Introduction
Heat shock protein 70 (Hsp70), used herein to denote
HSP70A1A, is a molecular chaperone, approximately 70 kDa, that
plays a key role in protein homeostasis (1). Its expression is markedly induced by
increased environmental temperature (2-4).
Hsp70 usually acts together with co-chaperones, forming protein
molecular machines (5-7), and its function is carried out by its
monomeric form (8). At the
molecular level, Hsp70 participates in protein folding (9), degradation (10) and translocation (11), as well as in single-strand DNA
repair mechanisms, both in the nucleus and the nucleolus (12). At the cellular level, Hsp70 has
been associated with cell viability (13,14)
as well as apoptosis (15,16). Finally, at the organism level,
Hsp70 has been linked to several diseases and pathological states,
such as neurodegenerative diseases (17,18),
cancer (19,20), PTZ kindling (21), cardiovascular conditions (22-24),
spinal cord ischemia (25) and
inner ear protection from exposure to inaudible low-frequency noise
(LFN) (26).
The upregulation of Hsp70 is relatively common in
human tumors, and it is often associated with an enhanced
resistance to chemotherapy and a poor patient prognosis (27). Indeed, over the past decade,
several proposed strategies have documented that chemotherapy
sensitizes cells to death via the selective inhibition of Hsp70.
Heat shock proteins, such as Hsp70, inhibit apoptosis by direct
physical interaction with apoptotic molecules, which are also
overexpressed in several tumor cells (28). The selective depletion of the
70-kDa heat shock protein activates a specific tumor cell death
pathway (29-31). This cell death, referred to as
anoikis, is a special type of apoptosis: It occurs in response to
the lack of cell attachment or inappropriate attachment to the
extracellular matrix (ECM) and neighboring cells (32). The property of cancer cells to act
independently of survival signals and lack of the ability to adhere
efficiently are key mechanisms for the transformation of neoplastic
into metastatic cells, since it allows malignant cells to detach
and migrate from the primary tumor by escaping cell death (33-35).
The ability of Hsp70 to suppress apoptosis by interfering with cell
pathways is a field of great interest. Significant results were
initially provided by a scientific group suggesting that Hsp70
prevents recruitment of procaspase-9 to the apaf-1apoptosome
(36).
Epithelial-to-mesenchymal transition (EMT) is a
biological process that allows a polarized epithelial cell to
undergo biochemical changes that render it capable of acquiring a
mesenchymal phenotype, which includes enhanced migration capacity,
invasiveness, an increased resistance to apoptosis and the markedly
increased production of ECM components (37). EMT is a critical event in the
process of cancer metastasis. In the present study, EMT was
considered to be a cellular process that mimics a cancer metastatic
step in real tumors. The series of events that occur during
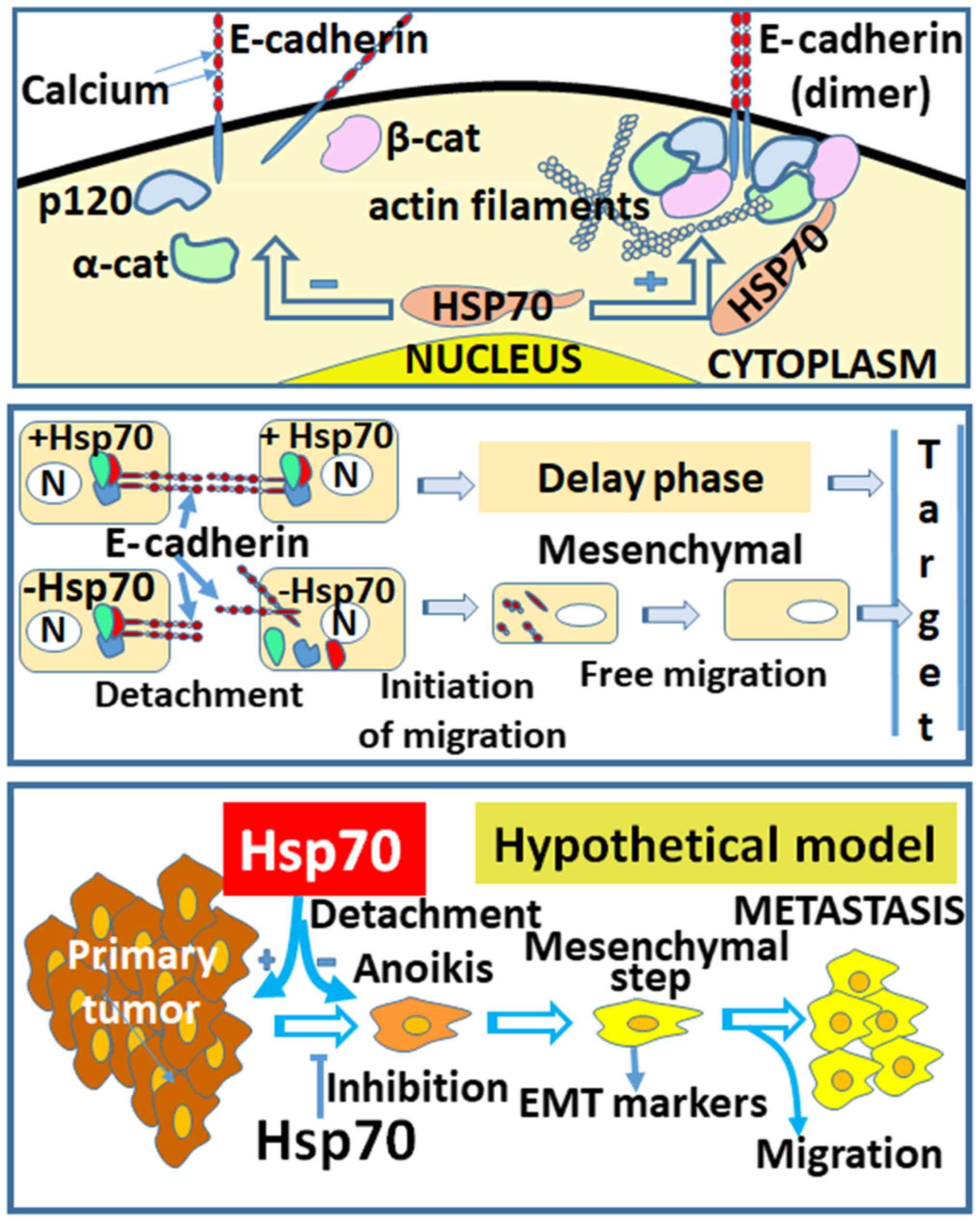
metastasis and the implication of Hsp70 are shown in the proposed
model of Fig. 8 (lower panel). The
model begins with the creation of the primary tumor, followed by
cell detachment/anoikis, the acquisition of the mesenchymal cell
phenotype, cell migration and, finally, attachment to a new
location distant from the primary tumor. The questions posed at
this stage are the following: the reasons as to why Hsp70 is highly
accumulated in cancer cells, and the potential roles of this
protein that render it an important protein in cancer. Based on
these questions, it may be hypothesized that the increased
expression of Hsp70 is associated with the particular stage of
tumor metastasis.
During EMT, the expression of several epithelial
proteins, including those that are part of the ligand complexes
(38,39), is downregulated. In addition, the
whole gene expression pattern is reprogrammed, thus promoting
changes in cytoskeletal architecture, mesenchymal cell adhesion and
cell interaction with the ECM (40). At the onset of EMT, components of
cadherin junctions, tight junctions, desmosomes and gap junctions
are destroyed. In addition, several other proteins are relocated to
other parts of the cell or are degraded. During the destabilization
of adhesions, epithelial E-cadherin undergoes degradation and
disappears from the cell membranes (41). Moreover, due to the loss of
E-cadherin from the cell junctions, β-catenin, a well-documented
cell adhesion protein, is released from the cell adhesions,
degraded, and a small part of it translocates to the nucleus where
it transactivates several growth-promoting and antiapoptotic genes
(42). In addition, the reduction
of E-cadherin during EMT leads to the release of catenin p120 from
the junctions and its translocation to the nucleus, where it
transactivates several genes (43). The above-mentioned changes increase
the motility of individual cells and promote the development of a
more invasive phenotype.
During EMT, several proteins are downregulated, such
as E-cadherin, or upregulated, such as N-cadherin, vimentin, Slug
and occludin, which are also used as EMT markers. It is well-known
that E-cadherin is an adhesion molecule whose reduction in
expression leads to loosening of cell-to-cell adhesion and
increased cell migration (44).
Another protein, occludin, stabilizes the dissolution of narrow
ligands and desmosomes (39). Due
to E-cadherin downregulation, the epithelial cells lose their
interactions and transform into mesenchymal cells. Subsequently,
new interactions are formed between mesenchymal cells mediated by
the N-cadherin protein, which is upregulated in mesenchymal cells.
These interactions are weaker compared with the homophilic
interactions between E-cadherins, and facilitate cell migration and
cancer spread (45,46).
The role of vimentin in cell migration and EMT of
epithelial carcinomas is of great interest (47). It is known that the suppression of
vimentin expression inhibits carcinoma cell migration and adhesion
(48). The intermediate filament
protein, vimentin, is an important marker of EMT and a requisite
regulator of mesenchymal cell migration (49).
Slug is a transcriptional repressor of E-cadherin
and its overexpression promotes the metastasis of cancer cells
(50). Finally, occludin is
another marker that is important for the induction and
establishment of EMT. Moreover, the dissolution of watertight
junctions is accompanied by reduced expression of clonidine and
occludin proteins and the diffusion of ZO1 (39).
The aim of the present study was to elucidate the
role of Hsp70 in the different stages of the metastatic process. It
is well known that Hsp70 renders cells resistant to apoptosis by
forming a more tumorigenic environment (15,35).
In addition, evidence suggests the involvement of Hsp70 in cancer
metastasis. Therefore, it is crucial to elucidate whether the
absence of Hsp70 promotes cell metastasis by enabling the cancer
cells to detach from the tumor and favoring their migration to
other parts of the body. In this study, we constructed cancer cell
lines in which Hsp70 was stably knocked down, aiming to demonstrate
the crucial role of Hsp70 in anoikis, the acquisition of the
mesenchymal phenotype and cell migration.
Materials and methods
Cell culture and heat shock
The cell lines used in the study were MCF7 (acronym
of Michigan Cancer Foundation-7, a human breast adenocarcinoma
cancer cell line, ATCC® HTB-22™), A549 (adenocarcinomic
human alveolar basal epithelial cells, ATCC® CCL-185™)
and HeLa (human epithelioid cervical carcinoma cell line, no. CCL
185; ATCC, Rockville, MD, USA). The human MCF7, A549 and HeLa cells
were cultured in Dulbecco's modified Eagle's medium with 4.5 g
glucose, 110 mg sodium pyruvate, L-glutamine (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) supplemented with 10% fetal bovine serum
and 1% penicillin-streptomycin. The cells were grown and passaged
by trypsinization. The cells were grown in a monolayer, as
previously described (51). In the
case of heat shock, subconfluent cells were exposed to immersing
dishes in a water bath set at 43.5°C for 90 min and a recovery
period of another 90 min at 37°C.
Plasmid construction and stably
transfected cellular clones
The DNA expression vectors pcDNA3 (Invitrogen;
Thermo Fisher Scientific, Waltham, MA, USA) and pSuper-siRNA-Hsp70
(52) were used for
pcDNA3-siRNA-Hsp70 plasmid preparation. Specifically, the
BamHI-KpnI (343 bp) DNA fragment of
pSuper-siRNA-Hsp70 was ligated to the BglII (generating
compatible ends with the BamHI)-KpnI DNA fragment
(4,559 bp) of the pcDNA3 plasmid, creating the 5,902 bp
pcDNA3-siRNA-Hsp70. This DNA plasmid contains a siRNA insert,
targeting and downregulating Hsp70, under the control of the human
H1 promoter. It also contains an ampicillin and neomycin selection
marker, located after the SV40 promoter and ending at poly-A SV40.
This construct (2 µg) was stably transfected into MCF7, A549
and HeLa cells (5×106) in 400 µl serum-free
medium, with electroporation (GenePulser, Xcell; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Transfected and stable
clones were selected, subcloned by limiting dilution and maintained
in 500 µg/ml of G418. The selection of cells containing
stably integrated copies of the transfected plasmid was
accomplished by the addition of Geneticin (G-418: BRL-Gibco; Thermo
Fisher Scientific, Inc.; 11811-031) to the medium at a
concentration of 700-1,000 µg/ml.
Antibodies
The antibodies used in our experiments were as
follows: Anti-Hsp70 antibody (a mouse monoclonal antibody that
specifically binds to human HSP70A1A; dilution, 1:2,500; cat. no.
SPA-810; StressGen Biotechnologies Corporation, San Diego, CA,
USA); anti-α-tubulin antibody (dilution, 1:10,000; cat. no. T5168;
Sigma-Aldrich; Merck KGaA); anti-Annexin V-fluorescein
isothiocyanate (FITC) antibody (dilution, as proposed by the
manufacturer in the staining protocol; cat. no. 556420, BD
Pharmigen™; BD Biosciences, Franklin Lakes, NJ, USA); and
anti-occludin antibody (dilution, 1:1,000; cat. no. ab31721, Abcam,
Cambridge, UK). The EMT kit (Epithelial-Mesenchymal Transition
Antibody Sampler kit #9782; Cell Signaling Technology, Inc.,
Danvers, MA, USA,) containing antibodies against β-catenin
(dilution, 1:1,000), E-cadherin (dilution, 1:1,000), N-cadherin
(dilution, 1:1,000), vimentin (dilution, 1:1,000) and Slug
(dilution, 1:1,000).
Cell extract and western blot
analysis
Control and heat-treated cells were harvested,
washed with phosphate-buffered saline (PBS; 139 mM NaCl, 5.4 mM
KCl, 0.37 mM Na2HPO4, 0.44 mM
KH2PO4 and 4.16 mM NaHCO3) and
resuspended in RIPA buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl,
1% (v/v) Triton X-100, 1% (w/v) sodium deoxycolate, 0.1% (w/v)
sodium dodecyl phosphate (SDS)] supplemented with 1 mM PMSF, 1
µg/ml leupeptin and 1 µg/ml pepstatin. Lysates were
prepared as previously described (24) and mixed with SDS sample buffer
until 1X final concentration [62.5 mM Tris-HCl, 3% (w/v) SDS, 10%
(v/v) glycerol, 5% (v/v) 2-mercaptoethanol, 0.01% (w/v) bromophenol
blue]. Prior to use, protein extracts (20 µg/sample) were
boiled for 3 min at 95°C and analyzed by 10% SDS-polyacrylamide gel
electrophoresis and subsequently by western blot analysis using
nitrocellulose membranes (Hybond C, Amersham; GE Healthcare Life
Sciences, Little Chalfont, UK). The blots were blocked overnight at
4°C and then incubated with the primary antibodies for 1 h at room
temperature, rinsed with Tris-buffered saline/Tween-20 and
incubated with peroxidase-conjugated secondary antibodies,
specifically with anti-mouse IgG-HRP (cat. no. 31430; dilution,
1:5,000) and anti-rabbit IgG-HRP (cat. no. 31460; dilution,
1:5,000; Pierce/Thermo Fisher Scientific) for 45 min at room
temperature. Protein expression was detected with the enhanced
chemiluminescence's method (SuperSignal West Pico, Chemiluminescent
Substrate CA 47079; Pierce; Thermo Fisher Scientific, Inc.).
Immunofluorescence-confocal
microscopy
The control or heat-shocked (43.5°C, 90 min) parent
and stably transfected MCF7, A549 and HeLa cells were grown on
coverslips and subjected to indirect immunofluorescence (12). In brief, the cells were washed with
PBS, fixed with 2% formaldehyde for 10 min and permeabilized for
3-5 min with absolute methanol. The cells were then washed with
PBS, incubated in 3% BSA to prevent non-specific staining and
subjected to incubation with the appropriate antibodies. For the
Hsp70 scoring, we used 1.5-µg anti-Hsp70/ml sample (2%
paraformaldehyde in PBS) containing 1×106 cells. Images
were captured using a Leica TCS-SP confocal microscope, equipped
with an argent-crypt laser and Leica TCS software (Leica
Microsystems GmbH, Wetzlar, Germany). The stimulation of FITC and
TRITC was achieved using wavelengths of 488 and 568 nm,
respectively.
Detection of Hsp70 and apoptosis by flow
cytometry
To detect Hsp70 expression in the cell lines, a
specific antibody was used that only detects inducible Hsp70
(HSP70A1A). The measurement of apoptosis was performed by flow
cytometry and propidium iodide/Annexin V-FITC staining. The cells
were processed as previously described (24) and cytometric analysis was performed
in a Partec ML flow cytometer (CyFlow ML, Partec, Munster,
Germany); the results were analyzed using Partec FloMax
software.
Activation of anoikis by
poly-2-hydroxyethyl methacrylate (poly-hema)
The cells were grown in suspension on plates coated
with polyhema, which prevents cellular attachment, in order to
induce anoikis apoptosis in vitro. The lack of cell
anchorage leads to decreased cellular development and, eventually,
cell death. More specifically, a poly-hema solution 20 mg/ml in 95%
EtOH, was prepared by stirring for several hours at 50°C. The empty
Petri dishes (10 cm) were covered by the addition of 4 ml of the
poly-hema solution and were allowed to dry. Prior to use, the
dishes were sterilized by UV irradiation and washed twice with 5 ml
PBS (1X).
Migration and wound healing assays
The wound healing assay is used to measure the
metastatic ability of cells in vitro. The cells were seeded
in 6-well plates (0.25×106 cells/well). After becoming
confluent, the cell monolayer was scratched using a p200 tip. The
cells were washed twice with 1X PBS, new culture medium was added
and wound healing was recorded at 12, 24, 48 and 72 h of culture.
For quantification, images of 4 random fields along the scratch
were captured. Identical rectangles with a width corresponding to
the width of the original scratch were designed in these fields.
Migrated cells were counted under an inverted microscope (Eclipse
TS100; Nikon Europe B.V., Amsterdam, The Netherlands) and data were
normalized to the number of cells migrated in the control (53).
Statistical analysis
Data are expressed as the means ± standard deviation
(SD) and all experiments were performed in triplicate. The
determination of statistical significance of the differences in the
comparisons of our results was performed using Student's t-tests.
For the statistical evaluation of differences between groups, were
performed using the one-way analysis of variance model (ANOVA) and
Tukey's or Dunn's tests for post hoc comparisons. P-values <0.05
were considered to indicate statistically significant
differences.
Results
Downregulation of Hsp70 in cancer cell
lines by siRNA technology leads to phenotypic alterations
Stably transfected MCF7, A549 and HeLa cell lines
with Hsp70 downregulation were generated by transfection with the
electroporation of parent cells with the plasmid pcDNA-Hsp70-siRNA
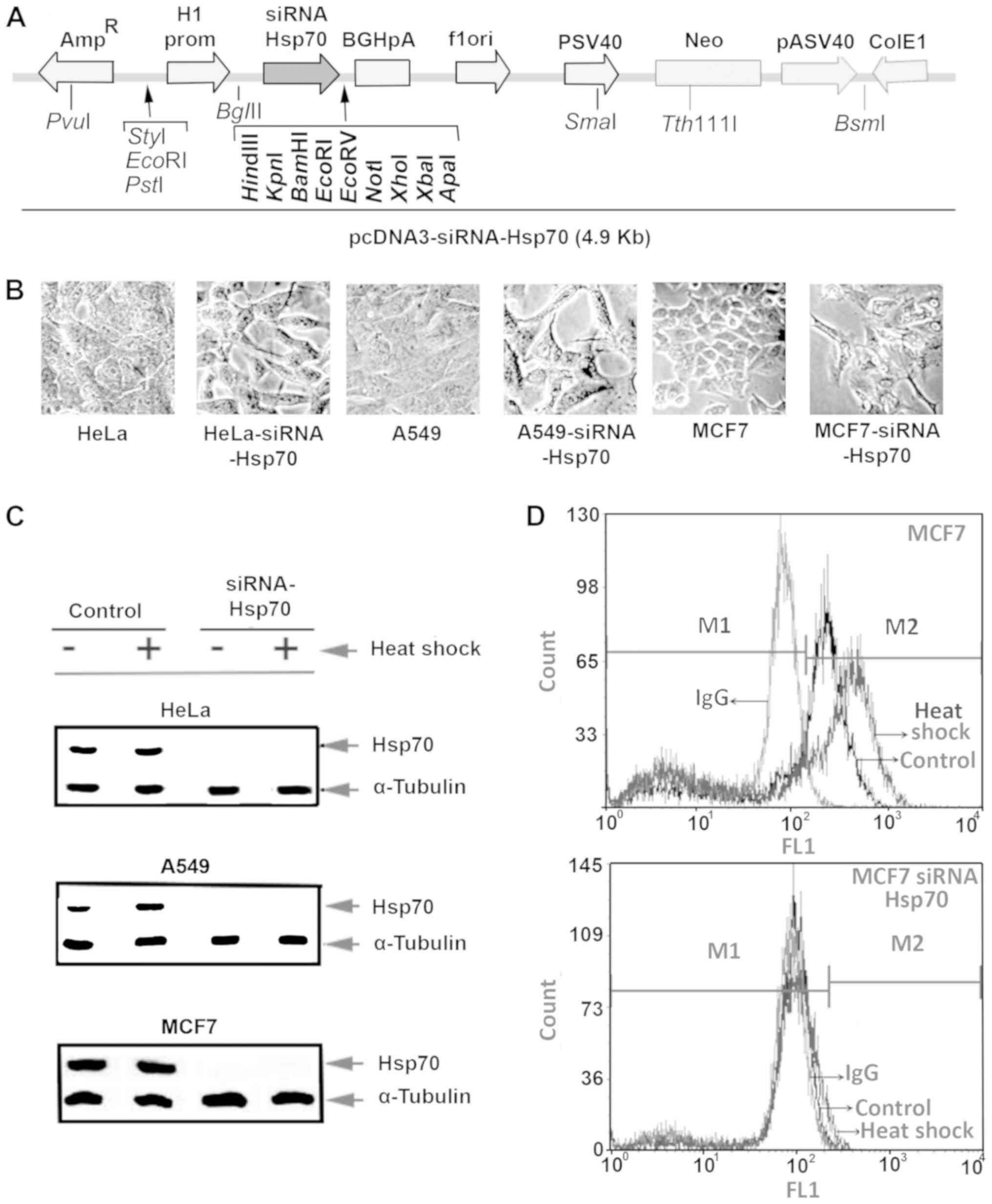
carrying hsp70-siRNA sequences (Fig.
1A). Several clones were selected after culturing the cells in
the presence of the appropriate antibiotic, as described in the
Materials and methods. Initially, we detected changes in the cell
phenotype, as well as the inability of cells to form tight
connections. Furthermore, the cells were elongated and the spaces
between them were enlarged. Identical changes were observed in all
3 cell lines under investigation (Fig.
1B).
Half of the plates from each cell line were
subjected to heat shock, and the cell extracts were examined by
western blot analysis (Fig. 1C)
using specific antibody for Hsp70 identification. This analysis
revealed that none of the 3 cell lines in the knockdown experiments
expressed Hsp70, not even following exposure to heat shock.
Flow cytometric analysis of intracellular Hsp70,
using the same anti-Hsp70 antibody and mouse IgG-FITC as an isotype
control, was applied to confirm the absence of Hsp70. The absence
of Hsp70 expression following Hsp70 gene silencing was confirmed in
all 3 cell lines, and representative results in MCF7 cells are
illustrated in Fig. 1D.
After the first observations, we aimed to identify
factors associated with EMT by the downregulation of Hsp70, the
destabilization of the cadherin dimer molecules and the
deregulation of the β-catenin protein, which are known to be
involved in the Wnt signaling pathway.
Hsp70 is essential for the assembly of
intercellular complexes among cadherins and catenins in cancer
cells
Several studies have suggested that the β-catenin
signaling pathway plays an important role in EMT. It has already
been demonstrated that E-cadherin downregulation releases β-catenin
from the junctions; subsequently, β-catenin translocates to the
nucleus where, in a complex with the TCF cofactor, transactivates
several growth-promoting and antiapoptotic genes (54-56).
However, there is no direct evidence suggesting a possible
crosstalk between Hsp70 and the β-catenin pathway to be essential
for the establishment of EMT.
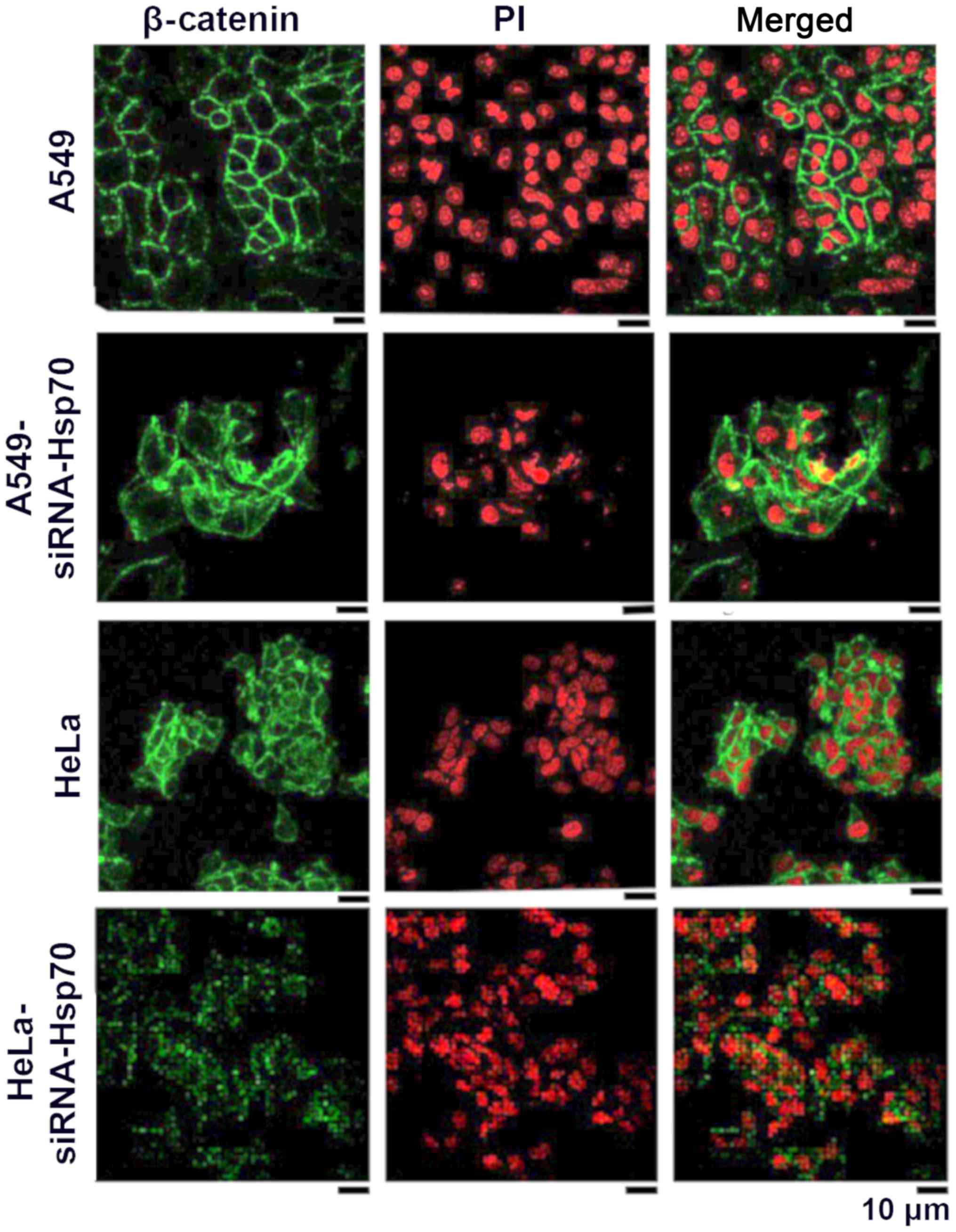
In this study, to elucidate whether the absence of
Hsp70 affects β-catenin and induces EMT, indirect
immunofluorescence was applied to detect β-catenin in A549 and HeLa
control cells, as well as in the corresponding Hsp70-silenced cell
lines. In normal cells, β-catenin is expected to be in a complex
with intracellular E-cadherin in epithelial cells. In cells that
have acquired mesenchymal-like characteristics, the expression of
E-cadherin is downregulated, and the free β-catenin either
accumulates in the cytoplasm, where it is degraded by the
proteasomal pathway, or the free β-catenin translocates to the
nucleus where it engages in transcription.
As shown in the HeLa cells (Fig. 2), β-catenin is mainly located
peripherally in the cell, but a small part of it is located within
the cytoplasm. This may explain why the absence of E-cadherin
results in the release of β-catenin. In the A549 cells, β-catenin
is only confined to the periphery of the cell, where it combines
with the intracellular E-cadherin. Following the gene silencing of
Hsp70, free β-catenin accumulates and diffuses into the cytosol.
This indicates that β-catenin is released from the complexes and
diffuses into the surrounding environment. In none of the
above-mentioned cases was β-catenin translocated in the nucleus to
act as a transcriptional factor for the induction of EMT under our
conditions (Fig. 2).
In the HeLa cells, β-catenin appeared to accumulate
somewhat further away from the membrane, indicating that, in these
cells, cadherin-catenin complexes have been diminished, resulting
in the loss of interactions between adjacent cells.
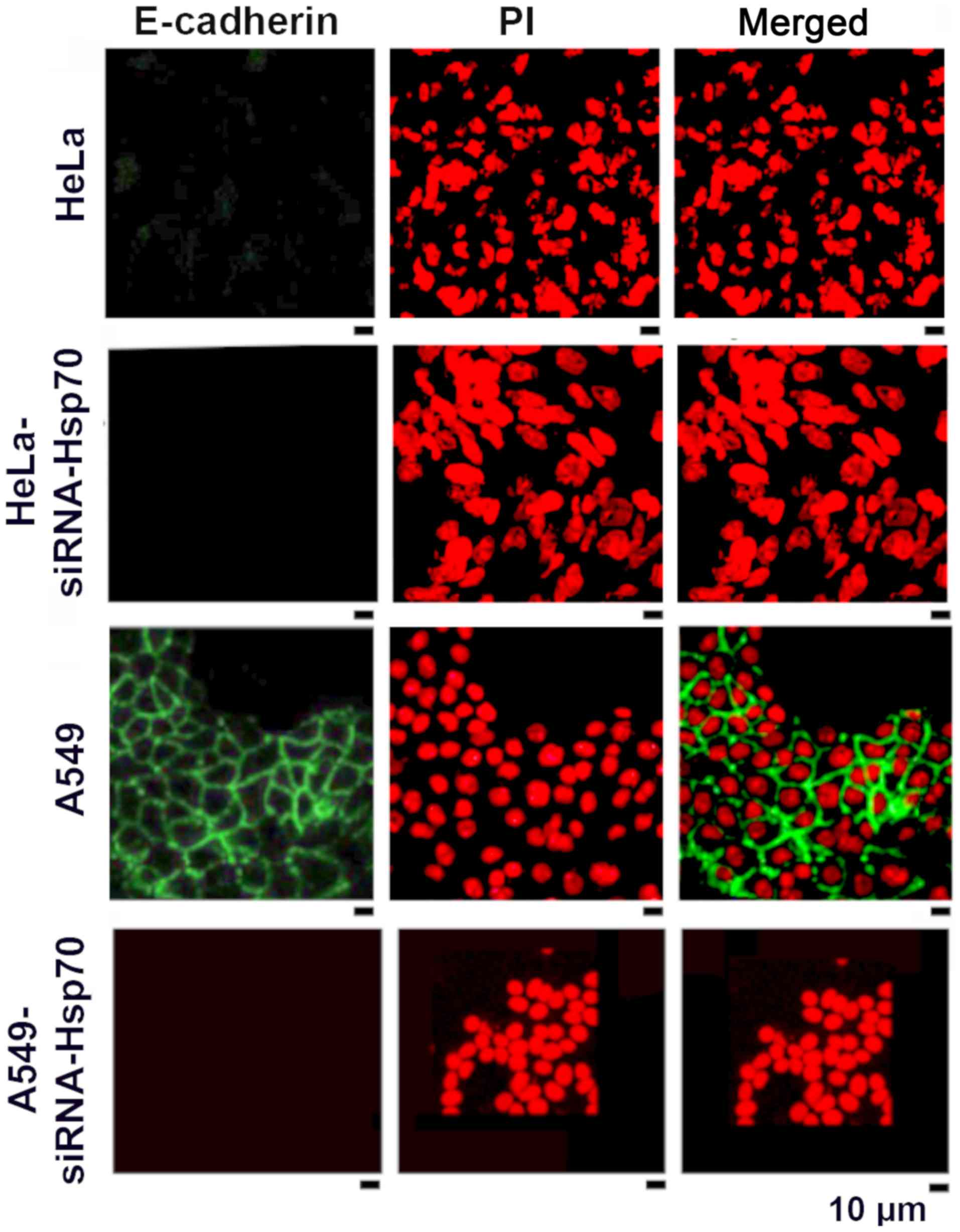
HSP70 gene silencing reduces the
accumulation of E-cadherin
As mentioned above, the loss of Hsp70 led to the
disruption of intercellular junctions in various cancer cell lines.
One exception among the studied cell lines was the HeLa cells, in
which E-cadherin expression and accumulation was absent in the
parental cells (Fig. 3). In the
literature, E-cadherin has been reported to be an intercellular
adhesion molecule that participates in homotypic, calcium-dependent
interactions to form adhesion bonds between adjacent cells. The
loss of E-cadherin expression is often correlated with the early
stages of EMT (57).
We noted that the absence of Hsp70 was accompanied
by the deregulation of E-cadherin expression in the tested cell
lines, further confirming our hypothesis that the destruction of
the cadherin-catenin complexes induces the affected cells to
acquire a mesenchymal phenotype. In the HeLa cells, however, this
hypothesis could not be confirmed, as the parental HeLa cells did
not express E-cadherin (Fig.
3).
As confirmed by the literature, EMT is a process
with several intermediate steps (58). In fact, the absence of E-cadherin
in the parental HeLa cells suggests that these cells 'escape' from
the epithelial phenotype and proceed directly towards the
mesenchymal one. On the contrary, E-cadherin accumulates on the
membranes of A549 cells (green fluorescence) and disappears in
A549-siRNA-Hsp70 cells (Fig. 3).
Similar results were obtained in MCF 7 cells (data not shown).
Hsp70 is involved in the anoikis of
cancer cells
It is well known that the detachment of a tumor cell
from the tumor, or from the neighboring cells in culture, leads to
anoikis. The mechanism underlying anoikis is initiated by the
destruction of the cadherin-catenin complexes. Of note, our results
predicted that the presence of Hsp70 stabilizes the
E-cadherin-catenin complexes, as its loss results in the detachment
of adjacent cells, as it is shown in Figs. 2 and 3.
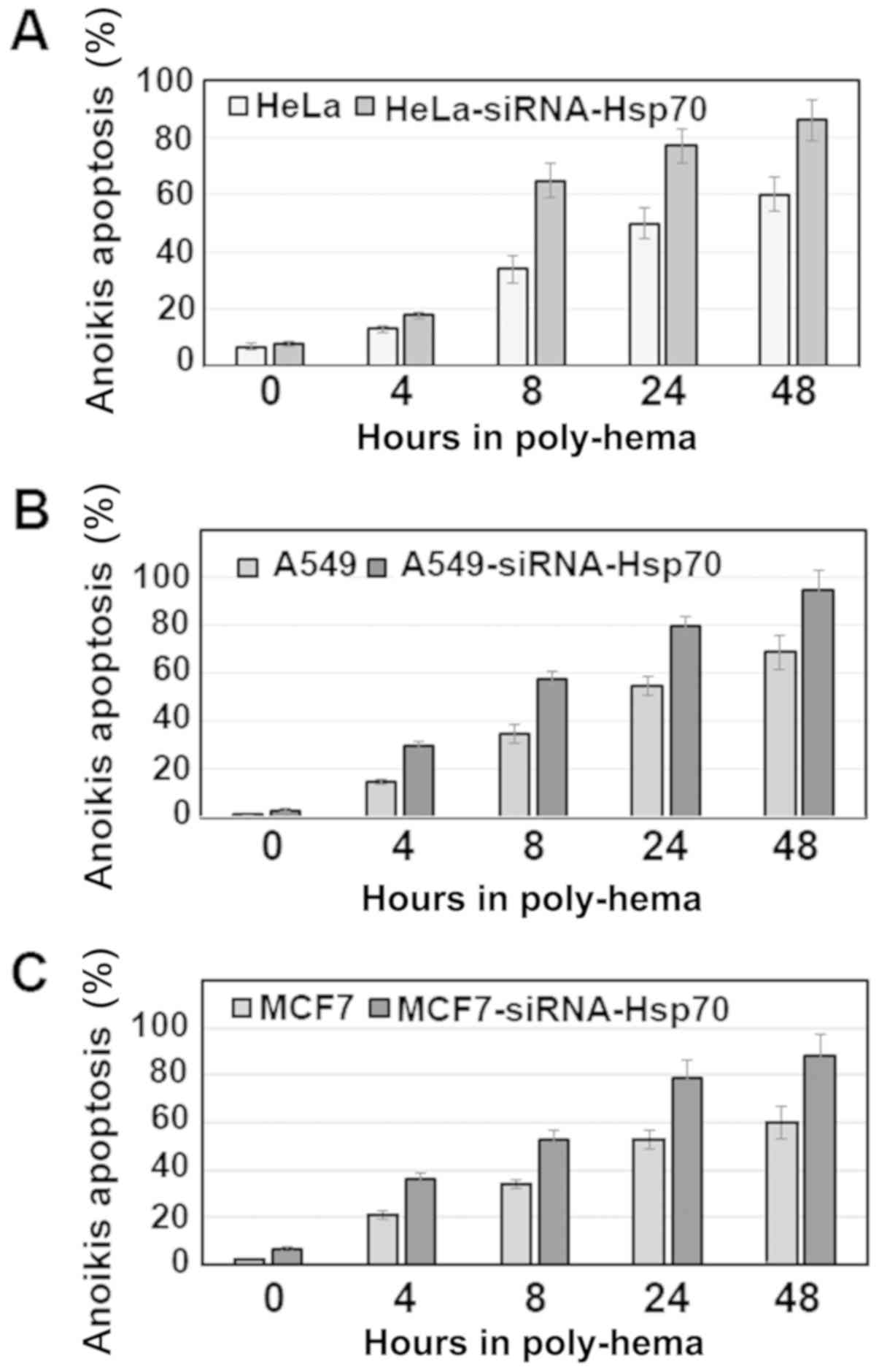
Anoikis was investigated in the same cancer cell
lines by activating the apoptotic pathway with poly-hema (see
Materials and methods). It was observed that, in the cell lines
without Hsp70 expression, there was a higher percentage of cell
death compared with that in the parental cells (Fig. 4). Of note, the percentage of cells
undergoing anoikis was >50%, even after 8 h. On the contrary,
the parental cells, which retained Hsp70 expression, were more
resistant and exhibited a significantly lower percentage of
apoptosis.
Therefore, if we accept that the cells cultured in
the presence of poly-hema proceed directly into the second stage of
anoikis, which occurs following their isolation from the rest of
the cells, we can then predict that these cells escape death,
acquire a mesenchymal phenotype and become migratory and
metastatic. The role of Hsp70 until the cells reach their final
destination was examined in subsequent experiments.
Absence of Hsp70 causes the cells to
acquire a mesenchymal-like phenotype
Furthermore, the probability of cell transition to a
mesenchymal phenotype due to the absence of Hsp70 was investigated.
Known markers, such as E-cadherin, occludin, N-cadherin, Vimentin
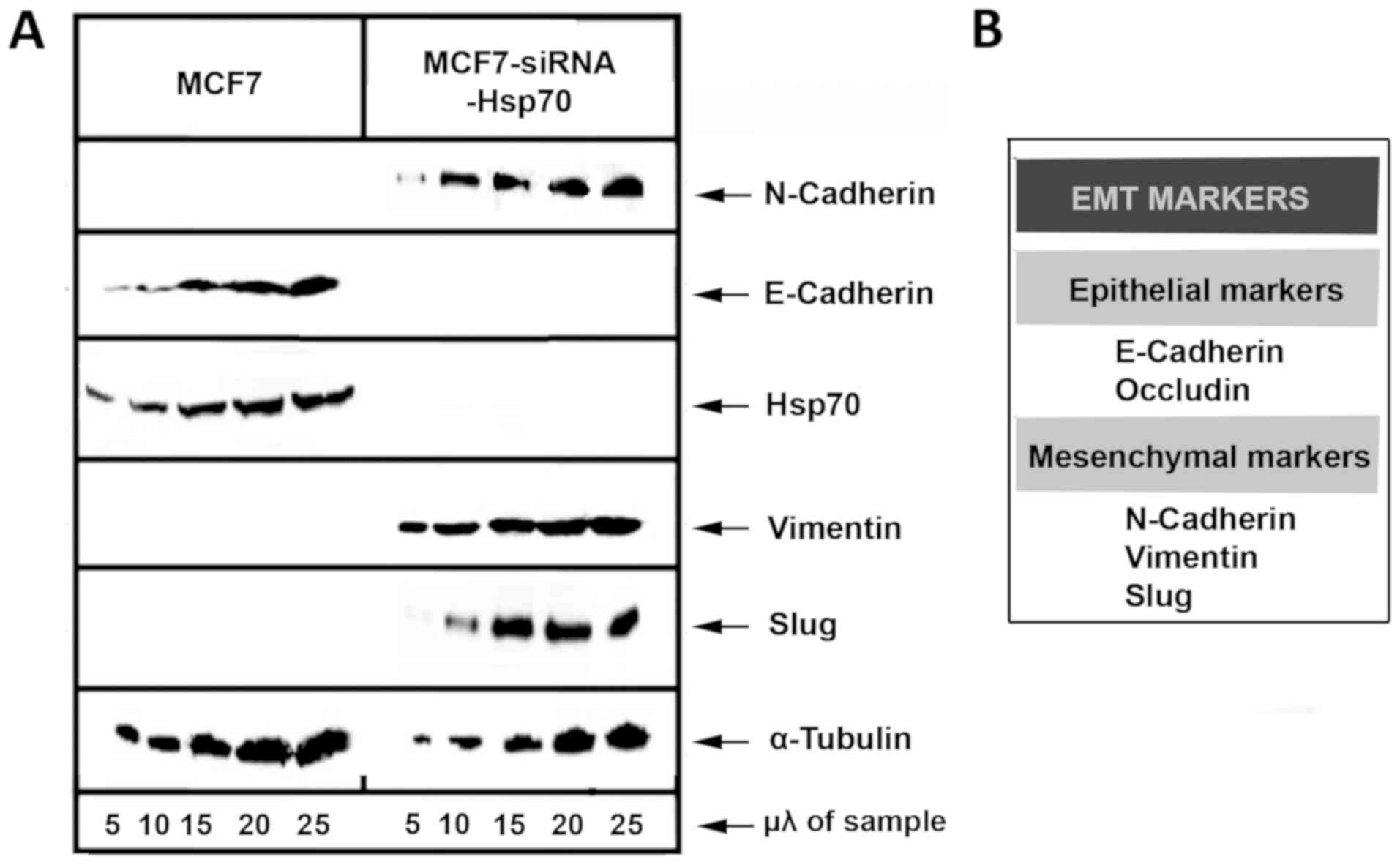
and Slug, were used to describe EMT (Fig. 5).
Initially, MCF7 and stably transfected
MCF7-siRNA-Hsp70 cancer cells were phenotypically screened and then
lysed and subjected to western blot analysis to detect the
expression levels of E-cadherin. A substantial reduction of
E-cadherin expression, a marker used to indicate the epithelial
cell origin, was observed in the MCF7-siRNA-Hsp70 cells compared
with parental MCF7 cells. In addition, the cells in which Hsp70 was
knocked down had lost their ability to adhere to each other, as
observed at the onset of the experimental procedure. However, the
MCF7-siRNA-Hsp70 cells, which had lost cell-cell adhesions after
Hsp70 knockdown, did not express E-cadherin (Fig. 5). Another indication that the
MCF7-siRNA-Hsp70 cells had acquired a mesenchymal phenotype was the
increased expression of several mesenchymal markers, such as
N-cadherin, vimentin and Slug (Fig.
5). All the above-mentioned results lead to the conclusion that
the absence of Hsp70 induces cell transition from an epithelial to
a mesenchymal phenotype. To generalize our observations, we
conducted similar experiments using the A549 and HeLa cell lines,
obtaining identical results. Indeed, the combination of
accumulation of N-cadherin, vimentin and Slug proteins, and the
decrease in E-cadherin and occludin expression confirmed the
transition of cells from epithelial to mesenchymal due to the
absence of Hsp70 (Fig. 6). We used
α-tubulin to ensure that equal protein amounts were loaded from
each sample. Therefore, it is clear that Hsp70 silencing converts
the cells to mesenchymal and increases cell motility, which are
features characterizing metastatic cells.
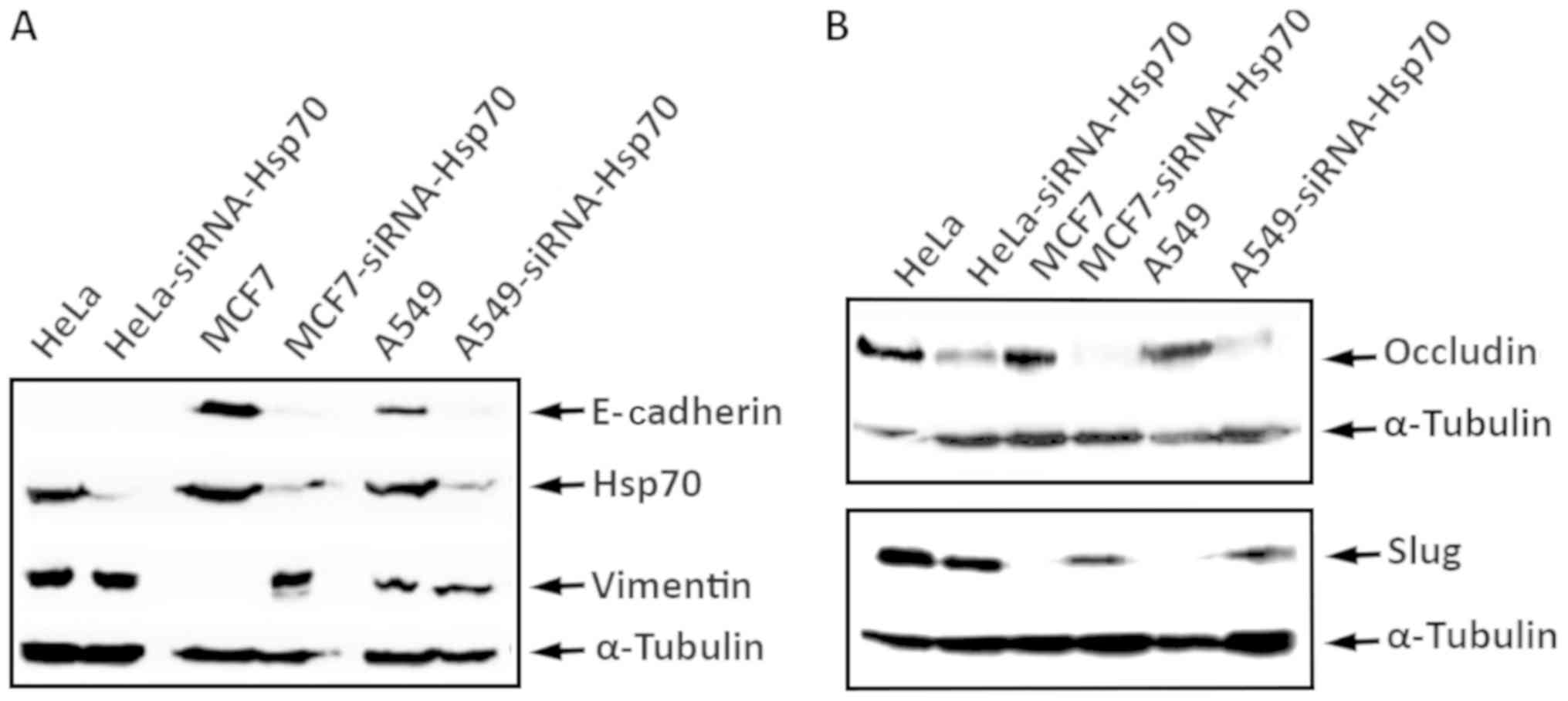 | Figure 6HeLa, HeLa-siRNA-Hsp70, MCF7,
MCF7-siRNA-Hsp70, A549, and A549-siRNA-Hsp70 cells were cultured in
60 mm plates. RIPA cell extracts were prepared, analyzed by
SDS-PAGE and then subjected to western blot analysis, using
specific antibodies for (A) E-cadherin, Hsp70, Vimentin and
α-tubulin, or (B) against occludin, Slug and α-tubulin, and the
enhanced chemiluminescence method. |
Finally, the EMT process, which occurred due to the
absence of Hsp70, was broadly applied to all the cancer cell lines
that have been investigated.
Hsp70 downregulation enhances cancer cell
migration ability
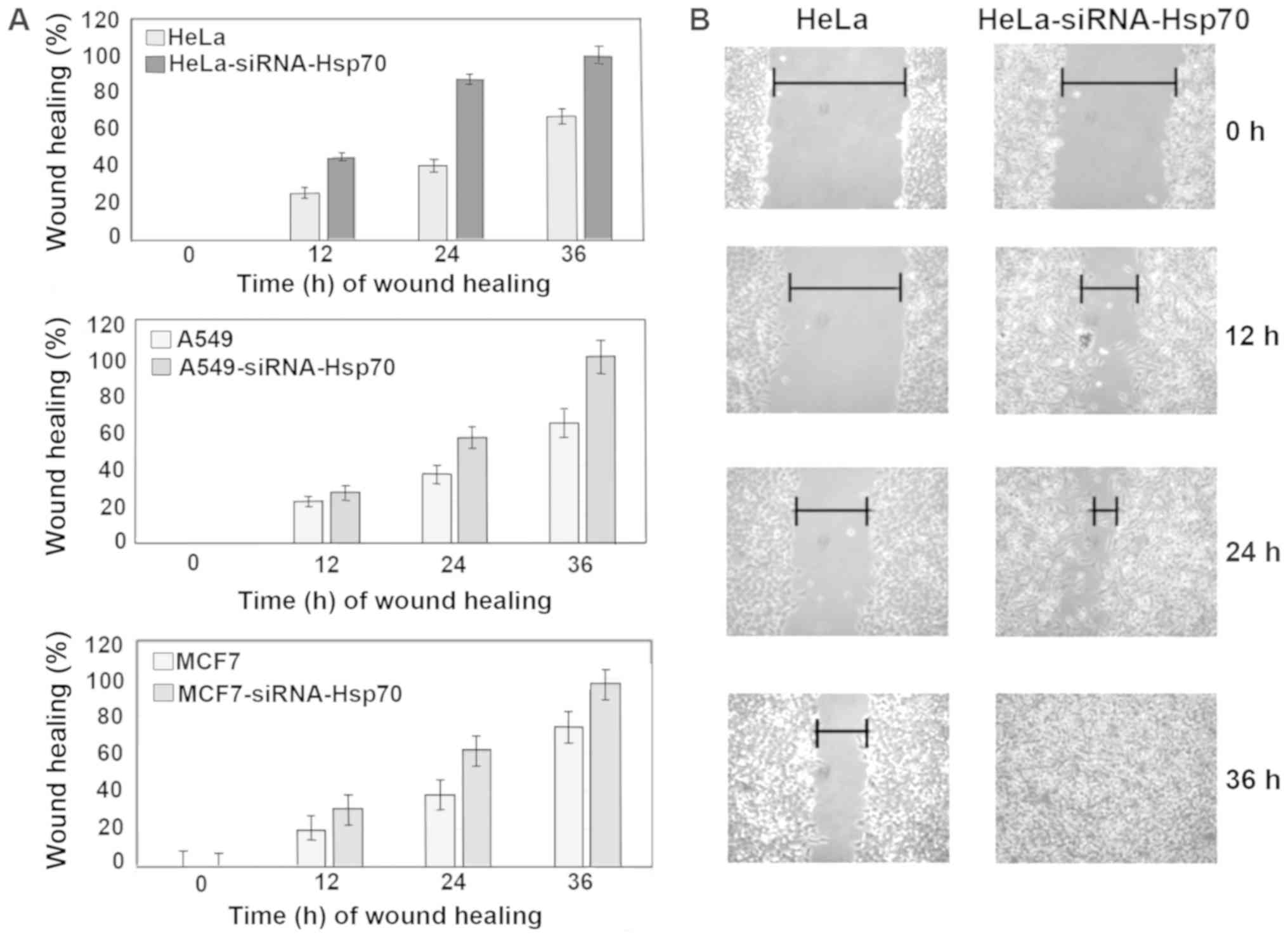
The wound healing assay was used to evaluate the
migratory capacity of the studied cells (59). Images were captured, cell migration
was calculated in the selected areas and data between cell cultures
were compared. On plates with confluent cell monolayer, scratches
of the same size were created. Cell migration was evaluated by the
motility of cells located in the area of scratch. In cell cultures
lacking Hsp70, healing was completed more rapidly compared with the
control cell cultures (Fig. 7). In
particular, HeLa-siRNA-Hsp70 healing occurred within 24 h, as
opposed to HeLa cells, in which complete healing was observed after
at least 36 h (Fig. 7A and B). The
same experiments were performed in the other cell lines, obtaining
similar results. As expected, wound healing occurred more rapidly
in the cells lacking Hsp70 (Fig.
7A).
Finally, at 36 h after scratching, the wound
remained open in the Hela cells that expressed Hsp70 at high levels
(Fig. 7A and B). Although this
result may initially appear contradictory to part of the
international bibliography, this is actually not the case, as will
be explained in the Discussion below. In order to compare the
migration ability of the cancer cell lines under our experimental
conditions, the integrity of the junctions between cells formed by
adhesion molecules, which are negative factors of migration, should
also be taken into consideration (Fig.
8, upper and middle panels). Thus, it may be suggested that the
absence of Hsp70 enhances the migratory ability of cells compared
with those that retain high expression levels of Hsp70.
Therefore, it may be concluded that Hsp70 gene
silencing promotes cancer cells to acquire a mesenchymal phenotype.
This change confers an increased migratory ability, as proposed in
the hypothetical model (Fig. 8,
lower panel).
Discussion
Hsp70 is known to be a powerful chaperone, whose
expression is induced in response to a wide variety of
physiological and environmental insults, allowing the cell to
survive under lethal conditions. Hsp70 has long been considered to
be involved in tumorigenesis and cancer metastasis. Its
overexpression is observed in a wide range of human cancers and is
implicated in tumor cell proliferation, differentiation, invasion,
metastasis, death and recognition by the immune system (60). Furthermore, it has been proven that
the knockdown of Hsp70 in cervical, bladder, breast and endometrial
cancer cell lines reduces invasiveness and propensity for
metastasis (61,62).
Although Hsp70 overexpression is considered to
contribute to a pre-cancerous environment (35), and that its presence allows the
onset and progression of a number of types of cancer (53), there is currently no direct
evidence that it causes cancer. In addition, mice overexpressing
Hsp70 (14) do not exhibit
increased carcinogenesis and survive longer compared with wild-type
mice (24). We herein present the
dual function of Hsp70 which, on the one hand, contributes to
maintaining the integrity of the tumors and, on the other hand,
acts against the metastasis of tumor cells in the tumor.
Although available data indicate the important role
of Hsp70 in tumorigenesis, the underlying mechanisms remain
unclear. The same uncertainty characterizes the involvement of
Hsp70 in metastasis. In the present study, existing or newly
prepared cancer cell lines were used. The main objective was to
investigate the involvement of Hsp70 in metastasis and specifically
its role during EMT, a subject that has not been adequately
explored. Therefore, morphological and molecular characteristics
were selected to indicate EMT status, including the loss of
intercellular connections, cell elongation, increased migration and
resistance to anoikis (63).
Initially, the appropriate sequence derived from the
human Hsp70 gene was used to generate the efficient gene silencing
of Hsp70 mRNAs. This sequence was selected by an Oligo Engine
program and inserted into a suitable expression vector creating the
pSuper-siRNA-Hsp70 plasmid. α plasmid vector carrying a DNA insert
was then constructed to express siRNA for gene silencing of Hsp70
named pcDNA3-siRNA-Hsp70. Using this plasmid, stably transfected
cell lines were created, named A549-siRNA-Hsp70, MCF7-siRNA-Hsp70
and HeLa-siRNA-Hsp70. In these cell lines, the downregulation of
Hsp70 was achieved in >90% of the total cell population. The
next step would be to discover and comprehend the unique
characteristics of the new cell line. Initially, we noted that
Hsp70 silencing led to profound changes in the morphology of the
cells, which became more elongated and lost their connection to
neighboring cells, features typically observed during EMT. EMT is a
cellular process that mimics a metastatic step in vivo.
Bearing in mind the significant changes in the shape of the cells,
we were interested in studying in detail the role of Hsp70 during
EMT. It is well known that the sequence of events occurring during
metastasis include the dispersion of a cancer cell from the primary
to a metastatic site, the dislocation and destruction of
intracellular connections, the overcoming of anoikis and the
ability of the metastatic cell to migrate from the primary tumor to
distant locations.
E-cadherin is classified among the intermediate cell
junction proteins associated with the actin filament network via
the catenins (64). In
immunofluorescence experiments (Fig.
3) using confocal microscopy, it was demonstrate that the
absence of Hsp70 caused the disorganization of E-cadherin. As shown
in Fig. 2, β-catenin was markedly
concentrated and focused in the membranes of A549 cells. Alongside,
in the A549-siRNA-Hsp70 cells, its focus was overturned and
exhibited a scattered distribution. Moreover, our findings support
the hypothesis depicted in our model (Fig. 8), in which the loss of Hsp70
disrupts the complexes of cadherins with p120, α- and β-catenins
and their association with actin filaments.
In addition, to the best of our knowledge, the
present study is the first to provide evidence that loss of Hsp70
sensitizes cells to anoikis, a well-characterized type of cell
death, due to their detachment from the cell surface and their
isolation from the neighboring cells. It has been well documented
that, when epithelial cells are detached from each other and from
the ECM, they undergo anoikis. However, further molecular
modifications render the cells resistant to anoikis and induce the
expression of metastatic characteristics. Previous studies have
demonstrated the important contribution of Hsp70 to DNA damage
repair, oxidative stress, heat shock and other cellular processes
(12,16). In this study, investigating the
role of Hsp70 in anoikis in the 3 cancer cell lines demonstrated
that Hsp70 gene silencing sensitized cells to death by anoikis, as
shown in Fig. 4.
In order to confirm the conversion of epithelial to
mesenchymal cells, specific markers of the epithelial and
mesenchymal phenotype were selected. In particular, E-cadherin and
occludin, which are epithelial markers, and N-cadherin, Vimentin
and Slug, which are mesenchymal markers, were selected. The cancer
cell lines used in the present study acquired a mesenchymal-like
phenotype. It was investigated whether the modified cell lines had
acquired new characteristic mesenchymal cell properties, such as a
higher migratory capacity. As shown in Fig. 7, cancer cells lacking Hsp70
exhibited accelerated migration ability. A possible explanation of
cells migrating rapidly was their loss of intercellular connections
isolation from the rest of the cell colony. This appears to be
achieved by the absence of Hsp70, which contributes to the
formation of complexes that serve cellular adhesion. The observed
greater mobility when Hsp70 was absent was not due to the different
degree of proliferation. Experiments conducted in parallel during
the same wound healing period have demonstrated that, at least for
the time of wound healing (~36 h after creating the wound), the
cells exhibited a similar proliferative capacity (data not
shown).
It has heretofore been hypothesized that the ability
of cancer cells to promote EMT and subsequent metastasis is
increased when Hsp70 is downregulated. A previously published
article supporting a similar hypothesis indicate specific
mechanisms for the effect of Hsp70 on EMT, which are activated via
different stimuli and, more specifically, that Hsp70 can play a
protective role against TGF-β2-induced EMT-enhancing cell survival
(65). Additional articles support
the involvement of Hsp70 in the TGF-β-mediated inhibition of EMT
(66). It has also been suggested
that HSP70 inhibits the MAPK/EKK and TGF-β/Smad signaling pathways,
thus protecting peritoneal mesothelial cells (PMCs) from EMT caused
by the advanced glycation end products (AGEs) (67). Furthermore, there are indications
that HSP70 decreases the receptor-dependent phosphorylation of
Smad2, blocking TGF-β-induced EMT (68), while inhibiting EMT of PMCs
primarily by attenuating Smad3 and Smad4 activation and reducing
the release of reactive oxygen species (69). In addition to the aforementioned
findings, it may be hypothesized that the inability of cells to
construct E-cadherin linkages with neighboring cells, due to the
absence of Hsp70, allows cells to migrate more freely. By contrast,
as shown in Fig. 8 (middle panel),
cells in the presence of Hsp70 retain E-cadherin linkages intact
and, therefore, enter a delay phase. This suggestion is enhanced by
the previous observation that correlates E-cadherin downregulation
with increasing cell migration (44).
The factors contributing to EMT are known to be
implicated in several processes, such as occurrence and progression
(70-74). In the Hsp70-EMT model, as shown in
Fig. 8 (lower panel), Hsp70
appears to be strongly implicated in the regulation of the EMT
pathway. In the absence of Hsp70, the cancer cells transitioned to
a mesenchymal state, which is known to be highly migratory.
Therefore, Hsp70 may represent a promising therapeutic target
against cancer metastasis; however, more extensive research is
required to confirm the findings of our model.
Finally, given the fact that metastasis is the main
cause of treatment failure in cancer and the leading cause of
cancer-related mortality (75),
further research is required to identify novel inhibitors of
metastasis and decode their mechanisms of action to further prolong
cancer patient survival (76).
Funding
This research was partially co-funded by the
European Union and the Hellenic Ministry of Education (program
'Herakleitos') within the 'Operational Program for Education and
Initial Vocational Training'. It was also partially supported by a
grant to PVe from the Empeirikio Institution, Athens, Greece.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Authors' contributions
CA and CK were involved in the conceptualization of
the study. CA, PK, PVr, AD, SZ and PVe were involved in the
methodology. CA, SZ, PVe, PK and PVr were involved in analysis
using software. CA, CK, PVe, SZ, PK and PVr were involved in data
validation. CA, SZ, CK, PVr, PVe and PK were responsible for formal
analysis. CA, PVr, PVe, SZ, AD and PK were involved in the
investigative part of the study. PK, PVr, SZ and AD performed the
experiments. CA, CK and PVe provided resources. CA, PVe and PK were
involved in data curation. CA, PK, SZ, PVr, PVe, AD and CK were
involved in the writing of the manuscript and original draft
preparation. CA, PK, SZ, PVr, PVe, AD and CK were involved in the
writing, reviewing and editing of the manuscript. CA, PVr, PK and
PVe were involved in visualization. CA, PK and PVe supervised the
study. CA was involved in project administration. CA, PK and PVe
were involved in funding acquisition. All the above-mentioned
authors participated in the conception and design of the study. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors are very thankful to Professor
Gerassimos Pagoulatos for his support with the reagents or useful
discussions. The authors would also like to thank Dr G. Markopoulos
for his technical support.
Abbreviations:
|
Hsp70
|
heat shock protein 70 (also known as
HSP70A1A)
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
poly-hema
|
poly(2-hydroxyethyl methacrylate)
|
References
|
1
|
Murphy ME: The HSP70 family and cancer.
Carcinogenesis. 34:1181–1188. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lindquist S and Craig EA: The heat-shock
proteins. Annu Rev Genet. 22:631–677. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morimoto RI, Tissières A and Georgopoulos
C: The stress response, function of the proteins, and perspectives.
Stress Proteins in Biology and Medicine. Morimoto RI, Tissières A
and Georgopoulos C: Cold Spring Harbor Laboratory Press, Cold
Spring Harbor; New York: pp. 1–36. 1990
|
|
4
|
Hightower LE: Heat shock, stress proteins,
chaperones, and proteotoxicity. Cell. 66:191–197. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Minami Y, Höhfeld J, Ohtsuka K and Hartl
F-U: Regulation of the heat-shock protein 70 reaction cycle by the
mammalian DnaJ homolog, Hsp40. J Biol Chem. 271:19617–19624. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang HC, Sherman MY, Kandror O and
Goldberg AL: The molecular chaperone DnaJ is required for the
degradation of a soluble abnormal protein in Escherichia coli. J
Biol Chem. 276:3920–3928. 2001. View Article : Google Scholar
|
|
7
|
Bozidis P, Lazaridis I, Pagoulatos GN and
Angelidis CE: Mydj2 as a potent partner of hsc70 in mammalian
cells. Eur J Biochem. 269:1553–1560. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Angelidis CE, Lazaridis I and Pagoulatos
GN: Aggregation of hsp70 and hsc70 in vivo is distinct and
temperature-dependent and their chaperone function is directly
related to non-aggregated forms. Eur J Biochem. 259:505–512. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beckmann RP, Mizzen LE and Welch WJ:
Interaction of Hsp 70 with newly synthesized proteins: Implications
for protein folding and assembly. Science. 248:850–854. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Saliba RS, Munro PM, Luthert PJ and
Cheetham ME: The cellular fate of mutant rhodopsin: Quality
control, degradation and aggresome formation. J Cell Sci.
115:2907–2918. 2002.PubMed/NCBI
|
|
11
|
Chirico WJ, Waters MG and Blobel G: 70K
heat shock related proteins stimulate protein translocation into
microsomes. Nature. 332:805–810. 1988. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kotoglou P, Kalaitzakis A, Vezyraki P,
Tzavaras T, Michalis LK, Dantzer F, Jung JU and Angelidis C: Hsp70
translocates to the nuclei and nucleoli, binds to XRCC1 and PARP-1,
and protects HeLa cells from single-strand DNA breaks. Cell Stress
Chaperones. 14:391–406. 2009. View Article : Google Scholar :
|
|
13
|
Angelidis CE, Lazaridis I and Pagoulatos
GN: Constitutive expression of heat-shock protein 70 in mammalian
cells confers thermoresistance. Eur J Biochem. 199:35–39. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Angelidis C, Nova C, Lazaridis I,
Kontoyiannis D, Kollias G and Pagoulatos GN: Overexpression of
HSP70 in transgenic mice results in increased cell thermotolerance.
Transgenics. 2:111–117. 1996.
|
|
15
|
Jäättelä M, Wissing D, Kokholm K, Kallunki
T and Egeblad M: Hsp70 exerts its anti-apoptotic function
downstream of caspase-3-like proteases. EMBO J. 17:6124–6134. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Damalas A, Velimezi G, Kalaitzakis A,
Liontos M, Papavassiliou AG, Gorgoulis V and Angelidis C: Loss of
p14(ARF) confers resistance to heat shock- and oxidative
stress-mediated cell death by upregulating β-catenin. Int J Cancer.
128:1989–1995. 2011. View Article : Google Scholar
|
|
17
|
Cummings CJ, Cummings CJ, Sun Y, Opal P,
Antalffy B, Mestril R, Orr HT, Dillmann WH and Zoghbi HY:
Overexpression of inducible HSP70 chaperone suppresses
neuropathology and improves motor function in SCA1 mice. Hum Mol
Genet. 10:1511–1518. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Adachi H, Katsuno M, Minamiyama M, Sang C,
Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu
M, et al: Heat shock protein 70 chaperone overexpression
ameliorates phenotypes of the spinal and bulbar muscular atrophy
transgenic mouse model by reducing nuclear-localized mutant
androgen receptor protein. J Neurosci. 23:2203–2211. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scott MD and Frydman J: Aberrant protein
folding as the molecular basis of cancer. Methods Mol Biol.
232:67–76. 2003.PubMed/NCBI
|
|
20
|
Mosser DD and Morimoto RI: Molecular
chaperones and the stress of oncogenesis. Oncogene. 23:2907–2918.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ammon-Treiber S, Grecksch G, Angelidis C,
Vezyraki P, Höllt V and Becker A: Emotional and learning behaviour
in mice over-expressing heat shock protein 70. Neurobiol Learn Mem.
90:358–364. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Plumier JC, Ross BM, Currie RW, Angelidis
CE, Kazlaris H, Kollias G and Pagoulatos GN: Transgenic mice
expressing the human heat shock protein 70 have improved
post-ischemic myocardial recovery. J Clin Invest. 95:1854–1860.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lysitsas DN, Katsouras CS, Papakostas JC,
Toumpoulis IK, Angelidis C, Bozidis P, Thomas CG, Seferiadis K,
Psychoyios N, Frillingos S, et al: Antirestenotic effects of a
novel polymer-coated d-24851 eluting stent. Experimental data in a
rabbit iliac artery model. Cardiovasc Intervent Radiol.
30:1192–1200. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Naka KK, Vezyraki P, Kalaitzakis A,
Zerikiotis S, Michalis L and Angelidis C: Hsp70 regulates the
doxorubicin-mediated heart failure in Hsp70-transgenic mice. Cell
Stress Chaperones. 19:853–864. 2014. View Article : Google Scholar
|
|
25
|
Kyrou IE, Papakostas JC, Ioachim E,
Koulouras V, Arnaoutoglou E, Angelidis C and Matsagkas MI: Early
ischaemic preconditioning of spinal cord enhanced the binding
profile of heat shock protein 70 with neurofilaments and promoted
its nuclear translocation after thoraco-abdominal aortic occlusion
in pigs. Eur J Vasc Endovasc Surg. 43:408–414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ninomiya H, Ohgami N, Oshino R, Kato M,
Ohgami K, Li X, Shen D, Iida M, Yajima I, Angelidis CE, et al:
Increased expression level of Hsp70 in the inner ears of mice by
exposure to low frequency noise. Hear Res. 363:49–54. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Morano KA: New tricks for an old dog: The
evolving world of Hsp70. Ann N Y Acad Sci. 1113:1–14. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dudeja V, Mujumdar N, Phillips P, Chugh R,
Borja-Cacho D, Dawra RK, Vickers SM and Saluja AK: Heat shock
protein 70 inhibits apoptosis in cancer cells through simultaneous
and independent mechanisms. Gastroenterology. 136:1772–1782. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wei YQ, Zhao X, Kariya Y, Teshigawara K
and Uchida A: Inhibition of proliferation and induction of
apoptosis by abrogation of heat-shock protein (HSP) 70 expression
in tumor cells. Cancer Immunol Immunother. 40:73–78. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nylandsted J, Rohde M, Brand K, Bastholm
L, Elling F and Jäättelä M: Selective depletion of heat shock
protein 70 (Hsp70) activates a tumor-specific death program that is
independent of caspases and bypasses Bcl-2. Proc Natl Acad Sci USA.
97:7871–7876. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nylandsted J, Wick W, Hirt UA, Brand K,
Rohde M, Leist M, Weller M and Jäättelä M: Eradication of
glioblastoma, and breast and colon carcinoma xenografts by Hsp70
depletion. Cancer Res. 62:7139–7142. 2002.PubMed/NCBI
|
|
32
|
Frisch SM and Francis H: Disruption of
epithelial cell-matrix interactions induces apoptosis. J Cell Biol.
124:619–626. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guan JL and Shalloway D: Regulation of
focal adhesion-associated protein tyrosine kinase by both cellular
adhesion and oncogenic transformation. Nature. 358:690–692. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ruoslahti E and Reed JC: Anchorage
dependence, integrins, and apoptosis. Cell. 77:477–478. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jäättelä M: Escaping cell death: Survival
proteins in cancer. Exp Cell Res. 248:30–43. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Beere HM, Wolf BB, Cain K, Mosser DD,
Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM and Green DR:
Heat-shock protein 70 inhibits apoptosis by preventing recruitment
of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2:469–475.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang RY, Guilford P and Thiery JP: Early
events in cell adhesion and polarity during epithelial-mesenchymal
transition. J Cell Sci. 125:4417–4422. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yilmaz M and Christofori G: Mechanisms of
motility in metastasizing cells. Mol Cancer Res. 8:629–642. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yilmaz M and Christofori G, Yilmaz M and
Christofori G: EMT, the cytoskeleton, and cancer cell invasion.
Cancer Metastasis Rev. 28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Niehrs C: The complex world of WNT
receptor signalling. Nat Rev Mol Cell Biol. 13:767–779. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kourtidis A, Ngok SP and Anastasiadis PZ:
p120 catenin: An essential regulator of cadherin stability,
adhesion-induced signaling, and cancer progression. Prog Mol Biol
Transl Sci. 116:409–432. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hajra KM and Fearon ER: Cadherin and
catenin alterations in human cancer. Genes Chromosomes Cancer.
34:255–268. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wheelock MJ, Shintani Y, Maeda M, Fukumoto
Y and Johnson KR: Cadherin switching. J Cell Sci. 121:727–735.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Theveneau E and Mayor R: Cadherins in
collective cell migration of mesenchymal cells. Curr Opin Cell
Biol. 24:677–684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Franke WW, Grund C, Kuhn C, Jackson BW and
Illmensee K: Formation of cytoskeletal elements during mouse
embryogenesis. III. Primary mesenchymal cells and the first
appearance of vimentin filaments. Differentiation. 23:43–59. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
McInroy L and Määttä A: Down-regulation of
vimentin expression inhibits carcinoma cell migration and adhesion.
Biochem Biophys Res Commun. 360:109–114. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Vuoriluoto K, Haugen H, Kiviluoto S,
Mpindi JP, Nevo J, Gjerdrum C, Tiron C, Lorens JB and Ivaska J:
Vimentin regulates EMT induction by Slug and oncogenic H-Ras and
migration by governing Axl expression in breast cancer. Oncogene.
30:1436–1448. 2011. View Article : Google Scholar
|
|
50
|
Sun Y, Song GD, Sun N, Chen JQ and Yang
SS: Slug overexpression induces stemness and promotes
hepatocellular carcinoma cell invasion and metastasis. Oncol Lett.
7:1936–1940. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Angelidis CE, Lazaridis I and Pagoulatos
GN: Specific inhibition of simian virus 40 protein synthesis by
heat and arsenite treatment. Eur J Biochem. 172:27–34. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Doulias P-T, Kotoglou P, Tenopoulou M,
Keramisanou D, Tzavaras T, Brunk U, Galaris D and Angelidis C:
Involvement of heat shock protein-70 in the mechanism of hydrogen
peroxide-induced DNA damage: The role of lysosomes and iron. Free
Radic Biol Med. 42:567–577. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gabai VL, Yaglom JA, Wang Y, Meng L, Shao
H, Kim G, Colvin T, Gestwicki J and Sherman MY: Anti-cancer effects
of targeting Hsp70 in tumor stromal cells. Cancer Res.
76:5926–5932. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Novak A and Dedhar S: Signaling through
beta-catenin and Lef/Tcf. Cell Mol Life Sci. 56:523–537. 1999.
View Article : Google Scholar
|
|
55
|
Chaw SY, Abdul Majeed A, Dalley AJ, Chan
A, Stein S and Farah CS: Epithelial to mesenchymal transition (EMT)
biomarkers - E-cadherin, beta-catenin, APC and Vimentin - in oral
squamous cell carcinogenesis and transformation. Oral Oncol.
48:997–1006. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mao J, Hu X, Xiao Y, Yang C, Ding Y, Hou
N, Wang J, Cheng H and Zhang X: Overnutrition stimulates intestinal
epithelium proliferation through β-catenin signaling in obese mice.
Diabetes. 62:3736–3746. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cowin P, Rowlands TM, Hatsell SJ and Cowin
P: Rowlands TM and Hatsell SJ: Cadherins and Catenins in breast
cancer. Curr Opin Cell Biol. 17:499–508. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wu CY, Tsai YP, Wu MZ, Teng SC and Wu KJ:
Epigenetic reprogramming and post-transcriptional regulation during
the epithelial-mesenchymal transition. Trends Genet. 28:454–463.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rodriguez LG, Wu X and Guan JL:
Wound-healing assay. Methods Mol Biol. 294:23–29. 2005.
|
|
60
|
Ciocca DR and Calderwood SK: Heat shock
proteins in cancer: Diagnostic, prognostic, predictive, and
treatment implications. Cell Stress Chaperones. 10:86–103. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Garg M, Kanojia D, Seth A, Kumar R, Gupta
A, Surolia A and Suri A: Heat-shock protein 70-2(HSP70-2)
expression in bladder urothelial carcinoma is associated with
tumour progression and promotes migration and invasion. Eur J
Cancer. 46:207–215. 2010. View Article : Google Scholar
|
|
62
|
Teng Y, Ngoka L, Mei Y, Lesoon L and
Cowell JK: HSP90 and HSP70 proteins are essential for stabilization
and activation of WASF3 metastasis-promoting protein. J Biol Chem.
287:10051–10059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Moreno-Bueno G, Peinado H, Molina P,
Olmeda D, Cubillo E, Santos V, Palacios J, Portillo F and Cano A:
The morphological and molecular features of the
epithelial-to-mesenchymal transition. Nat Protoc. 4:1591–1613.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Buxton RS and Magee AI: Structure and
interactions of desmosomal and other cadherins. Semin Cell Biol.
3:157–167. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Banh A, Deschamps PA, Vijayan MM, Sivak JG
and West-Mays JA: The role of Hsp70 and Hsp90 in TGF-β-induced
epithelial-to-mesenchymal transition in rat lens epithelial
explants. Mol Vis. 13:2248–2262. 2007.PubMed/NCBI
|
|
66
|
Yun CH, Yoon SY, Nguyen TT, Cho HY, Kim
TH, Kim ST, Kim BC, Hong YS, Kim SJ and Lee HJ: Geldanamycin
inhibits TGF-β signaling through induction of Hsp70. Arch Biochem
Biophys. 495:8–13. 2010. View Article : Google Scholar
|
|
67
|
Yang J, Zhu T, Liu X, Zhang L, Yang Y,
Zhang J and Guο M: Heat shock protein 70 protects rat peritoneal
mesothelial cells from advanced glycation end-products-induced
epithelial-to-mesenchymal transition through mitogen activated
protein kinases/extracellular signal-regulated kinases and
transforming growth factor-β/Smad pathways. Mol Med Rep.
11:4473–4481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Li Y, Kang X and Wang Q: HSP70 decreases
receptor-dependent phosphorylation of Smad2 and blocks
TGF-β-induced epithelial-mesenchymal transition. J Genet Genomics.
38:111–116. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu J, Bao J, Hao J, Peng Y and Hong F:
HSP70 inhibits high glucose-induced Smad3 activation and attenuates
epithelial-to-mesenchymal transition of peritoneal mesothelial
cells. Mol Med Rep. 10:1089–1095. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Guarino M, Rubino B and Ballabio G: The
role of epithelial-mesenchymal transition in cancer pathology.
Pathology. 39:305–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Klymkowsky MW and Savagner P:
Epithelial-mesenchymal transition: A cancer researcher's conceptual
friend and foe. Am J Pathol. 174:1588–1593. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Thiery JP: Epithelial-mesenchymal
transitions in cancer onset and progression. Bull Acad Natl Med.
193:1969–1979. 2009.In French.
|
|
75
|
Steeg PS: Targeting metastasis. Nat Rev
Cancer. 16:201–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Qian CN, Mei Y and Zhang J: Cancer
metastasis: Issues and challenges. Chin J Cancer. 36:382017.
View Article : Google Scholar : PubMed/NCBI
|