Introduction
Down syndrome (DS) is the most common form of
intellectual disability associated with central nervous system
abnormalities and results from an extra complete or partial copy of
human chromosome 21 (1,2). It is likely that most DS phenotypes
are associated with alterations in gene expression due to the
supernumerary copy of chromosome 21 (3). Previous studies have demonstrated the
upregulation of a certain subset of chromosome 21 genes,
accompanied by numerous transcriptional changes throughout the
genome (3,4). Among the possible causes, alterations
in epigenetic modifications may contribute to genome-wide changes
in gene expression patterns in DS.
Epigenetics is the study of DNA methylation,
patterns of histone modifications and non-coding RNAs that lead to
changes in gene expression that are not accompanied by alterations
in DNA sequence (5). Previous
findings have shown that perturbation of DNA methylation is
conserved in the peripheral blood leukocytes of adults with DS and
in the placental villi of women with DS pregnancies. Differentially
methylated genes have been identified on various autosomes in the
leukocytes of patients with DS, and global DNA hypermethylation has
been identified in the placenta of DS pregnancies (6,7),
suggesting that dysregulated methylation is an important cause of
disrupted gene expression in DS. Furthermore, DNA methylation is
perturbed to a greater extent in genes that are associated with DS
phenotypes (7).
Using methylated DNA immunoprecipitation microarray
(known as MeDIP-chip), 207 genes were identified with differential
DNA methylation between the DS and normal controls, which may
contribute to the clinical manifestations in DS (data not shown).
For example, PR domain containing 8 (PRDM8), one of the genes
hypermethylated in DS, participates in the development of the
nervous system (8,9). However, no correlation was identified
between the extent of methylation and expression of certain
differentially methylated genes in DS (6,7). One
explanation may be that DNA hydroxymethylation, an important
regulator of gene expression, is involved but has gone
undetected.
Five-hydroxymethylcytosine (5hmC) is an epigenetic
DNA modification produced through the enzymatic activity of ten
eleven translocation (TET) enzymes (10). TETs are 2-oxoglutarate- and Fe
(II)-dependent dioxygenases that catalyze the hydroxylation of
5-methylcytosine (5mC) to 5hmC in the DNA (11). Further oxidation of 5hmC produces
5fC and 5-carboxylcytosine (5caC) (12). 5hmC acts as an intermediate
involved in the DNA demethylation and as a functional epigenetic
marker involved in gene regulation (13). 5hmC is highly enriched in the adult
brain and accumulates across the lifespan and is markedly regulated
by neural activity. Thus, 5hmC promotes rapid behavioral adaptation
(14), suggesting that it has an
important function in neural development. However, changes in
hydroxylation of genes associated with the nervous system
development have not been investigated in DS patients.
The aim of the present study was to investigate
whether DNA hydroxymethylation is perturbed in a specific gene
related to DS phenotypes and examine the functional relevancy of
DNA hydroxymethylation to alterations of gene expression in DS.
Materials and methods
Samples from DS subjects and normal
controls
Peripheral blood samples were obtained from 16 DS
patients (age range, 2 days to 14 years) and 19 age-matched normal
controls (age range, 13 days to 14 years). Informed consent was
obtained from the parents of all the individuals. Mononuclear cells
were freshly isolated using Lymphoprep (Axis-Shield Density
Gradient Media; Alere Technologies AS, Oslo, Norway), and total RNA
was extracted using TRIzol reagent (Ambion; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). RNA purity (A260/280 nm) was
assessed using NanoDrop 2000 (Thermo Fisher Scientific, Inc.) and
2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA,
USA). DNA samples were extracted and purified using the Lab-Aid 820
Automated Blood DNA Extraction System (Xiamen Zeesan Biotech Co.,
Ltd., Fujian, China). The present study was approved by the Ethics
Committee of Shanghai Children's Hospital, Shanghai Jiao Tong
University (Shanghai, China).
Bisulfite pyrosequencing
(BS-pyrosequencing)
CGIs of PRDM8 were analyzed with MethPrimer
(http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi),
and 5hmC and 5mC levels were detected with BS pyrosequencing.
BS-pyrosequencing was carried out as previously described (15). Briefly, genomic DNA (500 ng) from
each sample was subjected to bisulfite treatment using the EpiTect
Plus DNA Bisulfite kit (Qiagen GmbH, Hilden, Germany) according to
the manufacturer's instructions. Pyrosequencing primers were
designed using the PyroMark Assay Design software, version 2.0
(Qiagen GmbH). Table I shows the
primer sequences and length of PCR products. In each PCR reaction,
one of the primers was labeled at the 5′ flanking region with
biotin (Invitrogen; Thermo Fisher Scientific. Inc.).
 | Table IPrimer sequences used for
pyrosequencing. |
Table I
Primer sequences used for
pyrosequencing.
| CGI | Primer name | Sequence
(5′→3′) | Product size
(bp) | CpG site (n) |
|---|
| P | PRDM8 P-1 F |
AGGGAGGAATAGTTTTTGGATTAGAG | 111 | 7 |
| PRDM8 P-1 RB |
ACAAAACCAACCCTATAACCC | | |
| PRDM8 P-1 S |
GGAATAGTTTTTGGATTAGAGTA | | |
| PRDM8 P-2 F |
GGAGGGAAGGGATATTGAAAG | 161 | 4 |
| PRDM8 P-2 RB |
AAACCTACTCTCTAAATCTAAAACCCA | | |
| PRDM8 P-2 S |
GGTAGTAGTGGTTGGTAAT | | |
| E1I1 | PRDM8 E1I1 F |
ATGTGTAAGGATAGAAGGGAAAT | 157 | 8 |
| PRDM8 E1I1 RB |
AATCCCCATCACTCACTTTAC | | |
| PRDM8 E1I1 S1 |
AGAAGGGAAATTGAGGA | | |
| PRDM8 E1I1 S2 |
GGTTTTGAAGTGGAGTAG | | |
| I1-1 | PRDM8 I1-1 F |
AGGAGGTTTAGAGTTTTGGTTAG | 244 | 13 |
| PRDM8 I1-1 RB |
ACCCAACTTACAAATTCTTTCTT | | |
| PRDM8 I1-1 S1 |
AGGTTTTTTTGTTTATTTTTAGA | | |
| PRDM8 I1-1 S2 |
TTTTTAATAGATTGA | | |
| I1-2 | PRDM8 I1-2 F |
GGAAGGTTAAAGAATATGGGAAATGT | 212 | 9 |
| PRDM8 I1-2 RB |
AAACCCTAACACAAAAAACTACC | | |
| PRDM8 I1-2 S |
GTTTAGGGTTTATTTGGAG | | |
| I5E6 | PRDM8 I5E6 FB |
GGTGGTTTTAGGGTTAGAGAAT | 156 | 9 |
| PRDM8 I5E6 R |
AACCTTTCCCCCTTTCACTAAACACTT | | |
| PRDM8 I5E6 S |
ACAAAAAACTAAACACCCA | | |
| I6 | PRDM8 I6 F |
AGAGGGTTAGAGTTTTAGGAGG | 168 | 15 |
| PRDM8 I6 RB |
CCCCTCCCTTTAACTCTTT | | |
| PRDM8 I6 S1 |
GGGTTAGAGTTTTAGGAGGA | | |
| PRDM8 I6 S2 |
GGGGTAGGAGTTTAGGATT | | |
| I7 | PRDM8 I7-1 F |
TGAGGGGTTGTTTATTGTTAGTAATAT | 221 | 13 |
| PRDM8 I7-1 RB |
ACCCCCCTCTAAACCCAAATTCTT | | |
| PRDM8 I7-1 S1 |
ATTGTTAGTAATATTGTATAAAAGG | | |
| PRDM8 I7-1 S2 |
GTTAGATAATGTTTGTTT | | |
| PRDM8 I7-2 F |
TTGGGGTATATTTTTAGGGTAGG | 144 | 10 |
| PRDM8 I7-2 RB |
CCTCAAACCCATCACAATAACC | | |
| PRDM8 I7-2 S |
GGATAAGAATTTGGGTTTAGA | | |
| PRDM8 I7-3 F |
TTAGAGGTTATTGTGATGGGTTTGAG | 215 | 9 |
| PRDM8 I7-3 RB |
CAATTTCTCTCTTCCTTTTAAAAATCTCT | | |
| PRDM8 I7-3 S |
ATTGTGATGGGTTTGAG | | |
| E9I9 | PRDM8 E9I9 F |
TTGGTAAGGGAAAGAATTGATTGAGT | 207 | 12 |
| PRDM8 E9I9 RB |
CCAAACTACATCTCAAAATCTTCCTATAA | | |
| PRDM8 E9I9 S |
ATAATAAAATGAATGGTAGGTT | | |
| I9E10 | PRDM8 I9E10 F |
AGGGGATGGTGGTAAATT | 337 | 14 |
| PRDM8 I9E10 RB |
AACTTAAAACCCCAACCTAAAAATACCTCC | | |
| PRDM8 I9E10 S1 |
GATGGTGGTAAATTGG | | |
| PRDM8 I9E10 S2 |
AGATTTTATAGAGTTGATATAAGT | | |
| E10 | PRDM8 E10-1 F |
GGAGGTAGTAGTTGTTTTTTAGTTTAGA | 139 | 14 |
| PRDM8 E10-1 RB |
CCTCCCAAAATTTCCTCTTTCCTTTAC | | |
| PRDM8 E10-1 S1 |
TGTTTTTTAGTTTAGAGTTTTAGTA | | |
| PRDM8 E10-2 F |
GGTTTGTTTAAGTAGAGTTTTTTT | 270 | 9 |
| PRDM8 E10-2 RB |
CAAAAACTCCATCCCATACTCCTTTTTA | | |
| PRDM8 E10-2 S1 |
GTTATAGTTTTTTGGTTTAAGAGT | | |
Bisulfite-treated DNA was amplified in the specific
region using the PyroMark PCR kit (Qiagen GmbH) according to the
manufacturer's instructions. The PCR cycling conditions were as
follows: 95°C for 15 min, followed by 50 cycles of 30 sec at 95°C,
30 sec at 55°C, and 30 sec at 72°C, with final extension at 72°C
for 10 min. The quality of the PCR products was evaluated by
agarose gel electrophoresis (Biowest Agarose G-10; Gene Co., Ltd.,
Chai Wan, Hong Kong).
Templates were prepared using the PyroMark Q24
Vacuum Prep Workstation (Qiagen GmbH) according to the
manufacturer's instructions. Each single-stranded template was
annealed to the sequencing primer (0.375 µM) at 80°C for 5
min. Pyrosequencing was performed with the PyroMark Q24 system
(Qiagen GmbH) using the PyroMark Q24 Advanced CpG Reagents (Qiagen
GmbH). The data were analyzed with the PyroMark Q24 Advanced
(Qiagen GmbH).
Standard sample synthesis
To determine the conversion efficiency of 5hmC to
uracil in oxidative bisulfite pyrosequencing (oxBS-pyrosequencing),
a 49-nt standard sample containing three different cytosines (C,
5mC, 5hmC) was designed and synthesized by GenScript (Nanjing City,
China). The forward, reverse and sequencing standard sample primers
were as follows: 5′-AGGAGGTTTAGAGTTTTGG-3′ (F),
5′-ACCCAACTTACAAATTCTTTCTT-3′ (RB) and 5′-AGGTTTAGAGTTTTGGT-3′ (S),
respectively (Invitrogen; Thermo Fisher Scientific. Inc.)
OxBS-pyrosequencing
The oxBS-pyrosequencing procedures were carried out
as previously described (16,17)
with minor modifications. To optimize oxidation conditions, the
49-nt standard sample (50 ng) was denatured in NaOH (0.05 M) at
37°C for 30 min at room temperature. The solution was cooled at 4°C
for 10 min, followed by the oxidation reaction with
KRuO4 (Sigma-Aldrich, St. Louis, MO, USA) at different
concentrations (0.6–2.5 mM) at 4°C for 1 h. Each sample was then
centrifuged at 14,000 × g for 10 min to pellet any black
precipitate. Subsequently, oxidized DNA was purified using the mini
Quick Spin Oligo columns (Roche Diagnostics GmbH, Mannheim,
Germany). BS-pyrosequencing was performed following the above
protocol using the EpiTect Fast DNA Bisulfite kit (Qiagen
GmbH).
To select the appropriate oxidation temperature,
oxidation reactions were performed with KRuO4 (2 mM) at
different temperatures (4, 16, 25 and 37°C) for 1 h. The duration
of bisulfite treatment was then optimized. Subsequent to the
treatment of each 49-nt standard sample with KRuO4 (2
mM) at 4°C for 1 h, BS-pyrosequencing was carried out following the
above bisulfite conversion with 1–4 amplification cycles.
To detect hydroxymethylation and methylation of
PRDM8 in DS and normal control samples, oxBS-pyrosequencing was
carried out following the above optimized protocol. In brief,
genomic DNA (500 ng) was incubated with KRuO4 (2 mM) for
1 h at 4°C. Following purification of oxidized DNA using the mini
Quick Spin Oligo column, oxidized and non-oxidized genomic DNA (500
ng) from the same sample were simultaneously subjected to 2 cycles
of bisulfite conversion amplification using the EpiTect Plus DNA
Bisulfite kit. The oxidized and non-oxidized DNA from each sample
were then subjected to pyrosequencing.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Reverse transcription was carried out with the RNase
H Minus M-MLV reverse transcriptase (Takara Biotechnology Co.,
Ltd., Shiga, Japan) using ~1 µg total RNA and 0.5 µg
oligo (dT)18 primer (Shanghai Generay Biotech Co., Ltd., Shanghai,
China). The reagent was incubated for 10 min at 70°C, then placed
in an ice bath for 2 min. Subsequently, the reaction solution,
including the M-MLV buffer, dNTPs, RNase inhibitor and M-MLV
reverse transcriptase (Takara Biotechnology Co., Ltd.), was
incubated at 42°C for 1 h and 70°C for 15 min.
The relative expression of the two transcripts of
PRDM8 was determined using the TaqMan Gene Expression Assay (Thermo
Fisher Scientific, Inc.). Hs01027634_g1 was used for transcript
variant 1 and a customized assay for variant 2, and Hs03929097_g1
was used for GAPDH as a control (Applied Biosystems; Thermo Fisher
Scientific, Inc.). qPCR analysis was performed using 25 ng cDNA and
a QuantiNova Probe PCR Kit (Qiagen GmbH), according to the
manufacturer's protocol, on a 7500 system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The thermal profile for quantitative PCR was 95°C for 10
min followed by 40 cycles of 95°C for 15 sec and 60°C for 60 sec.
The relative expression levels were calculated using the
2−ΔΔCq normalization method and the values of PRDM8 and
GAPDH (18).
Statistical analysis
Student's t-test (normally distributed data) and the
Mann-Whitney test (non-normally distributed data) were used for
comparing two groups. The Pearson's correlation was used for
correlation analysis. Statistical analysis was performed using the
SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Optimization of the oxBS-pyrosequencing
protocol
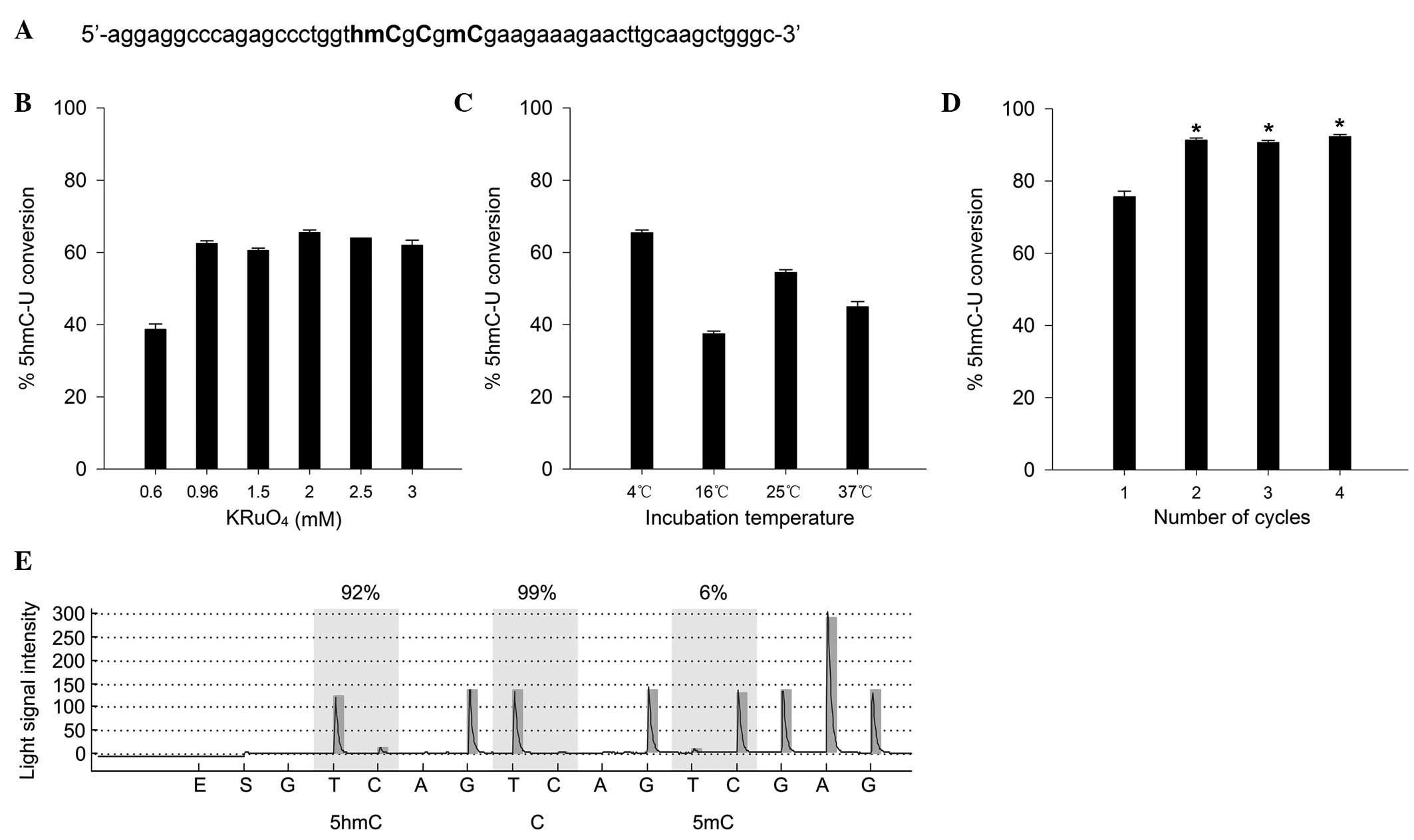
To achieve maximal conversion efficiency of 5hmC to
uracil (5hmC→5fC→U), the experimental conditions, including the
appropriate concentration of oxidant (KRuO4), oxidation
temperature and time of bisulfite treatment, were optimized. A
49-nt standard sample containing three different cytosines (C, 5mC,
5hmC) was designed and synthesized to determine the conversion
efficiency of 5hmC to uracil in oxBS-pyrosequencing (Fig. 1A). The results indicated that the
concentration of KRuO4 was an important factor affecting
the conversion efficiency. As shown in Fig. 1B, the conversion efficiency
improved from 38.7 to 62.5% following the change of concentration
of KRuO4 from 0.6 to 0.96 mM. The highest efficiency of
5hmC conversion was 65.5% at 2 mM KRuO4, and thus this
concentration was used in subsequent experiments (Fig. 1B).
To determine the appropriate oxidation temperature,
standard samples were incubated with KRuO4 (2 mM) at
different temperatures (4, 16, 25 and 37°C). The greatest
conversion was achieved at 4°C (Fig.
1C), which was then used in subsequent experiments.
Furthermore, to achieve a higher conversion efficiency, different
numbers of bisulfite conversion amplification cycles (1–4 cycles)
were attempted subsequent to the oxidation of the sample with
KRuO4 (2 mM). The conversion efficiency of cycles 2, 3
and 4 was significantly increased, as compared with cycle 1
(Fig. 1D; P<0.01); however, no
significant differences were detected between cycles 2, 3 and 4.
Although the highest conversion efficiency was achieved with 4
cycles, the incorrect conversion of 5mC to uracil was elevated with
the increasing cycle number (data not shown), and therefore 2
cycles were carried out in subsequent experiments. The optimized
oxBS-pyrosequencing protocol is summarized as follows: the
conversion efficiency of 5hmC to uracil reached 91.3% when the
standard sample was treated with 2 mM KRuO4 at 4°C,
followed by two cycles of bisulfite conversion amplification using
the EpiTect Plus DNA Bisulfite kit (Fig. 1E).
Observation of hypermethylation and
hyperhydroxymethylation in an internal promoter of PRDM8 in DS
Regions in PRDM8 containing different quantities of
5mC and 5hmC were identified with single-base resolution by
oxBS-pyrosequencing of peripheral blood samples from children with
DS and normal controls. Several CpG islands (CGIs) were detected in
PRDM8 and two transcripts were encoded (Fig. 2A). The results indicated that 5mC
and 5hmC were not evenly distributed across PRDM8, and the levels
of methylation and hydroxymethylation were low in the 5′ and 3′
flanking regions (Fig. 2B and C).
Hypermethylation was identified in 4 CGIs including I5E6
(represents the CGI across intron 5 and exon 6; 44.1 vs. 25.6%;
P=5.63×10−5), I6 (represents the CGI within intron 6;
30.9 vs. 12.2%; P=1.71×10−4), I7 (represents the CGI
within intron 7; 33.2 vs. 16.7%: P=3.49×10−5), and E9I9
(represents the CGI across exon 9 and intron 9; 82.9 vs. 78.2%;
P=0.003) in DS samples (Fig. 2B).
The most significant hypermethylation was identified in the
internal promoter region (I5E6, I6 and I7). Hyperhydroxymethylation
was observed in only one CGI of patients DS (I6; 7.4 vs. 2.0%;
Fig. 2C; P=0.0046).
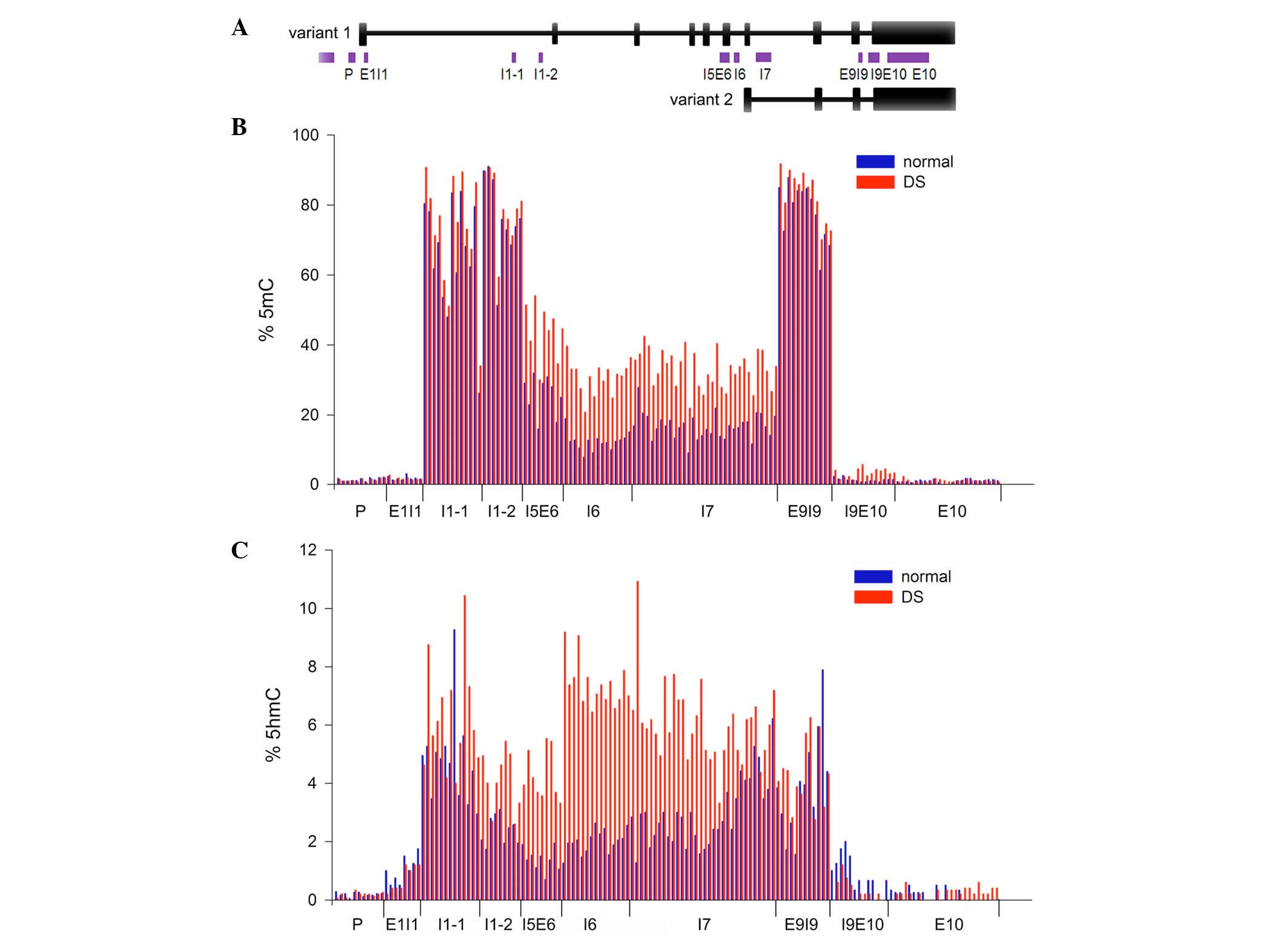 | Figure 2Levels of 5mC and 5hmC in PRDM8 from
peripheral blood samples. (A) Sketch map of CGIs and two
transcripts of PRDM8. CGIs of PRDM8 were analyzed with MethPrimer
and are represented by the purple bars. Location of CGIs was
denoted by P, E1I1, I1-1, I1-2, I5E6, I6, I7, E9I9, I9E10, E10,
where P is the promoter, E is the exon and I is the intron. Black
vertical bars represent exons of PRDM8. The long and short isoforms
are transcript variants 1 and 2, respectively. (B) Averaged 5mC
levels of PRDM8 in peripheral blood samples from children with DS
and normal controls. The number of CGI samples sequenced was 16
from DS and 19 from normal samples for P, I1-1, I1-2, I5E6, I6, I7
and E9I9. The number sequenced for the remaining CGIs was 5 for DS
and 4 for normal samples. (C) Averaged 5hmC levels of PRDM8 in
peripheral blood samples from children with DS and normal controls.
DS, Down syndrome; PRDM8, PR domain containing 8; 5hmC,
5-hydroxymethylcytosine; 5mC, 5-methylcytosine; CGI, CpG
island. |
Expression level of PRDM8 transcript
variant 2 is greater in DS
The expression levels of PRDM8 transcript variant 1
(long transcript) and transcript variant 2 (short transcript) were
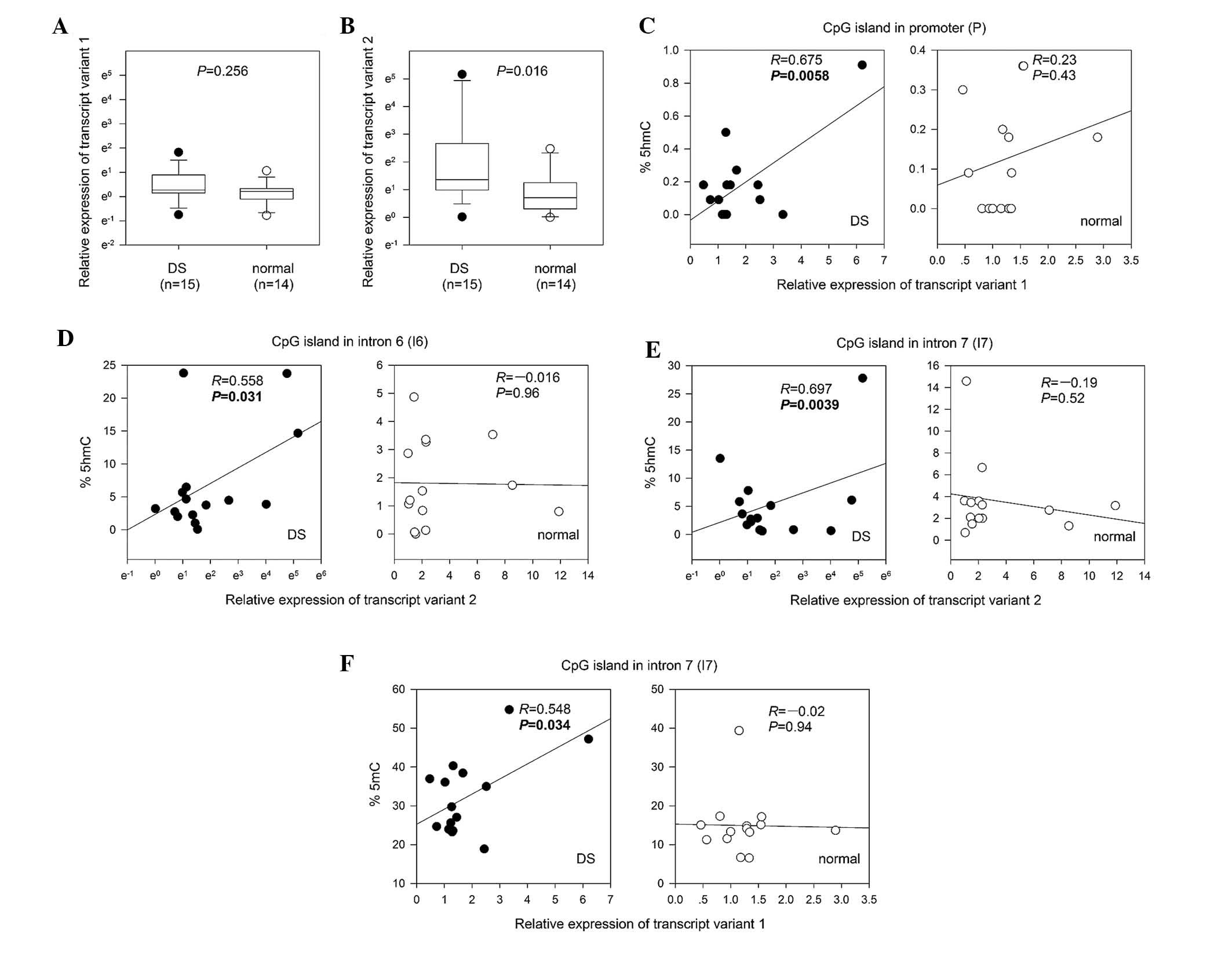
measured in 15 DS and 14 normal samples by qRT-PCR (Fig. 3A and B). The expression of
transcript variant 2 was significantly higher in the DS group
(median: 3.9 vs. 2.04; Fig. 3B;
P=0.016). The level of transcript variant 1 was modestly higher in
DS, but the difference was not statistically significant (median:
1.31 vs. 1.23; Fig. 3A;
P=0.256).
In DS, the expression levels of PRDM8 transcripts 1
and 2 were correlated with the external promoter and internal
promoter hydroxymethylation, respectively (Figs 3C–E).
To investigate the regulation of PRDM8 expression in
DS, the association of DNA methylation or hydroxymethylation of
several CGIs (P, I1-1, I1-2, I5E6, I6, I7 and E9I9) in various
genomic contexts (promoter and gene body), and the expression of
the two transcripts was analyzed in DS and normal samples. As
demonstrated in Fig. 3C, the
expression of PRDM8 transcript variant 1 correlated positively with
hydroxymethylation in the promoter region (R=0.675, P=0.0058) in
DS, but not in the normal samples. Furthermore, as shown in
Fig. 3D and E, a positive
correlation was identified only in the DS samples among the
expression of the PRDM8 transcript variant 2 and hydroxymethylation
of I6 (R= 0.558, P= 0.0305) and I7 (R= 0.697, P= 0.0039), located
in the internal promoter region. These results suggested that
hydroxymethylation in the promoter region is associated with higher
levels of PRDM8 expression in DS.
Expression of PRDM8 transcript 1
correlates with internal promoter methylation in DS
The expression of PRDM8 transcript variant 1
correlated positively with methylation of the internal promoter
region (I7; R=0.548, P=0.034) in the DS samples, but not in the
normal samples (Fig. 3F). These
data suggested that intragenic methylation serves an important role
in higher PRDM8 expression in DS.
Discussion
Changes in DNA methylation throughout the genome may
be a reason for genome-wide alteration of gene expression in DS
(3,4), conserved in different tissues,
particularly for genes associated with DS phenotypes (6,7).
Hydroxymethylation is a vital functional marker involved in
neurogenesis. However, in previous studies, methods for mapping 5mC
in DS have been based on bisulfite conversion of genomic DNA
(6,7), which cannot discriminate 5mC from
5hmC. Thus, 5hmC is read as C following PCR amplification (19). Oxidative bisulfite sequencing
(oxBS-Seq) is a novel method for quantitative mapping of 5hmC in
genomic DNA at a single-nucleotide resolution (17). Selective chemical oxidation of 5hmC
to 5-formylcytosine (5fC) allows for bisulfite conversion of 5fC to
uracil (17). Therefore, the
difference between 5hmC and 5mC is be discriminated accurately
through DNA sequencing. Due to the fundamental mechanism of
oxBS-Seq, the approach is compatible with any sequencing platform.
However, the reaction conditions of the oxidative bisulfite, that
is important for accurate quantization, were not discussed in
length in the previous studies. In the current study,
pyrosequencing was used to detect the 5mC and 5hmC quantitatively
through analyzing the ratio of C/T in oxidized and non-oxidized
genomic DNA from the same sample, and the oxBS-pyrosequencing
protocol was investigated. The results demonstrate that the
concentration of KRuO4 and the cycle of the bisulfite
conversion may be two of the most important factors for improving
conversion efficiency, as the conversion efficiency reached 91.3%
following optimized conditions of oxBS-pyrosequencing.
PRDM8 is a key mediator of development, including
the neurogenesis of the central nervous system (8,9).
PRDM8 has two alternative promoters that produce two transcripts,
and several CGIs are located in different regions, including the
promoter and gene body. In the current study, the expression of the
PRDM8 transcript variant 2 was significantly higher in patients
with DS. Therefore, PRDM8 is an optimal model gene for
investigating the effects of epigenetic modifications on gene
expression in DS.
Transcriptional regulation of DNA hydroxymethylation
is associated with genomic contexts and cell types (20). Gene expression is generally
suppressed by promoter hydroxymethylation in embryonic stem cells
(ES cells) of humans (21) and
mice (22,23), whereas it is usually upregulated
with gene body hydroxymethylation in mouse ES cells (22,23)
[but not in human ES cells (24)],
neurons (25,26), spermatogenic cells (27) and T cells (10). The results of the present study
show that the expression of variants 1 and 2 correlates positively
with the hydroxymethylation at the external and internal promoters,
including I6 and I7, respectively, in the peripheral blood samples
of patients with DS. This correlation was not apparent in the
normal samples. A similar correlation has been identified in the
human brain among the transcript levels and 5hmC content of
promoters with low CpG content (28). These results suggest that DNA
hyperhydroxymethylation may serve a critical role in the regulation
of abnormal overexpression of PRDM8 transcript in DS.
It is well known that promoter methylation
suppresses gene expression (29).
The association of the intragenic methylation and gene expression
is contradictory, varying with cell type and dependent on whether
the methylated cytosine exists in a CpG or non-CpG context
(20). Previous studies have
suggested that the intragenic methylation contributes to higher
gene expression in dividing cells, such as B lymphocytes,
peripheral white blood cells, placenta and fibroblasts (30,31).
The results of the present study show that the expression of PRDM8
transcript variant 1 correlates positively with the intragenic
methylation (I7) in DS, but not in normal samples, suggesting that
intragenic methylation may be another mechanism for regulating
PRDM8 expression in patients with DS.
In conclusion, the findings of the current study
have demonstrated that alteration of hydroxymethylation and
methylation of PRDM8 correlates with changes in its expression in
the peripheral blood of children with DS. Given the proposed
function of PRDM8 in cognitive disability in DS, we speculate that
the alteration of epigenetic modification leading to abnormal PRDM8
expression may affect the transcription of a series of downstream
genes that serve a critical role in abnormal central nervous system
neurogenesis and development in patients with DS. Further studies
of the epigenetic deregulation of PRDM8 using mouse models may aid
to further elucidate the influence of PRDM8 upregulation on the
nervous system development and to demonstrate the molecular
mechanism(s) underlying DS in individual patients.
Abbreviations:
|
OxBS-pyrosequencing
|
oxidative bisulfite
pyro-sequencing
|
|
BS-pyrosequencing
|
bisulfite pyrosequencing
|
|
CGI
|
CpG island
|
|
5mC
|
5-methylcytosine
|
|
5hmC
|
5-hydroxymethylcytosine
|
Acknowledgments
The present study was supported by the National
Basic Research Project of China (grant nos. 2010CB529502 and
2007CB511904), the National Natural Science Foundation of China
(grant no. 81471485), the key program for the fundamental research
of the Science and Technology Commission of Shanghai (grant no.
11JC1411000), the Shanghai Joint Research Project for Important
Disease (grant no. 2013ZYJB0015) and the Project from Shanghai
Municipal Commission of Health and Family Planning (grant no.
20144Y0178).
References
|
1
|
Dierssen M: Down syndrome: The brain in
trisomic mode. Nat Rev Neurosci. 13:844–858. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Haydar TF and Reeves RH: Trisomy 21 and
early brain development. Trends Neurosci. 35:81–91. 2012.
View Article : Google Scholar
|
|
3
|
Letourneau A, Santoni FA, Bonilla X,
Sailani MR, Gonzalez D, Kind J, Chevalier C, Thurman R, Sandstrom
RS, Hibaoui Y, et al: Domains of genome-wide gene expression
dysregulation in Down's syndrome. Nature. 508:345–350. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lockstone HE, Harris LW, Swatton JE,
Wayland MT, Holland AJ and Bahn S: Gene expression profiling in the
adult Down syndrome brain. Genomics. 90:647–660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rozek LS, Dolinoy DC, Sartor MA and Omenn
GS: Epigenetics: Relevance and implications for public health. Annu
Rev Public Health. 35:105–122. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kerkel K, Schupf N, Hatta K, Pang D, Salas
M, Kratz A, Minden M, Murty V, Zigman WB, Mayeux RP, et al: Altered
DNA methylation in leukocytes with trisomy 21. PLoS Genet.
6:e10012122010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jin S, Lee YK, Lim YC, Zheng Z, Lin XM, Ng
DP, Holbrook JD, Law HY, Kwek KY, Yeo GS, et al: Global DNA
hypermethylation in down syndrome placenta. PLoS Genet.
9:e10035152013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Inoue M, Kuroda T, Honda A,
Komabayashi-Suzuki M, Komai T, Shinkai Y and Mizutani K: Prdm8
regulates the morphological transition at multipolar phase during
neocortical development. PLoS One. 9:e863562014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ross SE, McCord AE, Jung C, Atan D, Mok
SI, Hemberg M, Kim TK, Salogiannis J, Hu L, Cohen S, et al: Bhlhb5
and Prdm8 form a repressor complex involved in neuronal circuit
assembly. Neuron. 73:292–303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsagaratou A, Äijö T, Lio CW, Yue X, Huang
Y, Jacobsen SE, Lähdesmäki H and Rao A: Dissecting the dynamic
changes of 5-hydroxymethylcytosine in T-cell development and
differentiation. Proc Natl Acad Sci USA. 111:E3306–3315. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tahiliani M, Koh KP, Shen Y, Pastor WA,
Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et
al: Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in
mammalian DNA by MLL partner TET1. Science. 324:930–935. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ito S, Shen L, Dai Q, Wu SC, Collins LB,
Swenberg JA, He C and Zhang Y: Tet proteins can convert
5-methylcytosine to 5-formyl-cytosine and 5-carboxylcytosine.
Science. 333:1300–1303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu H and Zhang Y: Reversing DNA
methylation: Mechanisms, genomics, and biological functions. Cell.
156:45–68. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li X, Wei W, Zhao QY, Widagdo J,
Baker-Andresen D, Flavell CR, D'Alessio A, Zhang Y and Bredy TW:
Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine
promotes rapid behavioral adaptation. Proc Natl Acad Sci USA.
111:7120–7125. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mikeska T, Felsberg J, Hewitt CA and
Dobrovic A: Analysing DNA methylation using bisulphite
pyrosequencing. Methods Mol Biol. 791:33–53. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Booth MJ, Ost TW, Beraldi D, Bell NM,
Branco MR, Reik W and Balasubramanian S: Oxidative bisulfite
sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nat
Protoc. 8:1841–1851. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Booth MJ, Branco MR, Ficz G, Oxley D,
Krueger F, Reik W and Balasubramanian S: Quantitative sequencing of
5-methyl-cytosine and 5-hydroxymethylcytosine at single-base
resolution. Science. 336:934–937. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
19
|
Huang Y, Pastor WA, Shen Y, Tahiliani M,
Liu DR and Rao A: The behaviour of 5-hydroxymethylcytosine in
bisulfite sequencing. PLoS One. 5:e88882010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pastor WA, Aravind L and Rao A: TETonic
shift: Biological roles of TET proteins in DNA demethylation and
transcription. Nat Rev Mol Cell Biol. 14:341–356. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stroud H, Feng S, Morey Kinney S, Pradhan
S and Jacobsen SE: 5-Hydroxymethylcytosine is associated with
enhancers and gene bodies in human embryonic stem cells. Genome
Biol. 12:R542011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang Y, Chavez L, Chang X, Wang X, Pastor
WA, Kang J, Zepeda-Martínez JA, Pape UJ, Jacobsen SE, Peters B, et
al: Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in
mouse embryonic stem cells. Proc Natl Acad Sci USA. 111:1361–1366.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu H, D'Alessio AC, Ito S, Wang Z, Cui K,
Zhao K, Sun YE and Zhang Y: Genome-wide analysis of
5-hydroxymethylcy-tosine distribution reveals its dual function in
transcriptional regulation in mouse embryonic stem cells. Genes
Dev. 25:679–684. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Szulwach KE, Li X, Li Y, Song CX, Han JW,
Kim S, Namburi S, Hermetz K, Kim JJ, Rudd MK, et al: Integrating
5-hydroxymethylcytosine into the epigenomic landscape of human
embryonic stem cells. PLoS Genet. 7:e10021542011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Colquitt BM, Allen WE, Barnea G and
Lomvardas S: Alteration of genic 5-hydroxymethylcytosine patterning
in olfactory neurons correlates with changes in gene expression and
cell identity. Proc Natl Acad Sci USA. 110:14682–14687. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wen L, Li X, Yan L, Tan Y, Li R, Zhao Y,
Wang Y, Xie J, Zhang Y, Song C, et al: Whole-genome analysis of
5-hydroxy-methylcytosine and 5-methylcytosine at base resolution in
the human brain. Genome Biol. 15:R492014. View Article : Google Scholar
|
|
27
|
Gan H, Wen L, Liao S, Lin X, Ma T, Liu J,
Song CX, Wang M, He C, Han C, et al: Dynamics of
5-hydroxymethylcytosine during mouse spermatogenesis. Nat Commun.
4:19952013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jin SG, Wu X, Li AX and Pfeifer GP:
Genomic mapping of 5-hydroxymethylcytosine in the human brain.
Nucleic Acids Res. 39:5015–5024. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ball MP, Li JB, Gao Y, Lee JH, LeProust
EM, Park IH, Xie B, Daley GQ and Church GM: Targeted and
genome-scale strategies reveal gene-body methylation signatures in
human cells. Nat Biotechnol. 27:361–368. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aran D, Toperoff G, Rosenberg M and
Hellman A: Replication timing-related and gene body-specific
methylation of active human genes. Hum Mol Genet. 20:670–680. 2011.
View Article : Google Scholar
|