Introduction
Chronic inflammation is the leading cause of
epithelial malignancies, such as prostate cancer (1). The progression of high-grade prostate
cancer is associated with chronic intraprostatic inflammation
(2). Previous studies reported a
key role for inflammatory responses in the pathogenesis of prostate
cancer (3,4). The molecular effects of inflammation
on carcinogenesis include the regulation of the tumor
microenvironment induced by altered levels of inflammatory
cytokines, reactive oxygen species and transcriptional factors
(5). Therefore, improved
understanding of chronic inflammation and its underlying mechanisms
may aid the development of therapeutic strategies in reducing
prostate cancer-associated mortality.
The adaptive immune system regulates antitumor
effects via immunosurveillance (6); however, certain tumors, such as
gastrointestinal cancer, are able to manipulate inflammatory
signals to their own advantage (7). In this context, nuclear factor
κ-light-chain-enhancer of activated B cells (NF-κB), a master
transcription factor, has been reported as the main mediator of
proinflammatory processes in the pathogenesis of prostate cancer
(8). During the progression of
prostate cancer, NF-κB promotes tumor invasion, metastasis, cell
survival and chemoresistance (9).
Constitutive activation of NF-κB has been demonstrated in primary
prostate cancer, and is associated with castration-resistant
phenotypes and the loss of androgen receptor function (10). Therefore, suppression of the NF-κB
pathway may regulate chronic inflammatory responses and reduce
their oncogenic effects (11).
Estrogen receptor β (ERβ) was reported to be
expressed in prostate carcinoma cells; ERβ-regulated estrogen
signaling served to inhibit tumor progression in patients with
prostate cancer (12).
Furthermore, it was demonstrated that ERβ-selective agonists are
able to deactivate microglia and suppress T cell activity via
downregulation of NF-κB (13). In
addition, it was reported that ERβ precludes NF-κB activation; the
loss of ERβ may be associated with chronic inflammation in prostate
cancer (14). Collectively, these
findings indicate that ERβ may reversibly regulate prostate
inflammation via suppression of the NF-κB signaling pathway. Thus,
in the present study, the mechanisms underlying the therapeutic
roles of ERβ in prostate inflammation and regulation of the NF-κB
signaling pathway were investigated.
Materials and methods
Cell culture
PC-3 and DU145, human prostate cancer cell lines,
were obtained from the Chinese Academy of Sciences (Shanghai,
China). PC-3 and DU145 prostate cancer cells were cultured in
RPMI-1640 culture medium (BD Biosciences, San Jose, CA, USA) with
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), 100 µg/ml streptomycin (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) and 100 U/ml penicillin (Sigma-Aldrich;
Merck KGaA). The cell lines were maintained in humidified
incubators with 5% CO2 at 37°C. PC-3 and DU145 cells
were transfected with an ERβ expression plasmid and empty vector
was used as negative control using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). Construction of
receptor expression vectors was made as previously described
(15). Briefly, to construct the
human ERβ expression plasmid, ERβ/pcDNA3.1+Zero pcDNA3.1+Zero/ERβ,
A 2367 bp fragment including the human ERβ sequence was excised
from the ERβ/pcDNA3 plasmid (originally isolated from the ERβ/Pcmv5
plasmid) and inserted into the HindIII/Xbal sites in the vector
pCDNA3.1+Zero (Invitrogen; Thermo Fisher Scientific, Inc.). Then,
10 ng/ml of lipopolysaccharide (LPS; Sigma-Aldrich; Merck KGaA) or
dimethyl sulfoxide (DMSO; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) and PHTPP (a selective ERβ receptor inhibitor, Tocris
Bioscience, Bristol, UK) was added to cell cultures and incubated
for 24 h with 5% CO2 at 37°C. The time between
transfection and treatment with LPS/DMSO was 12 h.
ERβ-specific short interfering RNA
(siRNA) transfection
The sequences of the siRNAs targeting ERβ and
scramble RNA were as follows: siRNA-ERβ#1,
5′-GCCCUGCUGUGAUGAAUUAdTdT-3′; siRNA-ERβ#2,
5′-CCACCUUCCUUUCUAUUAUdTdT-3′; siRNA-ERβ#3,
5′-CGGGCUUCAUAAGCUAGAUdTdT-3′; and scramble,
5′-GAACUGAUGACAGGGAGGCTT-3′. Cells (5×104/well) were
seeded in 96-well plates. Since the expression level of ERβ was
most pronounced with siRNA-ERβ#1, this cell line was selected for
the following experiments. DU145 cells were transfected with siRNA
(150 pmol/ml) using Lipofectamine 2000® at 37°C
overnight. Prior to transfection, the culture medium was replaced
with Opti-MEM® Reduced Serum Medium (Gibco; Thermo
Fisher Scientific, Inc.). The resulting cell populations were
selected for further experiments. The levels of ERβ mRNA and
protein expression were analyzed via reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blotting, respectively.
RT-qPCR
Total RNA was extracted from cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
RNA purity and concentration were determined using a NanoDrop™ 2000
spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific,
Inc., Wilmington, DE, USA). Total RNA was reverse transcribed into
cDNA using a PrimeScript Master Mix kit (Takara Bio, Inc., Otsu,
Japan) according to the manufacturer's protocols. The cDNA was
amplified with SYBR Green Master Mix (Takara Bio, Inc.), which was
performed using a LightCycler® 96 system (Roche Applied
Science, Penzberg, Germany). The primers (5′→3′) used in qPCR were:
GAPDH, forward CGACCACTTTGTCAAGCTCA, reverse
AGGGGAGATTCAGTGTGGTG; ERβ, forward CAAGCTCATCTTTGCTCCAGA,
reverse GCCTTGACACAGAGATATTCTTTG. The thermocycling program was:
50°C for 2 min, 95°C for 2 min, followed by 40 cycles of 95°C for
15 sec and 60°C for 30 sec. At the end of each PCR run, a melting
curve analysis from 65 to 95°C was applied to all reactions to
ensure the specificity of the amplified product. GAPDH was used as
a reference gene, and the qPCR data was analyzed using LightCycler
96 Application Software version 1.1 (Roche Applied Science). The
2−ΔΔCq method (16) was
employed to determine the relative expression of the target
gene.
Cell viability assay
Cells cultured in RPMI-1640 medium were seeded into
96-well plates at a density of 5×103 cells/well and
incubated at 37°C for 24 h. PC-3 and DU145 parental cells (negative
vector transfected cells used as the control), and ERβ
plasmid-transfected cells were treated with LPS for 24, 48, 72 and
96 h. MTT assay solution (20 µl) was applied to the medium, which
was subsequently incubated for 4 h at 37°C. Then, 150 µl DMSO was
added to cells for 10 min to solubilize the formazan crystals.
Vehicle (0.1% DMSO) was used as the control. The optical density
values in each well were measured at 595 nm with an absorption
spectrophotometer (Olympus Corporation, Tokyo, Japan). All
treatments were performed in triplicate.
Transwell migration assay
Cells (5×104 cells/well) were cultured in
serum-free RPMI-1640 medium (500 µl) and plated in the upper
chambers of Transwell plates. RPMI-1640 with 15% FBS (600 ml;
Beijing Transgen Biotech Co., Ltd., Beijing, China) was plated in
the lower chambers. Plates were incubated in humidified conditions
with 5% CO2 for 24 h at 37°C. Then, the inserts were
washed with PBS three times, and cells on the upper surface were
removed with a cotton swab. The cells remaining on the bottom
surface of the inserts were treated with methanol (500 µl, 50
µg/ml) at 4°C for 10 min prior to staining with hematoxylin at room
temperature for 10 min. A total of five predetermined fields
(magnification, ×200) of each well were selected for cell counting.
Images were captured by a photomicroscope (Axiovert 200 M; Carl
Zeiss AG, Oberkochen, Germany).
Western blotting
Following rinsing in cold PBS, total protein was
extracted from the cells using lysis buffer (0.1% SDS, 50 mmol
Tris-base, 1% Triton X-100, 150 mmol sodium chloride, 1 mmol sodium
orthovanadate, 0.5% sodium deoxycholate, 1% protease inhibitor
cocktail and 10 mmol sodium fluoride) and quantified using the
Bradford protein assay. Proteins (20 µg) were separated via 10%
SDS-PAGE at 100 V and electrotransferred onto 0.45 µm
polyvinylidene difluoride membranes. The membranes were blocked
with 5% non-fat milk in TBS-Tween 20 (10%) at room temperature for
2 h, and then incubated with primary antibodies against ERβ (1:500;
cat. no. ab3576), p65 (1:100; cat. no. ab16502), NF-κB inhibitor α
(IκBα, 1:200; cat. no. ab32518), phosphorylated (p)-IκBα (1:200;
cat. no. ab133462), proliferating cell nuclear antigen (PCNA; 1:50;
cat. no. ab92552), E-cadherin (1:100; cat. no. ab40772), B-cell
lymphoma 2-associated X protein (Bax; 1:200; cat. no. 32503),
caspase-3 (1:200; cat. no. ab32351), matrix metalloproteinase-2
(MMP-2; 1:200; cat. no. ab37150) and β-actin (1:50; cat. no.
ab8226; Abcam, Cambridge, UK) overnight at 4°C. Anti-rabbit
HRP-conjugated secondary antibody (Thermo Fisher Scientific, Inc.)
was then added at room temperature for 1 h, and membranes were
incubated for 20 min. Protein bands were visualized using an
enhanced chemiluminescence kit (Pierce; Thermo Fisher Scientific,
Inc.). Protein expression was quantified using Total Lab Nonlinear
Dynamic Image (version 2009) analysis software (MathWorks, Natick,
MA, USA).
ELISA
High binding microtiter plates were coated overnight
at 4°C with 100 µl per well of III-BSA coating antigen (0.05 µg/ml
in coating buffer). Cells were then rinsed with PBS and blocked
with 3% bovine serum albumin (BSA; Beijing Transgen Biotech Co.,
Ltd.) in PBS for 1 h at room temperature. Anti-tumor necrosis
factor-α (TNF-α; cat. no. ab91235), anti-interleukin (IL)-1β (cat.
no. ab242234), anti-monocyte chemoattractant protein-1 (MCP-1; cat.
no. ab9669) and anti-IL-6 (cat. no. ab178013) monoclonal antibodies
were purchased from Abcam, and were diluted in PBS + 3% BSA, added
to cells and incubated at room temperature for 2 h. Following
washing with PBS, cells were incubated with goat anti-mouse
secondary antibodies (1:100; cat. no. BMS2007INST; Thermo Fisher
Scientific, Inc.), and incubated for 60 min at room temperature.
Cells expressing TNF-α, IL-1β, MCP-1 and IL-6 were detected using
an ELISA Kit according to the manufacturer's protocols; protein
concentrations were determined using a Spectramax M5 plate reader
(Molecular Devices, LLC, Sunnyvale, CA, USA).
Annexin V/propidium iodide (PI)
assay
Apoptotic cell death was determined using the
Annexin V (conjugated to Alexa Fluor 594; excitation/emission:
590/617 nm) Apoptosis Detection Kit (BioVision, Inc., Milpitas, CA,
USA) and PI. Following transfection, cells were incubated for 48 h,
and then collected and stained with Annexin V/PI according to the
manufacturer's protocols. The percentage of early apoptotic, late
apoptotic and necrotic cells were determined using a flow cytometer
(BD Bioscience) and analyzed with guavaSoft 3.1.1 software (EMD
Millipore, Billerica, MA, USA). The occurrence of apoptosis in each
group was investigated in ≥3 independent experiments.
Statistical analysis
SPSS 19.0 (IBM Corp., Armonk, NY, USA) was used for
all statistical analyses. Data are presented as the mean ± standard
deviation. Comparisons across three or more groups were performed
using one-way analyses of variance followed by Tukey's test, while
a Student's t-test was used to determine significant differences
between two groups. P<0.05 was considered to indicate a
statistically significant difference. All tests were investigated
in ≥3 independent experiments.
Results
Elevated expression of proinflammatory
genes in prostate cancer cells following LPS treatment
Following incubation with LPS for 24 h, the
expression levels of proinflammatory cytokines in the supernatants
of the prostate cancer cell lines PC-3 and DU145 were determined
using ELISA. Stimulation with LPS led to significantly elevated
expression levels of TNF-α, MCP-1, IL-1β and IL-6 in PC-3 and DU145
cells compared with controls treated with DMSO (P<0.05; Fig. 1A). These results indicated that LPS
successfully induced inflammation in prostate cancer cells.
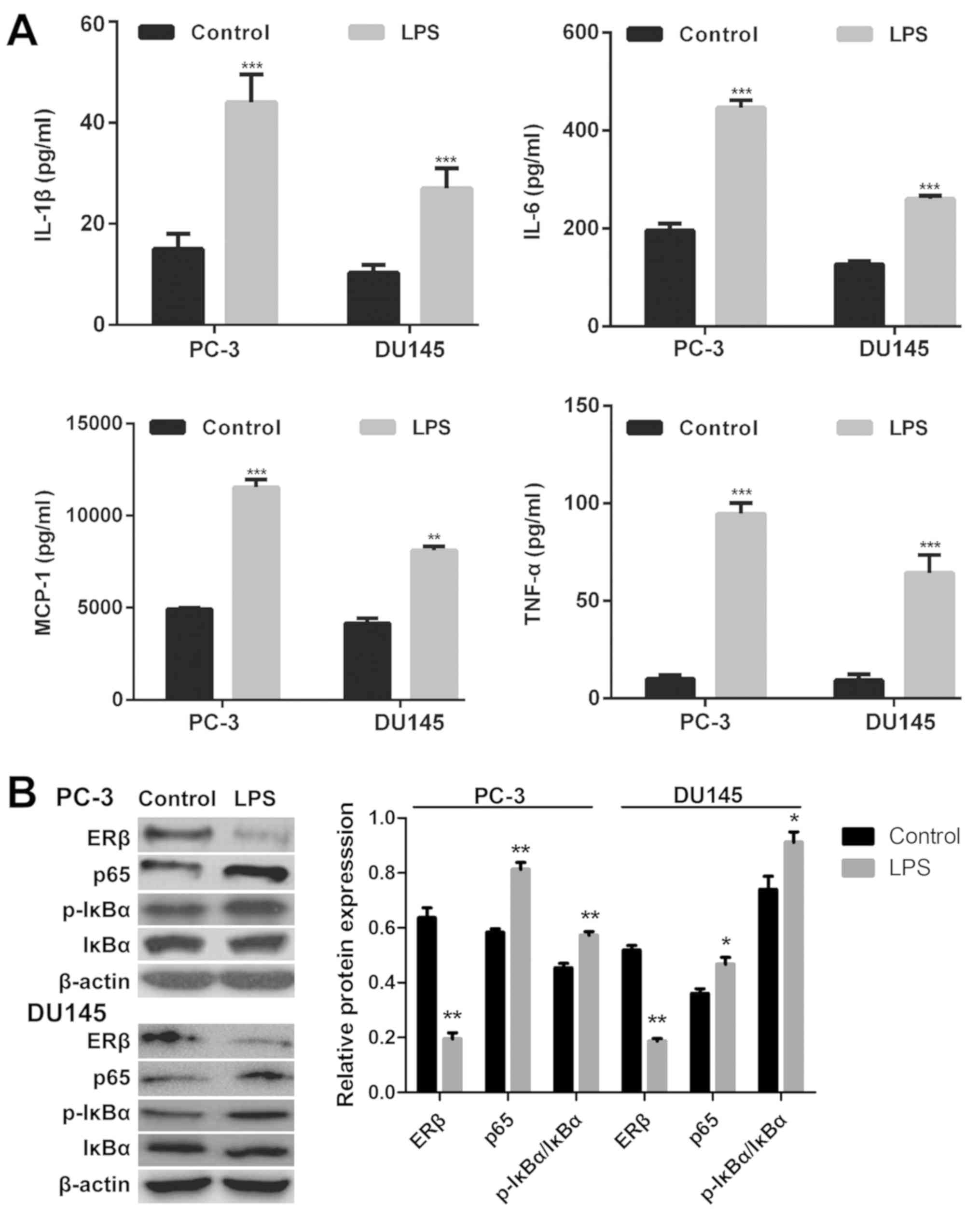 | Figure 1.Elevated expression of proinflammatory
genes and activation of the NF-κB pathway in prostate cancer cells
following LPS treatment. (A) Expression of proinflammatory
cytokines was determined using ELISA in LPS-treated and control
PC-3 and DU145 prostate cancer cells. (B) Expression of NF-κB
pathway-associated proteins was determined via western blot
analysis in LPS- and control-treated PC-3 and DU145 cells. Data are
presented as the mean ± standard deviation; *P<0.05, **P<0.01
and ***P<0.001 vs. the control group. Control, treatment with
dimethyl sulfoxide; ERβ, estrogen receptor β; IL, interleukin;
NF-κB, nuclear factor-κ-light-chain-enhancer of activated B cells;
IκBα, NF-κB inhibitor α; LPS, lipopolysaccharide; MCP-1, monocyte
chemoattractant protein 1; p, phosphorylated; TNF-α, tumor necrosis
factor α. |
To investigate whether p65 is regulated by ERβ
signaling, the expression levels of proteins involved in the NF-κB
pathway were determined. As presented in Fig. 1B, increased p65 and p-IκBα
expression, and decreased ERβ expression were observed in
LPS-stimulated PC-3 and DU145 cells compared with the controls;
however, no significant differences were reported for total IκBα
expression.
ERβ reduces the LPS-induced release of
inflammatory cytokines via suppression of the NF-κB pathway in
vitro
To investigate the role of the NF-κB pathway in the
regulation of ERβ-induced inflammation, western blot analysis was
performed to determine the expression of NF-κB pathway-associated
proteins in PC-3 and DU145 cells transfected with an ERβ expression
vector or treated with the ERβ antagonist PHTPP. It was revealed
that ERβ overexpression suppressed the expression of p65 and
p-IκBα, but not total IκBα expression in LPS-stimulated PC-3 and
DU145 cells compared with control cells. This expression profile
was reversed following treatment with PHTPP (Fig. 2A).
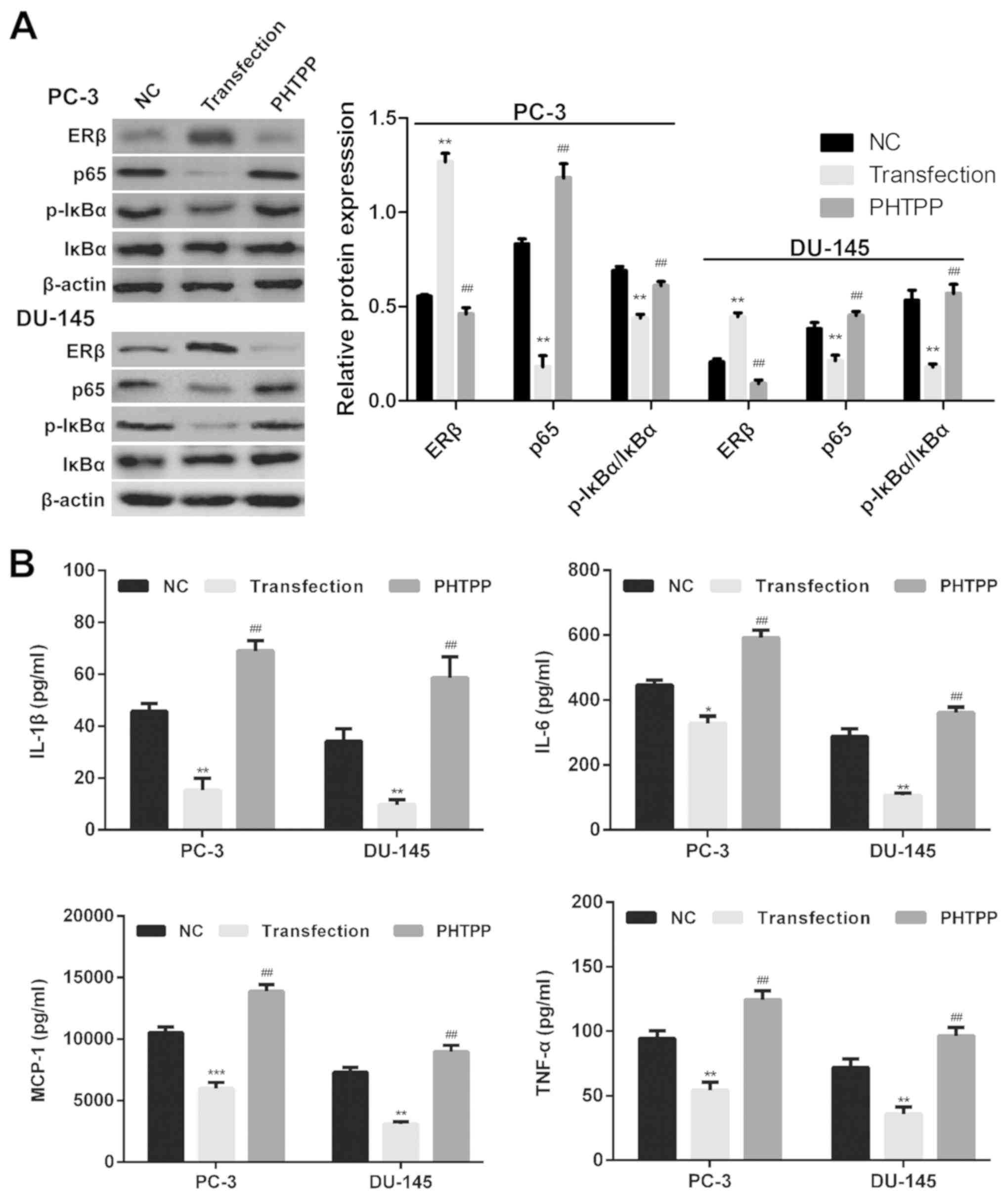 | Figure 2.ERβ-induced reduction in
proinflammatory cytokine release via suppression of the NF-κB
pathway. (A) Expression of NF-κB pathway-associated proteins in NC
empty vector-transfected, ERβ expression vector-transfected and
PHTPP-treated PC-3 and DU145 cells was determined via western blot
analysis. (B) Production of inflammatory cytokines was determined
using ELISA in NC, ERβ-transfected and PHTPP-treated PC-3 and DU145
cells. Data are presented as the mean ± standard deviation;
*P<0.05, **P<0.01, ***P<0.001 vs. NC;
#P<0.05, ##P<0.01, vs. transfection
group. ERβ, estrogen receptor β; IL, interleukin; NF-κB, nuclear
factor κ-light-chain-enhancer of activated B cells; IκBα, NF-κB
inhibitor α; LPS, lipopolysaccharide; MCP-1, monocyte
chemoattractant protein 1p, phosphorylated; TNF-α, tumor necrosis
factor-α. |
Providing ERβ is a suppressor of prostate
inflammation, ERβ activation may suppress the expression of certain
proinflammatory genes. To investigate the involvement of ERβ
signaling in LPS-induced inflammation, the expression of the
proinflammatory cytokines TNF-α, IL-1β, MCP-1 and IL-6, was
determined in cells overexpressing ERβ and transfected cells
treated with PHTPP via ELISA. The results demonstrated that the
LPS-induced production of TNF-α, MCP-1, IL-1β and IL-6 was
significantly reduced in prostate cancer cells following
overexpression of ERβ compared with negative control vector cells
(P<0.05; Fig. 2B). This
expression profile was significantly reversed in PHTPP-treated
transfected cells compared with ERβ-overexpressing cells
(P<0.05). These data suggested that ERβ signaling regulated
LPS-induced prostate cancer cell inflammation.
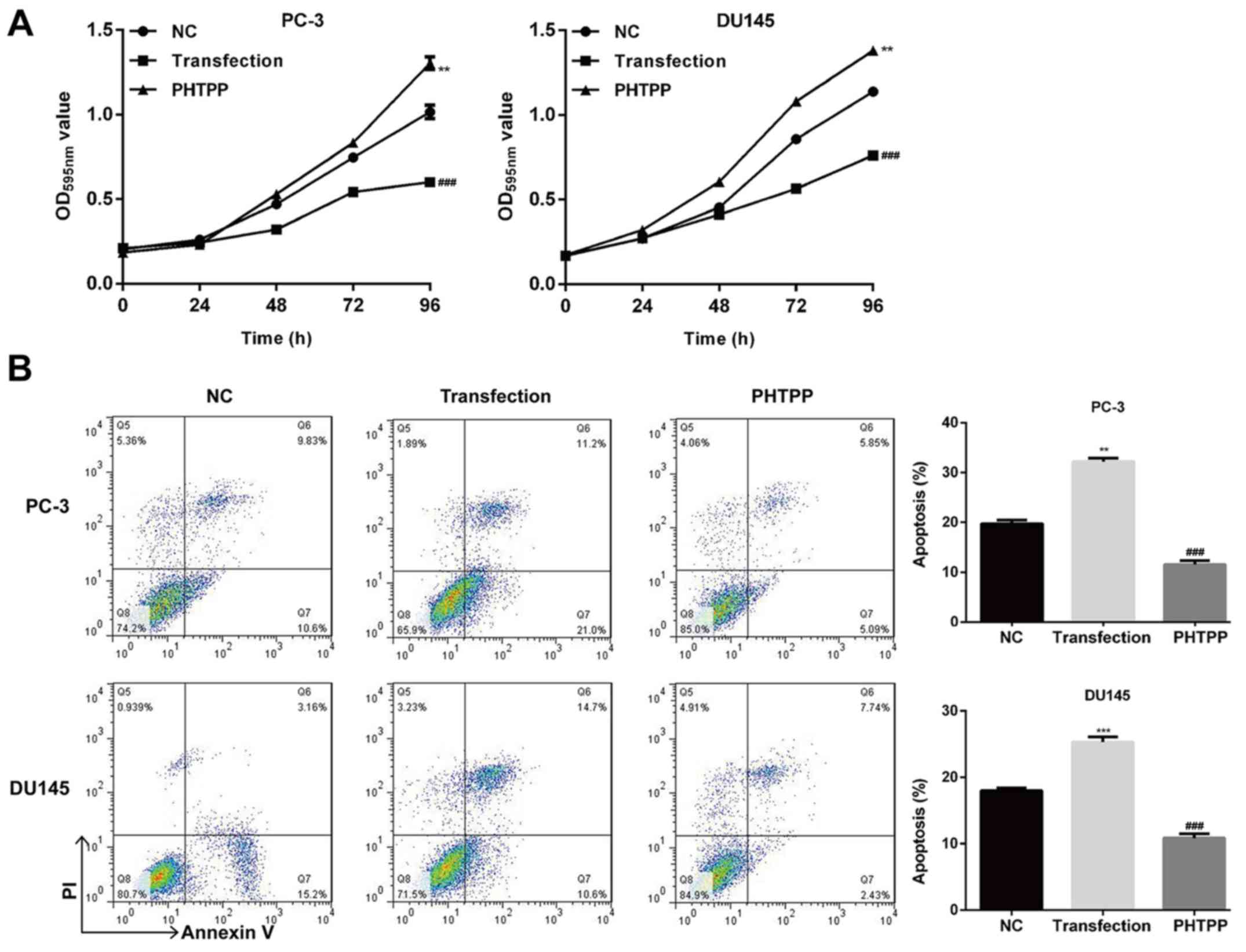
ERβ activation suppresses viability
and migration, and promotes the apoptosis of prostate cancer
cells
To determine the effects of ERβ overexpression on
prostate cancer cell viability, apoptosis and migration, MTT,
Annexin V/PI and Transwell assays were performed using control,
ERβ-overexpressing, and ERβ-inhibited cells. As presented in
Fig. 3A, ERβ overexpression
significantly suppressed the viability of PC-3 and DU145 cells
compared with control and PHTPP-treated transfected cells
(P<0.01). When cells were treated with PHTPP, their viability
was markedly reduced compared with ERβ-overexpressed cells.
Furthermore, the number of apoptotic cells among those treated with
ERβ transfection was significantly higher compared with control and
ERβ-inhibited cells (P<0.01; Fig.
3B). The cell migration assay revealed that upregulation of ERβ
significantly decreased migration compared with control and
PHTPP-treated transfected PC-3 and DU145 cells (P<0.01; Fig. 4A).
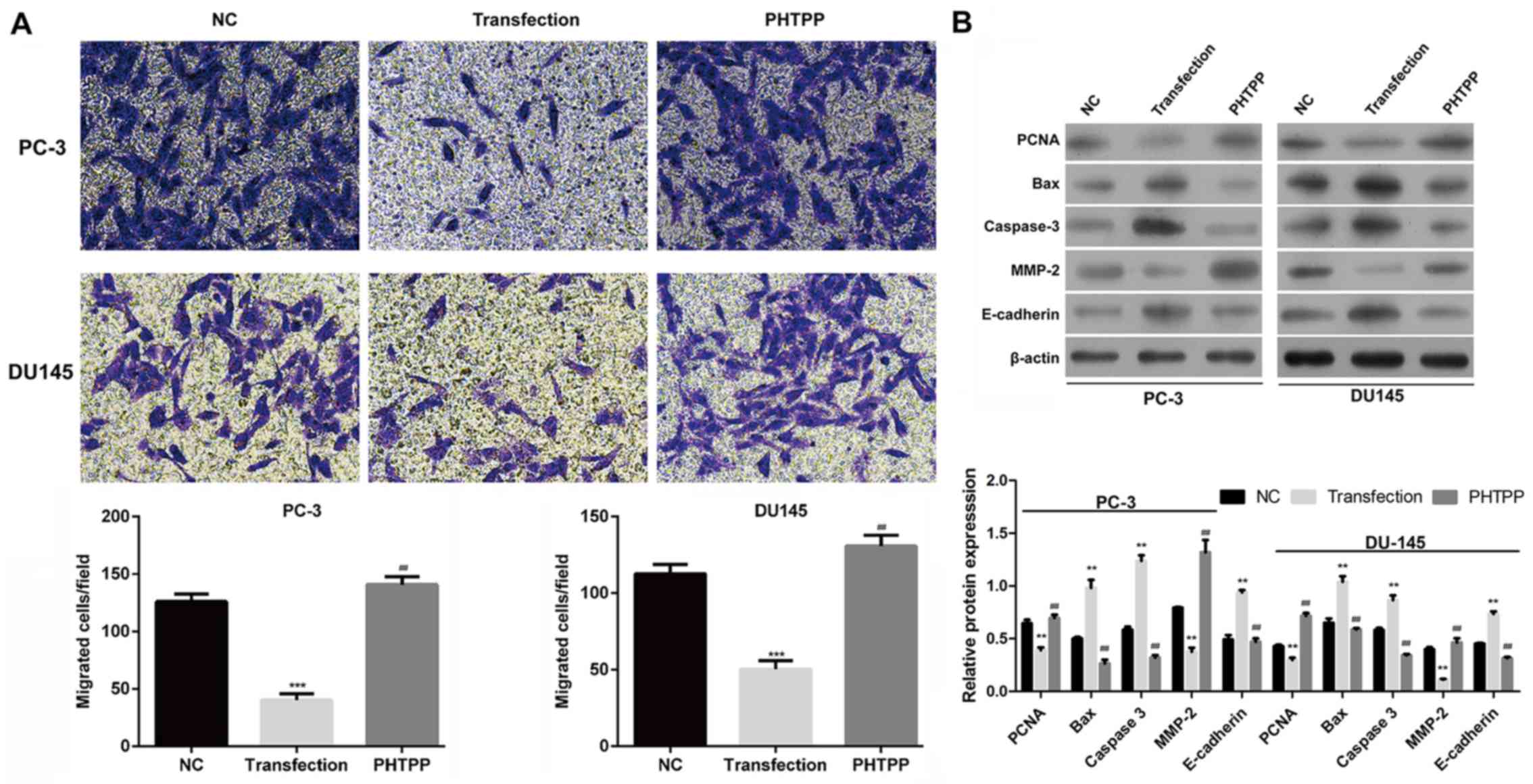 | Figure 4.ERβ activation inhibits the migration
of prostate cancer cells and alters the expression of
proliferation-, apoptosis- and migration-associated proteins. (A)
Cell migration was determined using a Transwell assay in NC,
ERβ-transfected and PHTPP-treated transfected PC-3 and DU145 cells.
(B) Expression of PCNA, Bax, caspase-3, MMP-2 and E-cadherin was
evaluated via western blot analysis in NC, ERβ-transfected and
PHTPP-treated transfected PC-3 and DU145 cells. Data are presented
as the mean ± standard deviation; **P<0.01, ***P<0.001 vs.
NC; ##P<0.01, vs. transfection group. Bax, B-cell
lymphoma 2-associated X protein; ERβ, estrogen receptor β; MMP-2,
matrix metalloproteinase-2; NC, negative control; PCNA,
proliferating cell nuclear antigen. |
As ERβ signaling appeared to be involved in the
regulation of cell viability, apoptosis and migration, and the
expression of proteins associated with these properties was
determined. PCNA, Bax and caspase-3, and MMP-2 and E-cadherin were
selected as markers of proliferation (17), apoptosis (18) and epithelial-mesenchymal
transition-associated migration (19), respectively. As presented in
Fig. 4B, overexpression of ERβ
suppressed the expression of PCNA and MMP-2, but promoted the
expression of Bax, caspase-3 and E-cadherin in PC-3 and DU145
cells. These alterations in expression were reversed by treatment
with PHTPP.
To further investigate the effects of ERβ on the
progression of prostate cancer, ERβ-targeting siRNAs were employed
to downregulate ERβ expression in DU145 prostate cancer cells. The
silencing effects of three siRNAs compared with a negative control
scramble siRNA were reported via RT-qPCR analysis (Fig. 5A); siRNA-ERβ#1 was selected for
subsequent experiments, as it exhibited a notable effect on ERβ
expression compared with siRNAs-ERβ#2 and 3. Decreased levels of
ERβ protein expression in prostate cancer cells following
transfection with siRNA-ERβ#1 were demonstrated via western blot
analysis (Fig. 5B). ERβ knockdown
induced similar effects to treatment with PHTPP; increased
expression of p65 and p-IκBα was observed, but not of total IκBα
compared with scramble-transfected cells (Fig. 5B). Furthermore significantly
increased cell viability (Fig.
5C), decreased number of apoptotic cells (Fig. 5D), upregulated expression of
proinflammatory cytokines (Fig.
5E), enhanced cell migration (Fig.
5F) and altered expression of associated proteins (Fig. 5G) were reported in DU145 prostate
cancer cells following transfection with siRNA-ERβ#1 compared with
scramble-transfected cells. Collectively, these findings indicated
that upregulation of ERβ reduced the viability and migration, and
increased the apoptosis of human prostate cancer cells, which may
suppress the progression of prostate cancer.
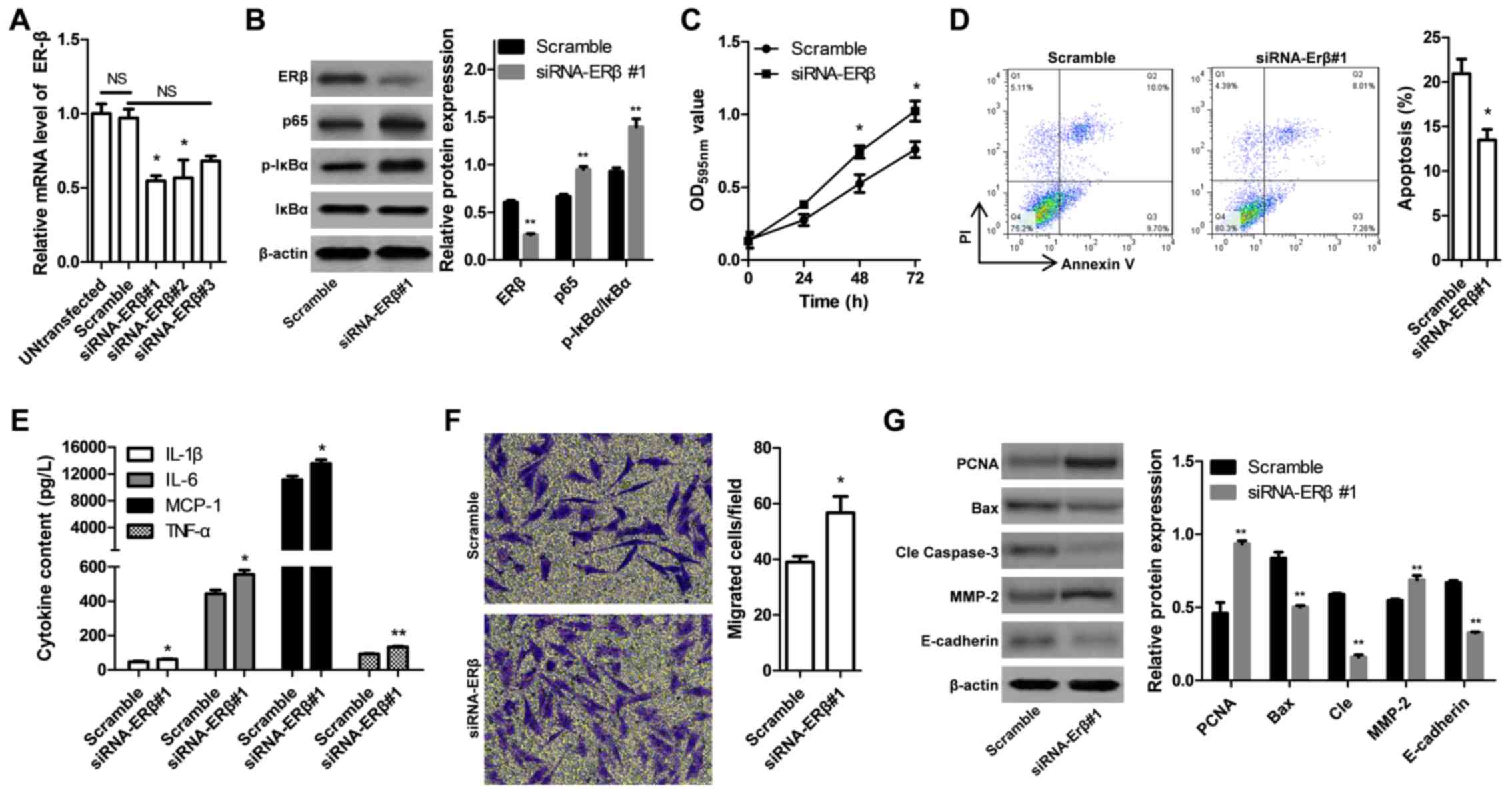 | Figure 5.Effects of ERβ-specific siRNA on
inflammation, proliferation, apoptosis and migration of DU145
prostate cancer cells. (A) Expression levels of ERβ mRNA following
treatment with siRNAs were determined via reverse
transcription-quantitative polymerase chain reaction. (B)
Expression of proteins associated with the nuclear factor
κ-light-chain-enhancer of activated B cells pathway was determined
via western blot analysis. (C) Cell viability of DU145 was measured
with a MTT assay, with absorbance at 595 nm used as an index. (D)
Cell apoptosis was determined via flow cytometry. (E) Secretion of
proinflammatory cytokines was measured using ELISA. (F) Migration
of DU145 cells was evaluated using a Transwell migration assay. (G)
Expression levels of proliferation-, apoptosis- and
migration-associated proteins in DU145 cells following transfection
with siRNA-ERβ#1 or Scramble were determined via western blot
analysis. Data are presented as the mean ± standard deviation;
*P<0.05, **P<0.01 vs. Scramble group. Bax, B-cell lymphoma
2-associated X protein; cle, cleaved; ERβ, estrogen receptor β; IL,
interleukin; IκBα, nuclear factor of κ light polypeptide gene
enhancer in B-cells inhibitor α; MCP-1, monocyte chemoattractant
protein 1; MMP-2, matrix metalloproteinase-2; NC, negative control;
NS, not significant; OD, optical density; p, phosphorylated; PCNA,
proliferating cell nuclear antigen; PI, propidium iodide; siRNA,
short interfering RNA; TNF-α, tumor necrosis factor-α. |
Discussion
In the present study, it was revealed that ERβ
overexpression reduced the inflammation induced by LPS treatment
via regulating the levels of proinflammatory cytokines, including
TNF-α, MCP-1, IL-1β and IL-6. Furthermore, it was demonstrated that
the ERβ antagonist PHTPP increased the expression of
proinflammatory cytokines. Additionally, it was observed that ERβ
overexpression suppressed the viability and migration of PC-3 and
DU145 prostate cancer cells, and promoted apoptosis. These findings
may improve understanding of the possible mechanisms by which ERβ,
a tumor suppressor, may contribute to inhibition of the NF-κB
pathway.
LPS is a common inducer of inflammation; exposure
leads to the activation of a number of components involved in
chronic inflammation processes, such as altered cytokine levels
(20,21). Carcinogenesis was studied in cells
treated with LPS, as determined by tumor cell survival,
proliferation, invasion and metastasis (1,22,23).
LPS induces PC-3 cell migration (24) by stimulating the production of
TNF-α and IL-6 (25), and the
serum levels of these cytokines are associated with stages of
prostate cancer (26). A study
investigated the effects of treatment in patients with localized
aggressive periodontitis, and observed that improvement in clinical
parameters correlated with reductions in responsiveness to LPS, as
determined by the release of cytokines, including TNF-α, IL-1β,
MCP-1 and IL-6 (27). In the
present study, LPS-induced inflammatory responses were observed in
PC-3 and DU145 prostate cancer cells, with elevated serum levels of
TNF-α, IL-1β, IL-6 and MCP-1.
ERβ has been associated with the differentiation of
prostatic epithelial cells, and was reported to exhibit
antiproliferative effects on prostate cancer cells (28). The regulatory effects of estrogen
on PC-3 cell proliferation are mediated by ERβ, and can be
inhibited by the ERβ-selective antagonist PHTPP (29). The role of ERβ in oppressing
androgen receptor signaling, and suppressing proliferation and
inflammation, suggests that ERβ may be a potential therapeutic
target to prevent the progression of prostate cancer (30). Differentiation of prostatic
epithelial cells, in which ERβ serves a significant role, is a
defensive mechanism required for the protection against
inflammation in the prostate of FGF8b transgenic mice (31). The findings of the present study
revealed that ERβ overexpression reduced the upregulated expression
levels of TNF-α, MCP-1, IL-6 and IL-1β in PC-3 cells induced by
LPS, indicating a potential therapeutic role of ERβ in the
reduction of inflammation in prostate cancer.
It was reported that, under physiological
conditions, estrogen signaling via ERβ inhibited prostate gland
growth (32,33). Dihydrotestosterone inhibits
prostate cancer cell migration and proliferation via conversion to
3β-Androstanediol, a metabolite that selectively interacts with ERβ
(34,35). It was previously demonstrated that
treatment with four ligands of ERβ (tamoxifen, raloxifene, curcumin
and genistein) inhibited prostate cancer cell proliferation and
migration, suggesting a potential protective effect against the
progression of prostate cancer (36). Consistent with these findings, it
was observed in the present study that overexpression of ERβ
reduced the viability and migration of PC-3 and DU145 cells, and
promoted apoptosis.
NF-κB is activated by various signals via
phosphorylation and degradation of the inhibitory IκBα protein
(37). NF-κB functions via
canonical and non-canonical pathways. A recent study proposed that
ERβ suppresses the activation of NF-κB activation, due to the
negative correlation between ERβ and p65 expression (14). In the present study, it was
reported that the expression of p65 and p-IκBα was increased, and
that of ERβ decreased in LPS-stimulated cells. Furthermore, ERβ
overexpression reduced the expression of p65 and p-IκBα, but not
total IκBα expression; this effect was reversed by PHTPP. These
findings suggested that the underlying mechanisms involved in the
potential therapeutic effects of ERβ may be associated with
suppressed NF-κB activation in prostate cancer cells.
In conclusion, the present study reported that
activation of ERβ suppressed the NF-κB signaling pathway, and
suggested that ERβ may be a potential anti-inflammatory and
anticancer agent in the treatment of prostate cancer.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Internal
Research Institutions Fund of Health and Technology Planning
Commission of Yunnan Province (grant no. 2016NS209).
Availability of data and materials
All data generated or analyzed during this study are
included in this manuscript.
Authors' contributions
LX and YL made substantial contributions to data
acquisition and analysis, and were major contributors in drafting
the manuscript. RT was responsible for interpretation of data; NZ
conceived and designed the study, and revised the manuscript
critically for important intellectual content. All authors have
read and agreed with the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Winchester DA, Till C, Goodman PJ, Tangen
CM, Santella RM, Johnson-Pais TL, Leach RJ, Xu J, Zheng SL,
Thompson IM, et al: Variation in genes involved in the immune
response and prostate cancer risk in the placebo arm of the
Prostate Cancer Prevention Trial. Prostate. 75:1403–1418. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sfanos KS, Isaacs WB and De Marzo AM:
Infections and inflammation in prostate cancer. Am J Clin Exp Urol.
1:3–11. 2013.PubMed/NCBI
|
|
4
|
Haverkamp J, Charbonneau B and Ratliff TL:
Prostate inflammation and its potential impact on prostate cancer:
A current review. J Cell Biochem. 103:1344–1353. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kanda Y, Osaki M and Okada F:
Chemopreventive strategies for inflammation-related carcinogenesis:
Current status and future direction. Int J Mol Sci. 18:E8672017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McKown RL, Orle KA, Chen T and Craig NL:
Sequence requirements of Escherichia coli attTn7, a specific site
of transposon Tn7 insertion. J Bacteriol. 170:352–358. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun B and Karin M: The therapeutic value
of targeting inflammation in gastrointestinal cancers. Trends
Pharmacol Sci. 35:349–357. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nguyen DP, Li J, Yadav SS and Tewari AK:
Recent insights into NF-κB signalling pathways and the link between
inflammation and prostate cancer. BJU Int. 114:168–176. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang JJ, Chu HX, Jiang ZY, Zhang XJ, Sun
HP and You QD: Recent advances in the structure-based and
ligand-based design of IKKβ inhibitors as anti-inflammation and
anti-cancer agents. Curr Med Chem. 21:3893–3917. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gasparian AV, Yao YJ, Kowalczyk D, Lyakh
LA, Karseladze A, Slaga TJ and Budunova IV: The role of IKK in
constitutive activation of NF-kappaB transcription factor in
prostate carcinoma cells. J Cell Sci. 115:141–151. 2002.PubMed/NCBI
|
|
11
|
Verzella D, Fischietti M, Capece D,
Vecchiotti D, Del Vecchio F, Cicciarelli G, Mastroiaco V, Tessitore
A, Alesse E and Zazzeroni F: Targeting the NF-κB pathway in
prostate cancer: A promising therapeutic approach? Curr Drug
Targets. 17:311–320. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakamura Y, Felizola SJ, Kurotaki Y,
Fujishima F, McNamara KM, Suzuki T, Arai Y and Sasano H: Cyclin D1
(CCND1) expression is involved in estrogen receptor beta (ERβ) in
human prostate cancer. Prostate. 73:590–595. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu WF, Tan XJ, Dai YB, Krishnan V, Warner
M and Gustafsson JA: Targeting estrogen receptor β in microglia and
T cells to treat experimental autoimmune encephalomyelitis. Proc
Natl Acad Sci USA. 110:3543–3548. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mak P, Li J, Samanta S and Mercurio AM:
ERβ regulation of NF-κB activation in prostate cancer is mediated
by HIF-1. Oncotarget. 6:40247–40254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Soshilov A and Denison MS: Role of the
Per/Arnt/Sim domains in ligand-dependent transformation of the aryl
hydrocarbon receptor. J Biol Chem. 283:32995–33005. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao H, Lo YH, Ma L, Waltz SE, Gray JK,
Hung MC and Wang SC: Targeting tyrosine phosphorylation of PCNA
inhibits prostate cancer growth. Mol Cancer Ther. 10:29–36. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shen H, Zeng G, Sun B, Cai X, Bi L, Tang G
and Yang Y: A polysaccharide from Glycyrrhiza inflata Licorice
inhibits proliferation of human oral cancer cells by inducing
apoptosis via mitochondrial pathway. Tumour Biol. 36:4825–4831.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang S, Qiu M, Xia W, Xu Y, Mao Q, Wang J,
Dong G, Xu L, Yang X and Yin R: Glypican-5 suppresses
epithelial-mesenchymal transition of the lung adenocarcinoma by
competitively binding to Wnt3a. Oncotarget. 7:79736–79746.
2016.PubMed/NCBI
|
|
20
|
Schetter AJ, Heegaard NH and Harris CC:
Inflammation and cancer: Interweaving microRNA, free radical,
cytokine and p53 pathways. Carcinogenesis. 31:37–49. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu P, Cai F, Liu X and Guo L: Sesamin
inhibits lipopolysaccharide-induced proliferation and invasion
through the p38-MAPK and NF-κB signaling pathways in prostate
cancer cells. Oncol Rep. 33:3117–3123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mantovani A: Cancer: Inflammation by
remote control. Nature. 435:752–753. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hsu RY, Chan CH, Spicer JD, Rousseau MC,
Giannias B, Rousseau S and Ferri LE: LPS-induced TLR4 signaling in
human colorectal cancer cells increases beta1 integrin-mediated
cell adhesion and liver metastasis. Cancer Res. 71:1989–1998. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li J, Chong T, Wang Z, Chen H and Li H,
Cao J, Zhang P and Li H: A novel anticancer effect of resveratrol:
Reversal of epithelialmesenchymal transition in prostate cancer
cells. Mol Med Rep. 10:1717–1724. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Im HJ, Park NH, Kwon YJ, Shin S, Kim D and
Chun YJ: Bacterial lipopolysaccharides induce steroid sulfatase
expression and cell migration through IL-6 pathway in human
prostate cancer cells. Biomol Ther (Seoul). 20:556–561. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Michalaki V, Syrigos K, Charles P and
Waxman J: Serum levels of IL-6 and TNF-alpha correlate with
clinicopathological features and patient survival in patients with
prostate cancer. Br J Cancer. 90:2312–2316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shaddox LM, Gonçalves PF, Vovk A, Allin N,
Huang H, Hou W, Aukhil I and Wallet SM: LPS-induced inflammatory
response after therapy of aggressive periodontitis. J Dent Res.
92:702–708. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Christoforou P, Christopoulos PF and
Koutsilieris M: The role of estrogen receptor beta in prostate
cancer. Mol Med. 20:427–434. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lombardi AP, Pisolato R, Vicente CM,
Lazari MF, Lucas TF and Porto CS: Estrogen receptor beta (ERβ)
mediates expression of β-catenin and proliferation in prostate
cancer cell line PC-3. Mol Cell Endocrinol. 430:12–24. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu WF, Maneix L, Insunza J, Nalvarte I,
Antonson P, Kere J, Yu NY, Tohonen V, Katayama S, Einarsdottir E,
et al: Estrogen receptor β, a regulator of androgen receptor
signaling in the mouse ventral prostate. Proc Natl Acad Sci USA.
114:E3816–e3822. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Elo T, Yu L, Valve E, Mäkelä S and
Härkönen P: Deficiency of ERβ and prostate tumorigenesis in FGF8b
transgenic mice. Endocr Relat Cancer. 21:677–690. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weihua Z, Lathe R, Warner M and Gustafsson
JA: An endocrine pathway in the prostate, ERbeta, AR,
5alpha-androstane-3beta, 17beta-diol, and CYP7B1, regulates
prostate growth. Proc Natl Acad Sci USA. 99:13589–13594. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Prins GS and Korach KS: The role of
estrogens and estrogen receptors in normal prostate growth and
disease. Steroids. 73:233–244. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dondi D, Piccolella M, Biserni A, Della
Torre S, Ramachandran B, Locatelli A, Rusmini P, Sau D, Caruso D,
Maggi A, et al: Estrogen receptor beta and the progression of
prostate cancer: Role of 5alpha-androstane-3beta, 17beta-diol.
Endocr Relat Cancer. 17:731–742. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guerini V, Sau D, Scaccianoce E, Rusmini
P, Ciana P, Maggi A, Martini PG, Katzenellenbogen BS, Martini L,
Motta M and Poletti A: The androgen derivative
5alpha-androstane-3beta, 17beta-diol inhibits prostate cancer cell
migration through activation of the estrogen receptor beta subtype.
Cancer Res. 65:5445–5453. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Piccolella M, Crippa V, Messi E, Tetel MJ
and Poletti A: Modulators of estrogen receptor inhibit
proliferation and migration of prostate cancer cells. Pharmacol
Res. 79:13–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huong PT, Moon DO, Kim SO, Kim KE, Jeong
SJ, Lee KW, Lee KS, Jang JH, Erikson RL, Ahn JS and Kim BY:
Proteasome inhibitor-I enhances tunicamycin-induced
chemosensitization of prostate cancer cells through regulation of
NF-κB and CHOP expression. Cell Signal. 23:857–865. 2011.
View Article : Google Scholar : PubMed/NCBI
|