability during storage was determined by
monitoring PTX content within the liposome suspension. Blank
liposomes and Rh-DPPE-containing liposomes served as controls and
were fabricated with SPC:cholesterol:mPEG-DSPE and SPC:
cholesterol:mPEG-DSPE:Rh-DPPE at molar ratios of 97:2:1 and
97:2:0.8:0.2 to match lipo-PTX in each experiment.
For the preparation of non-extruded lissamine
rhodamine B anchored liposomes containing OG488-PTX, Rh-DPPE and
OG488-PTX were added at the same time as lipid materials. The dry
lipid film was hydrated in PBS, without homogenization by
freeze-and-thaw cycles and extrusion.
Characterization of liposomes
The mean size and zeta potential of lipo-PTX were
detected by dynamic light scattering (Malvern Zetasizer Nano ZS;
Malvern Instruments Ltd., Malvern, UK). The lipo-PTX samples were
submitted to cryo-transmission election microscopy (Cryo-TEM) at
the Core Facility, Department of Academic Affairs and Instrument
Service at Academia Sinica (Taipei, Taiwan), to observe structural
features (30).
In vitro drug release
The PTX release profiles from Taxol and lipo-PTX
formulations were investigated according to a dialysis bag
diffusion method. Briefly, 1 ml Taxol or lipo-PTX (1 mg/ml) was
placed in a dialysis tube (molecular weight cut-off 6–8 kDa;
71509-3; Merck KGaA) and immersed into 40 ml release medium (PBS,
pH 7.4 or 5.0). The entire system was incubated at 37°C with
orbital agitation at 150 rpm. The concentration of PTX remaining in
the dialysis tube at predetermined time intervals (0, 0.5, 1, 2, 4,
6, 8, 24, 48, 72, 96 and 168 h) was measured by HPLC.
Pharmacokinetic assay
NOD/SCID mice were intravenously injected with Taxol
or lipo-PTX at 20 mg/kg (n=6/group). Blood samples were withdrawn
into BD Microtainer® blood collection tubes with EDTA
(365974; BD Biosciences, Franklin Lakes, NJ, USA) at various time
points after injection (0, 0.25, 0.5, 1, 2, 4, 6, 8, 24 and 30 h).
Total blood samples were then centrifuged at 3,000 × g for 10 min
at 4°C to obtain plasma samples. To remove the interfering
substances, plasma protein was precipitated with 10X acetonitrile.
The solution was agitated for 1 h to extract PTX from plasma
completely. PTX dissolved in acetonitrile was quantified by HPLC
(Waters Corporation).
Cell culture
Human breast cancer cell lines (MDA-MB-231, SKBR3
and MCF-7), human lung cancer cell lines (A549, H460 and CL1-5) and
a human ovarian cancer cell line (SKOV3) were purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA). Human
liver cancer cell lines, Mahlavu and SK-HEP-1, were kindly provided
by Dr M. Hsiao (Genomic Research Center, Academia Sinica).
MDA-MB-231 cells were grown in DMEM/F12 medium supplemented with
10% FBS. SKBR3, MCF-7, A549, H460 and CL1-5 cells were cultured in
RPMI-1640 supplemented with 10% FBS. SKOV3, Mahlavu and SK-HEP-1
cells were maintained in DMEM supplemented with 10% FBS. All cells
were maintained in the appropriate medium containing 100 U/ml P/S
and were incubated in a humidified atmosphere containing 5%
CO2 at 37°C, according to the ATCC protocols.
Cellular uptake
The cellular internalization of liposomal contents
was determined by quantifying the fluorescence intensity of
lissamine rhodamine B within cells, which was conjugated to DPPE
(Rh-DPPE) and inserted in the liposomes. MDA-MB-231 and SKOV3 cells
were seeded onto 24-well plates at a density of 8×104
cells/well and were cultured for 24 h at 37°C. The cells were then
treated with Rh-DPPE-containing liposomes at a final Rh-DPPE
concentration of 0, 2, 5, 10 and 20 µM for 2 h. After
washing with cold PBS twice, cells were incubated with 0.1 M
glycine (pH 2.2) (50046; Sigma-Aldrich; Merck KGaA) on ice for 5
min to remove residual liposomes from the cell surface. The cells
were harvested by scraping with 1% Triton-X 100/PBS after washing
with PBS twice. The fluorescence intensity in the cells was
measured using a spectrofluorometer (SpectraMax M5, Molecular
Devices) at excitation 520 nm/emission 570 nm.
In vitro cellular cytotoxicity
Cells were seeded onto a 96-well culture plate at
5×103 cells/well and were incubated with culture medium
containing 5% FBS overnight at 37°C. Cells were then treated with
two-fold serially diluted Taxol and lipo-PTX at doses ranging
between 0.195 and 50 nM for 48 h. The equal amount of phosphorus in
empty liposome suspension was used as a lipid toxicity control.
After being washed with PBS twice, cells were incubated with
serum-free medium containing 0.5 mg/ml MTT for 4 h at 37°C.
Subsequently, the medium was removed, and purple crystals within
the cells were dissolved with dimethyl sulfoxide. Cell viability
was determined by measuring absorbance at 540 nm.
In vivo antitumor efficacy
The flanks of NOD/SCID mice were subcutaneously
(s.c.) injected with 2×106 cells (MDA-MB-231, SKOV3 or
SK-HEP-1). The tumor volume was calculated as follows: Length x
(width)2 × 0.52. When the average tumor volume reached
70–100 mm3, mice were randomized into different groups:
Taxol, Abraxane, lipo-PTX and PBS (n=5–11/group). The drugs (15 or
30 mg/kg PTX formulations) were intravenously administered every 4
days for a total of four injections. PBS was injected into the
negative control group. The tumor inhibition rate was defined as
follows: 100% - (tumor volume upon PTX injection/tumor volume of
control group) × 100%.
In vivo biodistribution of PTX
The flanks of NOD/SCID mice were injected (s.c.)
with 107 MDA-MB-231 cells. Tumor volume was calculated as follows:
Length x (width)2 × 0.52. When the average tumor volume
reached 200–300 mm3, mice were randomized into four
groups: PBS, Taxol, Abraxane and lipo-PTX (n=3/group). Mice were
intravenously injected once with 15 mg/kg Taxol, Abraxane or
lipo-PTX. Vital organs, including the brain, heart, liver, kidney
and spleen, as well as tumors were harvested 2 h post-injection,
and were homogenized in distilled water using a MagNA Lyser
Instrument (Roche Diagnostics, Basel, Switzerland). PTX in the
organs was extracted using 40% acetonitrile in distilled water and
the samples were evaluated by HPLC (Waters Corporation).
In vivo toxicity assay
ICR mice were randomized into four groups
(n=12/group), and were intravenously injected with 15 mg/kg Taxol,
Abraxane, lipo-PTX or PBS every 4 days for a total of four
injections. A total of 1 h after the last injection, whole blood,
which was collected in BD Microtainer® blood collection
tubes (BD Biosciences) was supplied to Taiwan Mouse Clinic (Taipei,
Taiwan) for blood cell count using the IDEXX ProCyte Dx Hematology
Analyzer (IDEXX Laboratories, Inc., Westbrook, MA, USA).
For blood biochemistry examination, 24 h after the
last injection, whole blood was collected in BD
Microtainer® tubes (BD Biosciences) and was centrifuged
at 3,000 × g for 10 min at 4°C to obtain plasma samples. Plasma
samples were supplied to Taiwan Mouse Clinic to determine blood
Ca2+ concentration, alanine aminotransferase (ALT),
aspartate aminotransferase (AST), total bilirubin (TBIL), blood
urea nitrogen (BUN) and uric acid (UA) using the Fuji Dri-Chem
4000i (Fujifilm, Tokyo, Japan).
For histological analysis, 24 h after the last
intravenous treatment, major organs, including the brain, heart,
liver, kidney and spleen were harvested, fixed in 10%
neutrally-buffered formalin (3.7% formaldehyde, 0.029 M
KH2PO4, and 0.037 M K2HPO4 in
distilled-deionized water) overnight at room temperature, and were
then dehydrated in an ascending series of alcohol. The specimens
were then immersed in xylene (Merck KGaA) and embedded in paraffin
(39601006; Leica Biosystems Richmond, Inc., Richmond, IL, USA).
Sections (size, 4 µm) were cut with a microtome (Olympus Cut
4055; Olympus Corporation, Tokyo, Japan) and mounted on slides. The
slides then were subjected to hematoxylin and eosin staining.
Briefly, the sections were deparaffinized in xylene and rehydrated
in a descending series of alcohol. Subsequently, the sections were
stained with Gill's hematoxylin (GHS132; Sigma-Aldrich; Merck KGaA)
for 15 min. After washing under running water for 15 min, the
sections were stained with 0.5% eosin Y (230251; Sigma-Aldrich;
Merck KGaA) for 2 min. Finally, the stained sections were
dehydrated and mounted with Permount (Thermo Fisher Scientific,
Inc.), and examined by light microscopy. All histopathological
images were captured with an Olympus BX53 microscope (Olympus
Corporation) and DP73-BSW image acquisition software (Olympus
Corporation).
ECG recordings
The ECG recordings were performed non-invasively on
anesthetized mice in lead II using PowerLab 8/30 and Animal Bio Amp
(ADInstruments Pty Ltd., New South Wales, Australia) at the Taiwan
Mouse Clinic Center (Taipei, Taiwan). The ECG was recorded
continuously from 5 min before to 15 min after intravenous PTX
administration in all individual ICR mice (n=9/group). Heart rate,
PR interval, RR interval, QRS interval and QT interval were
analyzed using LabChart software version 8.1 (ADInstruments Pty
Ltd.). The corrected QT (QTc) interval represented the QT interval
normalized for heart rate using Bazett's formula: QTc = measured QT
interval / √RR interval.
Statistical analysis
Data are presented as the means ± standard deviation
or standard error of the mean of at least three independent
experiments. GraphPad Prism software version 5 (GraphPad Software,
Inc., La Jolla, CA, USA) was used for statistical analysis.
Two-tailed unpaired Student's t-test was used for comparisons
between two groups. One-way analysis of variance (ANOVA) followed
by Student-Newman-Keuls test and two-way ANOVA followed by
Bonferroni post hoc test were used for comparisons among three or
more groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
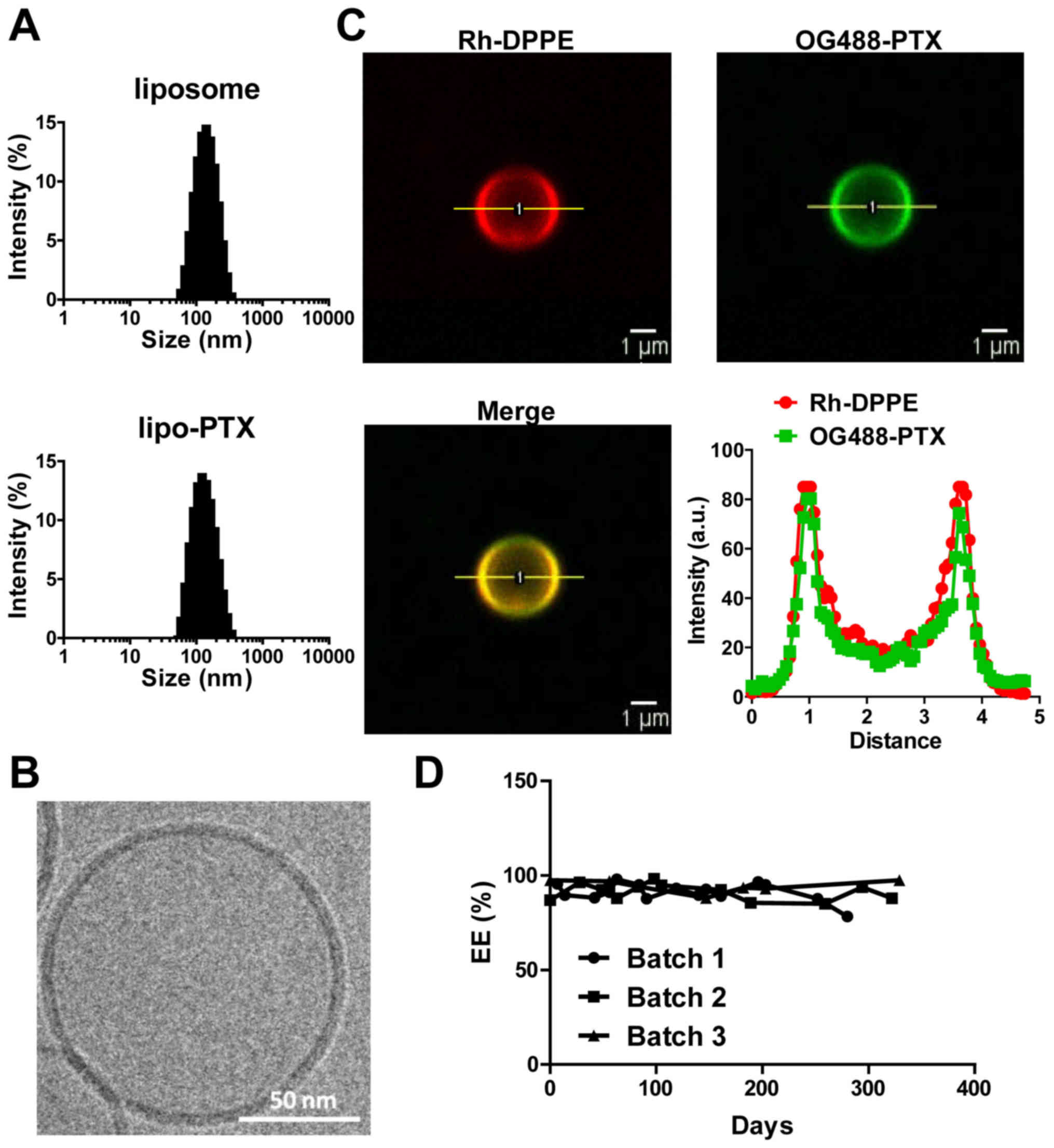
Development of a homogenous and stable
lipo-PTX formulation
Physical characteristics of formulations, including
particle size and surface charge, have a marked impact on the
bioavailability and solubility of therapeutics. The mean particle
size of liposomes and lipo-PTX used in the present study was ~125
nm and the polydispersity index was 0.12. No substantial
differences in size distribution or mean size were detected between
the control liposomes and lipo-PTX. The nanoparticles were mildly
negatively charged (surface potential: -15.42±1.40 mV, Table I and Fig. 1A). These measurements indicated
that this liposomal preparation was homogeneous and suitable for
penetration through leaky tumor vasculature, while evading
clearance by phagocytic systems. With regards to therapeutic
payload, EE was 94%, and 2×104 molecules of PTX were
estimated to be loaded per vesicle (Table I).
 | Table ICharacteristics of liposomal
formulation. |
Table I
Characteristics of liposomal
formulation.
| Variable | Liposome | lipo-PTX |
|---|
| Average particle
size (nm) | 127.98±3.35 | 123.12±4.18 |
| Polydispersity
index | 0.12±0.02 | 0.12±0.01 |
| Zeta potential
(mV) | −17.52±0.64 | −15.42±1.40 |
| PTX encapsulation
efficiency (%) | – | 94.16±1.85 |
| No. of PTX
molecules in a liposome | – |
2.30×104 |
Cryo-TEM imaging demonstrated that the nanoparticles
were spherical liposomes, composed of lipid bilayers (Fig. 1B). Co-localization of OG488-PTX and
Rh-DPPE on non-extruded liposomes was confirmed by confocal
microscopy. Furthermore, the PTX payload was harbored within the
lipid membrane and not the aqueous core of liposomes (Fig. 1C). Subsequently, the stability of
lipo-PTX was evaluated over the course of 300 days. The PTX
concentration in the liposome suspension was not significantly
altered within 250 days at 4°C (Fig.
1D). These results indicated that the liposomes were stable and
possessed a reasonable shelf life for future clinical
application.
Lipo-PTX is bioequivalent to Taxol in
vivo
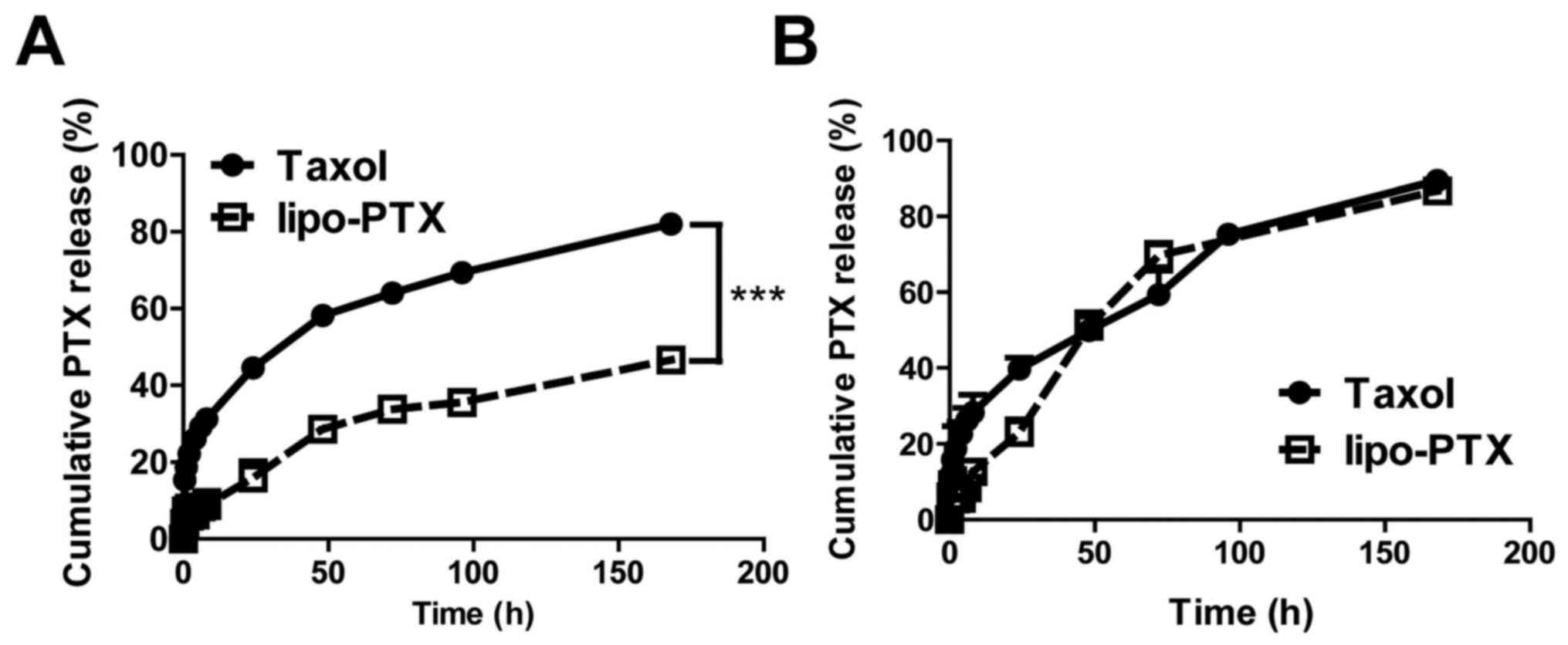
Premature drug release in serum and tissue fluid may
hamper the therapeutic effects of a drug and cause undesirable
toxicity to normal tissues. The present study monitored the release
profile of lipo-PTX and Taxol at physiological pH (Fig. 2A). At pH 7.4, only 16% of PTX was
released from lipo-PTX within 24 h, whereas 45% of PTX was released
from Taxol in the same time period. Taxol achieved 50% PTX release
at 34 h; conversely, the amount of PTX released from lipo-PTX was
only 47% after 168 h incubation in PBS at pH 7.4. These results
suggested that lipo-PTX is significantly more stable than the Taxol
formulation and that collateral toxicity to normal tissue might be
alleviated due to reduced premature release of PTX.
Nanoparticles taken up by cells are expected to fuse
with endosomes and lysosomes, where the contents will be degraded
in an acidic microenvironment with a pH value near 5.0 (32,33).
The profile of drug release from Taxol at pH 5.0 was comparable to
that at pH 7.4. However, PTX release from lipo-PTX exhibited a 1.5-
to 2-fold increase following 48 h in an acidic environment (pH 5.0)
when compared with at pH 7.4 (48 h, 51 vs. 29%; 72 h, 70 vs. 34%;
168 h, 87 vs. 47%) (Fig. 2B).
These results indicated that lipo-PTX is a pH-sensitive drug
delivery platform, which is stable at physiological pH. However,
the efficiency of drug release is enhanced in acidic environments,
such as those that exist in intracellular lysosomes and
intratumoral stroma.
The present study also compared the pharmacokinetics
of Taxol and lipo-PTX. Both PTX formulations were administered
intravenously to NOD/SCID mice, and the time-dependent plasma PTX
concentration profiles were determined by HPLC. As shown in
Table II, PTX concentration in
the plasma was continually and quickly diminished following Taxol
administration, and dropped to the basal level after 10 h. The area
under the curve (AUC)0→24 for Taxol injection was 53.24
mg·h/l, and the half-life (t1/2) was 1.10 h. Total
clearance was 0.38 l/h/kg, and the elimination constant for Taxol
was 0.63 h-1 (Table II). All of
the pharmacokinetic parameters were consistent with the results
from a previous study in mice (34). Although the blood PTX concentration
and AUC0→24 (89.12 mg·h/l) in the lipo-PTX-administered
group were slightly higher than those in the Taxol group, lipo-PTX
only slightly extended the circulation time for PTX in the blood
(t1/2 = 1.73 h) compared with Taxol (t1/2 =
1.10 h). Consequently, there were no significant differences in
pharmacokinetic parameters between lipo-PTX and Taxol (Table II), thus indicating that lipo-PTX
was bioequivalent to Taxol in vivo.
| Table IIPharmacokinetic parameters of PTX
observed in NOD/SCID mice after intravenous injection
administration of 20 mg/kg of Taxol and lipo-PTX. |
Table II
Pharmacokinetic parameters of PTX
observed in NOD/SCID mice after intravenous injection
administration of 20 mg/kg of Taxol and lipo-PTX.
| Therapy | Dose (mg/kg) | AUC0→24
(mg·h/l) | K
(h−1) | t1/2
(h) | CL (l/h/kg) |
|---|
| Taxol | 20 | 53.17 | 0.63 | 1.10 | 0.38 |
| lipo-PTX | 20 | 89.12 | 0.40 | 1.73 | 0.22 |
Antineoplastic efficacy of lipo-PTX is
comparable to that of commercial PTX formulations in vitro and in
vivo
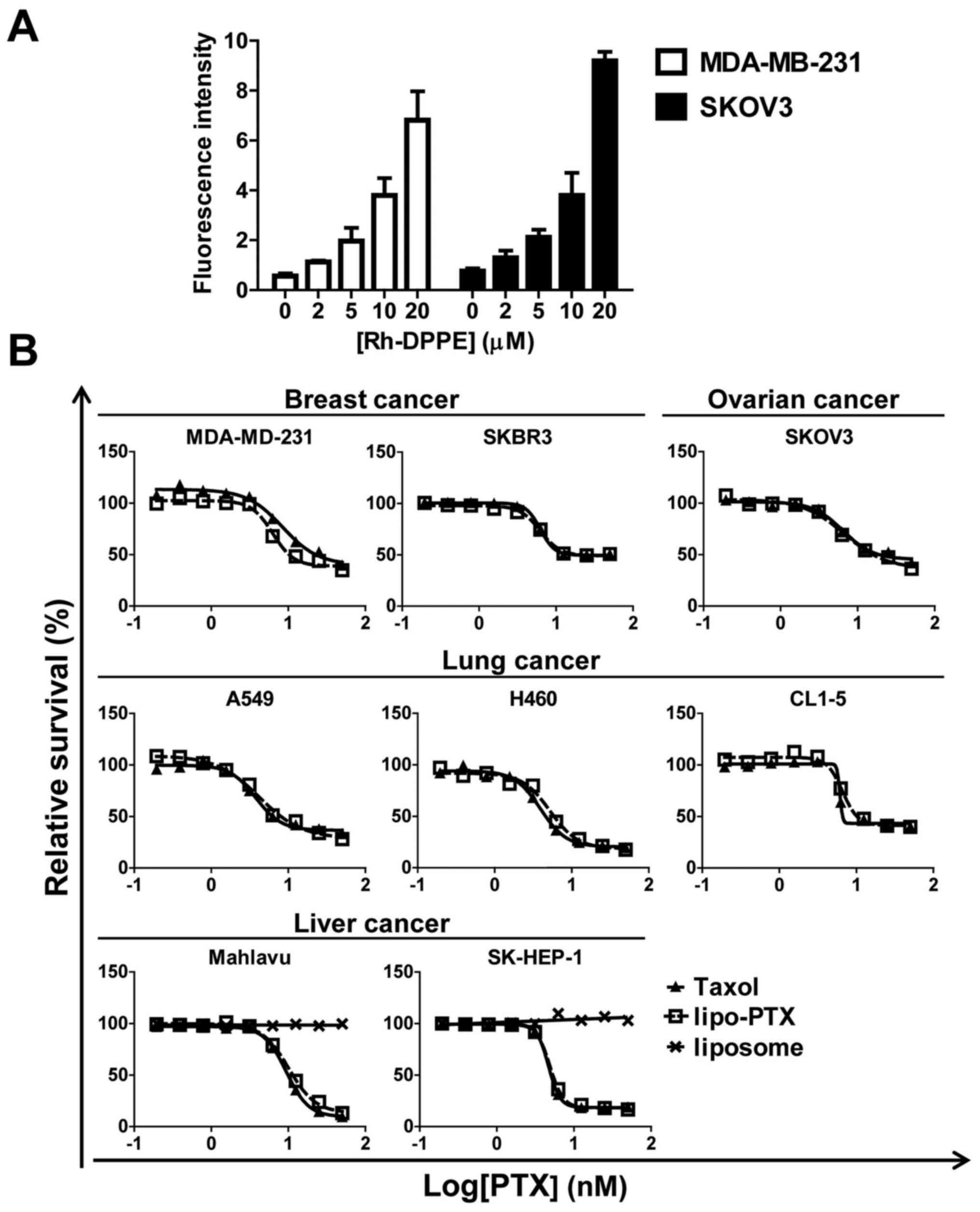
To confirm that lipo-PTX would be taken up by cancer
cells, Rh-DPPE was used to quantify the level of cellular
internalization. The intracellular fluorescence in MDA-MB-231 and
SKOV3 cells was dose-dependently increased following incubation
with liposomes (Fig. 3A), thus
indicating that the cells took up the liposomal nanoparticles. In
addition, the antineoplastic action of lipo-PTX was detected in
cell cultures. The results indicated that lipo-PTX markedly
suppressed cell proliferation in various cancer cell lines. The
half maximal inhibitory concentration (IC50) values of
lipo-PTX ranged between 4. and 13.2 nM, and were similar to those
of Taxol in each corresponding cell line (Fig. 3B). No cytotoxicity was discernible
when liver cancer cells were treated with empty liposomes as a
negative control. Taken together, these data suggested that the
therapeutic effects of lipo-PTX resulted from the PTX payload,
instead of the liposomal vehicle, and that its anti-neoplastic
action was comparable to that of CrEL-containing Taxol.
The present study further compared the therapeutic
efficacy of lipo-PTX to Taxol in various subcutaneous xenograft
murine models (Fig. 4A–H). Body
weight was recorded and served as a gross indicator of systemic
toxicity. As shown in Fig. 4A,
MDA-MB-231 xenografts were sensitive to treatment with both Taxol
and lipo-PTX, and tumor size was markedly reduced following PTX
infusion. Lipo-PTX administration suppressed the tumor burden of
MDA-MB-231 xenograft mice slightly more efficiently than Taxol did
(inhibition rate on day 20, 76 vs. 72%). In SKOV3 xenograft mice,
lipo-PTX exhibited a comparable therapeutic effect to Taxol
(inhibition rate on day 26, 82 vs. 84%) (Fig. 4C). However, lipo-PTX exhibited a
slightly inferior antineoplastic effect compared with Taxol on
SK-HEP-1 xenografts (inhibition rate on day 20, 92 vs. 98%)
(Fig. 4E). In addition, the
therapeutic effects of lipo-PTX were compared with those of
Abraxane in MDA-MB-231 xenografts. The inhibitory rate of lipo-PTX
(15 mg/kg) was slightly lower than that of Abraxane (inhibition
rate on day 20, 76 vs. 81%, Fig.
4A); however, administration with 30 mg/kg lipo-PTX suppressed
tumor growth more efficiently than Abraxane (inhibitory rate, 94
vs. 77%, Fig. 4G). The effects of
empty liposomes were not detected in vivo, because the
intention was to compare the novel liposome formulation with
existing formulations. Overall, lipo-PTX exhibited a therapeutic
effect that was superior or equivalent to that of Taxol and
Abraxane in numerous murine subcutaneous xenograft models. No
significant systemic toxicity was observed in any group, as the
body weight of the mice was stable (Fig. 4B, D, F and H).
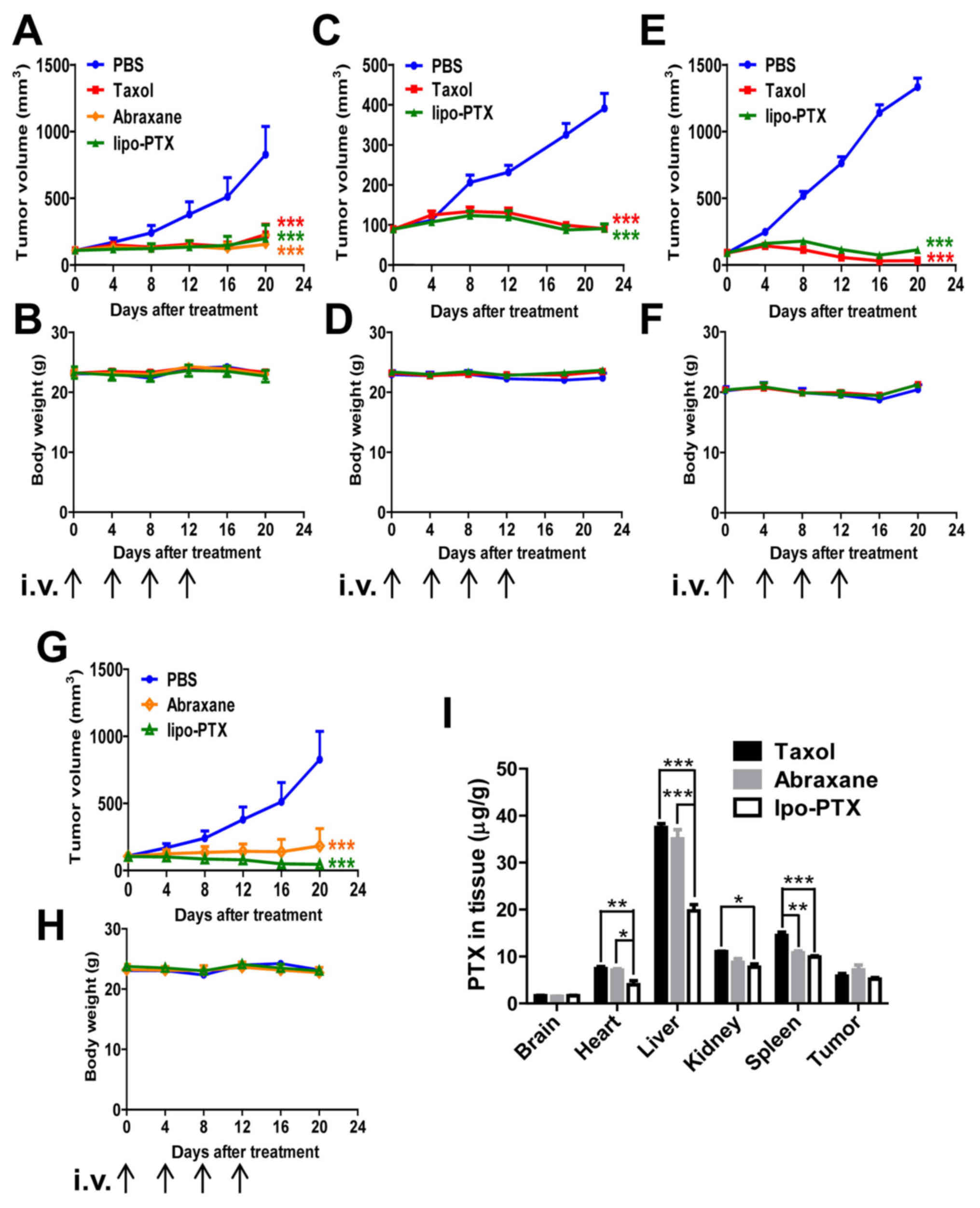 | Figure 4Therapeutic efficacy of liposomal PTX
in xenograft models. (A-H) Mouse xenograft models of (A, B, G and
H) MDA-MB-231 human breast cancer cells, (C and D) SKOV3 human
ovarian cancer cells or (E and F) SK-HEP-1 human liver cancer
cells; mice were intravenously injected four times with Taxol,
Abraxane or lipo-PTX at 15 or 30 mg/kg every 4 days. PBS was used
as a negative control. (A, C, E and G) Tumor volume and (B, D, F
and H) body weight within each group were determined. (A and B, 15
mg/kg, n=6; C and D, 15 mg/kg, n=11; E and F, 15 mg/kg, n=8; G and
H, 30 mg/kg, n=5). Data are presented as the means ± standard error
of the mean. ***P<0.001 compared with the PBS group,
two-way analysis of variance followed by Bonferroni post hoc test.
(I) PTX distribution in MDA-MB-231-bearing mice 2 h after treatment
with 15 mg/kg Taxol, Abraxane or lipo-PTX. Data are presented as
the means ± standard error of the mean from three independent mice
in each group. *P<0.05, **P<0.01,
***P<0.001, two-way analysis of variance followed by
Bonferroni post hoc test. lipo-PTX, novel liposomal PTX; PTX,
paclitaxel. |
Lipo-PTX has reduced side effects
compared with Taxol or Abraxane
Although no significant alterations in body weight
were detected in mice treated with Taxol, Abraxane or lipo-PTX
(Fig. 4B, D, F and H), HSRs were
observed upon administration of Taxol, but not lipo-PTX (data not
shown). The reactions were characterized by loss of motility,
angioedema, dyspnea, syncope and even mortality. To further
investigate the systemic toxicity of lipo-PTX, PTX accumulation in
major organs was examined 2 h after the injection of PTX
formulations. If the tumor is too small, the blood supply to the
tumor will be limited, making it difficult to measure the precise
dosage and time for drug accumulation. When the tumor is too large,
the center of the tumor will undergo necrosis, which will also
impact the drug/tissue ratio (w/w) (30). Therefore, tumor volume at 200–300
mm3 was used to avoid problems associated with limited
blood supply or necrosis. As shown in Fig. 4I, Taxol, Abraxane and lipo-PTX
mainly accumulated in the liver, spleen and kidney. No evident
histological abnormalities were noted in the vital organs,
including the brain, heart, liver, kidney and spleen of any of the
four groups (Fig. 5A). In
addition, no significant elevation of liver damage indicators
(serum ALT, AST, and TBIL) and kidney function markers (BUN and UA)
was detected in any of the treatment groups (Fig. 5B). Taken together, these data
indicated that no evident hepatic damage or renal impairment was
present in any treatment group.
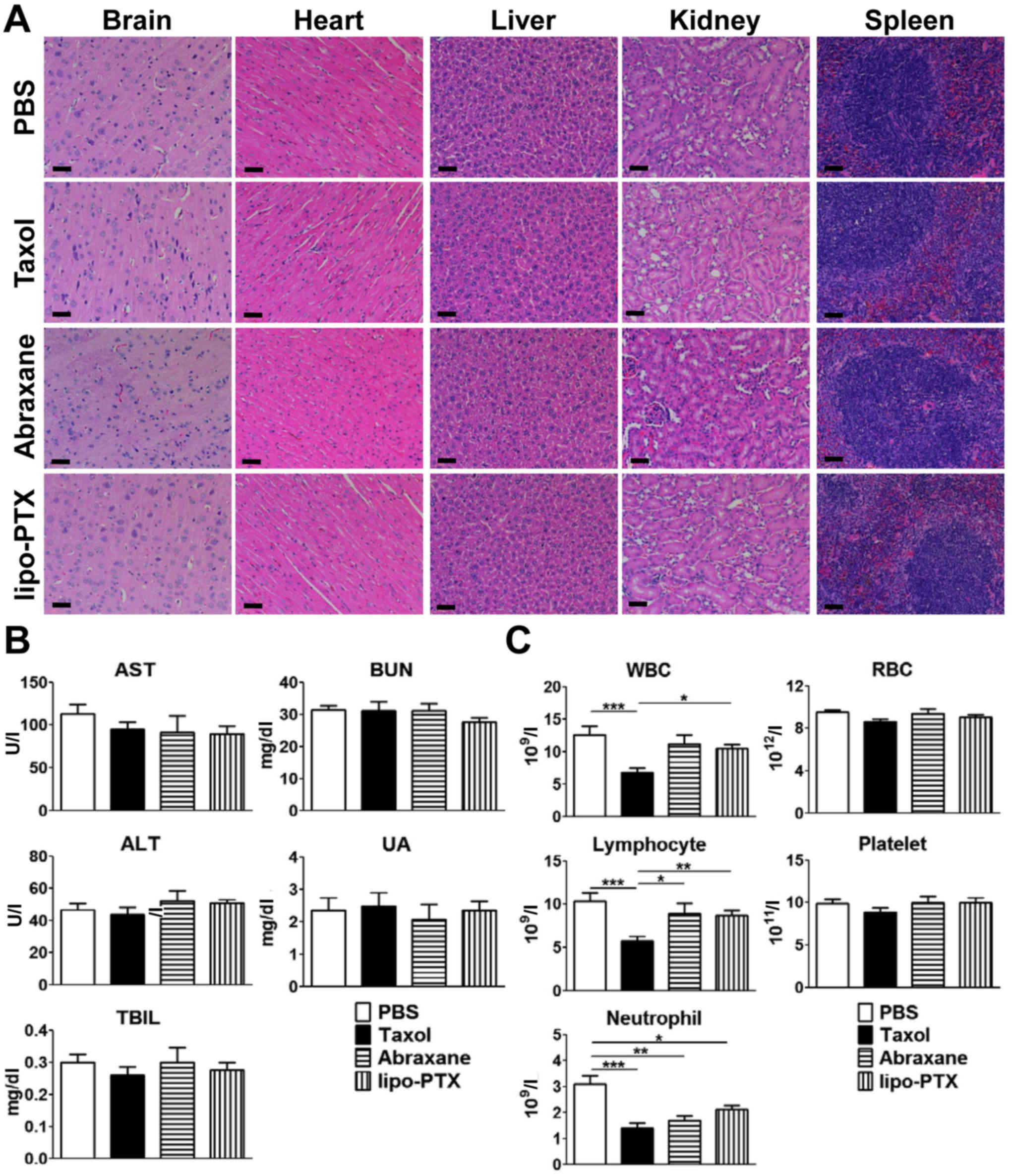 | Figure 5Evaluation of potential toxicity of
lipo-PTX. Healthy tumor-free ICR mice were intravenously injected
four times with Taxol or lipo-PTX (15 mg/kg) every 4 days. PBS was
used as a negative control. Blood and organs were harvested 1 h
after the last treatment. (A) Histopathology of vital organs, (B)
serum chemistry levels and (C) blood counts are shown. Scale bars,
50 nm. Data are presented as the means ± standard deviation (n=12).
*P<0.05, **P<0.01,
***P<0.001, one-way analysis of variance followed by
Student-Newman-Keuls test. ALT, alanine aminotransferase; AST,
aspartate aminotransferase; BUN, blood urea nitrogen; lipo-PTX,
novel liposomal PTX; PTX, paclitaxel; RBC, red blood cells; TBIL,
total bilirubin; UA, uric acid; WBC, white blood cells. |
With regards to the suppression of hematopoiesis,
Taxol administration markedly decreased the number of white blood
cells, lymphocytes and neutrophils compared with in the PBS-treated
group (Fig. 5C). Conversely, no
significant myelosuppression was noted in groups treated with
lipo-PTX or Abraxane, except for a consistent decrease in
neutrophils in all three treatment groups. Nevertheless, the
reduction in neutrophils was not as apparent in the lipo-PTX group
as in the Taxol group. Furthermore, the lymphocyte and white blood
cell populations of the lipo-PTX group were significantly higher
compared with in the Taxol group, whereas the differences between
lipo-PTX and Abraxane were not significant. Therefore, it may be
concluded that lipo-PTX has lower hematopoietic toxicity than
Taxol.
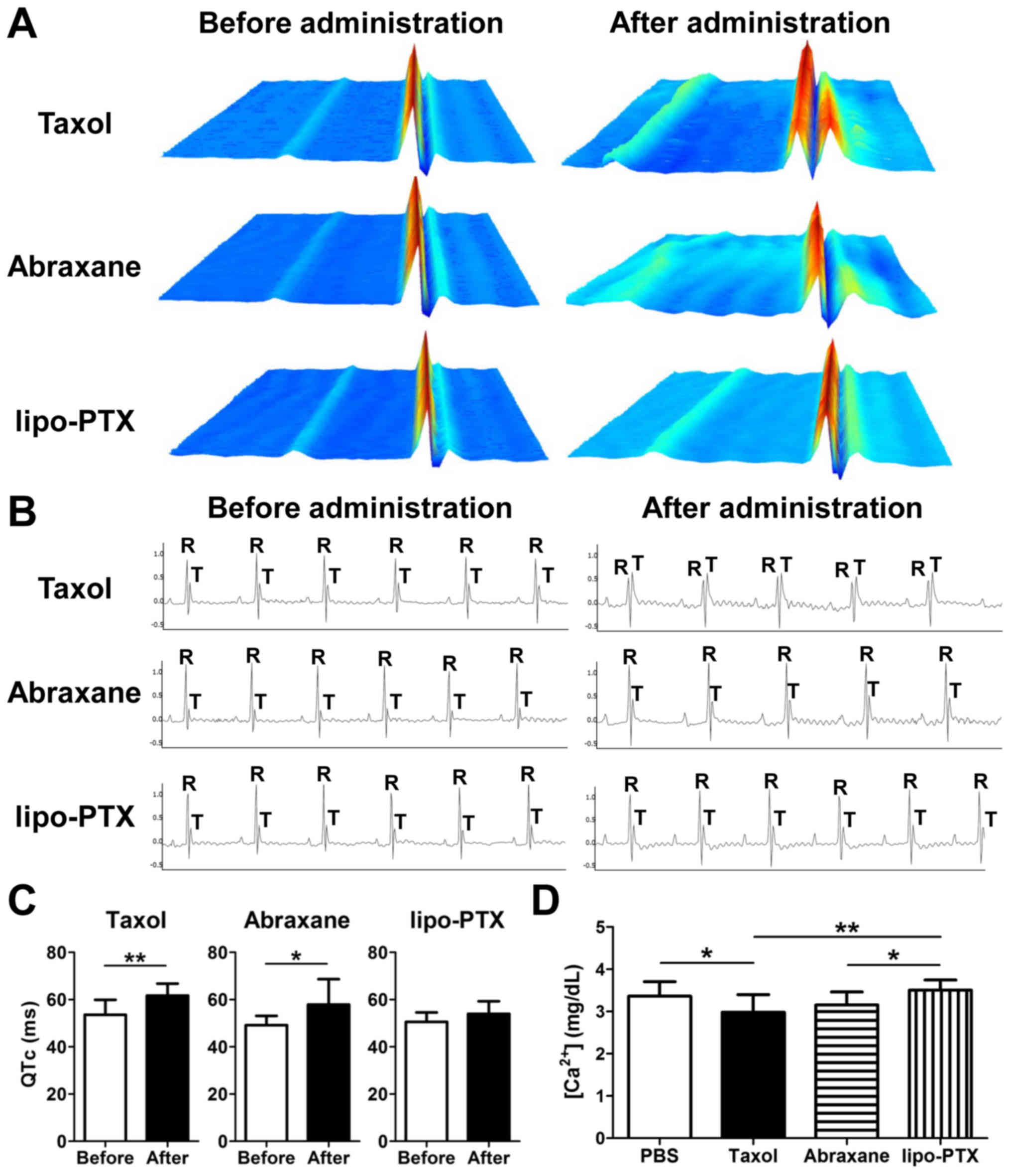
Taxol, Abraxane and lipo-PTX deposited similar
amounts of PTX in the tumor tissues following treatment (Fig. 4I). Notably, there was more Taxol
and Abraxane accumulation in heart than that of lipo-PTX. In a
previous study, Taxol infusion was revealed to induce sinus
bradycardia and other myocardial disturbances (10). To further elucidate the
cardiovascular impacts of drug administration, ECG traces were
recorded and analyzed (Fig. 6A–C).
Overall, ECG abnormalities were present in 100% (9/9) of mice
injected with Taxol, whereas this phenomenon was only seen in 66%
(6/9) of mice treated with Abraxane and 22% (2/9) of mice treated
with lipo-PTX. Notably, the irregularities in lipo-PTX-treated mice
were transient and sustained for less than 3 min, which was
approximately one-fourth the duration observed in the Taxol group
during the first 15 min after administration. Mice treated with
Taxol and Abraxane exhibited abnormal ECG recordings, which
included an irregular heartbeat and apparent QTc prolongation
(Fig. 6A and C), indicating
cardiac arrhythmia. Furthermore, a reduction in R-wave amplitude
and an increase in T-wave amplitude were detected in the
Taxol-treated mice (Fig. 6B).
These irregularities indicated that Taxol may impair the strength
of contractions, as previously reported (35). Although no apparent alterations
were observed in the R- and T-wave amplitude following Abraxane
infusion, an irregular RR interval and an increase in QTc, which
suggested long QT syndrome, were detected (Fig. 6A and C). Conversely, there were no
obvious aberrations in ECG recordings following lipo-PTX
treatment.
Previous studies have demonstrated that Taxol
impairs cardiac contraction via the acceleration of L-type calcium
incurrent and a reduction in calcium signaling through the
phosphoinositide signaling pathway (36–38).
Therefore, calcium concentration in the serum was further measured.
As shown in Fig. 6D, calcium
concentration was significantly reduced in Taxol-treated mice.
Lipo-PTX-treated mice, but not Abraxane-treated mice, exhibited a
significantly increased calcium concentration compared with in the
Taxol-treated group. These results suggested that lipo-PTX inflicts
less collateral damage to normal tissue compared with
CrEL-containing Taxol or albumin-associated Abraxane; therefore,
lipo-PTX may be more suitable for clinical use. The potential
benefits of lipo-PTX compared with Taxol and Abraxane are
summa-rized in Table III.
| Table IIIComparative effectiveness of
lipo-PTX. |
Table III
Comparative effectiveness of
lipo-PTX.
| Variable | Taxol | Abraxane | lipo-PTX |
|---|
| Therapeutic
efficacya | +++ | +++ | +++ |
| Body weight
changea | − | − | − |
| Histological
damagea | − | − | − |
| Liver and kidney
function lossa | − | − | − |
|
Lymphocytopeniaa | +++ | − | − |
| Neutropeniaa | ++ | + | − |
| Anemiaa | ++ | − | − |
|
Thrombocytopeniaa | − | − | − |
| ECG
abnormalities | +++ | ++ | + |
| QTc,
prolongedb | ++ | + | − |
| Decreased blood
Ca2+ | +a; ##c | −a; #c | −a |
Discussion
One primary goal of successful cancer treatment
regimens is to deliver sufficient amounts of drug to tumors while
minimizing damage to normal tissues. Therefore, novel drug delivery
systems are often generated from ongoing efforts to improve the
selectivity and efficacy of antineoplastic drugs. Recently, many of
these novel drug delivery systems comprise nanoparticles (14,15),
including lipid-based carriers, such as liposomes. Compared with
conventional administration methods for chemotherapeutic agents,
lipid- or polymer-based nanomedicines have the advantage of
improving the pharmacological and therapeutic properties of
cytotoxic drugs (39). The present
study developed and optimized a novel liposome formulation of PTX,
which exhibited high drug EE and liposome stability, and resulted
in fewer side effects compared with commercial PTX
formulations.
Numerous factors in the manufacturing of liposomes
may affect the stability of the end product. These include the type
of constitutive lipid, the presence of cholesterol and/or
polyethylene glycol (PEG), and the ratio of lipid to therapeutic
payload. PTX, which is hydrophobic, is not water-soluble and
precipitates in aqueous solution. The present study used SPC,
cholesterol and mPEG-DSPE in the liposome formulation. These
materials are all amphipathic fatty acids, which were used due to
their preferable lipophilic characteristics and ability to
encapsulate PTX in the lipid bilayer. The outer layer of the
liposome is hydrophilic, allowing suspension in aqueous solution.
The bulky PEG head with highly hydrated groups on the liposome
surface serves to sterically inhibit hydrophobic and electrostatic
interactions with plasma proteins, decreasing recognition by the
mononuclear phagocyte system (MPS). PTX, which is encapsulated in
the lipid bilayer of the liposome, does not come into contact with
the aqueous solution.
To determine the optimal formulation for desirable
liposome properties, liposomes derived from saturated lipid acid
(DSPC), monounsaturated lipid acid (egg PC) and polyunsaturated
lipid acid (SPC) were analyzed. It was demonstrated that the
loading efficiency of liposomes composed of DSPC was only 50% that
of SPC liposomes, and the vesicles broke up within 7 days. The
stability of liposomes derived from monounsaturated lipid acid was
slightly better, since nanoparticle rupture occurred on ~day 14
(data not shown). Conversely, the liposomes synthesized from SPC
were stable for 328 days when stored at 4°C, and they also
possessed better loading efficiency for PTX. Sufficient space in
the hydrophobic region of the lipid bilayers is necessary for high
loading capacity of PTX in liposomal platforms (25). Addition of cholesterol can expand
the inter-membrane space within the liposome; however, this
supplement also increases the stiffness of the liposomes and
diminishes their stability (25,40,41).
The present study revealed that the integrity of liposomes was
sustained for only 90 days when the molar ratio of cholesterol was
elevated from to 4 to 6%, whereas the increase in loading
efficiency was minimal (84 and 85%, respectively) (data not shown).
Such a short duration of stability is unsuitable for clinical
application. Surface PEGylation of liposomes is known to help
particles evade premature clearance by the MPS, in order to prolong
half-life in serum. However, these functions are gained at the
expense of decreasing loading efficiency and stability of
hydrophobic drugs (24,25,42).
In the present study, the PEG-containing (1% molar ratio)
formulation exhibited a shelf life of 328 days, whereas its EE of
PTX was 94%, which was far superior to previous PEGylated liposome
formulations (22,24). Although it is tempting to load the
delivery system with as much drug as possible, the present study
observed that the stability of the liposome was compromised if PTX
was loaded at a molar ratio more than 2%. The liposomes ruptured
within 60 days when loaded at 3% and 1 day after encapsulation of
4% PTX (data not shown). Compared with previously published
liposomal PTX formulations (43,44),
the newly developed lipo-PTX formulation exhibited much higher PTX
loading efficiency. Notably, to the best of our knowledge, no
publication has reported a liposomal formulation of PTX with such a
long shelf life (23,45–47).
The potential benefits of the lipo-PTX formulation
compared with two commercial PTX formulations, Taxol and Abraxane.
At present, there are two liposomal PTX formulations (EndoTAG-1 and
LEP-ETU) in clinical use. EndoTAG-1, which targets the tumor
vasculature instead of tumor cells (48,49),
is currently in phase III clinical trials. LEP-ETU was approved by
the FDA in 2015 as an orphan drug for use in ovarian cancer
treatment. However, its pharmacokinetic profile and therapeutic
efficacy exhibits no significant benefits over Taxol (50,51).
PTX administration has been associated with
myocardial ischemia, infarction, arrhythmias and ECG abnormalities
in patients; however, the mechanisms underlying cardiotoxicity are
not well defined. It has previously been suggested that
Taxol-induced arrhythmia is caused either directly by the effects
of PTX on the Purkinje system or indirectly via CrEL-mediated
histamine release (52). However,
since ECG abnormalities are also common in patients treated with
CrEL-free Abraxane infusion, it is most likely that the direct
effects of PTX on cardiomyocytes are clinically important. In the
case of Abraxane, the instability of the formulation in circulation
may be considered a major risk for inducing ECG abnormalities. In a
previous study, the albumin-PTX complex and unbound PTX were
monitored in plasma following Abraxane injection. The unbound form
comprised 6.4% of total drug in human plasma and this percentage
did not vary with time (53).
Notably, it has also been shown that Abraxane injection leads to
greater PTX accumulation in the heart than Taxol, both 1 and 5 days
after dosing (54). The present
results regarding drug distribution indicated that there were no
significant differences in PTX accumulation between Taxol- and
Abraxane-treated groups; however, lipo-PTX treatment reduced PTX
accumulation in the heart compared with either Taxol or Abraxane
administration. These results suggested that, compared with both
Taxol and Abraxane, lipo-PTX exhibits reduced PTX leakage in the
circulation, as well as diminished uptake and cardiac toxicity. A
prior study demonstrated that the frequency of spontaneous calcium
oscillations is significantly increased in mice several hours
following Taxol injection. Furthermore, Taxol is known to increase
the expression of neuronal calcium sensor-1 in cardiomyocytes,
leading to increased inositol trisphosphate receptor-dependent
calcium oscillations (36). Based
on the present experimental data, it was indicated that
Taxol-induced arrhythmia may be directly associated with calcium
concentration in cardiomyocytes. In this study, Taxol-treated mice
exhibited decreased blood calcium concentrations. This finding is
possibly due to the accumulation of PTX, which enhances calcium
influx and induces arrhythemia in mice. In addition, calcium
signaling may also be affected by modulation of ion channels, such
as the sodium-potassium pump.
In previous studies, Abraxane was reported to
significantly elevate antitumor activity, intratumor PTX
concentration and response rate, thus resulting in decreased tumor
progression, when compared with Taxol in human and mouse models
(18,55). However, in those studies, the PTX
dose in the Abraxane-treated groups was greater than in the
Taxol-treated groups. Therefore, it is unclear how Abraxane and
Taxol compare, in terms of therapeutic efficacy, when the same
concentration of PTX is administered. In the present study,
lipo-PTX exhibited comparable antitumor effects, but limited side
effects, when compared with Taxol. In conclusion, the novel
formulation, lipo-PTX, diminished systemic side effects that are
associated with CrEL and possessed a high therapeutic index
compared with clinical PTX formulations in mouse models. Therefore,
lipo-PTX may represent a feasible solution to safely increase the
dose of PTX whilst reducing the limitations of CrEL-induced side
effects. Based on these observations, lipo-PTX may be consider for
evaluation in clinical trials to evaluate safety and efficacy in
patients with breast, ovarian and liver cancer.
Acknowledgments
The authors would like to thank Dr Y.-C. Chang and
Miss H.-J. Huang for assistance in Cryo-TEM imaging and also for
the use of the Tecnai F20 in the Cryo-TEM Core Facility, Department
of Academic Affairs and Instrument Service at Academia Sinica. In
addition, the authors thank the Taiwan Animal Consortium (MOST
106-2319-B-001-004)-Taiwan Mouse Clinic, which is funded by the
Ministry of Science and Technology (MOST) of Taiwan for technical
support in complete blood count, blood chemistry and ECG recording.
Authors also thank Y.-C. Su in the Contract Breeding and Research
Division of NLAC at NARLabs for assistance in the analysis of ECG
traces, and the Core Facility of the Institute of Cellular and
Organismic Biology for their technical assistance.
References
|
1
|
Kampan NC, Madondo MT, McNally OM, Quinn M
and Plebanski M: Paclitaxel and its evolving role in the management
of ovarian cancer. BioMed Res Int. 2015.413076:2015.
|
|
2
|
Rowinsky EK and Donehower RC: Paclitaxel
(taxol). N Engl J Med. 332:1004–1014. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Javeed A, Ashraf M, Riaz A, Ghafoor A,
Afzal S and Mukhtar MM: Paclitaxel and immune system. Eur J Pharm
Sci. 38:283–290. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hadzic T, Aykin-Burns N, Zhu Y, Coleman
MC, Leick K, Jacobson GM and Spitz DR: Paclitaxel combined with
inhibitors of glucose and hydroperoxide metabolism enhances breast
cancer cell killing via H2O2-mediated
oxidative stress. Free Radic Biol Med. 48:1024–1033. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alexandre J, Hu Y, Lu W, Pelicano H and
Huang P: Novel action of paclitaxel against cancer cells: Bystander
effect mediated by reactive oxygen species. Cancer Res.
67:3512–3517. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Adams JD, Flora KP, Goldspiel BR, Wilson
JW, Arbuck SG and Finley R: Taxol: A history of pharmaceutical
development and current pharmaceutical concerns. J Natl Cancer Inst
Monogr. 15:141–147. 1993.
|
|
7
|
Goldspiel BR: Clinical overview of the
taxanes. Pharmacotherapy. 17:110S–125S. 1997.PubMed/NCBI
|
|
8
|
Gelderblom H, Verweij J, Nooter K and
Sparreboom A: Cremophor EL: The drawbacks and advantages of vehicle
selection for drug formulation. Eur J Cancer. 37:1590–1598. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Szebeni J, Alving CR, Savay S, Barenholz
Y, Priev A, Danino D and Talmon Y: Formation of
complement-activating particles in aqueous solutions of Taxol:
Possible role in hypersensitivity reactions. Int Immunopharmacol.
1:721–735. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
U.S. Department of Health and Human
Services: TAXOL® (paclitaxel) injection label.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020262s049lbl.pdf.
|
|
11
|
Weiss RB, Donehower RC, Wiernik PH, Ohnuma
T, Gralla RJ, Trump DL, Baker JR Jr, Van Echo DA, Von Hoff DD and
Leyland-Jones B: Hypersensitivity reactions from taxol. J Clin
Oncol. 8:1263–1268. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sparreboom A, van Tellingen O, Nooijen WJ
and Beijnen JH: Nonlinear pharmacokinetics of paclitaxel in mice
results from the pharmaceutical vehicle Cremophor EL. Cancer Res.
56:2112–2115. 1996.PubMed/NCBI
|
|
13
|
Sparreboom A, van Zuylen L, Brouwer E,
Loos WJ, de Bruijn P, Gelderblom H, Pillay M, Nooter K, Stoter G
and Verweij J: Cremophor EL-mediated alteration of paclitaxel
distribution in human blood: Clinical pharmacokinetic implications.
Cancer Res. 59:1454–1457. 1999.PubMed/NCBI
|
|
14
|
Surapaneni MS, Das SK and Das NG:
Designing Paclitaxel drug delivery systems aimed at improved
patient outcomes: Current status and challenges. ISRN Pharmacol.
2012.623139:2012.
|
|
15
|
Meng Z, Lv Q, Lu J, Yao H, Lv X, Jiang F,
Lu A and Zhang G: Prodrug strategies for Paclitaxel. Int J Mol Sci.
17:7962016. View Article : Google Scholar :
|
|
16
|
Yardley DA: nab-Paclitaxel mechanisms of
action and delivery. J Control Release. 170:365–372. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ibrahim NK, Desai N, Legha S, Soon-Shiong
P, Theriault RL, Rivera E, Esmaeli B, Ring SE, Bedikian A,
Hortobagyi GN, et al: Phase I and pharmacokinetic study of ABI-007,
a Cremophor-free, protein-stabilized, nanoparticle formulation of
paclitaxel. Clin Cancer Res. 8:1038–1044. 2002.PubMed/NCBI
|
|
18
|
Gradishar WJ, Tjulandin S, Davidson N,
Shaw H, Desai N, Bhar P, Hawkins M and O'Shaughnessy J: Phase III
trial of nanoparticle albumin-bound paclitaxel compared with
polyethylated castor oil-based paclitaxel in women with breast
cancer. J Clin Oncol. 23:7794–7803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
US Department of Health and Human
Services: ABRAXANE® label. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/021660s041lbl.pdf.
|
|
20
|
eHealthMe: Abraxane and myocardial
infarction. https://www.ehealthme.com/ds/abraxane/myocardial-infarction/.
|
|
21
|
Feng L and Mumper RJ: A critical review of
lipid-based nanoparticles for taxane delivery. Cancer Lett.
334:157–175. 2013. View Article : Google Scholar
|
|
22
|
Hong SS, Choi JY, Kim JO, Lee MK, Kim SH
and Lim SJ: Development of paclitaxel-loaded liposomal nanocarrier
stabilized by triglyceride incorporation. Int J Nanomedicine.
11:4465–4477. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feng Q, Yu MZ, Wang JC, Hou WJ, Gao LY, Ma
XF, Pei XW, Niu YJ, Liu XY, Qiu C, et al: Synergistic inhibition of
breast cancer by co-delivery of VEGF siRNA and paclitaxel via
vapreotide-modified core-shell nanoparticles. Biomaterials.
35:5028–5038. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Immordino ML, Brusa P, Arpicco S, Stella
B, Dosio F and Cattel L: Preparation, characterization,
cytotoxicity and pharma-cokinetics of liposomes containing
docetaxel. J Control Release. 91:417–429. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Crosasso P, Ceruti M, Brusa P, Arpicco S,
Dosio F and Cattel L: Preparation, characterization and properties
of sterically stabilized paclitaxel-containing liposomes. J Control
Release. 63:19–30. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. The National Academies Press; Washington, DC: 2011
|
|
27
|
Gregoriadis G: Drug entrapment in
liposomes. FEBS Lett. 36:292–296. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Allen TM and Chonn A: Large unilamellar
liposomes with low uptake into the reticuloendothelial system. FEBS
Lett. 223:42–46. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee TY, Lin CT, Kuo SY, Chang DK and Wu
HC: Peptide-mediated targeting to tumor blood vessels of lung
cancer for drug delivery. Cancer Res. 67:10958–10965. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu CH, Kuo YH, Hong RL and Wu HC:
α-Enolase-binding peptide enhances drug delivery efficiency and
therapeutic efficacy against colorectal cancer. Sci Transl Med.
7:290ra912015. View Article : Google Scholar
|
|
31
|
Bartlett GR: Phosphorus assay in column
chromatography. J Biol Chem. 234:466–468. 1959.PubMed/NCBI
|
|
32
|
Fiandaca MS, Berger MS and Bankiewicz KS:
The use of convection-enhanced delivery with liposomal toxins in
neuroon-cology. Toxins (Basel). 3:369–397. 2011. View Article : Google Scholar
|
|
33
|
Hillaireau H and Couvreur P: Nanocarriers'
entry into the cell: Relevance to drug delivery. Cell Mol Life Sci.
66:2873–2896. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Eiseman JL, Eddington ND, Leslie J,
MacAuley C, Sentz DL, Zuhowski M, Kujawa JM, Young D and Egorin MJ:
Plasma pharmacokinetics and tissue distribution of paclitaxel in
CD2F1 mice. Cancer Chemother Pharmacol. 34:465–471. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Howarth FC, Calaghan SC, Boyett MR and
White E: Effect of the microtubule polymerizing agent taxol on
contraction, Ca2+ transient and L-type Ca2+
current in rat ventricular myocytes. J Physiol. 516:409–419. 1999.
View Article : Google Scholar
|
|
36
|
Zhang K, Heidrich FM, DeGray B, Boehmerle
W and Ehrlich BE: Paclitaxel accelerates spontaneous calcium
oscillations in cardiomyocytes by interacting with NCS-1 and the
InsP3R. J Mol Cell Cardiol. 49:829–835. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Boehmerle W, Splittgerber U, Lazarus MB,
McKenzie KM, Johnston DG, Austin DJ and Ehrlich BE: Paclitaxel
induces calcium oscillations via an inositol 1,4,5-trisphosphate
receptor and neuronal calcium sensor 1-dependent mechanism. Proc
Natl Acad Sci USA. 103:18356–18361. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Boehmerle W, Zhang K, Sivula M, Heidrich
FM, Lee Y, Jordt SE and Ehrlich BE: Chronic exposure to paclitaxel
diminishes phosphoinositide signaling by calpain-mediated neuronal
calcium sensor-1 degradation. Proc Natl Acad Sci USA.
104:11103–11108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Drummond DC, Meyer O, Hong K, Kirpotin DB
and Papahadjopoulos D: Optimizing liposomes for delivery of
chemotherapeutic agents to solid tumors. Pharmacol Rev. 51:691–743.
1999.PubMed/NCBI
|
|
40
|
McIntosh TJ: The effect of cholesterol on
the structure of phosphatidylcholine bilayers. Biochim Biophys
Acta. 513:43–58. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Deniz A, Sade A, Severcan F, Keskin D,
Tezcaner A and Banerjee S: Celecoxib-loaded liposomes: Effect of
cholesterol on encapsulation and in vitro release characteristics.
Biosci Rep. 30:365–373. 2010. View Article : Google Scholar
|
|
42
|
Sharma A, Mayhew E, Bolcsak L, Cavanaugh
C, Harmon P, Janoff A and Bernacki RJ: Activity of paclitaxel
liposome formulations against human ovarian tumor xenografts. Int J
Cancer. 71:103–107. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu Y, Ran R, Chen J, Kuang Q, Tang J, Mei
L, Zhang Q, Gao H, Zhang Z and He Q: Paclitaxel loaded liposomes
decorated with a multifunctional tandem peptide for glioma
targeting. Biomaterials. 35:4835–4847. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li XY, Zhao Y, Sun MG, Shi JF, Ju RJ,
Zhang CX, Li XT, Zhao WY, Mu LM, Zeng F, et al: Multifunctional
liposomes loaded with paclitaxel and artemether for treatment of
invasive brain glioma. Biomaterials. 35:5591–5604. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ruttala HB and Ko YT: Liposomal
co-delivery of curcumin and albumin/paclitaxel nanoparticle for
enhanced synergistic antitumor efficacy. Colloids Surf B
Biointerfaces. 128:419–426. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Barbosa MV, Monteiro LO, Carneiro G,
Malagutti AR, Vilela JM, Andrade MS, Oliveira MC, Carvalho-Junior
AD and Leite EA: Experimental design of a liposomal lipid system: A
potential strategy for paclitaxel-based breast cancer treatment.
Colloids Surf B Biointerfaces. 136:553–561. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Assanhou AG, Li W, Zhang L, Xue L, Kong L,
Sun H, Mo R and Zhang C: Reversal of multidrug resistance by
co-delivery of paclitaxel and lonidamine using a TPGS and
hyaluronic acid dual-functionalized liposome for cancer treatment.
Biomaterials. 73:284–295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Schmitt-Sody M, Strieth S, Krasnici S,
Sauer B, Schulze B, Teifel M, Michaelis U, Naujoks K and Dellian M:
Neovascular targeting therapy: Paclitaxel encapsulated in cationic
liposomes improves antitumoral efficacy. Clin Cancer Res.
9:2335–2341. 2003.PubMed/NCBI
|
|
49
|
Strieth S, Eichhorn ME, Werner A, Sauer B,
Teifel M, Michaelis U, Berghaus A and Dellian M: Paclitaxel
encapsulated in cationic liposomes increases tumor microvessel
leakiness and improves therapeutic efficacy in combination with
Cisplatin. Clin Cancer Res. 14:4603–4611. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fetterly GJ, Grasela TH, Sherman JW, Dul
JL, Grahn A, Lecomte D, Fiedler-Kelly J, Damjanov N, Fishman M,
Kane MP, et al: Pharmacokinetic/pharmacodynamic modeling and
simulation of neutropenia during phase I development of
liposome-entrapped paclitaxel. Clin Cancer Res. 14:5856–5863. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Slingerland M, Guchelaar HJ, Rosing H,
Scheulen ME, van Warmerdam LJ, Beijnen JH and Gelderblom H:
Bioequivalence of Liposome-Entrapped Paclitaxel Easy-To-Use
(LEP-ETU) formulation and paclitaxel in polyethoxylated castor oil:
A randomized, two-period crossover study in patients with advanced
cancer. Clin Ther. 35:1946–1954. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yeh ET and Bickford CL: Cardiovascular
complications of cancer therapy: Incidence, pathogenesis,
diagnosis, and management. J Am Coll Cardiol. 53:2231–2247. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gardner ER, Dahut W and Figg WD:
Quantitative determination of total and unbound paclitaxel in human
plasma following Abraxane treatment. J Chromatogr B Analyt Technol
Biomed Life Sci. 862:213–218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sparreboom A, Scripture CD, Trieu V,
Williams PJ, De T, Yang A, Beals B, Figg WD, Hawkins M and Desai N:
Comparative preclinical and clinical pharmacokinetics of a
cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and
paclitaxel formulated in Cremophor (Taxol). Clin Cancer Res.
11:4136–4143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Desai N, Trieu V, Yao Z, Louie L, Ci S,
Yang A, Tao C, De T, Beals B, Dykes D, et al: Increased antitumor
activity, intratumor paclitaxel concentrations, and endothelial
cell transport of cremophor-free, albumin-bound paclitaxel,
ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res.
12:1317–1324. 2006. View Article : Google Scholar : PubMed/NCBI
|