Acute promyelocytic leukemia (APL) is a rare
leukemia characterized by the balanced reciprocal translocation
between the promyelocytic leukemia (PML) gene on chromosome 15 and
the retinoic acid receptor α (RARα) gene on chromosome 17, and
accounts for between 10 and 15% of newly diagnosed acute myeloid
leukemia (AML) cases each year (1).
Potentially life-threatening coagulopathy, and distinct morphologic
and cytogenetic aberrations define APL as a unique subtype of AML
(2). Early studies have demonstrated
a median survival time of 1 week, ranging from 1 day to 1 month
(3–6)
in patients who were untreated or only received corticosteroids.
The combined administration of all-trans retinoic acid receptor α
(ATRA) and arsenic trioxide (ATO) as primary therapy has notably
improved the survival rate and decreased toxicity in patients.
Early death (ED; mortality in the first 30 days following therapy)
remains a major contribution to treatment failure (7). A previous study analyzed the data from
surveillance, epidemiology and an end result program of 1,400 APL
patients, revealing an ED rate (EDR) of 17.3% (8). Due to delayed diagnosis, delayed
administration of ATRA and/or inadequate supportive care, the EDR
of APL did not change substantially (9); if high-risk patients could be identified
earlier and provided with better supportive care, such as the
hemostatic targets protocol (e.g., platelets >30×109/l, normal
prothrombin time, normal activated partial thromboplastin time,
fibrinogen >1.5 g/l) (10), the
EDR is expected to decrease.
In the present study, published data is reviewed
with a focus on the factors that may contribute to the ED of
patients with APL, in order to improve the identification of
high-risk patients.
The FLT3 gene, a class III tyrosine kinase receptor,
is located on chromosome 13q12 in humans (11). Somatic mutations in AML are common,
including missense mutations in the activation loop domain (ALM) of
the tyrosine kinase domain (FLT3/ALM) and internal tandem
duplications of the juxtamembrane domain coding sequence
[FLT3-internal tandem duplication (ITD)] (12–14). It is
the most frequent genetic event in APL that may coincide with
t(15;17) translocation. Several studies have demonstrated that
20–40% of APL patients possess the FLT3-ITD mutation and another
10–20% carry an FLT3/ALM mutation (15,16). Thus,
Souza et al (17) suggested
that FLT3-ITD positive APL patients may be classified as a new
sub-type.
Previous studies have identified that FLT3-ITD is
associated with a high white blood cell count (WBC) (17–28), the
microgranular variant (M3V) type (17–20,23,25–27),
short type PML/RARα [break cluster region 3 (BCR3)] (17,19,20,23,24,26,27),
sex (28), low-fibrinogen
concentration (22), hemoglobin
levels (17,26) and high lactate dehydrogenase (LDH)
level (22). However, when discussed
in the context of ED, the prognostic significance of this
association remains unclear.
In studies that used ATO as one of the introductory
chemotherapeutic drugs for APL, the clinical outcomes presented
were more favorable. For example, the study by Mathews et al
(24) reported an ITD mutation rate
of 33% in 98 APL patients and identified no impact on outcome. A
study by the Shanghai Group (33)
also suggested that the status of FLT3 did not associate with low
EDR in 85 patients with APL receiving ATRA/ATO, and suggested that
ATO may inhibit the negative effect of ITD. In a different study,
4/124 patients succumbed during primary therapy and no adverse
outcomes influenced by FLT3 mutation status were identified
(10). It has been demonstrated that
inferior OS and DFS were significantly associated with FLT3-ITD
(24). Therefore, any adverse effect
of FLT3 mutations appears to be neutralized by the addition of ATO
during primary therapy and consolidation. Furthermore, the authors
suggest that FLT3 inhibitor therapy will serve no function in the
future management of APL despite the occurrence of FLT3 mutations
in APL. Poiré et al (27)
analyzed 245 newly diagnosed adult patients with APL treated on
intergroup trial C9710 and identified that FLT3 status had no
association with EDR. The same result was also examined by Stock
et al (16) in their study of
78 adults with newly diagnosed APL entered onto CALGB 9710, a North
American Intergroup phase III trial. However, the authors suggested
that targeted therapy with FLT3 inhibitors may improve relapse free
survival for patients with FLT3 + APL.
The ITD mutation may have a relatively reduced
function in the progression of APL and early mortality in patents
that did not receive ATO as the initial chemotherapy. However, when
ATO was used as a primary therapy, the inferior outcome was
observed to be reversed. Continued study may resolve the mechanism
of this phenomenon. Due to limitations in patient numbers and
selection, more retrospective or prospective studies should focus
on FLT3-ITD and early mortality in patients that received ATO as a
first induction chemotherapy regimen.
CD56, which is known as a neural cell adhesion
molecule and associated with unfavorable clinical outcome in AML
with t(8;21) (56), is expressed in
11–15% of patients with APL (57–59). It
has been associated with CD2+ (57),
CD34+ (57,59), CD7+ (57), HLA-DR+ (57), CD15+ (57), CD117+ (57), BCR3 isoform (57,60,61),
fibrinogen range (60), and a trend
toward M3v, CD11b+ and CD9+ has been demonstrated (57). Murray et al (60) first described a decreased CR rate
associated with expression of CD56 and noticed the association
between CD56 and natural killer and T cell markers. The association
between CD56+ and immaturity-associated markers (CD117) and natural
killer and T-cell antigens, including CD2 and CD7 has been
identified and it is hypothesized that these sub-groups of APL may
have arisen in progenitors that have not undergone lineage
restriction (57). The immature,
undifferentiated and pluripotent leukemic stem cells are less
sensitive to the primary therapy, which may explain why the CD56+
group experienced a decreased CR rate and an increased EDR
(57). Ito et al (59) did not identify any impact of CD56+ on
EDR in their study of 28 patients with APL. Similar results were
also obtained by Ferrara et al (58), however, the authors observed that CD56
is an independent prognostic impact on survival that includes WBC
count which indicated that the poor outcome of CD56+ APL is not
associated with high WBC count. However, when drugs including ATO
and gemtuzumab ozogamicin were administered as primary treatment,
the predicted value of CD56 is waiting to be verified in the future
studies (62).
The balanced reciprocal translocation t(15;17)
(q22;q11-21) leading to PML gene and RARΑ gene fusion is the
genetic characteristic of APL. The classical t(15;17) (q22;q12) is
observed in between 70 and 90% of APL cases (69), and a number of patients exhibit
complex translocations, involving chromosomes 15, 17 and other
chromosomes (70,71). The most common abnormality is trisomy
8 (72,73). A number of the complex mutations were
sensitive for the prediction of ED in APL. In a study by De Botton
et al (73) and the Southwest
Oncology Group (74), additional
cytogenetic changes in patients with t(15;17) had no effect on the
CR rate, EFS, relapse and overall survival at 2 years, which is in
accordance with the findings of Grimwade et al (72). Mi et al (75) identified that a complex karyotype may
contribute to an improved prognosis. Other reports have suggested
the presence of complex karyotype changes adversely affects
prognosis (76). A study of C9710
analyzed 245 newly diagnosed adult patients with APL (27). The authors identified a significantly
lower CR rate in the complex karyotype [two or more additional
chromosomal abnormalities (ACAs)] subgroup compared with one ACA or
normal karyotype (P=0.001) independent of ATO treatment. This may
be due to the reduced sensitivity to ATRA/ATO and the delay in
administering ATO, which has been demonstrated by an
Italian/German/Austrian cooperative group study that suggested
earlier administration of ATO may overcome the negative effect of
complex karyotypes (77). The natural
resistance to primary therapy is also a factor contributing to ED
in patients with APL.
Co-expression of t(8;21) and t(15;17) is rare in
APL, with only 6 patients reported at present (79–84). Neto
et al (79) reported a case of
APL-M3V which was sensitive to ATRA treatment, and detected a novel
t(8;21) chromosomal aberration between 3 and 18 months after
initial treatment. The authors noted the intermittent detection of
t(8;21) during periods without ATRA suggested there was an
antitumor effect of ATRA on M2 leukemic cells. Charrin et al
(80) and Bonomi et al
(81) reported two cases and
identified that the t(15;17) may be acquired subsequent to t(8;21).
A total of 5 patients received the ATRA-based treatment and
achieved CR. No ED occurred during primary therapy, despite a high
rate of relapse. A favorable response to chemotherapeutic induction
indicated that the ATRA and idarubicin and Ara-C induction
treatment was suitable for this complex karyotype (84). Another study also suggested that at
the time of diagnosis the rate of M2 leukemic cells could be tested
using polymerase chain reaction detection and the alteration of
bone marrow cell kinetics may trigger t(8;21) via complex
mechanisms following chemotherapy (79).
Ider(17), which has been reported in only 72 APL
cases globally, is a relatively rare variant cytogenetic
abnormality among patients with APL (83,85–97). This
isochromosomal abnormality may occur following initial reciprocal
translocation of t(15;17), and is formed from the short arm and
duplication of the long arm of ider(17)t(15;17) (85). Clinical outcome data were available
for 36 patients with a CR rate of 77.8% and the response to ATRA
and EDR were observed to be similar to that in normal APL. A total
of 19 patients succumbed to the disease; however, the prognostic
significance of this abnormal karyotype remains unclear due to the
limited number of cases. The proportion of cells with the
ider(17)(q10)t(15;17) is higher, in 9/12 cases. Since tumor protein
p53 (TP53) is located on 17p, the ider(17)(q10)t(15;17) may provide
a growth advantage to the relevant clone (85). The long type PMA/RARα is prevalent in
this type (57%). Bcr1 splicing PML exon 5–6 was associated with
decreased sensitivity to ATRA (98),
which may explain the ATRA resistance of ider(17) patients. Since
the loss of TP53 and absence of PML exon 5 may occur in this rare
subtype and present data have identified a trend of poor outcome,
ider(17) is a candidate for further study.
The nuclear receptor binding SET domain protein 3,
lysine acetyltransferase 6A and fibroblast growth factor receptor 1
which regulate cell transcription (99) and are associated with the stem cell
myeloproliferative disorder are candidate genes involved with the
loss of 8p (100). Otero et
al (101) reported a patient
with APL with dicentric t(8;13)(q10;q10) who succumbed due to a
central nervous system hemorrhage on day 3 with ATRA based primary
therapy. The authors hypothesized that the additional chromosomal
changes were directly associated with the patient's prognosis, and
that the novel chromosomal abnormalities may predict a poor outcome
of APL.
Between 1 and 2% of APL cases are due to abnormal
translocations including zinc finger and BTB domain containing 16
(ZBTB16)/RARα, nucleophosmin (NPM)/RARα, nuclear matrix
associated/RARα, signal transducer and activator of transcription
5B (STAT5B)/RARα, protein kinase CAMP-dependent type 1 regulatory
subunit α/RARα, BCL6 corepressor/RARα and factor interacting with
PAPOLA and SPSF1/RARα, and all these translocations involve RARA
(70). A review by Adams and Nassiri
(102) discussed the various
translocations of APL and identified certain features. t(5;17)
NPM/RARα has been diagnosed in patients younger than 10 years which
is uncommon to normal APL (103). It
responds well to ATRA but has higher risk of relapse. Diagnosis of
ZBTB16/RARα t(11;17) APL can be difficult. This translocation is
more commonly associated with CD56 expression (104). Patients had an increased number of
hypogranular pelgeroid neutrophils and a more regular nucleus
compared with the bilobed nucleus typically found in APL (97). The majority of the translocations in
APL can be successfully treated with ATRA/ATO, while patients with
ZBTB16/RARα and STAT5B/RARα are resistant to ATRA and experienced a
poor prognosis (105). There is
currently limited data regarding the prognosis of patients with
abnormal translocations. ZBTB16/RARα and STAT5B/RARα are associated
with ED, while other variant translocations may result in a similar
outcome to cases with t(15;17) APL.
As an important member of the LEF/T-cell factor
(TCF) family, LEF1 regulates cellular proliferation and cell cycle
regulation (106). It is
traditionally regarded as a central mediator of the wingless-type
(Wnt) signaling pathway (106,107)
and may serve a function in development and cancerogenesis, control
self-renewal, cell proliferation and differentiation (108). Previous data reveal that it may
serve a vital function in early hematopoiesis and leukemic
transformation in murine models (109). However, certain functions
independent of wnt signaling have also been reported (107,110). A
study of 78 adult patients with APL (111) suggests a novel mechanism whereby
LEF1 serves a specific function in the Notch signaling pathway and
draws the conclusion that patients with APL overexpressing LEF1 are
more likely to experience a favorable outcome.
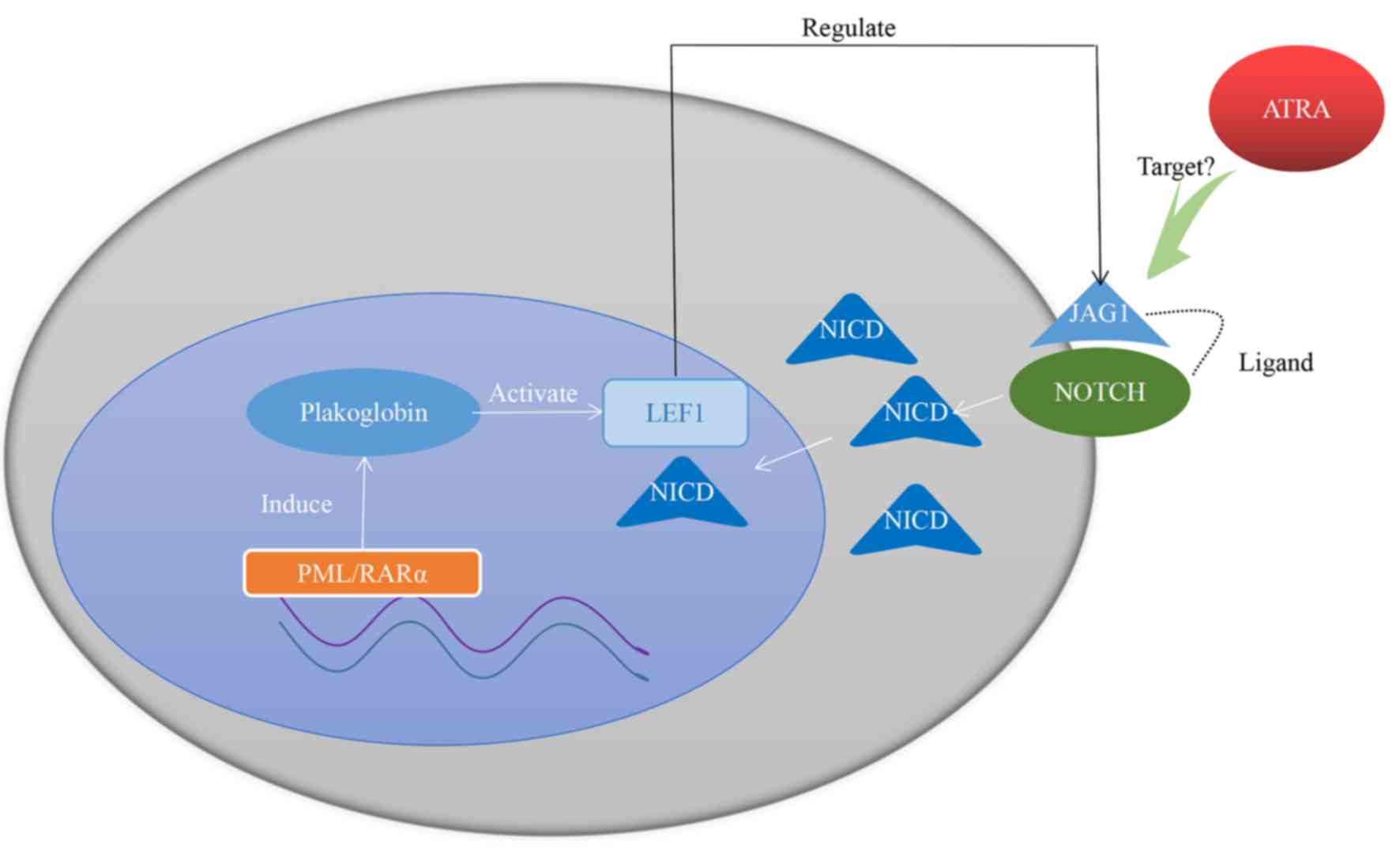
In the nucleus, PML/RARα fusion gene is able to
induce plakoglobin (γ-catenin) expression in primary patient
samples as well as in cell lines, leading to transcriptional
activation of LEF1 (112). LEF1
itself is a coactivator of Notch intracellular domain (107). Jagged1 (JAG1) is a downstream target
gene of LEF1 and is also the ligand of Notch (113). LEF1 is able to crosstalk with the
Notch signaling pathway by regulating the expression of JAG1 on the
cytomembrane (112). Furthermore,
JAG1 is more frequently expressed in APL and, when receiving ATRA
therapy, JAG1 is downregulated in the NB4 cell line (114,115).
Taken together, these studies indicate that JAG1 may be a
therapeutic target of ATRA, with high expression of LEF1 promoting
the curative effect of ATRA. The hypothesized mechanism is
presented in Fig. 1.
In trials, 103 newly diagnosed APL patients were
observed and treated with the AIDA-0493 (116) and AIDA-2000 (117) protocols between January 1996 and
December 2012. The median follow-up time was 5.7 years. Patients
were divided in two groups according to the expression level of
LEF1: A low LEF1 group with LEF1 values below the median value
(<2.1 fold-change) and a high LEF1 group with LEF1 values above
the median value (>2.1 fold-change). Fisher's exact test for
categorical data and the nonparametric Mann-Whitney U test for
continuous variables were used to identify the difference between
two groups. Survival curves and influence factors of survival
endpoints were measured by the Kaplan-Meier method and multivariate
Cox proportional hazards models accordingly. They demonstrated that
the LEF-high group exhibited lower WBC counts (P<0.0001),
trended towards a younger age (P=0.08), and presented more frequent
FLT3-ITD mutations (P=0.02). ED only occurred in the LEF-low group
(n=9; P=0.002). This suggests that the expression of LEF may be
studied as a novel marker of risk in APL if similar results can be
confirmed by further studies.
In conclusion, published data has been reviewed
with a focus on the factors associated with ED. When treated with
ATO as primary treatment, the FLT3-ITD has no impact on ED. Low LEF
expression may be a reliable marker of ED and a therapeutic target
if it can be proven by further studies. CD56+ and CD34+/CD2+ may be
candidates to select high-risk patients. High-risk patients still
cannot be identified via the cell surface makers, despite a number
of studies analyzing their prognostic significance. Complex
translocations did not reduce the EDR in APL; however, if an
abnormal karyotype [e.g., Ide(17), ZBTB16/RARα and STAT5B/RARα]
appeared singularly or as part of a complex mutation, there is a
high possibility of early mortality if clinicians are unable to
identify or monitor it.
|
1
|
Tallman MS and Altman JK: How I treat
acute promyelocytic leukemia. Blood. 114:5126–5135. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bennett JM, Catovsky D, Daniel MT,
Flandrin G, Galton DA, Gralnick HR and Sultan C: A variant form of
hypergranular promyelocytic leukaemia (M3). Br J Haematol.
44:169–170. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cooperberg AA and Neiman GM:
Fibrinogenopenia and fibrinolysis in acute myelogenous leukemia.
Ann Intern Med. 42:706–711. 1955. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Van Creveld S and Mochtar IA: Fibrinolysis
in acute leukemia. Maandschr Kindergeneeskd. 27:133–44. 1959.(In
Dutch). PubMed/NCBI
|
|
5
|
Ghitis J: Acute promyelocytic leukemia?
Blood. 21:237–240. 1963.PubMed/NCBI
|
|
6
|
Larson RA, Kondo K, Vardiman JW, Butler
AE, Golomb HM and Rowley JD: Evidence for a 15;17 translocation in
every patient with acute promyelocytic leukemia. Am J Med.
76:827–841. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tallman MS, Andersen JW, Schiffer CA,
Appelbaum FR, Feusner JH, Woods WG, Ogden A, Weinstein H, Shepherd
L, Willman C, et al: All-trans retinoic acid in acute promyelocytic
leukemia: Long-term outcome and prognostic factor analysis from the
North American Intergroup protocol. Blood. 100:4298–4302. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park JH, Qiao B, Panageas KS, Schymura MJ,
Jurcic JG, Rosenblat TL, Altman JK, Douer D, Rowe JM and Tallman
MS: Early death rate in acute promyelocytic leukemia remains high
despite all-trans retinoic acid. Blood. 118:1248–1254. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Watts JM and Tallman MS: Acute
promyelocytic leukemia: What is the new standard of care? Blood
Rev. 28:205–212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Iland HJ, Bradstock K, Supple SG, Catalano
A, Collins M, Hertzberg M, Browett P, Grigg A, Firkin F, Hugman A,
et al: All-trans-retinoic acid, idarubicin, and IV arsenic trioxide
as initial therapy in acute promyelocytic leukemia (APML4). Blood.
120:1570–1580. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matthews W, Jordan CT, Wiegand GW, Pardoll
D and Lemischka IR: A receptor tyrosine kinase specific to
hematopoietic stem and progenitor cell-enriched populations. Cell.
65:1143–1152. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meshinchi S, Alonzo TA, Stirewalt DL,
Zwaan M, Zimmerman M, Reinhardt D, Kaspers GJ, Heerema NA, Gerbing
R, Lange BJ and Radich JP: Clinical implications of FLT3 mutations
in pediatric AML. Blood. 108:3654–3661. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thiede C, Steudel C, Mohr B, Schaich M,
Schäkel U, Platzbecker U, Wermke M, Bornhäuser M, Ritter M,
Neubauer A, et al: Analysis of FLT3-activating mutations in 979
patients with acute myelogenous leukemia: Association with FAB
subtypes and identification of subgroups with poor prognosis.
Blood. 99:4326–4335. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kottaridis PD, Gale RE, Frew ME, Harrison
G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett
AK, et al: The presence of a FLT3 internal tandem duplication in
patients with acute myeloid leukemia (AML) adds important
prognostic information to cytogenetic risk group and response to
the first cycle of chemotherapy: Analysis of 854 patients from the
United Kingdom Medical Research Council AML 10 and 12 trials.
Blood. 98:1752–1759. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shih LY, Kuo MC, Liang DC, Huang CF, Lin
TL, Wu JH, Wang PN, Dunn P and Lai CL: Internal tandem duplication
and Asp835 mutations of the FMS-like tyrosine kinase 3 (FLT3) gene
in acute promyelocytic leukemia. Cancer. 98:1206–1216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stock W, Najib K, Moser BK, Powell BL,
Holowka N, Gulati K, Bloomfield CD, Larson RA and Sher D: High
incidence of FLT3 mutations in adults with acute promyelocytic
leukemia (APL): Correlation with diagnostic features and treatment
outcome (CALGB 9710). J Clin Oncol. 26 15 Suppl:S70022008.
View Article : Google Scholar
|
|
17
|
Souza Melo CP, Campos CB, Dutra ÁP, Neto
JC, Fenelon AJ, Neto AH, Carbone EK, Pianovski MA, Ferreira AC and
Assumpcão JG: Correlation between FLT3-ITD status and clinical,
cellular and molecular profiles in promyelocytic acute leukemias.
Leuk Res. 39:131–137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Au WY, Fung A, Chim CS, Lie AK, Liang R,
Ma ES, Chan CH, Wong KF and Kwong YL: FLT-3, aberrations in acute
promyelocytic leukaemia: Clinicopathological associations and
prognostic impact. Br J Haematol. 125:463–469. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Callens C, Chevret S, Cayuela JM, Cassinat
B, Raffoux E, de Botton S, Thomas X, Guerci A, Fegueux N, Pigneux
A, et al: Prognostic implication of FLT3 and Ras gene mutations in
patients with acute promyelocytic leukemia (APL): A retrospective
study from the European APL Group. Leukemia. 19:1153–1160. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gale RE, Hills R, Pizzey AR, Kottaridis
PD, Swirsky D, Gilkes AF, Nugent E, Mills KI, Wheatley K, Solomon
E, et al: Relationship between FLT3 mutation status, biologic
characteristics, and response to targeted therapy in acute
promyelocytic leukemia. Blood. 106:3768–3776. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chillón MC, Santamaría C, García-Sanz R,
Balanzategui A, Sarasquete ME, Alcoceba M, Marín L, Caballero MD,
Vidriales MB, Ramos F, et al: Long FLT3 internal tandem
duplications and reduced PML-RARα expression at diagnosis
characterize a high-risk subgroup of acute promyelocytic leukemia
patients. Haematologica. 95:745–751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kiyoi H, Naoe T, Yokota S, Nakao M, Minami
S, Kuriyama K, Takeshita A, Saito K, Hasegawa S, Shimodaira S, et
al: Internal tandem duplication of FLT3 associated with
leukocytosis in acute promyelocytic leukemia. Leukemia study group
of the ministry of health and welfare (Kohseisho). Leukemia.
11:1447–1452. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Noguera NI, Breccia M, Divona M, Diverio
D, Costa V, De Santis S, Avvisati G, Pinazzi MB, Petti MC, Mandelli
F and Lo Coco F: Alterations of the FLT3 gene in acute
promyelocytic leukemia: Association with diagnostic characteristics
and analysis of clinical outcome in patients treated with the
Italian AIDA protocol. Leukemia. 16:2185–2189. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mathews V, Thomas M, Srivastava VM, George
B, Srivastava A and Chandy M: Impact of FLT3 mutations and
secondary cytogenetic changes on the outcome of patients with newly
diagnosed acute promyelocytic leukemia treated with a single agent
arsenic trioxide regimen. Haematologica. 92:994–995. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schnittger S, Bacher U, Haferlach C, Kern
W, Alpermann T and Haferlach T: Clinical impact of FLT3 mutation
load in acute promyelocytic leukemia with t(15;17)/PML-RARA.
Haematologica. 96:1799–1807. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lucena-Araujo AR, Kim HT, Jacomo RH, Melo
RA, Bittencourt R, Pasquini R, Pagnano K, Fagundes EM, Chauffaille
Mde L, Chiattone CS, et al: Internal tandem duplication of the FLT3
gene confers poor overall survival in patients with acute
promyelocytic leukemia treated with all-trans retinoic acid and
anthracycline-based chemotherapy: An international consortium on
acute promyelocytic leukemia study. Ann Hematol. 93:2001–2010.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Poiré X, Moser BK, Gallagher RE, Laumann
K, Bloomfield CD, Powell BL, Koval G, Gulati K, Holowka N, Larson
RA, et al: Arsenic trioxide in front-line therapy of acute
promyelocytic leukemia (C9710): Prognostic significance of FLT3
mutations and complex karyotype. Leuk Lymphoma. 55:1523–1532. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Daver N, Kantarjian H, Marcucci G, Pierce
S, Brandt M, Dinardo C, Pemmaraju N, Garcia-Manero G, O'Brien S,
Ferrajoli A, et al: Clinical characteristics and outcomes in
patients with acute promyelocytic leukaemia and hyperleucocytosis.
Br J Haematol. 168:646–653. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kainz B, Heintel D, Marculescu R,
Schwarzinger I, Sperr W, Le T, Weltermann A, Fonatsch C, Haas OA,
Mannhalter C, et al: Variable prognostic value of FLT3 internal
tandem duplications in patients with de novo AML and a normal
karyotype, t(15;17), t(8;21) or inv(16). Hematol J. 3:283–289.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Barragán E, Montesinos P, Camos M,
González M, Calasanz MJ, Román-Gómez J, Gómez-Casares MT, Ayala R,
López J, Fuster Ó, et al: Prognostic value of FLT3 mutations in
patients with acute promyelocytic leukemia treated with all-trans
retinoic acid and anthracycline monochemotherapy. Haematologica.
96:1470–1477. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Breccia M, Loglisci G, Loglisci MG, Ricci
R, Diverio D, Latagliata R, Foà R and Lo-Coco F: FLT3-ITD confers
poor prognosis in patients with acute promyelocytic leukemia
treated with AIDA protocols: Long-term follow-up analysis.
Haematologica. 98:e161–e163. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Beitinjaneh A, Jang S, Roukoz H and
Majhail NS: Prognostic significance of FLT3 internal tandem
duplication and tyrosine kinase domain mutations in acute
promyelocytic leukemia: A systematic review. Leuk Res. 34:831–836.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hu J, Liu YF, Wu CF, Xu F, Shen ZX, Zhu
YM, Li JM, Tang W, Zhao WL, Wu W, et al: Long-term efficacy and
safety of all-trans retinoic acid/arsenic trioxide-based therapy in
newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci
USA. 106:pp. 3342–3347. 2009; View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shaft D, Shtalrid M, Berebi A, Catovsky D
and Resnitzky P: Ultrastructural characteristics and lysozyme
content in hypergranular and variant type of acute promyelocytic
leukaemia. Br J Haematol. 103:729–739. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mandelli F, Diverio D, Avvisati G, Luciano
A, Barbui T, Bernasconi C, Broccia G, Cerri R, Falda M, Fioritoni
G, et al: Molecular remission in PML/RAR alpha-positive acute
promyelocytic leukemia by combined all-trans retinoic acid and
idarubicin (AIDA) therapy. Gruppo Italiano-Malattie Ematologiche
Maligne dell'Adulto and Associazione Italiana di Ematologia ed
Oncologia Pediatrica Cooperative Groups. Blood. 90:1014–1021.
1997.PubMed/NCBI
|
|
36
|
McKenna RW, Parkin J, Bloomfield CD,
Sundberg RD and Brunning RD: Acute promyelocytic leukaemia: A study
of 39 cases with identification of a hyperbasophilic microgranular
variant. Br J Haematol. 50:201–214. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tallman MS, Kim HT, Montesinos P,
Appelbaum FR, de la Serna J, Bennett JM, Deben G, Bloomfield CD,
Gonzalez J, Feusner JH, et al: Does microgranular variant
morphology of acute promyelocytic leukemia independently predict a
less favorable outcome compared with classical M3 APL? A joint
study of the North American Intergroup and the PETHEMA Group.
Blood. 116:5650–5659. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kutny MA, Moser BK, Laumann K, Feusner JH,
Gamis A, Gregory J, Larson RA, Powell BL, Stock W, Willman CL, et
al: FLT3 mutation status is a predictor of early death in pediatric
acute promyelocytic leukemia: A report from the Children's Oncology
Group. Pediatr Blood Cancer. 59:662–667. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Biondi A, Luciano A, Bassan R, Mininni D,
Specchia G, Lanzi E, Castagna S, Cantù-Rajnoldi A, Liso V, Masera
G, et al: CD2 expression in acute promyelocytic leukemia is
associated with microgranular morphology (FAB M3v) but not with any
PML gene breakpoint. Leukemia. 9:1461–1466. 1995.PubMed/NCBI
|
|
40
|
Foley R, Soamboonsrup P, Carter RF, Benger
A, Meyer R, Walker I, Wan Y, Patterson W, Orzel A, Sunisloe L, et
al: CD34-positive acute promyelocytic leukemia is associated with
leukocytosis, microgranular/hypogranular morphology, expression of
CD2 and bcr3 isoform. Am J Hematol. 67:34–41. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Paietta E, Goloubeva O, Neuberg D, Bennett
JM, Gallagher R, Racevskis J, Dewald G, Wiernik PH and Tallman MS;
Eastern Cooperative Oncology Group, : A surrogate marker profile
for PML/RAR alpha expressing acute promyelocytic leukemia and the
association of immunophenotypic markers with morphologic and
molecular subtypes. Cytometry B Clin Cytom. 59:1–9. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Maslak P, Miller WH Jr, Heller G,
Scheinberg DA, Dmitrovsky E and Warrell RP Jr: CD2 expression and
PML/RAR-alpha transcripts in acute promyelocytic leukemia. Blood.
81:16661993.PubMed/NCBI
|
|
43
|
Reading CL, Estey EH, Huh YO, Claxton DF,
Sanchez G, Terstappen LW, O'Brien MC, Baron S and Deisseroth AB:
Expression of unusual immunophenotype combinations in acute
myelogenous leukemia. Blood. 81:3083–3090. 1993.PubMed/NCBI
|
|
44
|
Claxton DF, Reading CL, Nagarajan L,
Tsujimoto Y, Andersson BS, Estey E, Cork A, Huh YO, Trujillo J and
Deisseroth AB: Correlation of CD2 expression with PML gene
breakpoints in patients with acute promyelocytic leukemia. Blood.
80:582–586. 1992.PubMed/NCBI
|
|
45
|
Albano F, Mestice A, Pannunzio A, Lanza F,
Martino B, Pastore D, Ferrara F, Carluccio P, Nobile F, Castoldi G,
et al: The biological characteristics of CD34+ CD2+ adult acute
promyelocytic leukemia and the CD34 CD2 hypergranular (M3) and
microgranular (M3v) phenotypes. Haematologica. 91:311–316.
2006.PubMed/NCBI
|
|
46
|
Guglielmi C, Martelli MP, Diverio D, Fenu
S, Vegna ML, Cantù-Rajnoldi A, Biondi A, Cocito MG, Del Vecchio L,
Tabilio A, et al: Immunophenotype of adult and childhood acute
promyelocytic leukaemia: Correlation with morphology, type of PML
gene breakpoint and clinical outcome. A cooperative Italian study
on 196 cases. Br J Haematol. 102:1035–1041. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gallagher RE, Willman CL, Slack JL,
Andersen JW, Li YP, Viswanatha D, Bloomfield CD, Appelbaum FR,
Schiffer CA, Tallman MS and Wiernik PH: Association of PML-RAR
alpha fusion mRNA type with pretreatment hematologic
characteristics but not treatment outcome in acute promyelocytic
leukemia: An intergroup molecular study. Blood. 90:1656–1663.
1997.PubMed/NCBI
|
|
48
|
Davey FR, Davis RB, MacCallum JM, Nelson
DA, Mayer RJ, Ball ED, Griffin JD, Schiffer CA and Bloomfield CD:
Morphologic and cytochemical characteristics of acute promyelocytic
leukemia. Am J Hematol. 30:221–227. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bassan R, Battista R, Viero P, d'Emilio A,
Buelli M, Montaldi A, Rambaldi A, Tremul L, Dini E and Barbui T:
Short-term treatment for adult hypergranular and microgranular
acute promyelocytic leukemia. Leukemia. 9:238–243. 1995.PubMed/NCBI
|
|
50
|
Cunningham I, Gee TS, Reich LM, Kempin SJ,
Naval AN and Clarkson BD: Acute promyelocytic leukemia: Treatment
results during a decade at Memorial Hospital. Blood. 73:1116–1122.
1989.PubMed/NCBI
|
|
51
|
Kuchenbauer F, Schoch C, Kern W, Hiddemann
W, Haferlach T and Schnittger S: Impact of FLT3, mutations and
promyelocytic leukaemia-breakpoint on clinical characteristics and
prognosis in acute promyelocytic leukaemia. Br J Haematol.
130:196–202. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sohal J, Phan VT, Chan PV, Davis EM, Patel
B, Kelly LM, Abrams TJ, O'Farrell AM, Gilliland DG, Le Beau MM and
Kogan SC: A model of APL with FLT3 mutation is responsive to
retinoic acid and a receptor tyrosine kinase inhibitor, SU11657.
Blood. 101:3188–3197. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Haferlach T, Kohlmann A, Schnittger S,
Dugas M, Hiddemann W, Kern W and Schoch C: AML M3 and AML M3
variant each have a distinct gene expression signature but also
share patterns different from other genetically defined AML
subtypes. Genes Chromosomes Cancer. 43:113–127. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Marasca R, Maffei RP, Zucchini P, Castelli
I, Saviola A, Martinelli S, Ferrari A, Fontana M, Ravanetti S and
Torelli G: Gene expression profiling of acute promyelocytic
leukaemia identifies two subtypes mainly associated with flt3
mutational status. Leukemia. 20:103–114. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R,
Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C,
et al: Activating mutation of D835 within the activation loop of
FLT3 in human hematologic malignancies. Blood. 97:2434–2439. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Baer MR, Stewart CC, Lawrence D, Arthur
DC, Byrd JC, Davey FR, Schiffer CA and Bloomfield CD: Expression of
the neural cell adhesion molecule CD56 is associated with short
remission duration and survival in acute myeloid leukemia with
t(8;21)(q22;q22). Blood. 90:1643–1648. 1997.PubMed/NCBI
|
|
57
|
Montesinos P, Rayón C, Vellenga E, Brunet
S, González J, González M, Holowiecka A, Esteve J, Bergua J,
González JD, et al: Clinical significance of CD56 expression in
patients with acute promyelocytic leukemia treated with all-trans
retinoic acid and anthracycline-based regimens. Blood.
117:1799–1805. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ferrara F, Morabito F, Martino B, Specchia
G, Liso V, Nobile F, Boccuni P, Di Noto R, Pane F, Annunziata M, et
al: CD56 expression is an indicator of poor clinical outcome in
patients with acute promyelocytic leukemia treated with
simultaneous all-trans-retinoic acid and chemotherapy. J Clin
Oncol. 18:1295–1300. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ito S, Ishida Y, Oyake T, Satoh M, Aoki Y,
Kowata S, Uchiyama T, Enomoto S, Sugawara T, Numaoka H, et al:
Clinical and biological significance of CD56 antigen expression in
acute promyelocytic leukemia. Leuk Lymphoma. 45:1783–1789. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Murray CK, Estey E, Paietta E, Howard RS,
Edenfield WJ, Pierce S, Mann KP, Bolan C and Byrd JC: CD56
expression in acute promyelocytic leukemia: A possible indicator of
poor treatment outcome? J Clin Oncol. 17:293–297. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Breccia M, De Propris MS, Minotti C,
Stefanizzi C, Raponi S, Colafigli G, Latagliata R, Guarini A and
Foà R: Aberrant phenotypic expression of CD15 and CD56 identifies
poor prognostic acute promyelocytic leukemia patients. Leuk Res.
38:194–197. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hills RK, Castaigne S, Appelbaum FR,
Delaunay J, Petersdorf S, Othus M, Estey EH, Dombret H, Chevret S,
Ifrah N, et al: Addition of gemtuzumab ozogamicin to induction
chemotherapy in adult patients with acute myeloid leukaemia: A
meta-analysis of individual patient data from randomised controlled
trials. Lancet Oncol. 15:986–996. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Breccia M, De Propris MS, Stefanizzi C,
Raponi S, Molica M, Colafigli G, Minotti C, Latagliata R, Diverio
D, Guarini A and Foà R: Negative prognostic value of CD34 antigen
also if expressed on a small population of acute promyelocitic
leukemia cells. Ann Hematol. 93:1819–1823. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Paietta E: Expression of cell-surface
antigens in acute promyelocytic leukaemia. Best Pract Res Clin
Haematol. 16:369–385. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Xu F, Yin CX, Wang CL, Jiang XJ, Jiang L,
Wang ZX, Yi ZS, Huang KK and Meng FY: Immunophenotypes and immune
markers associated with acute promyelocytic leukemia prognosis. Dis
Markers. 2014:4219062014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ahmad EI, Akl HK, Hashem ME and Elgohary
TA: The biological characteristics of adult CD34+ acute
promyelocytic leukemia. Med Oncol. 29:1119–1126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Grimwade D, Outram SV, Flora R, Ings SJ,
Pizzey AR, Morilla R, Craddock CF, Linch DC and Solomon E: The
T-lineage-affiliated CD2 gene lies within an open chromatin
environment in acute promyelocytic leukemia cells. Cancer Res.
62:4730–4735. 2002.PubMed/NCBI
|
|
68
|
Sunter NJ, Scott K, Hills R, Grimwade D,
Taylor S, Worrillow LJ, Fordham SE, Forster VJ, Jackson G, Bomken
S, et al: A functional variant in the core promoter of the CD95
cell death receptor gene predicts prognosis in acute promyelocytic
leukemia. Blood. 119:196–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Rowley JD, Golomb HM and Dougherty C:
15/17 translocation, a consistent chromosomal change in acute
promyelocytic leukaemia. Lancet. 1:549–550. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Grimwade D, Biondi A, Mozziconacci MJ, et
al: Characterisation of acute promyelocytic leukaemia (APL) cases
lacking the classical t(15;17). Results of the European working
party. 92:677A. 1998.
|
|
71
|
Schoch C, Haferlach T, Haase D, Fonatsch
C, Löffler H, Schlegelberger B, Staib P, Sauerland MC, Heinecke A,
Büchner T, et al: Patients with de novo, acute myeloid leukaemia
and complex karyotype aberrations show a poor prognosis despite
intensive treatment: A study of 90 patients. Br J Haematol.
112:118–126. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Grimwade D, Howe K, Langabeer S, Davies L,
Oliver F, Walker H, Swirsky D, Wheatley K, Goldstone A, Burnett A
and Solomon E: Establishing the presence of the t(15;17) in
suspected acute promyelocytic leukaemia: Cytogenetic, molecular and
PML immunofluorescence assessment of patients entered into the
M.R.C. ATRA trial. M.R.C. Adult Leukaemia Working Party. ATRA
trial. Br J Haematol. 94:557–573. 1996.PubMed/NCBI
|
|
73
|
De Botton S, Chevret S, Sanz M, Dombret H,
Thomas X, Guerci A, Fey M, Rayon C, Huguet F, Sotto JJ, et al:
Additional chromosomal abnormalities in patients with acute
promyelocytic leukaemia (APL) do not confer poor prognosis: Results
of APL 93 trial. Br J Haematol. 111:801–806. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Slack JL, Arthur DC, Lawrence D, Mrózek K,
Mayer RJ, Davey FR, Tantravahi R, Pettenati MJ, Bigner S, Carroll
AJ, et al: Secondary cytogenetic changes in acute promyelocytic
leukemia-Prognostic importance in patients treated with
chemotherapy alone and association with the intron 3 breakpoint of
the PML gene: A cancer and leukemia group B study. J Clin Oncol.
15:1786–1795. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mi Y, Xue Y, Yu W, Liu S, Zhao Y, Meng Q,
Bian S and Wang J: Therapeutic experience of adult acute myeloid
leukemia in a single institution of China and its relationship with
chromosome karyotype. Leuk Lymphoma. 49:524–530. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Pantic M, Novak A, Marisavljevic D,
Djordjevic V, Elezovic I, Vidovic A and Colovic M: Additional
chromosome aberrations in acute promyelocytic leukemia:
Characteristics and prognostic influence. Med Oncol. 17:307–313.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lo-Coco F, Avvisati G, Vignetti M, Thiede
C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona
E, et al: Retinoic acid and arsenic trioxide for acute
promyelocytic leukemia. N Engl J Med. 369:112–21. 2013. View Article : Google Scholar
|
|
78
|
Samir MD, Pedro H and Ling Z: Tetraploidy
acute promyelocytic leuemia with double t(15;17)/PML-RARA, a case
report with review of literature. Genes Chromosomes Cancer.
46:635–643. 2007.PubMed/NCBI
|
|
79
|
Neto WK, Serpa M, Sanabani SS, Bueno PT,
Velloso ED, Dorlhiac-Llacer PE and Bendit I: Early detection of
t(8;21) chromosomal translocations during treatment of PML-RARA
positive acute promyelocytic leukemia: A case study. Clin Med
Insights Oncol. 4:163–170. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Charrin C, Ritouet D, Campos L, Devaux Y,
Archimbaud E, Fraisse J, Fiere D and Germain D: Association of
t(15;17) and t(8;21) in the initial phase of an acute promyelocytic
leukemia. Cancer Genet Cytogenet. 58:177–180. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Bonomi R, Giordano H, del Pilar Moreno M,
Bodega E, Landoni AI, Gallagher R and del Rosario Uriarte M:
Simultaneous PML/RARalpha and AML1/ETO expression with t(15;17) at
onset and relapse with only t(8;21) in an acute promyelocytic
leukemia patient. Cancer Genet Cytogenet. 123:41–43. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Varella-Garcia M, Brizard F, Roche J,
Flandrin G, Drabkin H and Brizard A: Aml1/ETO and Pml/RARA
rearrangements in a case of AML-M2 acute myeloblastic leukemia with
t(15;17). Leuk Lymphoma. 33:403–406. 2009. View Article : Google Scholar
|
|
83
|
Xu L, Zhao WL, Xiong SM, Su XY, Zhao M,
Wang C, Gao YR, Niu C, Cao Q, Gu BW, et al: Molecular cytogenetic
characterization and clinical relevance of additional, complex
and/or variant chromosome abnormalities in acute promyelocytic
leukemia. Leukemia. 15:1359–1368. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Uz B, Eliaçık E, Işık A, Aksu S, Büyükaşık
Y, Haznedaroğlu IC, Göker H, Sayınalp N and Ozcebe Oİ:
Co-expression of t(15;17) and t(8;21) in a case of acute
Promyelocytic leukemia: Review of the literature. Turk J Haematol.
30:400–404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Hu X, Ai G, Meng X, Hou J, Wei R, Tao Y,
Zhang Q, Han Y and Shi J: An ider(17)(q10)t(15;17) with spliced
long-type PML-RARA fusion transcripts in a case of acute
promyelocytic leukemia. Cancer Genet. 207:253–257. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lee GY, Christina S, Tien SL, Ghafar AB,
Hwang W, Lim LC and Lim TH: Acute promyelocytic leukemia with
PML-RARA fusion on i(17q) and therapy-related acute myeloid
leukemia. Cancer Genet Cytogenet. 159:129–136. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Im SA, Kim SH, Lee MA, Ahn JY, Yoo ES,
Choi DY, Lee JY, Lee S, Huh JW, Chung WS, et al: Identification of
ider[17q] in addition to t[15;17] in acute promyelocytic leukemia
using whole chromosome painting probes made by interspecies hybrid
using inter-Alu PCR. Cancer Genet Cytogenet. 118:169–170. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kim MJ, Cho SY, Lim G, Yoon HS, Lee HJ,
Suh JT, Lee J, Lee WI, Cho KS and Park TS: A rare case of
Microgranular acute Promyelocytic leukemia associated with
ider(17)(q10)t(15;17) in an old-age patient. Korean J Lab Med.
31:86–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kim M, Lee SA, Park HI, Oh EJ, Park CW,
Lim J, Han K and Kim Y: Two distinct clonal populations in acute
promyelocytic leukemia, one involving chromosome 17 and the other
involving an isochromosome 17. Cancer Genet Cytogenet. 197:185–188.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kim MJ, Yoon HS, Cho SY, Lee HJ, Suh JT,
Lee J, Yoon HJ, Lee WI and Park TS: ider(17)(q10)t(15;17)
associated with relapse and poor prognosis in a pediatric patient
with acute promyelocytic leukemia. Cancer Genet Cytogenet.
201:116–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Manola KN, Karakosta M, Sambani C,
Terzoudi G, Pagoni M, Gatsa E and Papaioannou M: Isochromosome
der(17)(q10)t(15;17) in acute promyelocytic leukemia resulting in
an additional copy of the RARA-PML fusion gene: Report of 4 cases
and review of the literature. Acta Haematol. 123:162–170. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Okoshi Y, Akiyama H, Kono N, Matsumura T,
Mizuchi D, Mori S, Ohashi K and Sakamaki H: Effect of additional
chromosomal abnormalities in acute promyelocytic leukemia treated
with all-trans-retinoic acid: A report of 17 patients. Int J
Hematol. 73:496–501. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Qiu HR, Li JY, Miao KR, Wang R and Xu W:
Clinical and laboratory studies of an acute promyelocytic leukemia
patient with double ider(17q) chromosome aberration. Cancer Genet
Cytogenet. 184:74–75. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Schoch C, Haase D, Haferlach T, Freund M,
Link H, Lengfelder E, Löffler H, Büchner T and Fonatsch C:
Incidence and implication of additional chromosome aberrations in
acute promyelocytic leukaemia with translocation t(15;17)(q22;q21):
A report on 50 patients. Br J Haematol. 94:493–500. 2015.
View Article : Google Scholar
|
|
95
|
Tong H, Li K, Mei C, Wang H, Chen Z and
Jin J: Arsenic trioxide may improve the prognosis of APL with
ider(17)(q10): Report of a rare adult case of acute promyelocytic
leukemia with ider(17)(q10)t(15;17) showing poor response to
all-trans retinoic acid. Ann Hematol. 90:1493–1494. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Wan TS, So CC, Hui KC, Yip SF, Ma ES and
Chan LC: Diagnostic utility of dual fusion PML/RARalpha
translocation DNA probe (D-FISH) in acute promyelocytic leukemia.
Oncol Rep. 17:799–805. 2007.PubMed/NCBI
|
|
97
|
Sainty D, Liso V, Cantù-Rajnoldi A, Head
D, Mozziconacci MJ, Arnoulet C, Benattar L, Fenu S, Mancini M,
Duchayne E, et al: A new morphologic classification system for
acute promyelocytic leukemia distinguishes cases with underlying
PLZF/RARA gene rearrangements. Blood. 96:1287–1296. 2000.PubMed/NCBI
|
|
98
|
Tan Y, Bian S, Xu Z, Chen X, Qi X, Ren F,
Li L, Guo H, Xu A, Zhang L and Wang H: The short isoform of the
long-type PML-RARA, fusion gene in acute promyelocytic leukaemia
lacks sensitivity to all-trans-retinoic acid. Br J Haematol.
162:93–97. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Rosati R, La Starza R, Veronese A, Aventin
A, Schwienbacher C, Vallespi T, Negrini M, Martelli MF and Mecucci
C: NUP98 is fused to the NSD3 gene in acute myeloid leukemia
associated with t(8;11)(p11.2;p15). Blood. 99:3857–3860. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ågerstam H, Lilljebjörn H, Lassen C,
Swedin A, Richter J, Vandenberghe P, Johansson B and Fioretos T:
Fusion gene-mediated truncation of RUNX1, as a potential mechanism
underlying disease progression in the 8p11 myeloproliferative
syndrome. Genes Chromosomes Cancer. 46:635–643. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Otero L, Terra B, Diniz C, Abdelhay E and
Fernandez Tde S: Dicentric t(8;13)(q10;q10) as an additional
chromosomal abnormality in a case of acute promyelocytic leukemia
with very poor outcome. Leuk Lymphoma. 50:287–289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Adams J and Nassiri M: Acute promyelocytic
Leukemia: A review and discussion of variant translocations. Arch
Pathol Lab Med. 139:1308–1313. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Corey SJ, Locker J, Oliveri DR,
Shekhter-Levin S, Redner RL, Penchansky L and Gollin SM: A
non-classical translocation involving 17q12 (retinoic acid receptor
alpha) in acute promyelocytic leukemia (APML) with atypical
features. Leukemia. 8:1350–1353. 1994.PubMed/NCBI
|
|
104
|
Yamanouchi J, Hato T, Niiya T, Miyoshi K,
Azuma T, Sakai I and Yasukawa M: A new four-way variant
t(5;17;15;20)(q33;q12;q22;q11.2) in acute promyelocytic leukemia.
Int J Hematol. 94:395–398. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Qiu HR, Li JY, Miao KR, Wang R, Zhang JF
and Xu W: A case of acute promyelocytic leukemia with variant
t(5;17) and trisomy 22. Zhonghua Yi Xue Yi Chuan Xue Za Zhi.
25:430–433. 2008.(In Chinese). PubMed/NCBI
|
|
106
|
Messmer BT, Messmer D, Allen SL, Kolitz
JE, Kudalkar P, Cesar D, Murphy EJ, Koduru P, Ferrarini M, Zupo S,
et al: In vivo measurements document the dynamic cellular kinetics
of chronic lymphocytic leukemia B cells. J Clin Invest.
115:755–764. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ross DA and Kadesch T: The notch
intracellular domain can function as a coactivator for LEF-1. Mol
Cell Biol. 21:7537–7544. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Holland JD, Klaus A, Garratt AN and
Birchmeier W: Wnt signaling in stem and cancer stem cells. Curr
Opin Cell Biol. 25:254–264. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Petropoulos K, Arseni N, Schessl C,
Stadler CR, Rawat VP, Deshpande AJ, Heilmeier B, Hiddemann W,
Quintanilla-Martinez L, Bohlander SK, et al: A novel role for
Lef-1, a central transcription mediator of Wnt signaling, in
leukemogenesis. J Exp Med. 205:515–522. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Skokowa J, Cario G, Uenalan M, Schambach
A, Germeshausen M, Battmer K, Zeidler C, Lehmann U, Eder M, Baum C,
et al: LEF-1 is crucial for neutrophil granulocytopoiesis and its
expression is severely reduced in congenital neutropenia. Nat Med.
12:1191–1197. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
111
|
Albano F, Zagaria A, Anelli L, Orsini P,
Minervini CF, Impera L, Casieri P, Coccaro N, Tota G, Brunetti C,
et al: Lymphoid enhancer binding factor-1 (LEF1) expression as a
prognostic factor in adult acute promyelocytic leukemia.
Oncotarget. 5:649–658. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Müller-Tidow C, Steffen B, Cauvet T,
Tickenbrock L, Ji P, Diederichs S, Sargin B, Köhler G, Stelljes M,
Puccetti E, et al: Translocation products in acute myeloid leukemia
activate the Wnt signaling pathway in hematopoietic cells. Mol Cell
Biol. 24:2890–2904. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zhang Y, Yu J, Shi C, Huang Y, Wang Y,
Yang T and Yang J: Lef1 contributes to the differentiation of bulge
stem cells by nuclear translocation and cross-talk with the Notch
signaling pathway. Int J Med Sci. 10:738–746. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Payton JE, Grieselhuber NR, Chang LW,
Murakami M, Geiss GK, Link DC, Nagarajan R, Watson MA and Ley TJ:
High throughput digital quantification of mRNA abundance in primary
human acute myeloid leukemia samples. J Clin Invest. 119:1714–1726.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Alcalay M, Meani N, Gelmetti V, Fantozzi
A, Fagioli M, Orleth A, Riganelli D, Sebastiani C, Cappelli E,
Casciari C, et al: Acute myeloid leukemia fusion proteins
deregulate genes involved in stem cell maintenance and DNA repair.
J Clin Invest. 112:1751–1761. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Avvisati G, Lo-Coco F, Paoloni FP, Petti
MC, Diverio D, Vignetti M, Latagliata R, Specchia G, Baccarani M,
Di Bona E, et al: AIDA 0493 protocol for newly diagnosed acute
promyelocytic leukemia: Very long-term results and role of
maintenance. Blood. 117:4716–4725. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lo-Coco F, Avvisati G, Vignetti M, Breccia
M, Gallo E, Rambaldi A, Paoloni F, Fioritoni G, Ferrara F, Specchia
G, et al: Front-line treatment of acute promyelocytic leukemia with
AIDA induction followed by risk-adapted consolidation for adults
younger than 61 years: Results of the AIDA-2000 trial of the GIMEMA
Group. Blood. 116:3171–3179. 2010. View Article : Google Scholar : PubMed/NCBI
|