Introduction
Innate immune cells, such as monocytes/macrophages,
function in the defense against pathogens and the initiation and
maintenance of the inflammatory response (1,2). A
robust inflammatory response is triggered when innate cells detect
pathogens or their associated endotoxins, such as
lipopolysaccharide (LPS), through pattern recognition receptors,
including toll-like receptor 4 (TLR4), expressed on the cell
surface (3,4). However, macrophages are not able to
respond to a subsequent challenge with LPS following long-term or
repeated exposure to LPS. This phenomenon is termed ‘endotoxin
tolerance’ (5,6). The characterization of gene
transcription following endotoxin tolerance revealed downregulation
of certain genes upon LPS restimulation, including tumor necrosis
factor-α (TNF-α) (7), interleukin
(IL)-1β (8), C-C motif chemokine
ligand (CCL)17, CCL22 (9) and
nitric oxide synthase 2 (iNOS) (10), while the expression of other genes,
including chitinase-like 3 (Chil3) and arginase-1 (Arg1), was
upregulated (11). The mixed
transcriptional phenotype observed in tolerant cells indicates a
gene reprogramming mechanism rather than a simple downregulation of
LPS-induced gene expression (5,12,13).
The phenomenon of endotoxin tolerance has been observed in
vitro and in vivo (14–16).
In patients with sepsis, endotoxin tolerance has been reported to
occur following inflammatory hypercytokinemia (17). Therefore, researchers previously
hypothesized that endotoxin tolerance may be a mechanism used to
protect the host against excessive inflammatory damage, as an
uncontrolled inflammatory response leads to extensive tissue damage
and septic shock (2). However,
more recently, a different hypothesis has been formulated, which
suggests that the endotoxin tolerant state is associated with
secondary infection and may render the host more susceptible to
septic progression and death (18). Therefore, strategies for the
prevention of endotoxin tolerance may represent an effective
treatment for sepsis (19).
Although endotoxin tolerance has been observed for
>50 years (20), the mechanisms
underlying macrophage reprogramming remain unclear. Overexpression
of certain regulators in the TLR4 pathway, including IL-1
receptor-associated kinase-M (IRAK-M), SH2-containing
inositol-5′-phosphatase and IRAK-M inducer hypoxia-inducible
factor-1α, was previously reported to be implicated in the
pathological process of endotoxin tolerance (14,21,22).
Among these regulators, interferon-induced double-stranded
RNA-dependent protein kinase (PKR) was investigated in the present
study. PKR is a widely expressed serine/threonine protein kinase
(23). It is activated by multiple
stimuli, including the inflammatory cytokines interferon and TNF-α
(24), bacterial infection and
viral double-stranded RNA (25–27).
In addition to its antiviral properties, phosphorylated (p)-PKR
also affects multiple transcription factors by activating numerous
signaling pathways. These transcription factors, including
interferon regulatory factor 3 (28) and nuclear factor-κB (NF-κB)
(29,30), are required for the expression of
genes encoding inflammatory cytokines (25). However, the role of PKR in
macrophage reprogramming remains to be elucidated. In the present
study, the role of PKR in endotoxin tolerance was determined. In
addition, the associated signaling pathways through which PKR may
mediate macrophage reprogramming were also investigated.
Materials and methods
Cells and reagents
LPS (cat. no. L2654) and LY294002 (cat. no. L9908)
were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Rotenone (cat. no. 557368) was purchased from Millipore (Merck
KGaA). RAW264.7 cells were purchased from the Type Culture
Collection of the Chinese Academy of Sciences (Shanghai, China) and
maintained in Dulbecco's modified Eagle's medium (DMEM; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal
bovine serum (Thermo Fisher Scientific, Inc.). Cells were
maintained in a 5% CO2 humidified incubator at 37°C.
Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Molecular
Technologies, Inc. (Kumamoto, Japan). Primary antibodies against
AKT (cat. no. 4691S; rabbit), p-AKT (Thr308; cat. no. 13038S;
rabbit) and β-actin (cat. no. 4970S; rabbit), and the anti-rabbit
IgG, HRP-linked Antibody (cat. no. 7074S), were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). The PKR antibody
(cat. no. sc-708; rabbit) was purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). The p-PKR antibody (T446;
cat. no. ab32036; rabbit) was purchased from Abcam (Cambridge, UK).
The primers for IL-1β, CCL17, CCL22, Arg1, iNOS, TNF-α, suppressor
of cytokine signaling 3 (Socs3), C-X-C motif chemokine ligand 11
(CXCL11) and β-actin were supplied by Sangon Biotech Co., Ltd.
(Shanghai, China). The Eastep Super Total RNA Extraction kit,
GoScript Reverse Transcription System and GoTaq qPCR Master Mix
were purchased from Promega Corporation (Madison, WI, USA).
Cell viability assays
Cell viability was measured using the CCK-8 assay
according to the manufacturer's protocol. Briefly, RAW264.7 cells
were seeded in 96-well culture plates at a density of 5,000
cells/well in DMEM and incubated in a humidified incubator at 37°C
overnight. Cells were exposed to different concentrations of LPS
(0, 1, 10, 100, 500 and 1,000 ng/ml) for 24 h. After a 24 h
incubation with LPS, 10 µl CCK-8 reagent was added to each well and
incubated for 1 h. Subsequently, the optical density (OD) was
measured at a wavelength of 450 nm. The percentage of viable cells
was determined using the following formula: Ratio (%)=[OD
(treated)-OD (blank)/OD (control)-OD (blank)] ×100. Cell viability
data are presented as the mean ± standard error of the mean of
three independent experiments, each containing three
replicates.
Endotoxin tolerant model in RAW264.7
cells
The endotoxin tolerance model was established as
follows. RAW264.7 cells were seeded in 6-well culture plates at a
density of 5×105 cells/well in DMEM and incubated in a
humidified incubator at 37°C overnight. Subsequently, cells were
initially stimulated with medium alone or medium containing LPS
(100 ng/ml) for 20 h, washed with PBS twice and restimulated with
medium or LPS (100 ng/ml) for 4 h prior to Reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) or 2
h prior to western blot analysis. Different durations of the second
LPS stimulation were because expression of inflammatory cytokines
depended on the activation of regulators and signaling (9,31).
Rotenone (10 µM) was added 1 h before the second LPS stimulation
and remained until the cells were lysed. LY294002 was used 2 h
before the second LPS stimulation at a concentration of 10 µM when
necessary and lasted until the end of the second LPS stimulation.
Macrophages that were continually cultured in DMEM were designated
medium/medium (M/M), cells that were stimulated with LPS following
the incubation with DMEM were designated medium/LPS (M/L) and cells
that were restimulated with LPS following stimulation with the same
dose of LPS were designated LPS/LPS (L/L). The cells were incubated
in a humidified incubator at 37°C during the whole experimental
process.
ELISA
TNF-α levels in the supernatants were analyzed using
the TNF-α ELISA kit (F11630; Westang BioTechnology Corporation
Ltd., Shanghai, China), according to the manufacturer's protocol.
In brief, medium in the 6-well plate was pipetted into the 96-wells
plate directly. During the first incubation, TNF-α bound the
capture antibody. Following washing, a detection antibody was added
to the wells, which bound to the TNF-α immobilized during the first
incubation. Subsequently, a horseradish peroxidase (HRP) conjugate
was added to bind to the detection antibody. Finally, a substrate
solution was added and converted by the enzyme to a detectable
form. The intensity of the colored product reflected the
concentration of TNF-α.
Preparation of whole-cell protein
lysates
Cells were washed twice with ice-cold PBS and
suspended in RIPA lysis buffer (P0013B; Beyotime Institute of
Biotechnology, Haimen, China) containing 1 mM phenylmethanesulfonyl
fluoride and 1 mM phosphatase inhibitors, and were centrifuged at
16,000 × g for 10 min to remove nuclei and cell debris.
Supernatants were rapidly frozen at −80°C or immediately used in
western blot assays.
Western blot analysis
Protein concentrations were determined using the
Pierce BCA Protein Assay kit (Thermo Fisher Scientific, Inc.) and
15 µg cellular proteins were electroblotted onto polyvinylidene
difluoride membranes following separation with 10% SDS-PAGE. The
membranes were blocked for 15 min with QuickBlock Blocking Buffer
for Western Blot (Beyotime Institute of Biotechnology, Haimen,
China) at room temperature, followed by an overnight incubation at
4°C with primary antibodies against PKR, p-PKR, AKT, p-AKT and
β-actin at a 1:1,000 dilution. Blots were washed three times with
TBS/0.2% Tween-20 (TBST) prior to incubation with the
HRP-conjugated secondary antibody (1:5,000) for 1 h at room
temperature. Blots were washed three times with TBST prior to
development by enhanced chemiluminescence using the Immobilon
Western Chemiluminescent HRP Substrate (Merck KGaA). Band
intensities were quantified using Quantity One software version
4.6.2 (Bio-Rad Laboratories, Inc., Hercules, CA, USA). β-actin was
used as a loading control for whole-cell protein lysates.
RT-qPCR assays
Total RNA was extracted using the Eastep Super Total
RNA Extraction kit, according to the manufacturer's protocol. A
total of 1 µg RNA was reverse transcribed into cDNAs using the
GoScript Reverse Transcription System, including elongation at 42°C
for 15 min and inactivation of reverse transcriptase at 70°C for 15
min. qPCR was performed using GoTaq qPCR Master Mix. In brief,
denaturation was performed at 95°C for 10 min, annealing at 60°C
for 1 min, and elongation at 95°C for 15 sec for 40 cycles. PCR was
carried out in triplicate and using the Bio-Rad CFX96 instrument
(Bio-Rad Laboratories, Inc.). Data were processed using Bio-Rad CFX
manager version 3.1 (Bio-Rad Laboratories, Inc.). The housekeeping
gene β-actin was used as the internal control. The relative
expression levels were calculated using the 2−∆∆Cq
method (32). The primer pairs
used for qPCR are presented in Table
I.
 | Table I.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction.
|
| Primer sequence
(5′→3′) |
|---|
|
|
|
|---|
| Gene | Forward | Reverse |
|---|
| TNF-α |
GACGTGGAACTGGCAGAAGAG |
TTGGTGGTTTGTGAGTGTGAG |
| IL-1β |
GCAACTGTTCCTGAACTCAACT |
ATCTTTTGGGGTCCGTCAACT |
| CXCL11 |
GGCTTCCTTATGTTCAAACAGGG |
GCCGTTACTCGGGTAAATTACA |
| CCL17 |
GACGACAGAAGGGTACGGC |
GCATCTGAAGTGACCTCATGGTA |
| CCL22 |
ATTCTGTGACCATCCCCTCAT |
TGTATGTGCCTCTGAACCCAC |
| Socs3 |
TGCAGGAGAGCGGATTCTAC |
AGCTGTCGCGGATAAGAAAG |
| Arg1 |
CTCCAAGCCAAAGTCCTTAGAG |
AGGAGCTGTCATTAGGGACATC |
| iNOS |
GACGAGACGGATAGGCAGAG |
CTTCAAGCACCTCCAGGAAC |
| β-actin |
GTGCTATGTTGCTCTAGACTTCG |
ATGCCACAGGATTCCATACC |
Statistical analysis
Prism 6 software (GraphPad, La Jolla, CA, USA) was
used for statistical analysis. All data are presented as the mean ±
standard error of the mean (n=3 independent experiments). Data were
analyzed using an unpaired two-tailed Student's t-test or one-way
analysis of variance followed by a Tukey's multiple comparison
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
LPS promotes cell proliferation in a
dose-depended manner
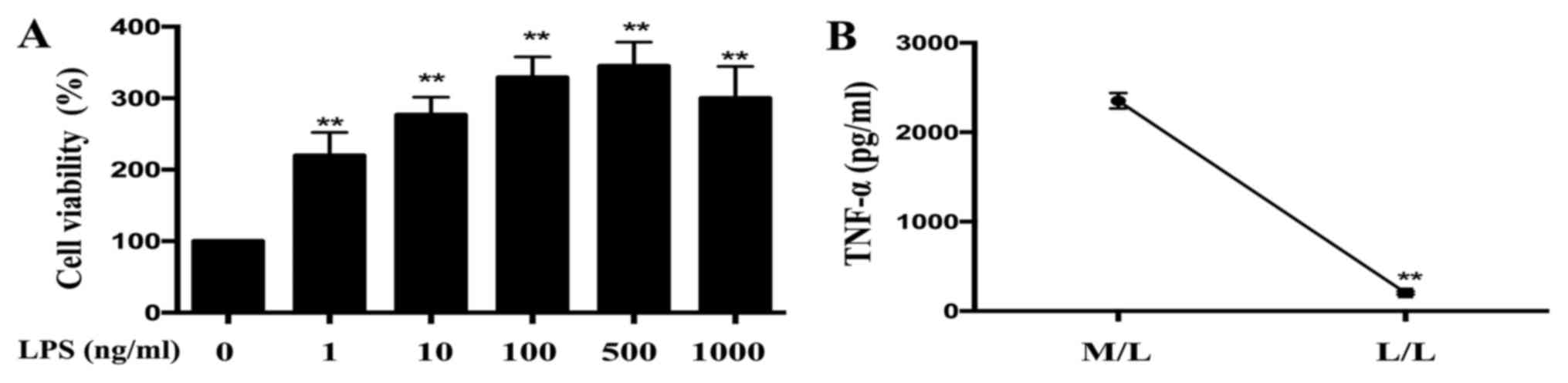
The viability of RAW264.7 cells was determined using
the CCK-8 assay. As demonstrated in Fig. 1A, treatments with different
concentrations of LPS (1, 10, 100, 500 and 1,000 ng/ml)
significantly promoted cell proliferation compared with the control
group. At LPS concentrations <500 ng/ml, cells proliferated in a
concentration-dependent manner (Fig.
1A). No obvious cytotoxicity was observed when cells were
treated with LPS at concentrations of 1–1,000 ng/ml (Fig. 1A).
TNF-α levels are decreased in L/L
macrophages compared with M/L macrophages
Cells were cultured and stimulated with LPS using
the methods described above. Supernatants were collected and
examined using ELISA. TNF-α levels were demonstrated to be
significantly reduced in LPS-tolerant L/L macrophages compared with
LPS-activated M/L macrophages (Fig.
1B).
Cytokine expression differs between
L/L macrophages and M/L macrophages
Cells were stimulated with or without LPS for 20 h,
washed twice with PBS and restimulated with LPS for 4 h. Cells were
subsequently lysed and RNA was isolated. The gene expression levels
in RAW264.7 cells were detected by RT-qPCR. Levels of TNF-α, IL-1β,
CXCL11, CCL17, CCL22 and Socs3 mRNA were markedly decreased in
LPS-tolerant L/L macrophages compared with LPS-activated M/L
macrophages (Fig. 2A). However,
elevated levels of Arg1 and iNOS mRNA were detected in the
LPS-tolerant L/L macrophages compared with LPS-activated M/L
macrophages.
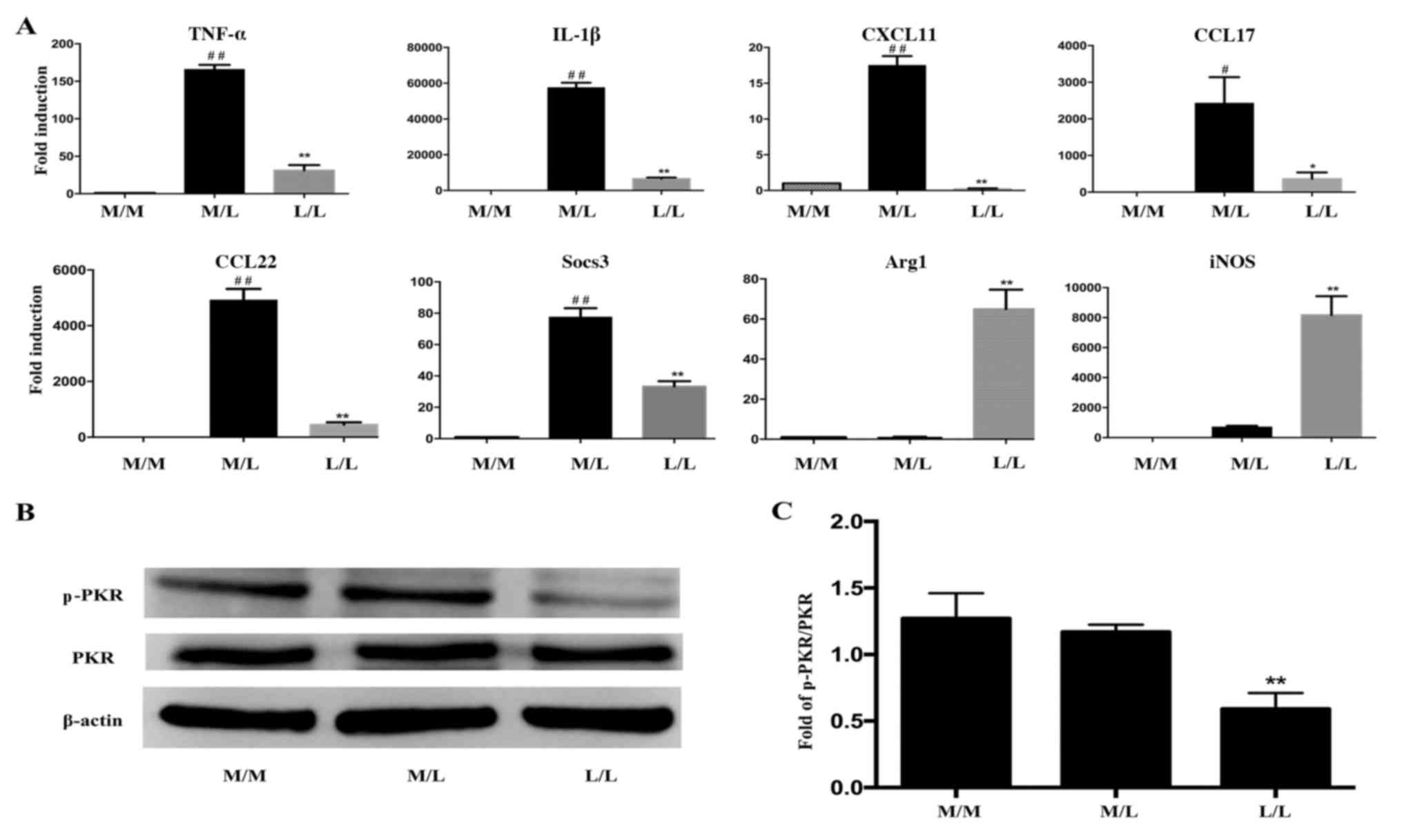 | Figure 2.PKR inactivation is involved in the
alterations in cytokine gene expression observed in LPS-tolerant
macrophages. (A) Reverse transcription-quantitative polymerase
chain reaction was performed to determine differences in the
expression of inflammatory cytokine genes in LPS-tolerant L/L
macrophages and LPS-activated M/L macrophages. The expression of
the TNF-α, IL-1β, CXCL11, CCL17, CCL22 and Socs3 mRNAs was markedly
downregulated, while the expression of the Arg1 and iNOS mRNAs was
upregulated, in LPS-tolerant L/L macrophages compared with
LPS-activated M/L macrophages. (B) Representative western blot
bands for the protein expression of p-PKR and PKR. β-actin was used
as a loading control. (C) Quantification of the ratio of the
intensities of the p-PKR/PKR bands by densitometry.
#P<0.05 and ##P<0.01 vs. M/M cells;
*P<0.05 and **P<0.01 vs. M/L cells. Data represent the
results from three independent experiments. PKR, interferon-induced
double-stranded RNA-dependent protein kinase; LPS,
lipopolysaccharide; TNF-α, tumor necrosis factor-α; IL-1β,
interleukin-1β; CXCL11, C-X-C motif chemokine ligand 11; CCL, C-C
motif chemokine ligand; Socs3, suppressor of cytokine signaling 3;
Arg1, arginase 1; iNOS, nitric oxide synthase 2; p-PKR,
phosphorylated-PKR; M/M, initial incubation with medium followed by
further incubation with medium; M/L, initial incubation with medium
followed by LPS stimulation; L/L, initial incubation with LPS
followed by restimulation with LPS. |
PKR inactivation is involved in the
altered cytokine gene expression observed in LPS-tolerant
macrophages
Macrophages were cultured and stimulated with LPS as
described above. Cells were lysed and protein levels were measured
by western blotting at 2 h following the LPS rechallenge. RAW264.7
macrophages that were restimulated with LPS for 2 h after the
initial 20 h challenge with LPS exhibited significant inactivation
of PKR compared with cells challenged with LPS for only 2 h
(Fig. 2B and C). However, the
level of p-PKR was not statistically significantly different
between M/M and M/L macrophages (Fig.
2C). In addition, total PKR levels were not altered among the
groups (Fig. 2B).
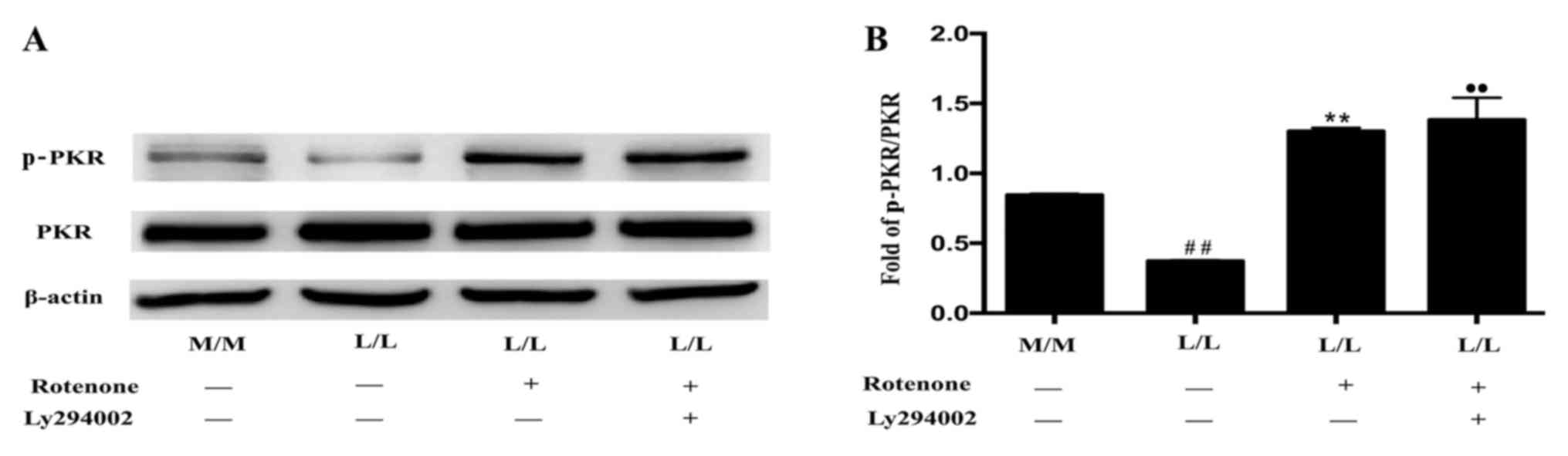
Rotenone alleviates endotoxin
tolerance by activating PKR in RAW264.7 cells
It has been previously demonstrated that rotenone
activates PKR (33). The level of
p-PKR was markedly increased following treatment with rotenone (10
or 20 µM) in LPS-tolerant L/L macrophages compared with untreated
LPS-tolerant L/L macrophages (Fig. 3A
and B). In addition, the level of p-PKR was not statistically
significantly different between LPS-tolerant L/L macrophages
treated with 10 and 20 µM rotenone. Furthermore, the mRNA levels of
IL-1β, CCL17 and CCL22 were increased, while the mRNA levels of the
Arg1 and iNOS were decreased, in rotenone-treated LPS-tolerant L/L
macrophages compared with untreated LPS-tolerant L/L macrophages
(Fig. 3C). The levels of TNF-α,
CXCL11 and Socs3 mRNA were not statistically significantly
different between rotenone-treated and untreated LPS-tolerant L/L
macrophage groups.
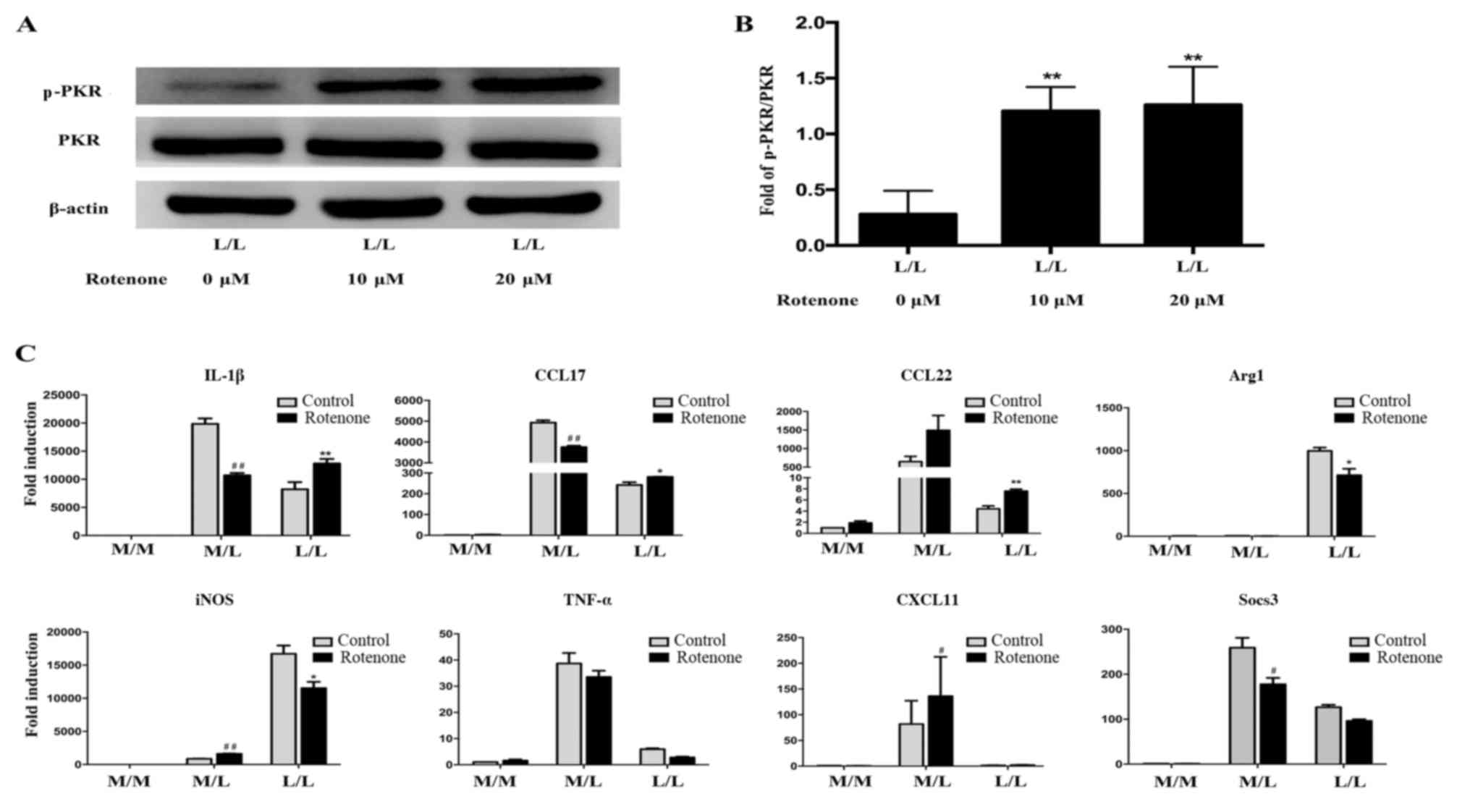 | Figure 3.Rotenone ameliorates endotoxin
tolerance by activating PKR. (A) Representative western blot bands
for the protein expression of p-PKR and PKR. β-actin was used as
the loading control. PKR activation was induced by 10 and 20 µM
rotenone in LPS-tolerant L/L RAW264.7 cells. (B) Quantification of
the ratio of the intensities of the p-PKR/PKR bands by
densitometry. The OD of the target protein is presented as a
proportion of the β-actin OD. (C) Rotenone at a concentration of 10
µM alleviated endotoxin tolerance by activating PKR. Reverse
transcription-quantitative polymerase chain reaction results
demonstrated increased levels of the IL-1β, CCL17 and CCL22 mRNAs,
and decreased levels of the Arg1 and iNOS mRNAs, in
rotenone-treated LPS-tolerant L/L macrophages compared with
untreated LPS-tolerant L/L macrophages. The expression of TNF-α,
CXCL11 and Socs3 mRNAs was not significantly different between the
rotenone-treated and untreated LPS-tolerant L/L macrophage groups.
#P<0.05 and ##P<0.01 vs.
untreated/control M/L macrophages; *P<0.05 and **P<0.01 vs.
untreated/control L/L macrophages. Data represent the results from
three independent experiments. PKR, interferon-induced
double-stranded RNA-dependent protein kinase; p-PKR,
phosphorylated-PKR; LPS, lipopolysaccharide; OD, optical density;
IL-1β, interleukin-1β; CCL, C-C motif chemokine ligand; Arg1,
arginase 1; iNOS, nitric oxide synthase 2; TNF-α, tumor necrosis
factor-α; CXCL11, C-X-C motif chemokine ligand 11; Socs3,
suppressor of cytokine signaling 3; M/M, initial incubation with
medium followed by further incubation with medium; M/L, initial
incubation with medium followed by LPS stimulation; L/L, initial
incubation with LPS followed by restimulation with LPS. |
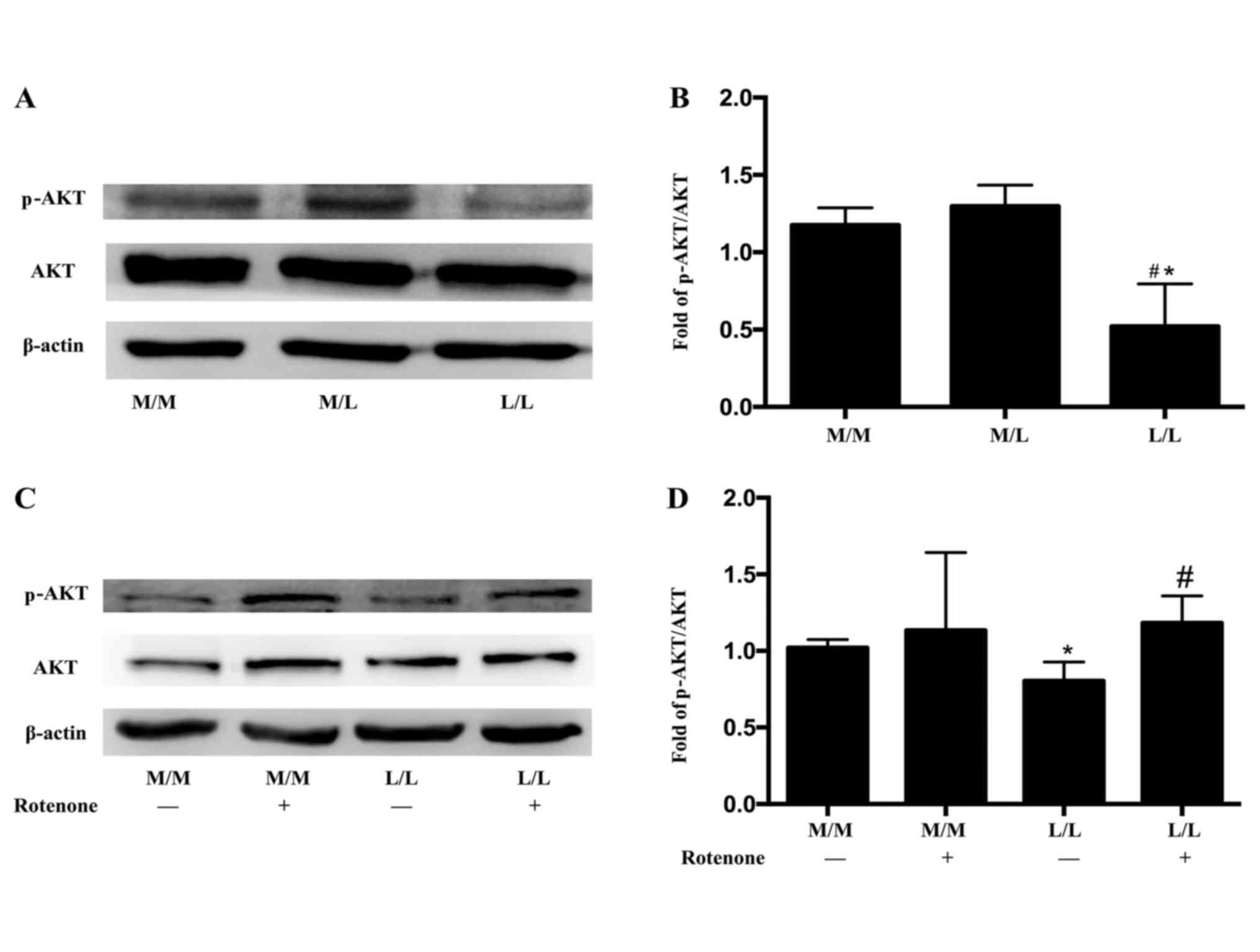
PKR mediates macrophage reprogramming
in LPS-tolerant RAW264.7 cells by inactivating AKT
RAW264.7 cells were cultured in DMEM and stimulated
with LPS as described above. Following a 2-h restimulation with
LPS, macrophages were lysed and levels of proteins were measured by
western blotting. AKT was activated in LPS-activated M/L
macrophages compared with M/M macrophages that received no
stimulation with LPS (Fig. 4A and
B). However, the levels of p-AKT were markedly decreased in
LPS-tolerant L/L macrophages compared with LPS-activated M/L
macrophages (Fig. 4A and B). The
total AKT levels were not altered among the groups (Fig. 4A). Rotenone induces PKR
phosphorylation. In the present study, AKT was activated in
rotenone-treated LPS-tolerant L/L macrophages compared with the
untreated L/L macrophages (Fig. 4C and
D). Ly294002, a phosphatidylinositol-4,5-bisphosphate 3-kinase
(PI3K)-AKT inhibitor, was added to LPS-tolerant L/L cells prior to
the 1 h rotenone treatment. Ly294002 (10 µM) did not affect the
activation of PKR in rotenone-treated LPS-tolerant L/L macrophages
(Fig. 5). However, AKT activation
in rotenone-treated LPS-tolerant L/L macrophages was inhibited by
Ly294002 (Fig. 6A and B).
Furthermore, Ly294002 partially reversed the rotenone-induced
variations in gene expression in LPS-tolerant L/L macrophages
(Fig. 6C). Specifically, Ly294002
downregulated IL-1β and CCL22 expression and upregulated Arg1 and
iNOS expression in the rotenone-treated LPS-tolerant L/L
macrophages (Fig. 6C).
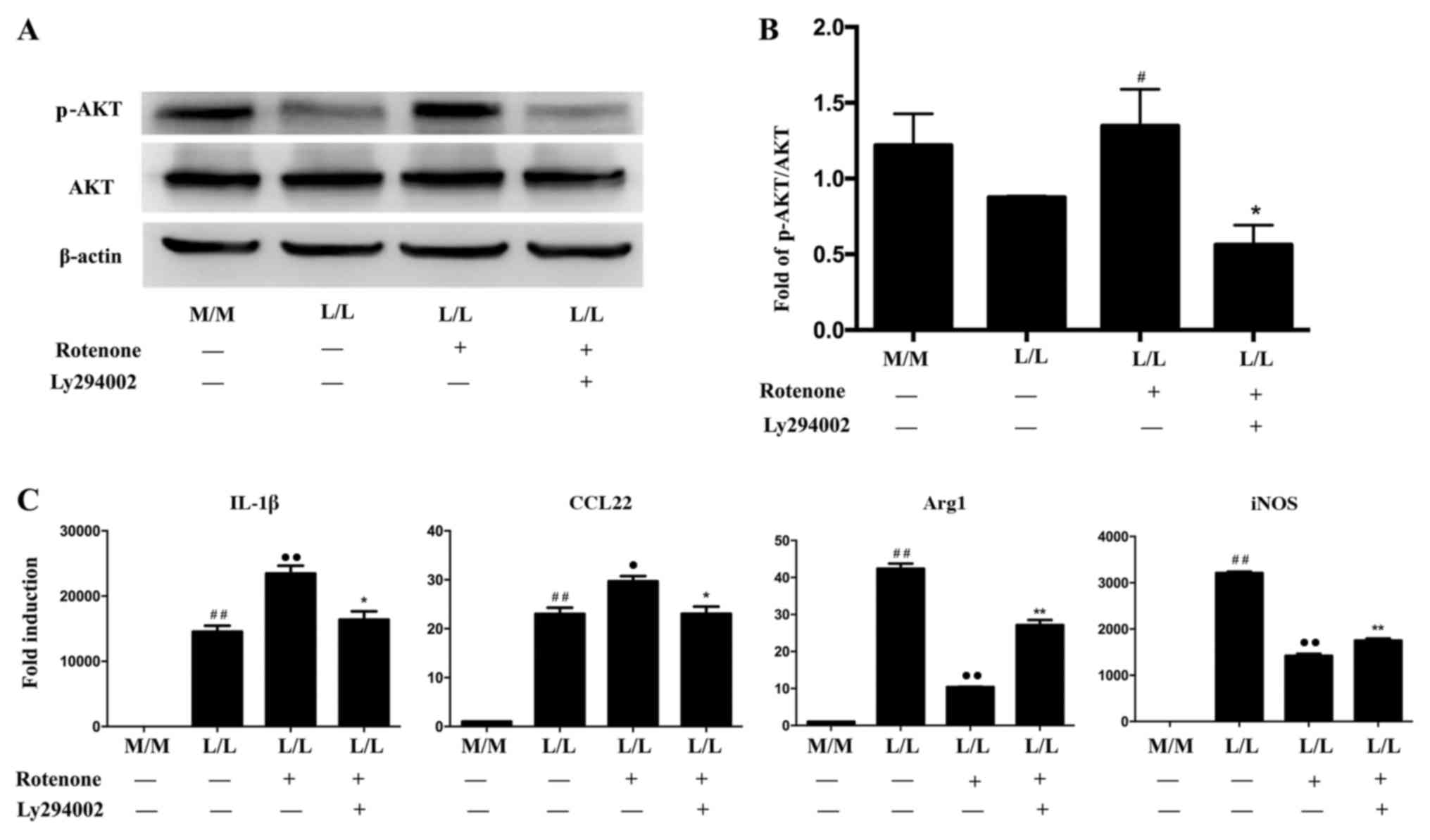 | Figure 6.Ly294002 partially prevents the
alterations in gene expression induced by rotenone in LPS-tolerant
L/L macrophages. (A) Representative western blot bands for p-AKT
and AKT protein expression following rotenone treatment with or
without Ly294002 in LPS-tolerant L/L macrophages. β-actin was used
as the loading control. (B) Quantification of the ratio of the
intensities of the p-AKT/AKT bands by densitometry.
#P<0.05 vs. L/L (−/−) macrophages, *P<0.05 vs. L/L
(+/-) macrophages. (C) Reverse transcription-quantitative
polymerase chain reaction results demonstrated that Ly294002
induced downregulation of IL-1β and CCL22 expression, and
upregulation of Arg1 and iNOS expression, in rotenone-treated
LPS-tolerant L/L macrophages. ##P<0.01 vs. M/M (−/−)
macrophages. •P<0.05 and ••P<0.01 vs.
L/L (−/−) macrophages; *P<0.05 and **P<0.01 vs. L/L (+/-)
macrophages. Data represent the results from three independent
experiments. LPS, lipopolysaccharide; p-AKT, phosphorylated-AKT;
IL-1β, interleukin-1β; CCL22, C-C motif chemokine ligand 22; Arg1,
arginase 1; iNOS, nitric oxide synthase 2; M/M, initial incubation
with medium followed by further incubation with medium; L/L,
initial incubation with LPS followed by restimulation with LPS. |
Discussion
Following long-term exposure to LPS, macrophages
enter an immunosuppressive state and are unable to respond to
further LPS challenges. The immunosuppressive or hyporesponsive
state that develops is termed endotoxin tolerance (5). Endotoxin tolerance has been
associated with various diseases, including sepsis, trauma,
pancreatitis and acute coronary syndrome (15,34,35).
The current hypothesis regarding the host immune response in
patients with sepsis indicates that it is characterized by an
initial hyperinflammatory phase that is sustained over several days
and progresses into a protracted immunosuppressive phase,
indicating that macrophages enter a tolerant state (18,36).
In patients with sepsis, mortality occurs primarily due to the
development of uncontrolled secondary infections as a result of
immunosuppression (37–39). Therefore, strategies that prevent
endotoxin tolerance have become a topic of interest in therapies
for sepsis (39).
In the present study, RAW264.7 macrophage cells were
stimulated with 100 ng/ml LPS for 20 h, washed twice with PBS and
restimulated with 100 ng/ml LPS for 2 or 4 h to establish an
LPS-tolerant model, as described previously (9,11).
TNF-α levels have been reported to be significantly decreased in
tolerant macrophages and are considered a reliable marker of
endotoxin tolerance (6,40,41).
In the present study, TNF-α secretion from LPS restimulated
tolerant macrophages was markedly decreased compared with
LPS-activated macrophages, indicating that the endotoxin tolerance
model was successfully established.
In LPS tolerant macrophages, the expression of
cytokine genes is reprogrammed rather than inhibited (5,12,13).
During macrophage reprogramming, the expression of certain genes is
downregulated, while other genes are upregulated (42). This phenomenon is similar to
macrophage polarization, in which macrophages undergo polarized
differentiation into classically activated macrophages (M1) or
alternatively activated macrophages (M2) in response to different
stimuli (43). M1 macrophages are
characterized by increased production of proinflammatory cytokines,
nitric oxide and reactive oxygen species that mediate antimicrobial
activities and induce cellular immunity (44,45).
M2 macrophages are characterized by intracellular expression of
Arg1 and secretion of chitinases, including Chil3, and
anti-inflammatory cytokines, including interleukin-10 (46). Therefore, M2 macrophages have been
associated with helminthic infection and tissue repair (47). Macrophage tolerance and M2
polarization are associated processes. It was previously reported
that the expression of M2-associated cytokines (CCL17, CCL22 and
Arg1) was upregulated, while the expression of M1-associated
cytokines (TNF-α, IL-1β, CXCL-11, Socs3 and iNOS) was
downregulated, in LPS-tolerant macrophages (31). In the present study, the mRNA
levels of the M1-associated cytokines TNF-α, IL-1β, CXCL-11 and
Socs3 were decreased and the levels of the M2-associated mediator
Arg1 was increased, similar to M2 polarization. However, the levels
of the M2-associated mediators CCL-17 and CCL-22 were decreased and
the level of the M1-associated mediator iNOS was increased in
LPS-tolerant macrophages, which differed from M2 polarization.
Variation in the expression of iNOS has been reported in
LPS-tolerant cells as certain studies have demonstrated that it was
elevated (48,49), while others detected decreased iNOS
levels, in LPS-tolerant cells (10,13).
These variations may depend on the cell type, duration of LPS
stimulation and the concentration of the LPS used in the different
studies.
In addition to its antiviral properties, PKR also
participates in the regulation of inflammatory cytokine and
chemokine expression, including IL-1β, IL-18 and high-mobility
group box 1, by affecting transcription factors (25–27,33).
Total PKR levels in tolerant macrophages were reported to be
decreased through differential K63/K48 ubiquitination (50). However, the role of PKR in
macrophage reprogramming remains to be elucidated. In the present
study, p-PKR levels were markedly decreased in LPS-tolerant
macrophages, whereas total PKR levels remained unaltered. Rotenone
is a plant extract that activates PKR (33). Administration of rotenone in the
present study regulated the mRNA expression of IL-1β, CCL17, CCL22,
Arg1 and iNOS in LPS-tolerant macrophages. Based the above data, it
may be hypothesized that PKR activation partially reverses
macrophage reprogramming in endotoxin tolerance. However, the
expression of the TNF-α, CXCL11 and Socs3 mRNAs was not
significantly different between rotenone-treated and untreated
LPS-tolerant cells. The expression of these cytokines may not be
regulated by PKR. However, the expression of these cytokines has
been previously demonstrated to be regulated by other proteins,
including p21 and p50 (11).
It has been demonstrated that several signaling
pathways, including NF-κb (51,52)
and mitogen-activated protein kinase (29,53)
pathways, are regulated by PKR to promote cytokine and chemokine
production. PKR has also been reported to participate in
physiological activities, including coordinating skeletal muscle
differentiation and choroidal neovascularization, via the PI3K/AKT
signaling pathway (54,55). However, to the best of our
knowledge, it has not been previously determined whether PKR
mediates macrophage reprogramming via the PI3K/AKT signaling
pathway. In the present study, AKT was inactivated in LPS-tolerant
macrophages. Rotenone-induced PKR activation was demonstrated to
increase the level of p-AKT in LPS-tolerant cells, reversing
endotoxin tolerance-induced inactivation of AKT. Furthermore,
inhibition of PI3K-AKT signaling with Ly294002, a PI3K/AKT
inhibitor, partially reversed the rotenone-induced alleviation of
endotoxin tolerance, which was supported by the alterations in the
expression of several endotoxin tolerance-associated genes,
including IL-1β, CCL22, Arg1 and iNOS.
In conclusion, the results of the current study
demonstrated that PKR inhibition induced endotoxin tolerance in
macrophages and these effects were partially mediated by the
PI3K/AKT signaling pathway. Therefore, PKR may be a potential
target for the treatment of endotoxin tolerance.
Acknowledgements
Not applicable.
Funding
The present study was funded by grants from the
Natural Science Foundation of Guangdong Province (grant no.
2016A030313269) and Fundamental Research Funds for the Central
Universities (grant no. 15ykpy14).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW and XG conceived and designed the experiments.
HX, JC, MC, FP and CQ performed the experiments. HX and XS analyzed
the data and produced the pictures. HX and CQ produced the
manuscript. HX submitted the manuscript and revised it. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gordon S and Taylor PR: Monocyte and
macrophage heterogeneity. Nat Rev Immunol. 5:953–964. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lawrence T and Natoli G: Transcriptional
regulation of macrophage polarization: Enabling diversity with
identity. Nat Rev Immunol. 11:750–761. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Beutler B: SHIP, TGF-beta, and endotoxin
tolerance. Immunity. 21:134–135. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Neill LA and Bowie AG: The family of
five: TIR-domain-containing adaptors in Toll-like receptor
signalling. Nat Rev Immunol. 7:353–364. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cavaillon JM and Adib-Conquy M:
Bench-to-bedside review: Endotoxin tolerance as a model of
leukocyte reprogramming in sepsis. Crit Care. 10:2332006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fan H and Cook JA: Molecular mechanisms of
endotoxin tolerance. J Endotoxin Res. 10:71–84. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
del Fresno C, García-Rio F, Gómez-Piña V,
Soares-Schanoski A, Fernández-Ruíz I, Jurado T, Kajiji T, Shu C,
Marín E, del Arroyo Gutierrez A, et al: Potent phagocytic activity
with impaired antigen presentation identifying
lipopolysaccharide-tolerant human monocytes: Demonstration in
isolated monocytes from cystic fibrosis patients. J Immunol.
182:6494–6507. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zuckerman SH and Evans GF: Endotoxin
tolerance: In vivo regulation of tumor necrosis factor and
interleukin-1 synthesis is at the transcriptional level. Cell
Immunol. 140:513–519. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rajaiah R, Perkins DJ, Polumuri SK, Zhao
A, Keegan AD and Vogel SN: Dissociation of endotoxin tolerance and
differentiation of alternatively activated macrophages. J Immunol.
190:4763–4772. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Piao W, Song C, Chen H, Diaz MA, Wahl LM,
Fitzgerald KA, Li L and Medvedev AE: Endotoxin tolerance
dysregulates MyD88- and Toll/IL-1R domain-containing adapter
inducing IFN-beta-dependent pathways and increases expression of
negative regulators of TLR signaling. J Leukoc Biol. 86:863–875.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rackov G, Hernández-Jiménez E, Shokri R,
Carmona-Rodríguez L, Mañes S, Álvarez-Mon M, López-Collazo E,
Martínez-A C and Balomenos D: p21 mediates macrophage reprogramming
through regulation of p50-p50 NF-κB and IFN-β. J Clin Invest.
126:3089–3103. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Foster SL and Medzhitov R: Gene-specific
control of the TLR-induced inflammatory response. Clin Immunol.
130:7–15. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Foster SL, Hargreaves DC and Medzhitov R:
Gene-specific control of inflammation by TLR-induced chromatin
modifications. Nature. 447:972–978. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shalova IN, Lim JY, Chittezhath M,
Zinkernagel AS, Beasley F, Hernández-Jiménez E, Toledano V,
Cubillos-Zapata C, Rapisarda A, Chen J, et al: Human monocytes
undergo functional re-programming during sepsis mediated by
hypoxia-inducible factor-1α. Immunity. 42:484–498. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pena OM, Hancock DG, Lyle NH, Linder A,
Russell JA, Xia J, Fjell CD, Boyd JH and Hancock RE: An endotoxin
tolerance signature predicts sepsis and organ dysfunction at
initial clinical presentation. EBioMedicine. 1:64–71. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hotchkiss RS, Levy JH and Levi M:
Sepsis-induced disseminated intravascular coagulation, symmetrical
peripheral gangrene, and amputations. Crit Care Med. 41:e290–e291.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moller K: Of cells and men: Ex vivo and in
vivo tolerance to lipopolysaccharide. Crit Care Med. 39:1997–1998.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Angus DC and van der Poll T: Severe sepsis
and septic shock. N Engl J Med. 369:20632013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Delano MJ and Ward PA: Sepsis-induced
immune dysfunction: Can immune therapies reduce mortality? J Clin
Invest. 126:23–31. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lopez-Collazo E and del Fresno C:
Pathophysiology of endotoxin tolerance: Mechanisms and clinical
consequences. Crit Care. 17:2422013. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sly LM, Rauh MJ, Kalesnikoff J, Song CH
and Krystal G: LPS-induced upregulation of SHIP is essential for
endotoxin tolerance. Immunity. 21:227–239. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiong Y and Medvedev AE: Induction of
endotoxin tolerance in vivo inhibits activation of IRAK4 and
increases negative regulators IRAK-M, SHIP-1, and A20. J Leukoc
Biol. 90:1141–1148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Meurs E, Chong K, Galabru J, Thomas NS,
Kerr IM, Williams BR and Hovanessian AG: Molecular cloning and
characterization of the human double-stranded RNA-activated protein
kinase induced by interferon. Cell. 62:379–390. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shen SJ, Zhang YH, Gu XX, Jiang SJ and Xu
LJ: Yangfei Kongliu Formula, a compound Chinese herbal medicine,
combined with cisplatin, inhibits growth of lung cancer cells
through transforming growth factor-β1 signaling pathway. J Integr
Med. 15:242–251. 2017.PubMed/NCBI
|
|
25
|
Balachandran S and Barber GN: PKR in
innate immunity, cancer, and viral oncolysis. Methods Mol Biol.
383:277–301. 2007.PubMed/NCBI
|
|
26
|
Williams BR: Signal integration via PKR.
Sci STKE. 2001:re22001.PubMed/NCBI
|
|
27
|
García MA, Gil J, Ventoso I, Guerra S,
Domingo E, Rivas C and Esteban M: Impact of protein kinase PKR in
cell biology: From antiviral to antiproliferative action. Microbiol
Mol Biol Rev. 70:1032–1060. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang P and Samuel CE: Induction of
protein kinase PKR-dependent activation of interferon regulatory
factor 3 by vaccinia virus occurs through adapter IPS-1 signaling.
J Biol Chem. 283:34580–34587. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zamanian-Daryoush M, Mogensen TH, DiDonato
JA and Williams BR: NF-kappaB activation by
double-stranded-RNA-activated protein kinase (PKR) is mediated
through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell
Biol. 20:1278–1290. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han BH, Lee YJ, Yoon JJ, Choi ES, Namgung
S, Jin XJ, Jeong DH, Kang DG and Lee HS: Hwangryunhaedoktang exerts
anti-inflammation on LPS-induced NO production by suppressing MAPK
and NF-κB activation in RAW264.7 macrophages. J Integr Med.
15:326–336. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Porta C, Rimoldi M, Raes G, Brys L, Ghezzi
P, Di Liberto D, Dieli F, Ghisletti S, Natoli G, De Baetselier P,
et al: Tolerance and M2 (alternative) macrophage polarization are
related processes orchestrated by p50 nuclear factor kappaB. Proc
Natl Acad Sci USA. 106:14978–14983. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lu B, Nakamura T, Inouye K, Li J, Tang Y,
Lundbäck P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, et al:
Novel role of PKR in inflammasome activation and HMGB1 release.
Nature. 488:670–674. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cavaillon JM, Adrie C, Fitting C and
Adib-Conquy M: Endotoxin tolerance: Is there a clinical relevance?
J Endotoxin Res. 9:101–107. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
del Fresno C, Gómez-Piña V, Lores V,
Soares-Schanoski A, Fernández-Ruiz I, Rojo B, Alvarez-Sala R,
Caballero-Garrido E, García F, Veliz T, et al: Monocytes from
cystic fibrosis patients are locked in an LPS tolerance state:
Down-regulation of TREM-1 as putative underlying mechanism. PLoS
One. 3:e26672008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Escoll P, del Fresno C, García L, Vallés
G, Lendínez MJ, Arnalich F and López-Collazo E: Rapid up-regulation
of IRAK-M expression following a second endotoxin challenge in
human monocytes and in monocytes isolated from septic patients.
Biochem Biophys Res Commun. 311:465–472. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hotchkiss RS, Monneret G and Payen D:
Immunosuppression in sepsis: A novel understanding of the disorder
and a new therapeutic approach. Lancet Infect Dis. 13:260–268.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hotchkiss RS and Karl IE: The
pathophysiology and treatment of sepsis. N Engl J Med. 348:138–150.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hotchkiss RS and Opal S: Immunotherapy for
sepsis-a new approach against an ancient foe. N Engl J Med.
363:87–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nahid MA, Satoh M and Chan EK: MicroRNA in
TLR signaling and endotoxin tolerance. Cell Mol Immunol. 8:388–403.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
West MA and Koons A: Endotoxin tolerance
in sepsis: Concentration-dependent augmentation or inhibition of
LPS-stimulated macrophage TNF secretion by LPS pretreatment. J
Trauma. 65:893–898. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Biswas SK and Lopez-Collazo E: Endotoxin
tolerance: New mechanisms, molecules and clinical significance.
Trends Immunol. 30:475–487. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mantovani A, Sozzani S, Locati M, Allavena
P and Sica A: Macrophage polarization: Tumor-associated macrophages
as a paradigm for polarized M2 mononuclear phagocytes. Trends
Immunol. 23:549–555. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mosser DM: The many faces of macrophage
activation. J Leukoc Biol. 73:209–212. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shirey KA, Pletneva LM, Puche AC, Keegan
AD, Prince GA, Blanco JC and Vogel SN: Control of RSV-induced lung
injury by alternatively activated macrophages is IL-4R alpha-,
TLR4-, and IFN-beta-dependent. Mucosal Immunol. 3:291–300. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gordon S: Alternative activation of
macrophages. Nat Rev Immunol. 3:23–35. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Castegren M, Skorup P, Lipcsey M, Larsson
A and Sjolin J: Endotoxin tolerance variation over 24 h during
porcine endotoxemia: Association with changes in circulation and
organ dysfunction. PLoS One. 8:e532212013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dias MB, Almeida MC, Carnio EC and Branco
LG: Role of nitric oxide in tolerance to lipopolysaccharide in
mice. J Appl Physiol (1985). 98:1322–1327. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Perkins DJ, Qureshi N and Vogel SN: A
Toll-like receptor-responsive kinase, protein kinase R, is
inactivated in endotoxin tolerance through differential K63/K48
ubiquitination. MBio. 1:e00239–e002310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Goh KC, deVeer MJ and Williams BR: The
protein kinase PKR is required for p38 MAPK activation and the
innate immune response to bacterial endotoxin. EMBO J.
19:4292–4297. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nakamura T, Furuhashi M, Li P, Cao H,
Tuncman G, Sonenberg N, Gorgun CZ and Hotamisligil GS:
Double-stranded RNA-dependent protein kinase links pathogen sensing
with stress and metabolic homeostasis. Cell. 140:338–348. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bonnet MC, Weil R, Dam E, Hovanessian AG
and Meurs EF: PKR stimulates NF-kappaB irrespective of its kinase
function by interacting with the IkappaB kinase complex. Mol Cell
Biol. 20:4532–4542. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Alisi A, Spaziani A, Anticoli S,
Ghidinelli M and Balsano C: PKR is a novel functional direct player
that coordinates skeletal muscle differentiation via p38MAPK/AKT
pathways. Cell Signal. 20:534–542. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhu M, Liu X, Wang S, Miao J, Wu L, Yang
X, Wang Y, Kang L, Li W, Cui C, et al: PKR promotes choroidal
neovascularization via upregulating the PI3K/Akt signaling pathway
in VEGF expression. Mol Vis. 22:1361–1374. 2016.PubMed/NCBI
|