Introduction
Leukoaraiosis (LA), a common form of cerebral white
matter lesion (WML), is characterized by punctuate or patchy
hyperintensities in the periventricular or subcortical white matter
observed via magnetic resonance imaging (MRI). Although it remains
asymptomatic, LA is not considered to be benign, and has been
demonstrated to be associated with poor clinical outcomes and
increased risk of disability, dementia, depression, stroke, and the
overall morbidity and mortality (1). LA is associated with increased age,
hypertension, diabetes mellitus, history of stroke and chronic
atherosclerotic diseases (2). As
hypertension is reported to be one of the major determinants of
WMLs, the present study only included subjects with a history of
hypertension. Despite intensive research, an understanding of the
pathological mechanism remains incomplete, and is likely to be
multifactorial. Therefore, identifying biomarkers is essential to
improve the early diagnosis of LA.
Circular RNAs (circRNAs) are a class of endogenous
RNAs that have a stable structure (3), which is a clear advantage of using
circRNAs as a diagnostic marker. Previous studies have demonstrated
that circRNAs are involved in the development of several types of
diseases, including nervous system disorders (4,5).
However, limited information is available regarding the association
of circRNAs with LA. The present study, based on the hypertension
population, compared the circRNA expression profiles of LA
sufferers and controls. By performing a circRNA array analysis and
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR), the present study demonstrated that certain circRNAs,
including has_circ_102533 and has_circ_103783, may have potential
as a novel type of biomarker for the diagnosis of LA. In addition,
the gene ontology and KEGG analysis of differentially expressed
(DE) genes may contribute to an improved understanding of the
pathogenesis of LA.
Materials and methods
Subjects and clinical specimens
The current study was performed in accordance with
the guidelines of the Helsinki Declaration. Written informed
consent was obtained from all subjects and the Ethics Committee of
Shandong Provincial Hospital Affiliated to Shandong University
(Jinan, China) approved all aspects of the present study. A total
of 30 subjects were recruited between August 2014 and September
2015 at Shandong Provincial Hospital Affiliated to Shandong
University. All cases had a history of hypertension for 5–15 years.
The groups were as follows: Test (LA sufferers; n=20; 7 males and
13 females) and control (no LA; n=10; 3 males and 7 females). The
mean age was 58±5.7 years. The subjects had a mean systolic
pressure of 150±7.1 mmHg and a mean diastolic pressure of 84±4.2
mmHg. LA is defined as punctuate or patchy hyperintensities in the
periventricular or subcortical white matter, observed by MRI.
Hypertension is defined as a history of high blood pressure
(≥140/90 mmHg) reported by the respondent, or by the current use of
antihypertensive medication. None of the cases suffered from
diabetes mellitus, heart diseases, dyslipidemia, hypotension,
immune system diseases, hematological disorders, malnutrition,
malignant tumors, serious liver and kidney diseases (including
severe hepatitis or nephritis), liver or kidney failure, or had a
history of heavy smoking (20 cigarettes daily) or alcoholism. Fresh
fasting peripheral blood samples (3 ml) were collected early in the
morning and subsequently stored at −80°C.
Microarray expression analysis
Based on random sampling, Arraystar Human circRNA
Microarray analysis version 2.0 (Arraystar, Inc., Rockville, MD,
USA) of 6 samples from LA cases and 6 samples from control cases
was performed. The sample preparation and microarray hybridization
were performed based on the Arraystar's standard protocols. Total
RNA from each sample was quantified using the NanoDrop®
ND-1000 (NanoDrop Technologies; Thermo Fisher Scientific, Inc.,
Wilmington, DE, USA). RNA Integrity and genomic DNA contamination
test was performed by denaturing agarose gel electrophoresis.
Sample labeling and array hybridization were performed according to
the manufacturer's protocol (Arraystar Inc.). Briefly, 2,600 ng/µl
total RNA (including 28s and 18s ribosomal RNA from each sample was
treated with RNase R (Epicentre, Inc.) to enrich circRNA. The
enriched RNA was subsequently amplified and transcribed into
fluorescent complementary RNA (cRNA) utilizing random primers
according to the Arraystar Super RNA Labeling kit protocol
(Arraystar, Inc.). The labeled cRNAs were hybridized onto the
Arraystar Human circRNA Mircoarrays (8×15K; Arraystar, Inc.) and
incubated for 17 h at 65°C in an Agilent hybridization oven. After
washing four times in hybridization buffer the labeled cRNAs,
slides were scanned with an Agilent G2505C Microarray Scanner
System (Agilent Technologies, Inc., Santa Clara, CA, USA).
Data collection and analysis
Scanned images were imported into Agilent Feature
Extraction software version 11.0.1.1 (Agilent Technologies, Inc.)
for raw data extraction. Quantile normalization of raw data and
subsequent data processing were performed using the R software
package (R Project for Statistical Computing, Vienna, Austria).
After quantile normalization of the raw data, low intensity
filtering was performed, and the circRNAs that were flagged as ‘P’
or ‘M’ (‘All Targets Value’) in at least 3 out of 6 samples were
retained for further analyses. When comparing profile differences
between two groups (including disease vs. control), the ‘fold
change’ (i.e., the ratio of the group mean averages) between the
groups for each circRNA was computed. The statistical significance
of the difference was determined by a t-test. circRNAs with fold
changes >1.5 and P<0.05 were selected as significantly
differentially expressed circRNAs. circRNA sequences were predicted
by bioinformatics methods, as described previously (6).
RT-qPCR detection of hsa_circ_102470,
hsa_circ_101396, hsa_circ_102533 and hsa_circ_103783
Total RNA in plasma was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and was quantified using the
NanoDrop® ND-1000, according to the manufacturer's
protocol. cDNA was synthesized by RT (Arraystar Super RNA Labeling
kit; Arraystar, Inc., Rockville, MD, USA) 3 times using random
primers and the Gene Amp PCR system 9700, according to the
manufacturer's protocol. A total of 1 µg RNA, 1 µl random (N9), 1.6
µl dNTP mix (2.5 mM dATP, dGTP, dCTP, and dTTP, provided by HyTest
Ltd) was combined to make the annealing mixture. This mixture was
incubated in a 65°C water bath for 5 min and then put on ice for 2
min. After brief centrifugation at 12,000 × g or 15 min at 4°C, the
RT reactant solution (4 µl 5X First-Strand Buffer, 1 µl 0.1 M DTT,
0.3 µl RNase inhibitor and 0.2 µl SuperScript III RT, provided by
Invitrogen; Thermo Fisher Scientific, Inc.) was added to a
centrifuge tube and incubated at 37°C in a water bath for 1 min.
The tube was then incubated at 50°C in a water bath for 60 min,
followed by 70°C for 15 min. Subsequently, cDNA was placed on ice.
qPCR was performed using Arraystar SYBR®-Green qPCR
Master Mix (ROX-), 5 ml on the ViiA™ 7 Real-time PCR system.
Cycling conditions were as follows: An initial predenaturation step
at 94°C for 20 sec, followed by 40 cycles of denaturation at 95°C
for 10 sec, annealing at 60°C for 60 sec and extension at 72°C for
15 sec. The primers used to quantify levels of the 4 circRNAs
(hsa_circ_102470, hsa_circ_101396, hsa_circ_102533 and
hsa_circ_103783) and GAPDH are presented in Table I. The data were analyzed using the
relative values following internal calibration, presented as
hsa_circ_102470/GAPDH, hsa_circ_101396/GAPDH, hsa_circ_102533/GAPDH
and hsa_circ_103783/GAPDH. Subsequently, the ratios (average
relative values in test group divided by the average relative
values in control group) of the 4 circRNAs were further analyzed.
The 2−∆∆Cq method was used for quantification (5).
 | Table I.Primers for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers for reverse
transcription-quantitative polymerase chain reaction.
|
| Sequence |
|
|
|---|
|
|
|
|
|
|---|
| Gene | Forward | Reverse | Annealing temperature
(°C) | Product length
(bp) |
|---|
| GAPDH |
5′GGGAAACTGTGGCGTGAT3′ |
5′GAGTGGGTGTCGCTGTTGA3′ | 60 | 299 |
|
hsa_circRNA_102533 |
5′GCTGCCAAAAGCATAACCAA3′ |
5′CCCCTTTTCTGCTAAATGAACTCT3′ | 60 | 198 |
|
hsa_circRNA_103783 |
5′AAGCTGTTAGCATGATCCCACC3′ |
5′GATGAAACTTTTCCAAGTGTGGC3′ | 60 | 133 |
|
hsa_circRNA_101396 |
5′AAAGGTCCACTTCGTATGCTG3′ |
5′ACTCTGTCATTGGAGCAACTGTAT3′ | 60 | 221 |
|
hsa_circRNA_102470 |
5′CCTAAATTTCACGACACCAG3′ |
5′ATTCAGATTGCTCAAGGTAACT3′ | 60 | 144 |
Prediction of DE genes and the
function analysis of DE genes
GO covers three domains, including biological
process, cellular component and molecular function. Fisher's exact
test is used by professionals to establish whether there is a
higher overlap between the DE list and the GO annotation list than
expected by chance. The P-value denotes the significance of GO term
enrichment in the DE genes. The lower the P-value, the higher the
significance of the GO Term (P<0.05 is recommended). Pathway
analysis is a functional analysis that maps genes to KEGG pathways.
The P-value (EASE-score, Fisher-P-value or Hypergeometric-P-value)
denotes the significance of the pathway associated with the
conditions; the lower the P-value, the higher the significance of
the pathway (The recommended P-value cut-off is 0.05).
Statistical analysis
Data are presented as the mean ± standard deviation
(continuous variables) or as a proportion (discrete variables). The
differences between groups were analyzed by Student's t-test
(continuous variables) or chi-squared test (discrete variables).
SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA) was used for
statistical analysis.
Results
Differential expression of
circRNAs
The characteristics of age- and sex-matched test and
control group subjects are presented in Table II. Compared with the controls,
certain circRNAs exhibited >1.5-fold change (P<0.05) in the
LA group; 32 were upregulated and 132 were downregulated. Among the
upregulated circRNAs, hsa_circ_103783, hsa_circ_101396 and
hsa_circ_102533 exhibited the highest statistically significant
difference in expression, while of the downregulated circRNAs,
hsa_circ_102470 was identified to exhibit the largest difference in
expression between the LA and control groups. The results of the
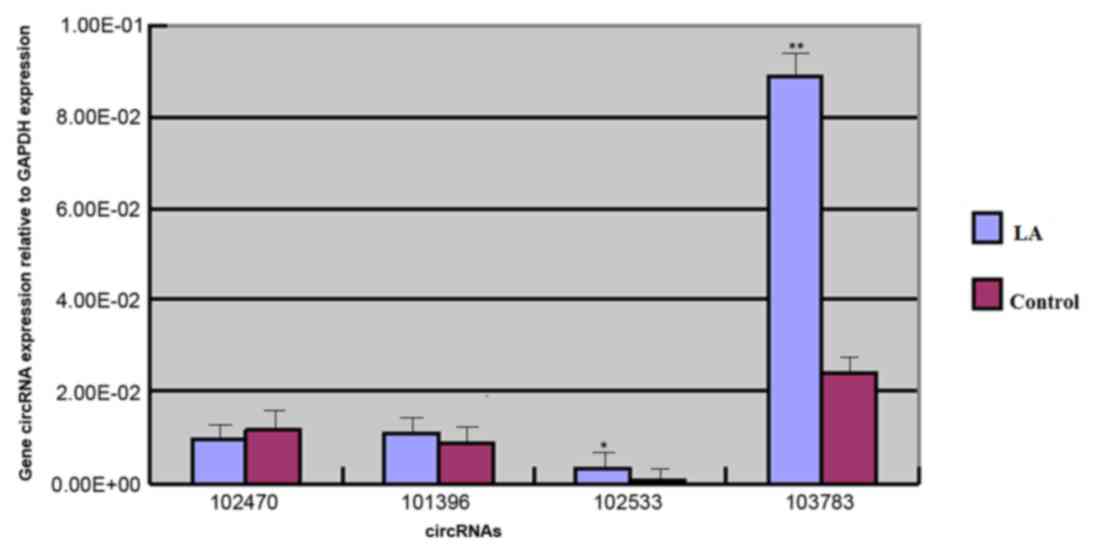
RT-qPCR detection of hsa_circ_102470, hsa_circ_101396,
hsa_circ_102533 and hsa_circ_103783 are presented in Fig. 1. The expression of hsa_circ_103783
and hsa_circ_102533 in the test group was significantly higher
compared with the control group, with fold increases in expression
of 3.67 (P<0.001) and 3.52 (P<0.05), respectively. The
expression of hsa_circ_101396 was 1.25-fold higher in the test
group compared with the control; however, this was not
statistically significant (P=0.05). Comparing the expression of
hsa_circ_102470 between the two groups, a 0.8-fold downregulation,
which was not statistically significant, was observed in the test
group (P=0.39).
| Table II.Subject characteristics. |
Table II.
Subject characteristics.
| Characteristic | Total | LA | Control | P-value |
|---|
| Age, years | 58±5.7 | 59±6.3 | 56±2.4 | 0.074 |
| Number of males
(%) | 10
(33.3) | 7
(35) | 3 (30) | 0.784 |
| Systolic pressure,
mmHg | 150 (±7.1) | 148 (±3.2) | 153
(±11.0) | 0.059 |
| Diastolic pressure,
mmHg | 84
(±4.2) | 82
(±3.9) | 85
(±4.6) | 0.162 |
GO analysis of DE genes
The top 10 most significant enrichment GO terms of
the upregulated DE genes are presented in Tables III–V. The biological process was primarily
enriched in metabolic processes, including nucleobase-containing
compounds, and heterocycle and cellular aromatic compound metabolic
processes. The major cellular components were organelles,
membrane-bounded organelles, intracellular membrane-bounded
organelles and intracellular organelles. The molecular function
predominantly enriched in the activity of histone acetyltransferase
and acetyl-CoA were L-lysine, N6-acetyltransferase and ion channel
activity. The top 10 most significantly enriched GO terms among the
downregulated DE genes are presented in Tables VI–VIII. The present study demonstrated
that cellular localization was the most enriched biological process
of DE genes, and the major cellular components involved were the
cytoplasm and cytosol. The molecular function was predominantly
enriched in kinase, enzyme and protein kinase binding.
| Table III.Top ten gene ontology BP terms that
upregulated genes were enriched in. |
Table III.
Top ten gene ontology BP terms that
upregulated genes were enriched in.
| BP term, metabolic
process | Enrichment score | P-value |
|---|
| Nucleobase-containing
compound | 2.66 | 0.002 |
| Heterocycle | 2.51 | 0.003 |
| Cellular aromatic
compound | 2.50 | 0.003 |
| Organic cyclic
compound | 2.33 | 0.004 |
| Cellular nitrogen
compound | 2.33 | 0.004 |
| Regulation of
nucleobase-containing compound | 2.33 | 0.004 |
| Regulation of
nitrogen compound | 0.52 | 0.005 |
| Nitrogen
compound | 2.24 | 0.009 |
| Single organismal
cell-cell adhesion | 2.03 | 0.013 |
| Nucleic acid | 1.86 | 0.014 |
| Table V.Top ten MF terms that upregulated
genes were enriched in. |
Table V.
Top ten MF terms that upregulated
genes were enriched in.
| MF term | Enrichment
score | P-value |
|---|
| Histone
acetyltransferase activity | 2.46 | 0.003 |
|
Acetyl-CoA:L-lysine | 2.46 | 0.003 |
|
N6-acetyltransferase |
|
|
| Ion channel
activity | 2.46 | 0.003 |
| Substrate-specific
channel activity | 2.42 | 0.004 |
| Channel
activity | 2.34 | 0.005 |
| Passive
transmembrane transporter activity | 2.34 | 0.005 |
| Enzyme activator
activity | 2.29 | 0.005 |
| Anion channel
activity | 2.12 | 0.008 |
| N-acetyltransferase
activity | 2.11 | 0.008 |
| GTPase activator
activity | 2.09 | 0.008 |
| Table VI.Top ten BP terms that downregulated
genes were enriched in. |
Table VI.
Top ten BP terms that downregulated
genes were enriched in.
| BP term | Enrichment
score | P-value |
|---|
| Cellular
localization | 4.97 | <0.001 |
| Establishment of
localization in cell | 4.92 | <0.001 |
| Intracellular
transport | 4.39 | <0.001 |
| Macromolecular
complex subunit organization | 4.26 | <0.001 |
| Cytoplasmic
transport | 4.18 | <0.001 |
|
Modification-dependent macromolecule
catabolic process | 4.10 | <0.001 |
| Organic substance
catabolic process | 3.98 | <0.001 |
| Catabolic
process | 3.97 | <0.001 |
| Cytoskeleton
organization | 3.84 | <0.001 |
| Organelle
localization | 3.83 | <0.001 |
| Table VIII.Top ten MF terms that downregulated
genes were enriched in. |
Table VIII.
Top ten MF terms that downregulated
genes were enriched in.
| MF term | Enrichment
score | P-value |
|---|
| Kinase binding | 6.17 | <0.001 |
| Enzyme binding | 5.05 | <0.001 |
| Protein kinase
binding | 4.53 | <0.001 |
| Protein
binding | 3.74 | <0.001 |
| Transferase
activity | 3.48 | <0.001 |
| Nucleotide
binding | 3.01 | <0.001 |
| Nucleoside
phosphate binding | 3.01 | <0.001 |
| Protein
homodimerization activity | 2.89 |
0.001 |
| ATP binding | 2.82 |
0.002 |
| Protein domain
specific binding | 2.81 |
0.002 |
KEGG analysis of DE genes
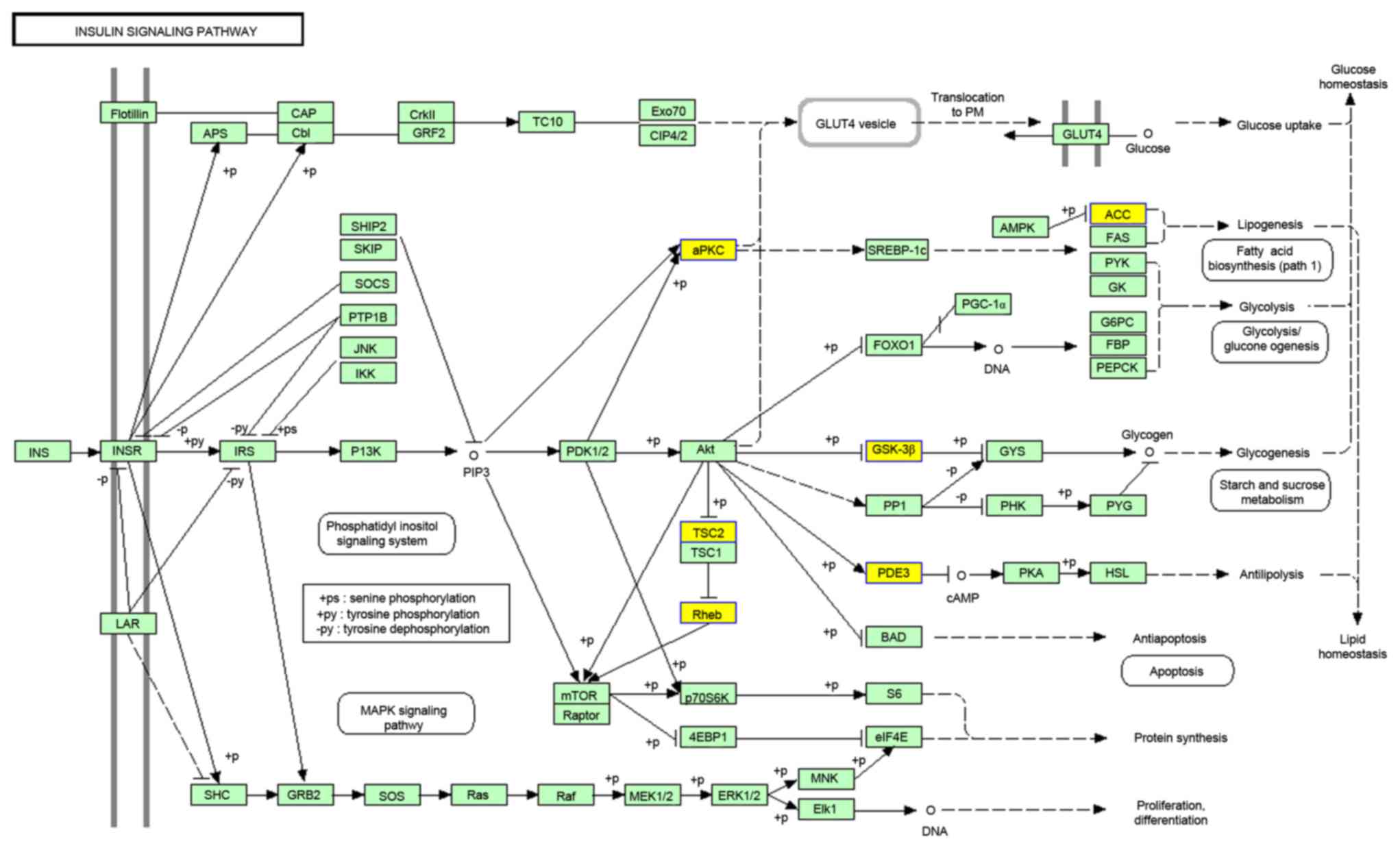
KEGG enrichment was identified for downregulated DE
genes; however, not for upregulated DE genes. The top 10 most
significant enrichment KEGG terms are listed in Table IX. The major KEGG item was the
insulin signaling pathway, with 10.3% of downregulated DE genes
involved in it (Fig. 2; http://www.genome.jp/kegg-bin/show_pathway?scale=1.0&query=&map=hsa04910&scale=0.67&auto_image=&show_description=hide&multi_query=
and http://www.kegg.jp/kegg/legal.html).
| Table IX.Top ten KEGG pathways that
downregulated genes were enriched in. |
Table IX.
Top ten KEGG pathways that
downregulated genes were enriched in.
| KEGG pathway | Enrichment
score | P-value |
|---|
| Insulin signaling
pathway, Homo sapiens | 2.97 | 0.001 |
| B cell receptor
signaling | 2.52 | 0.003 |
| Thyroid hormone
signaling | 2.51 | 0.003 |
| Axon guidance | 2.38 | 0.004 |
| mTOR signaling | 1.86 | 0.014 |
| Transcriptional
misregulation in cancer | 1.77 | 0.017 |
| Chemokine
signaling | 1.68 | 0.02 |
| Glyoxylate and
dicarboxylate metabolism | 1.64 | 0.02 |
| Propanoate
metabolism | 1.64 | 0.02 |
| Platelet
activation | 1.62 | 0.02 |
Discussion
At present, understanding of the pathogenesis of LA
remains to be fully elucidated, and there no reliable indicator for
the early diagnosis of LA in the hypertension population. Previous
studies have eported that LA may be associated with various
factors, including age, sex (7),
hypertension (2), small vessel
disease and atherosclerosis (8).
Hypertension is one of the most important established risk factors
for LA (9), and pulse pressure was
previously demonstrated to be independently associated with LA,
regardless of classical cardiovascular risk factors in elderly men
(10). However, patients with
hypertension do not necessarily suffer from LA. To predict the
potential for development of LA among patients with hypertension,
the present study investigated potential predictors at the circRNA
level.
Previously, circRNA was demonstrated to be a highly
prevalent RNA species in the human transcriptome (6), and they may have important roles in
the regulation of gene expression (11). circRNA expression levels can be
>10-fold higher compared with their linear isomers (6,12).
The two most important properties of circRNAs are highly conserved
sequences and have a high degree of stability in mammalian cells
(13). Certain circRNAs regulate
gene expression by serving as competing endogenous RNAs (14). circRNAs block the inhibitory effect
of microRNAs (miRNAs) on the target RNA by combining with miRNAs,
so as to regulate the expression level of the target RNA (15). Currently, the circRNA with the most
compelling evidence for a biological function is the miRNA-7
sponge, CDR1as (16). miRNA-7 was
previously demonstrated to serve a key role in Parkinson's and
Alzheimer's diseases (17,18).
Although an increasing number of studies have
investigated the potential functions of circRNAs in the brain, to
the best of our knowledge, the present study is the first to
investigate the association between circRNAs and LA. The current
study was the first to construct a spectrum of differentially
expressed circRNAs in patients with LA, and to identify potential
biomarkers for the diagnosis of LA and its pathogenesis. At
present, there is no circRNA database for LA. The present study
aimed to search a circRNA database for patients with LA and
hypertension, via an Arraystar Human circRNA Microarray analysis of
6 test samples and 6 control samples. Subsequently, 32 upregulated
and 132 downregulated circRNAs were screened out. Furthermore, 3
upregulated circRNAs (hsa_circ_101396, hsa_circ_102533 and
hsa_circ_103783) and 1 downregulated circRNA (hsa_circ_102470) were
selected based on a >1.5 fold-change (P<0.05), which were
further assessed by RT-qPCR. The results of the current study
demonstrated that the exonic RNAs, including hsa_circ_102533
(3.52-fold; P<0.05) and hsa_circ_103783 (3.67-fold; P<0.001),
were significantly upregulated in the LA patients compared with the
control group, indicating the potential diagnostic value of certain
circRNAs for LA.
GO enrichment analysis of the upregulated DE genes
indicated that DE genes were primarily associated with metabolic
processes, organelles and the activity of certain transferases.
However, it remains to be elucidated whether such gene products
have the potential to be novel detectors for LA. In addition, GO
and KEGG analysis of downregulated DE genes indicated that DE genes
were primarily enriched in the cytoplasm, kinase binding and the
insulin signaling pathway. The insulin signaling pathway was
revealed to be pleiotropic, and included c-Cbl-associated protein,
insulin receptor substrate-1, mitogen activated protein kinase,
plasma cell glycoprotein-1, phosphoinositide-dependent kinase,
protein kinase C and phosphatidylinositol 3-kinase (PI3-K). The
involvement of phosphatidylinositol 3-kinase/protein kinase B
(PI3-K/Akt) -dependent signaling pathways were found in normal
cellular functions. The dysfunction of this signaling pathway has a
pivotal role in atherosclerosis (19) and hypertension (20). Therefore, it was hypothesized that
the PI3-K/Akt-dependent signaling pathway may contribute to the
pathological changes of LA. However, this requires further
investigation.
In conclusion, DE circRNAs were identified between
patients with LA and controls among a hypertension population. The
present study indicated that the upregulated circRNAs,
hsa_circ_102533 and hsa_circ_103783, may have potential as
biomarkers for the diagnosis of LA. GO and KEGG enrichment analyses
of DE genes indicated that the DE genes were associated with the
pathogenesis of LA. However, due to the limited number of plasma
samples of LA, the present study only analyzed the expression of 4
circRNAs in 20 plasma samples with LA and 10 control samples.
Future studies should increase the sample size to further confirm
the diagnostic value of hsa_circ_102533 and hsa_circ_103783 for LA.
Importantly, the conclusions were based on a large number of early
experimental results; therefore, the specific mechanisms requires
further investigation.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 30970991) and the Key
Research and Development Program of Shandong Province (grant no.
2015GSF118069).
References
|
1
|
Lin Q, Huang WQ and Tzeng CM: Genetic
associations of leukoaraiosis indicate pathophysiological
mechanisms in white matter lesions etiology. Rev Neurosci.
26:343–358. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ben-Assayag E, Mijajlovic M,
Shenhar-Tsarfaty S, Bova I, Shopin L and Bornstein NM:
Leukoaraiosis is a chronic atherosclerotic disease.
ScientificWorldJournal. 2012:5321412012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Memczak S, Jens M, Elefsinioti A, Torti F,
Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer
M, et al: Circular RNAs are a large class of animal RNAs with
regulatory potency. Nature. 495:333–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen YT, Rettig WJ, Yenamandra AK, Kozak
CA, Chaganti RS, Posner JB and Old LJ: Cerebellar
degeneration-related antigen: A highly conserved neuroectodermal
marker mapped to chromosomes X in human and mouse. Proc Natl Acad
Sci USA. 87:3077–3081. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2 (−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Salzman J, Gawad C, Wang PL, Lacayo N and
Brown PO: Circular RNAs are the predominant transcript isoform from
hundreds of human genes in diverse cell types. PLoS One.
7:e307332012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Simoni M, Li L, Paul NL, Gruter BE, Schulz
UG, Küker W and Rothwell PM: Age- and sex-specific rates of
leukoaraiosis in TIA and stroke patients: Population-based study.
Neurology. 79:1215–1222. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Uh J, Yezhuvath U, Cheng Y and Lu H: In
vivo vascular hallmarks of diffuse leukoaraiosis. J Magn Reson
Imaging. 32:184–190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Avet J, Pichot V, Barthélémy JC, Laurent
B, Garcin A, Roche F and Celle S: Leukoaraiosis and ambulatory
blood pressure load in a healthy elderly cohort study: The PROOF
study. Int J Cardiol. 172:59–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim SH, Shim JY, Lee HR, Na HY and Lee YJ:
The relationship between pulse pressure and leukoaraiosis in the
elderly. Arch Gerontol Geriatr. 54:206–209. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo JU, Agarwal V, Guo H and Bartel DP:
Expanded identification and characterization of mammalian circular
RNAs. Genome Biol. 15:4092014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jeck WR, Sorrentino JA, Wang K, Slevin MK,
Burd CE, Liu J, Marzluff WF and Sharpless NE: Circular RNAs are
abundant, conserved, and associated with ALU repeats. RNA.
19:141–157. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hansen TB, Jensen TI, Clausen BH, Bramsen
JB, Finsen B, Damgaard CK and Kjems J: Natural RNA circles function
as efficient microRNA sponges. Nature. 495:384–388. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Danan M, Schwartz S, Edelheit S and Sorek
R: Transcriptome-wide discovery of circular RNAs in Archaea.
Nucleic Acids Res. 40:3131–3142. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Valdmanis PN and Kay MA: The expanding
repertoire of circular RNAs. Mol Ther. 21:1112–1114. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hentze MW and Preiss T: Circular RNAs:
Splicing's enigma variations. EMBO J. 32:923–925. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Junn E, Lee KW, Jeong BS, Chan TW, Im JY
and Mouradian MM: Repression of alpha-synuclein expression and
toxicity by microRNA-7. Proc Natl Acad Sci USA. 106:13052–13057.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lukiw WJ: Circular RNA (circRNA) in
Alzheimer's disease (AD). Front Genet. 4:3072013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Namgaladze D and Brüne B: Phospholipase
A2-modified low-density lipoprotein activates the
phosphatidylinositol 3-kinase-Akt pathway and increases cell
survival in monocytic cells. Arterioscler Thromb Vasc Biol.
26:2510–2516. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Northcott CA, Hayflick JS and Watts SW:
PI3-kinase upregulation and involvement in spontaneous tone in
arteries from DOCA-salt rats: Is p110delta the culprit?
Hypertension. 43:885–890. 2004. View Article : Google Scholar : PubMed/NCBI
|