The GEO is an international public repository that
stores and freely distributes microarray, second-generation
sequencing and other forms of high-throughput functional genomic
datasets (8). TCGA is a large-scale
cancer genome project that provides researchers with
multidimensional maps of the key genomic changes and
clinicopathological information in 33 types of cancer (9). GEPIA is a newly developed interactive
web server for analyzing RNA sequencing expression data based on
9,736 tumors and 8,587 normal samples from the TCGA and
Genotype-Tissue Expression databases. The gene expression dataset
GSE31552 (10) was downloaded from
GEO and included 25 nontumor tissues and 25 tumor tissues. The
GPL6244 Affymetrix Human Gene 1.0 ST Array (Affymetrix; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) was used. The TCGA
dataset was derived from the ‘Protein-coding Transcripts’ of LUSC
in the Cancer RNA-Seq Nexus (CRN; http://syslab4.nchu.edu.tw) database (11) and included 63 cancer samples and 51
normal samples. The GEPIA dataset was downloaded from the GEPIA
online database including 486 cancer samples and 338 normal
samples.
Significant DEGs between LUSC and normal samples
were screened using FunRich (version 3.1.3), which is an
open-access standalone functional enrichment and interaction
network analysis tool (15). A PPI
network of DEGs was constructed using the Gene MANIA database
(http://www.genemania.org). The Gene MANIA
database is a useful tool for generating hypotheses about gene
function, analyzing gene lists, prioritizing functionally analyzed
genes and reporting weights (16).
Subsequently, a hierarchical clustering of DEGs was constructed
using an online analysis database of University of California,
Santa Cruz Xena (UCSC Xena 2.0; http://xena.ucsc.edu/welcome-to-ucsc-xena) (17).
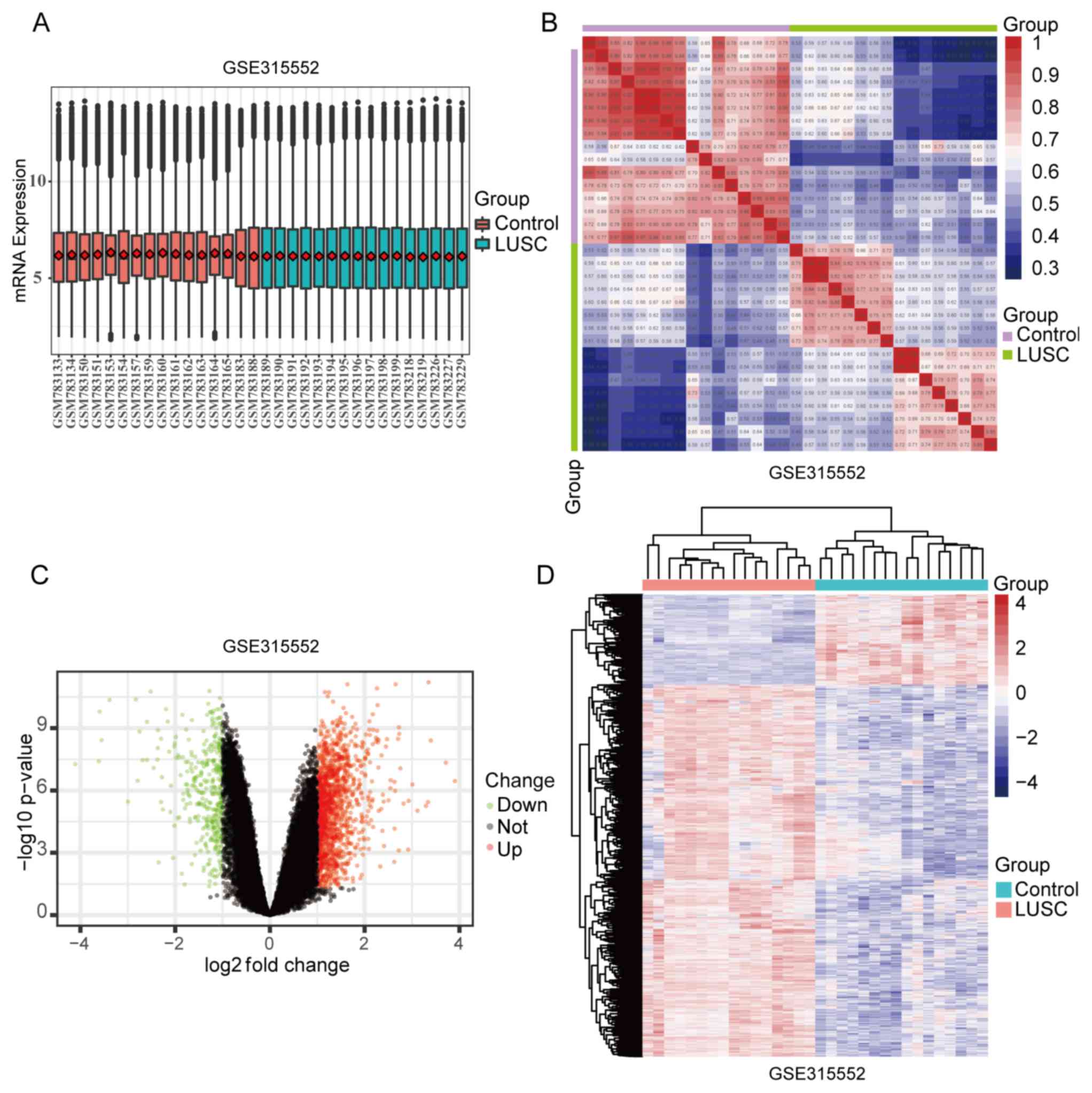
Following the evaluation of missing data and
normalization, the expression profiling data were plotted (Fig. 1A). The similar levels of data points
indicate high consistency showing high accuracy of classification
and comparison. The baseline level of Samples Clustering Analysis
(Fig. 1B) revealed that the sample
sources were reliable. For dataset GSE31552, a total of 1,712 DEGs
were identified at P<0.05 and |log2FC|>1, and the
rationality of the values was verified by volcano plots (Fig. 1C), in which red dots represent
upregulated genes, green dots represent downregulated genes, and
black dots represent unchanged genes. Hierarchical clustering
analysis revealed that the gene expression patterns of the DEGs
were similar among the array data of GSE31522 (Fig. 1D), indicating that the molecular
changes in LUSC are consistent and may represent a novel genetic
signature in LUSC.
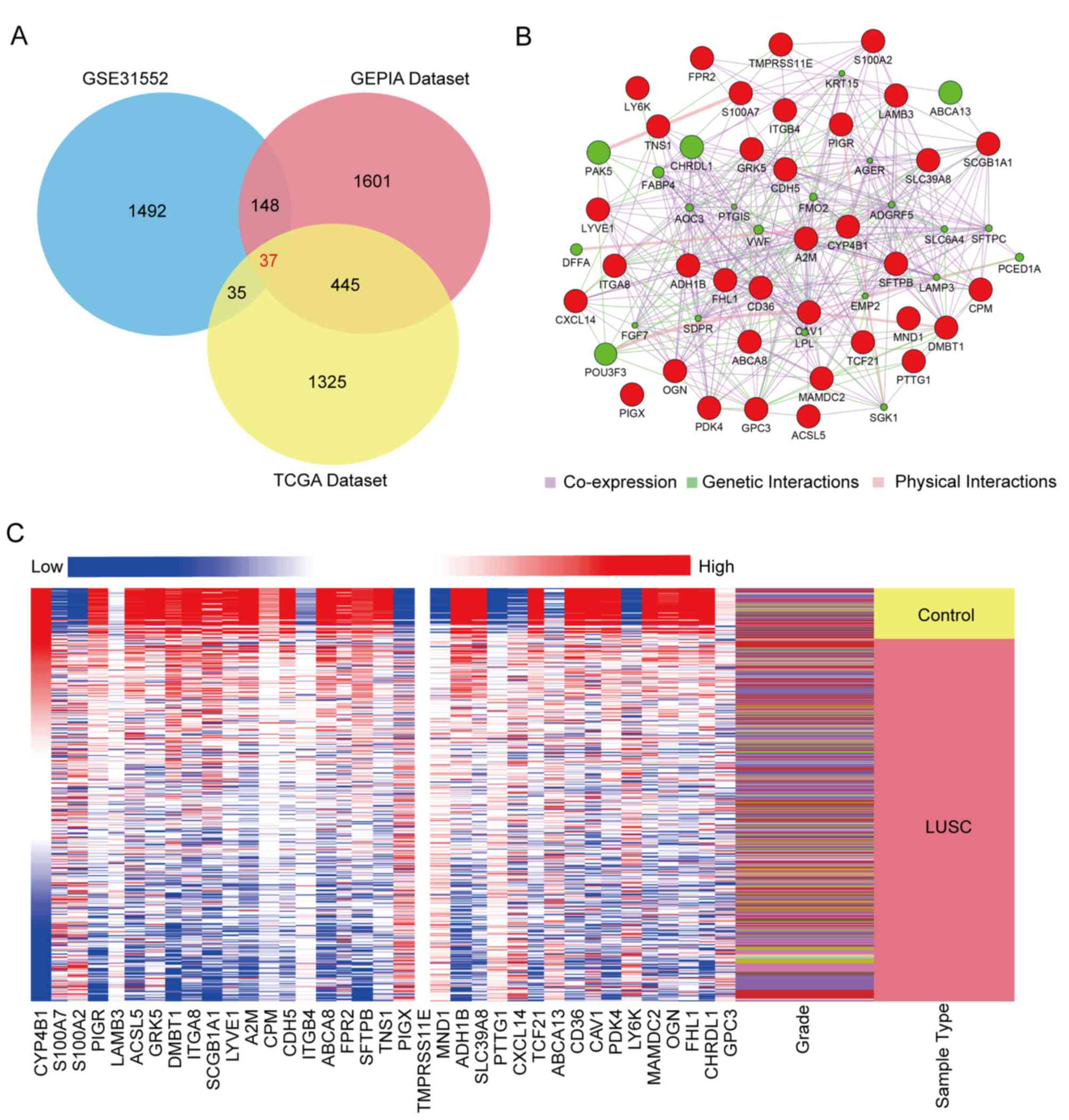
Following standardization of the microarray results,
DEGs (2,162 in GSE31552; 1,842 in TCGA database; and 1,691 in the
GEPIA database) were identified. The three datasets shared 37 genes
that were considered to be significant DEGs between LUSC and normal
tissues (Fig. 2A). These genes
included 26 downregulated and 11 upregulated genes.
To analyze the biological classifications of the
DEGs, DAVID was used for functional and pathway enrichment
analysis. The identified GO terms and pathways are presented in
Table I. The GO terms enriched by
DEGs were mainly associated with ‘cell adhesion’, ‘cell-matrix
adhesion’ and ‘anatomical structure morphogenesis’, whereas the
pathways enriched for DEGs were associated with ‘ECM-receptor
interaction’ and ‘focal adhesion’.
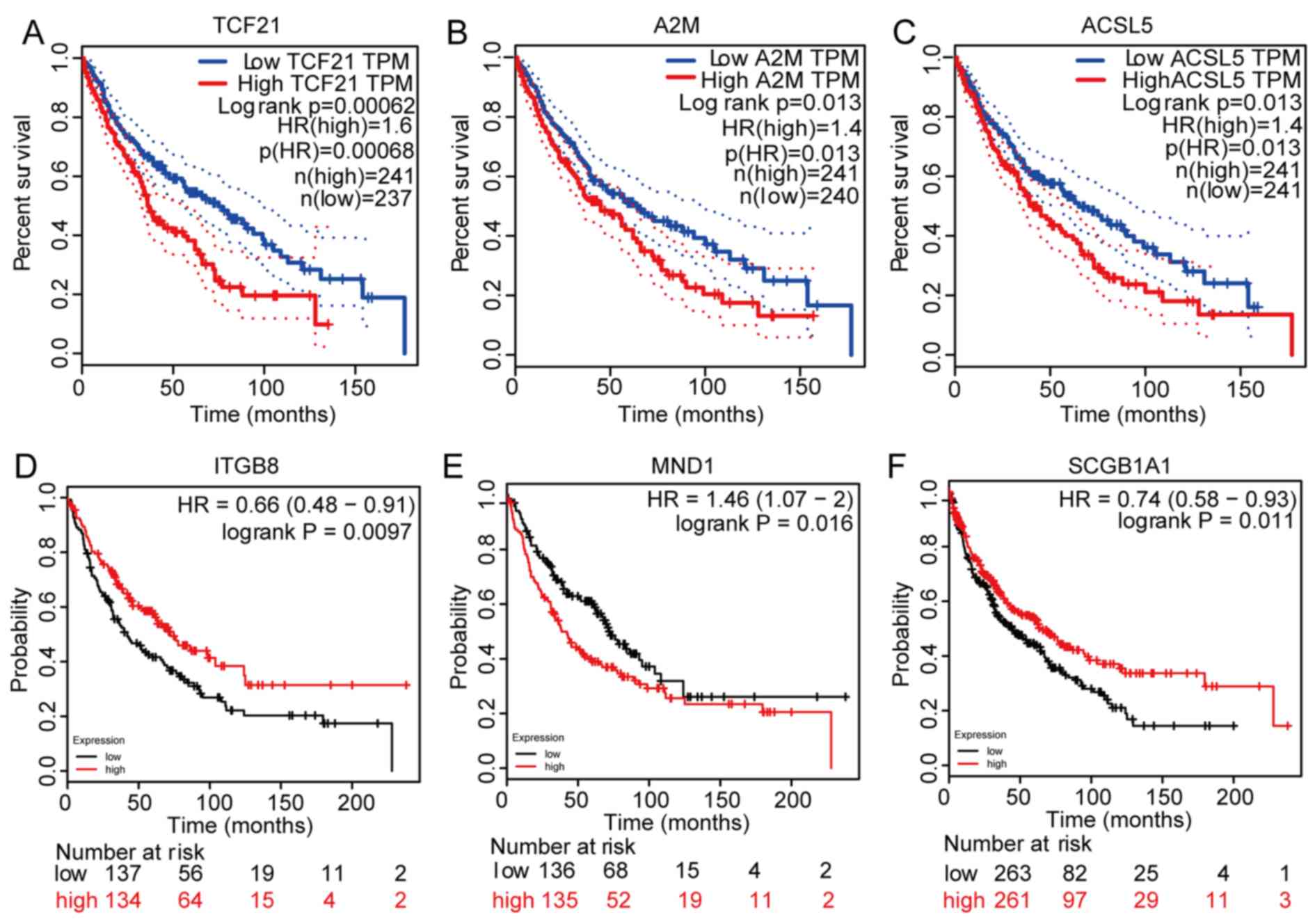
To ensure the accuracy of the analyses, overall
survival analysis of the DGEs was performed using GEPIA and
Kaplan-Meier plotters. Genes were selected if they are meaningful
(P<0.05) in both analyses. Six hub genes were ultimately
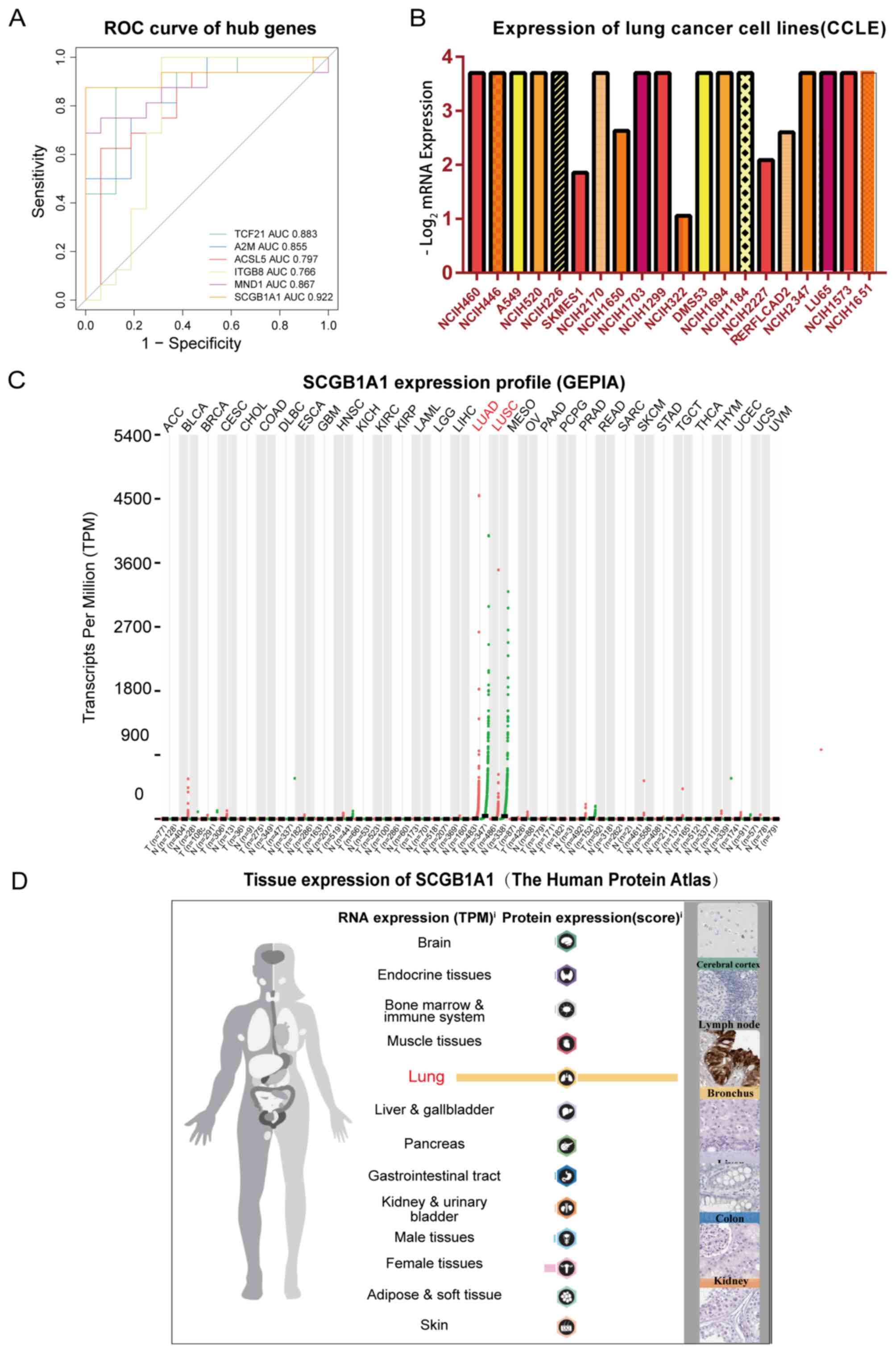
selected based on their significant effects on survival (Fig. 3). The survival curves indicated that
integrin subunit β8 (ITGB8) and SCGB1A1 were positively associated
with overall survival, whereas TCF21, A2M, ACSL5 and meiotic
nuclear divisions 1 (MND1) were negatively associated with overall
survival. The hub genes and their functions are presented in
Table II.
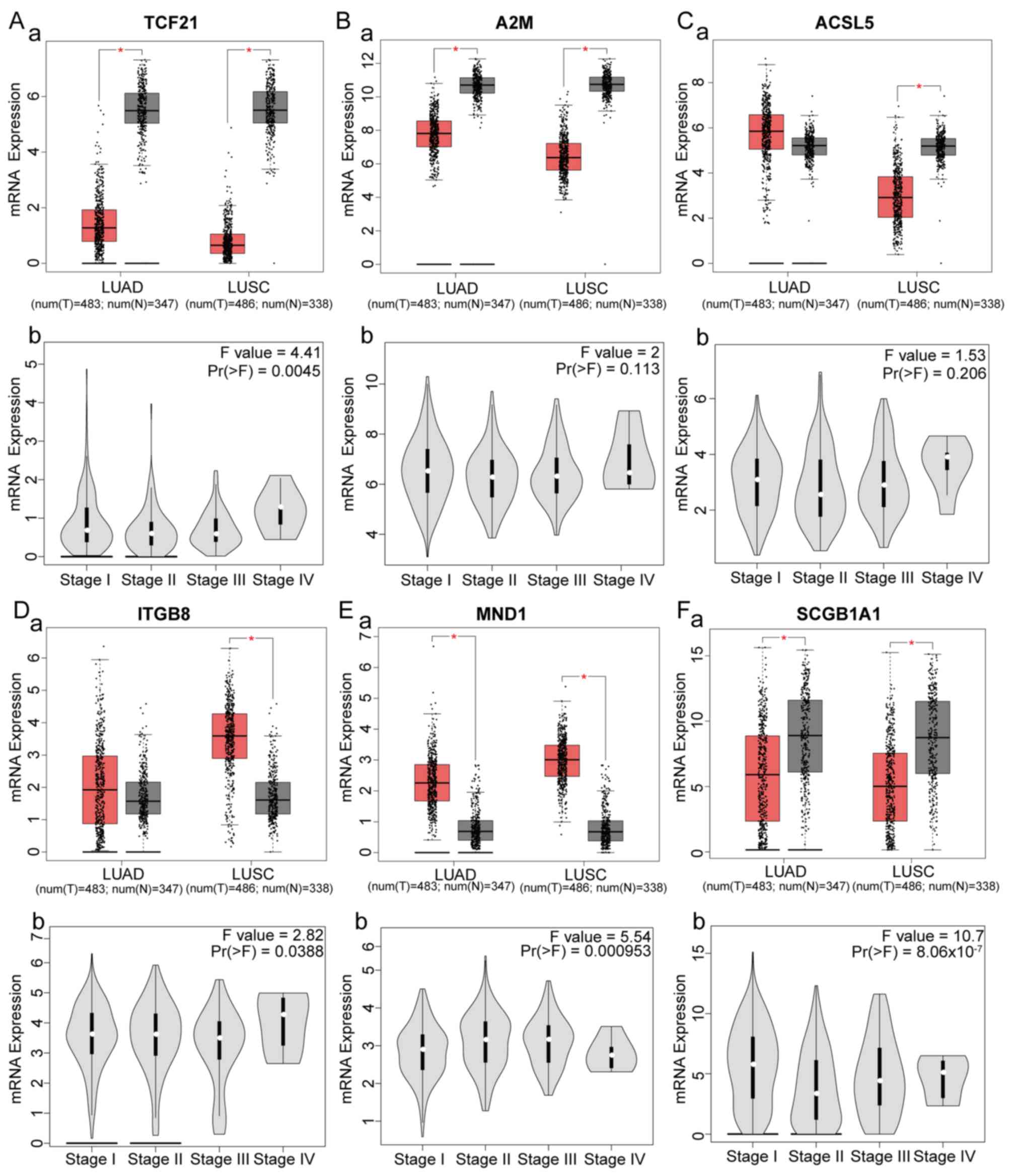
Six genes were identified as hub genes and had
significant effects on survival. The expression levels of the hub
genes in different subtypes of NSCLC and different stages of LUSC
were analyzed (Fig. 4). Compared
with healthy controls, TCF21, A2M and SCGB1A1 were downregulated in
LUSC and LUAD, ITGB8 and MND1 were upregulated in LUAD and LUSC,
and ACSL5 was downregulated in LUSC and upregulated in LUAD. In
addition, expression of TCF21, ITGB8, MND1 and SCGB1A1 was
significantly different among different stages of LUSC. SCGB1A1 was
closely associated with LUSC stage
[Pr(>F)=8.06×10−7]. ROC curves revealed that the hub
genes had a good diagnostic value for LUSC [area under the curve
(AUC)>0.7], particularly SCGB1A1 (AUC=0.922; Fig. 5A).
In summary, downregulation of SCGB1A1 was
significantly associated with overall survival of LUSC patients
(log-rank P=0.011; Fig. 3F), and
SCGB1A1 downregulation was significantly associated with LUSC
stages [Pr(>F)=8.06×10−7; Fig. 4C]. Additionally, ROC curve analysis
demonstrated that SCGB1A1 had high diagnostic value (AUC=0.922).
Thus, SCGB1A1 was screened as a target gene for LUSC. Further
analysis based on GEPIA Database revealed that SCGB1A1 expression
was lower in LUSC compared with normal lung tissue (Fig. 5C). The Human Protein Atlas database
analysis demonstrated that SCGB1A1 was overexpressed in normal lung
tissues compared with other tissues (Fig. 5D). The expression of SCGB1A1 in lung
cancer cell lines was analyzed using the CCLE online platform; the
results revealed that SCGBA1 was downregulated in the majority lung
cancer cell lines (Fig. 5B).
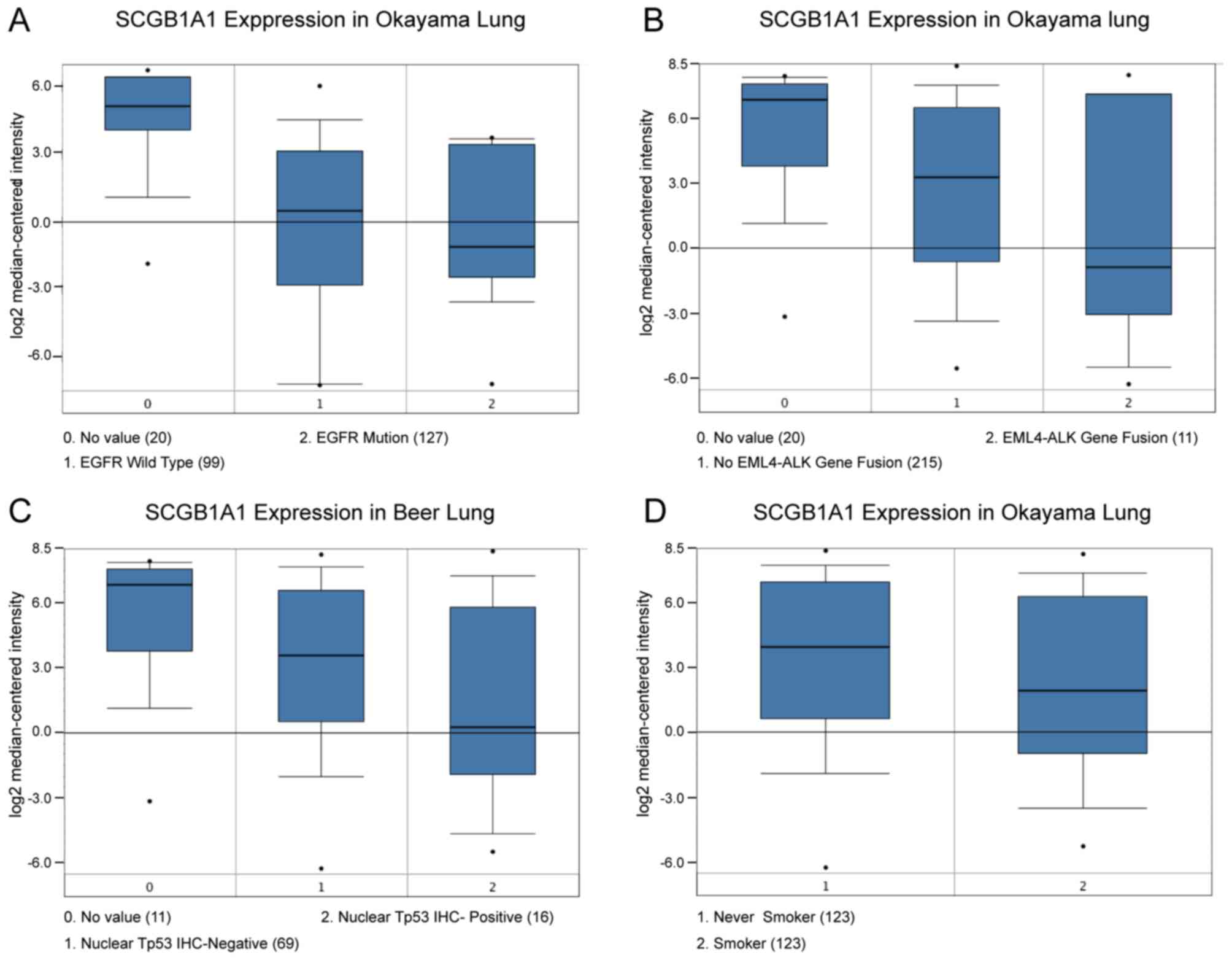
Oncomine analysis of Okayama and Beer datasets revealed that lower
mRNA levels of SCGB1A1 were associated with EGFR mutation, EML4-ALK
fusion, expression of TP53 and smoking (Fig. 6A-D).
The present study analyzed three datasets, including
GSE31552, and TCGA and GEPIA datasets, and revealed DEGs between
LUSC and normal tissues. A total of 37 DEGs were identified,
including 26 downregulated genes and 11 upregulated genes. To
understand the functions and associations of these DEGs, GO and
KEGG analyses were performed. The DEGs were mainly enriched in
‘cell adhesion’, ‘cell-matrix adhesion’, ‘anatomical structure
morphogenesis’, whereas KEGG pathway enrichment was mainly
concentrated in ‘ECM-receptor interaction’ and ‘focal adhesion’.
Previous studies have reported that cell adhesion, cell-matrix
adhesion and anoikis are closely associated with tumorigenesis and
development (35–37). Additionally, studies of anatomical
structure and morphogenesis have revealed cholesterol levels to be
associated with tumor metastasis and invasion (38–40).
These studies indicated that DEGs may be closely related to the
occurrence, development, metastasis and invasion of tumors. The
pathways of ECM-receptor interaction and focal adhesion are
important mediators of cell adhesion, growth, proliferation,
survival, angiogenesis and migration (41,42). The
results obtained in the aforementioned studies are consistent with
the results obtained in the current study. The enriched modules and
pathways identified in the current study may have genetic effects
on LUSC and the identified genes may interact within a network.
Survival analysis was performed using the DEGs and
six genes with significant impacts on overall survival were
identified as hub genes. SCGB1A1, also known as the Clara cell
secretory protein (43,44), had a good diagnostic value for LUSC
(AUC=0.922) and was significantly associated with poor overall
survival and tumor stage. Previous studies have demonstrated that
SCGB1A1 demonstrated anti-inflammatory, immunomodulatory and
antitoxin properties (45–49). SCGB1A1 mRNA expression is abundant
throughout the airway (50),
however, its expression level is very low in lung tumors (51), which is consistent with the results
obtained in the current study. Expression levels of SCGB1A1 are
decreased in diffuse goblet cell hyperplasia and squamous
metaplasia, whereas alveolar expression is elevated in alveolar
cell bronchitis (52). SCGB1A1
knockout mice are more susceptible to lung injury (by bacterial or
viral infection, ozone and cigarette smoke) or sensitization,
exhibit increased inflammation and remodeling reactions, have more
frequent lung tumors, and have a stronger T-helper 2-directed
immune response compared with control mice (53). In the present study, SCGB1A1 was
associated with tumor stage, EGFR mutation, ALK gene fusion and
smoking history. A previous study has revealed that EGFR tyrosine
kinase inhibitors have the same effect on the prognosis of patients
with LUSC as chemotherapy, with fewer complications and higher
quality of life (54). Therefore,
SCGB1A1 may serve a protective role in lung tissue. Inducing
SCGB1A1 expression may inhibit the expression of c-MYC and C-RAF,
which may further inhibit the metastasis of tumors (55). However, the mechanism remains
unclear, and future studies are required to elucidate the pathways
involved in SCGB1A1 and lung cancer.
TCF21, located on chromosome 6q23-q24, encodes a
basic helix-loop-helix transcription factor essential for
epithelial cell differentiation (56,57). It
can be readily methylated and subsequently cause tumorigenesis
(58,59). A previous study has indicated that
hypermethylation and decreased expression of TCF21 are
tumor-specific and are frequently observed in NSCLC (60). The protein encoded by A2M is a
protease inhibitor and cytokine transporter (61). A previous study has revealed that a
progressive A2M deficiency may promote tumor development in nude
mice (62). A2M regulated tumor cell
adhesion, migration and growth by inhibiting tumor-promoting
signaling pathways, including the phosphoinositide 3-kinase/protein
kinase B (PI3K/AKT) pathway and mothers against decapentaplegic
homolog (SMAD) and upregulating phosphatase and tensin homolog via
downregulation of microRNA-21 in vitro and in tumor
xenografts (62). The level of A2M
in human blood decreases with age (63). ACSL5, a mitochondria-localized enzyme
that catalyzes the synthesis of long-chain fatty acid thioesters,
is physiologically involved in the induction of apoptosis in
intestinal cells (64). Studies have
revealed that ACSL5 isozymes serve leading roles in the
biosynthesis of mitochondrial cardiolipin and may participate in
the survival of cancer cells (65–67).
ITGB8, a member of the integrin β chain family, is increased in
different types of cancer, including breast, lung, throat and
stomach cancer (68). High
expression of ITGB8 serves an important role in the metastasis of
human lung cancer cells. When ITGB8 is silenced, the expression of
E-cadherin and cystatin B is increased, whereas the expression of
C-X-C motif chemokine ligand CXCL1, CXCL2, CXCL5, matrix
metalloproteinase (MMP)-2 and MMP-9 is decreased (69). Furthermore, changes in the cell
cycle, the expression of metastasis-associated genes and metastatic
potential may be accompanied by decreased tumor cell signal
transduction and molecular activity (69,70). The
products of the MND1 gene bind to PSMC3 interacting protein to form
stable heterodimer complexes that bind to DNA and stimulate the
activities of RAD51 recombinase and DNA meiotic recombinase 1,
which are required for meiotic recombination (71). MND1 was significantly upregulated in
ovarian cancer compared with ovarian tissue samples from healthy
controls (72). However, to the best
of our knowledge, MND1 upregulation has not been previously
reported in human lung cancer.
The hub genes identified in the current study are
associated with the occurrence and development of tumors. The
involvement of TCF21 and ITGB8 in lung cancer has been previously
documented. However, there are fewer reports of SCGB1A1, A2M, ACSL5
and MND1 in lung cancer. In vitro overexpression of TCF21
may inhibit tumor growth and chemoresistance possibly through the
AKT signaling pathway (73,74). Upregulation of ITGB8 may promote the
expression of tumor metastasis genes and enhance the invasive
ability of tumor cells in LUSC by regulating the phosphorylation
levels of mitogen-activated protein kinase/extracellular
signal-regulated kinase and AKT. An increased incidence of lung
injury and lung tumors was reported following SCGB1A1 knockout
(52). A previous study has reported
that SCGB1A1 may serve an anti-inflammatory role by inhibiting
phospholipase A2 (75); therefore,
the downregulation of SCGB1A1 may lead to an imbalance of T cell
subsets, which in turn affects the antitumor activity of serum
peripheral blood mononuclear cells in patients with LUSC (76). Transcriptome analysis of A2M-treated
tumor cells, xenografts and mouse liver revealed that A2M modulates
tumor cell adhesion, migration and proliferation by inhibiting
tumor-promoting signaling pathways, such as PI3K/AKT and SMAD, and
by upregulating PTEN via downregulation of miR-21 in vitro
and in tumor xenografts (77). ASCL5
is closely associated with cancer cell apoptosis (64). The hub genes in the current study
were associated with poor overall survival rates, and ROC curves
revealed high diagnostic values (AUC>0.7). The results obtained
in the current study suggest that these genes may serve important
roles in the occurrence and development of LUSC and may be used as
biomarkers for the diagnosis of LUSC.
In conclusion, the purpose of the current study was
to identify genes that may be involved in the development or
progression of LUSC. A total of 37 DEGs were identified, of which 6
were identified as hub genes and may be used as biomarkers for the
diagnosis and prognosis evaluation of LUSC. However, the results of
this study were obtained through big data analysis, and validation
of the results via animal experiments and clinical trials is
required.
Not applicable.
The present study was financially supported by
Clinical Research Funds (grant no. CY2017-BJ11).
The dataset of GSE31522 analyzed during the present
study are available in the Gene Expression Omnibus repository
(https://www. ncbi.nlm.nih.gov/gds);
The dataset of TCGA analyzed during the current study are available
in Cancer RNA-Seq Nexus (https://syslab4.nchu.edu.tw); The dataset of GEPIA
analyzed during the current study are available in the Gene
Expression Profiling Interactive Analysis databases (https://gepia.cancer-pku.cn).
YXW, NNZ and QQX designed and conceived the study.
NNZ performed the bioinformatics analysis and wrote the manuscript.
HW, HC, FQW and DBDW contributed to data collection, data analysis
and revised the manuscript. All the authors read and approved the
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gandara DR, Hammerman PS, Sos ML, Lara PN
Jr and Hirsch FR: Squamous cell lung cancer: From tumor genomics to
cancer therapeutics. Clin Cancer Res. 21:2236–2243. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perezmoreno P, Brambilla E, Thomas R and
Soria JC: Squamous cell carcinoma of the lung: Molecular subtypes
and therapeutic opportunities. Clin Cancer Res. 18:2443–2451. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kuribayashi K, Funaguchi N and Nakano T:
Chemotherapy for advanced non-small cell lung cancer with a focus
on squamous cell carcinoma. J Cancer Res Ther. 12:528–534. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Morrey ME, Abdel MP, Riester SM, Dudakovic
A, van Wijnen AJ, Morrey BF and Sanchez-Sotelo J: Molecular
landscape of arthrofibrosis: Microarray and bioinformatic analysis
of the temporal expression of 380 genes during contracture genesis.
Gene. 610:15–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu C, Fei HD, Sun ZY and Tian JW:
Bioinformatic analysis of the microarray gene expression profile in
degenerative intervertebral disc cells exposed to TNF-α. Eur Rev
Med Pharmacol Sci. 19:3332–3339. 2015.PubMed/NCBI
|
|
7
|
Meltzer EB, Barry WT, D'Amico TA, Davis
RD, Shu SL, Onaitis MW, Morrison LD, Sporn TA, Steele MP and Noble
PW: Bayesian probit regression model for the diagnosis of pulmonary
fibrosis: Proof-of-principle. BMC Med Genomics. 4:702011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res 39 (Database Issue).
D991–D1005. 2013.
|
|
9
|
Tomczak K, Czerwińska P and Wiznerowicz M:
The cancer genome atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.PubMed/NCBI
|
|
10
|
Lin J, Marquardt G, Mullapudi N, Wang T,
Han W, Shi M, Keller S, Zhu C, Locker J and Spivack SD: Lung cancer
transcriptomes refined with laser capture microdissection. Am J
Pathol. 184:2868–2884. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li JR, Sun CH, Li W, Chao RF, Huang CC,
Zhou XJ and Liu CC: Cancer RNA-Seq Nexus: A database of
phenotype-specific transcriptome profiling in cancer cells. Nucleic
Acids Res. 44(D1): D944–D951. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Core Team R: R: A language and environment
for statistical computing. Computing. 1:12–21. 2015.
|
|
13
|
Troyanskaya O, Cantor M, Sherlock G, Brown
P, Hastie T, Tibshirani R, Botstein D and Altman RB: Missing value
estimation methods for DNA microarrays. Bioinformatics. 17:520–525.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hogan SJ, Feltz KT, Murdock DR, Goodman
TA, Vercande DJ, Tangeman MR, Busch EM, Kripakaran R, Jayasimha MG,
Smith KE, et al: Call-processing system and method. 2003.
|
|
15
|
Pathan M, Keerthikumar S, Ang CS, Gangoda
L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim
A, et al: FunRich: An open access standalone functional enrichment
and interaction network analysis tool. Proteomics. 15:2597–2601.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Warde-Farley D, Donaldson SL, Comes O,
Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT,
et al: The GeneMANIA prediction server: Biological network
integration for gene prioritization and predicting gene function.
Nucleic Acids Res 38 (Web Server Issue). W214–W220. 2010.
View Article : Google Scholar
|
|
17
|
Goldman M, Craft B, Zhu J and Haussler D:
Abstract 2584: The UCSC Xena system for cancer genomics data
visualization and interpretation. Cancer Res. 77:25842017.
|
|
18
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ogata H, Goto S, Sato K, Fujibuchi W, Bono
H and Kanehisa M: KEGG: Kyoto encyclopedia of genes and genomes.
Nucleic Acids Res. 27:29–34. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID gene functional classification tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res 45 (W1).
W98–W102. 2017. View Article : Google Scholar
|
|
22
|
Piao J, Sun J, Yang Y, Jin T, Chen L and
Lin Z: Target gene screening and evaluation of prognostic values in
non-small cell lung cancers by bioinformatics analysis. Gene.
647:306–311. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mitteer DR, Greer BD, Fisher WW and Cohrs
VL: Teaching behavior technicians to create publication-quality,
single-case design graphs in graphpad prism 7. J Appl Behav Anal.
51:998–1010. 2018. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pontén F, Jirström K and Uhlen M: The
human protein atlas-a tool for pathology. J Pathol. 216:387–393.
2010. View Article : Google Scholar
|
|
25
|
Rhodes DR, Yu J, Shanker K, Deshpande N,
Varambally R, Ghosh D, Barrette T, Pandey A and Chinnaiyan AM:
ONCOMINE: A cancer microarray database and integrated data-mining
platform. Neoplasia. 6:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Beer DG, Kardia SL, Huang CC, Giordano TJ,
Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, et al:
Gene-expression profiles predict survival of patients with lung
adenocarcinoma. Nat Med. 8:816–824. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Non-Small Cell Lung Cancer Collaborative
Group, . Chemotherapy and supportive care versus supportive care
alone for advanced non-small cell lung cancer. Cochrane Database
Syst Rev. 12:CD0073092010.
|
|
28
|
Blandin KS, Crosbie PA, Balata H, Chudziak
J, Hussell T and Dive C: Progress and prospects of early detection
in lung cancer. Open Biol. 7(pii): 1700702017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mayekar MK and Bivona TG: Current
landscape of targeted therapy in lung cancer. Clin Pharmacol Ther.
102:757–764. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jamal-Hanjani M, Wilson GA, McGranahan N,
Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R,
Rosenthal R, et al: Tracking the evolution of non-small-cell lung
cancer. N Engl J Med. 376:2109–2121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Youlden DR, Cramb SM and Baade PD: The
international epidemiology of lung cancer: Geographical
distribution and secular trends. J Thorac Oncol. 3:819–831. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng TY, Cramb SM, Baade PD, Youlden DR,
Nwogu C and Reid ME: The international epidemiology of lung cancer:
Latest trends, disparities, and tumor characteristics. J Thorac
Oncol. 11:1653–1671. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Raghavachari N: Microarray technology:
Basic methodology and application in clinical research for
biomarker discovery in vascular diseases. Methods Mol Biol.
1027:47–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li G, Li X, Yang M, Xu L, Deng S and Ran
L: Prediction of biomarkers of oral squamous cell carcinoma using
microarray technology. Sci Rep. 7:421052017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Albelda SM and Buck CA: Integrins and
other cell adhesion molecules. FASEB J. 4:2868–2880. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zaman MH, Trapani LM, Siemeski A,
Mackellar D, Gong H, Kamm RD, Wells A, Lauffenburger DA and
Matsudaira P: Migration of tumor cells in 3D matrices is governed
by matrix stiffness along with cell-matrix adhesion and
proteolysis. Proc Natl Acad Sci USA. 103:10889–10894. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Uccelli A, Moretta L and Pistoia V:
Mesenchymal stem cells in health and disease. Nat Rev Immunol.
8:726–736. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang D, Song Z and Wang Z: Common
mechanism of pathogenesis in various types of metastatic
osteosarcoma. Oncol Lett. 14:6307–6313. 2017.PubMed/NCBI
|
|
39
|
Laval S, Laklai H, FanJul M, Pucelle M,
Susini C, Pyronnet S and Bousquet C: Abstract 5181: Forced
hemidesmosome assembly blocks pancreatic cancer cell invasiveness.
Cancer Res. 70:51812011.
|
|
40
|
Zamanian-Daryoush M and DiDonato JA:
Apolipoprotein A-I and cancer. Front Pharmacol. 6:2652015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang H, Tao J, Sheng L, Hu X, Rong R, Xu
M and Zhu T: RETRACTED: Twist2 promotes kidney cancer cell
proliferation and invasion via regulating ITGA6 and CD44 expression
in the ECM-Receptor-Interaction pathway. Biomed Biomed
Pharmacother. 81:453–459. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Golubovskaya VM: Focal adhesion kinase as
a cancer therapy target. Anticancer Agents Med Chem. 10:735–741.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Randell SH: Airway epithelial stem cells
and the pathophysiology of chronic obstructive pulmonary disease.
Proc Am Thorac Soc. 3:718–725. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Giangreco A, Reynolds SD and Stripp BR:
Terminal bronchioles harbor a unique airway stem cell population
that localizes to the bronchoalveolar duct junction. Am J Pathol.
161:173–182. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Antico G, Lingen MW, Sassano A, Melby J,
Welch RW, Fiore S, Pilon AL and Miele L: Recombinant human
uteroglobin/CC10 inhibits the adhesion and migration of primary
human endothelial cells via specific and saturable binding to
fibronectin. J Cell Physiol. 207:553–561. 2010. View Article : Google Scholar
|
|
46
|
Singh G and Katyal SL: Clara cell
proteins. Ann N Y Acad Sci. 923:43–58. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang Y, Zhang Z, Mukherjee AB and Linnoila
RI: Increased susceptibility of mice lacking Clara cell 10-kDa
protein to lung tumorigenesis by
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a potent carcinogen
in cigarette smoke. J Biol Chem. 279:29336–29340. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang SZ, Rosenberger CL, Bao YX, Stark JM
and Harrod KS: Clara cell secretory protein modulates lung
inflammatory and immune responses to respiratory syncytial virus
infection. J Immunol. 171:1051–1060. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Watson TM, Reynolds SD, Mango GW, Boe IM,
Lund J and Stripp BR: Altered lung gene expression in CCSP-null
mice suggests immunoregulatory roles for Clara cells. Am J Physiol
Lung Cell Mol Physiol. 281:L1523–L1530. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Laucho-Contreras ME, Polverino F,
Tesfaigzi Y, Pilon A, Celli BR and Owen CA: Club cell protein 16
(CC16) augmentation: A potential disease-modifying approach for
chronic obstructive pulmonary disease (COPD). Expert Opin Ther
Targets. 20:869–883. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ludovini V, Bianconi F, Siggillino A,
Piobbico D, Vannucci J, Metro G, Chiari R, Bellezza G, Puma F,
Della Fazia MA, et al: Gene identification for risk of relapse in
stage I lung adenocarcinoma patients: A combined methodology of
gene expression profiling and computational gene network analysis.
Oncotarget. 7:30561–30574. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Linnoila RI, Szabo E, DeMayo F, Witschi H,
Sabourin C and Malkinson A: The role of CC10 in pulmonary
carcinogenesis: From a marker to tumor suppression. Ann N Y Acad
Sci. 923:249–267. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bourdin A, Kotsimbos T, Nguyen K, Vachier
I, Mainprice B, Farce M, Paganin F, Marty-Ané C, Vernhet H, Godard
P and Chanez P: Non-invasive assessment of small airway remodelling
in smokers. COPD. 7:102–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sun Y, Yin X, Wen MM, Zhang J, Wang XJ,
Xia JH, Zhang YN, Zhang ZP and Li XF: EGFR mutations subset in
Chinese lung squamous cell carcinoma patients. Mol Med Rep.
17:7575–7584. 2018.PubMed/NCBI
|
|
55
|
Thakur C, Rapp UR and Rudel T: Cysts mark
the early stage of metastatic tumor development in non-small cell
lung cancer. Oncotarget. 9:6518–6535. 2017.PubMed/NCBI
|
|
56
|
Robb L, Mifsud L, Hartley L, Biben C,
Copeland NG, Gilbert DJ, Jenkins NA and Harvey RP: Epicardin: A
novel basic helix-loop-helix transcription factor gene expressed in
epicardium, branchial arch myoblasts, and mesenchyme of developing
lung, gut, kidney, and gonads. Dev Dyn. 213:105–113. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Smith LT, Lin M, Brena RM, Lang JC,
Schuller DE, Otterson GA, Morrison CD, Smiraglia DJ and Plass C:
Epigenetic regulation of the tumor suppressor gene TCF21 on
6q23-q24 in lung and head and neck cancer. Proc Natl Acad Sci USA.
103:982–987. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Arab K, Smith LT, Gast A, Weichenhan D,
Huang JP, Claus R, Hielscher T, Espinosa AV, Ringel MD, Morrison
CD, et al: Epigenetic deregulation of TCF21 inhibits metastasis
suppressor KISS1 in metastatic melanoma. Carcinogenesis.
32:1467–1473. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Costa VL, Henrique R, Danielsen SA, Eknaes
M, Patrício P, Morais A, Oliveira J, Lothe RA, Teixeira MR, Lind GE
and Jerónimo C: TCF21 and PCDH17 methylation: An innovative panel
of biomarkers for a simultaneous detection of urological cancers.
Epigenetics. 6:1120–1130. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Richards KL, Zhang B, Sun M, Dong W,
Churchill J, Bachinski LL, Wilson CD, Baggerly KA, Yin G, Hayes DN,
et al: Methylation of the candidate biomarker TCF21 is very
frequent across a spectrum of early-stage nonsmall cell lung
cancers. Cancer. 117:606–617. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chen X, Kong X, Zhang Z, Chen W, Chen J,
Li H, Cao W, Ge Y and Fang S: Alpha-2-macroglobulin as a
radioprotective agent: A review. Chin J Cancer Res. 26:611–621.
2014.PubMed/NCBI
|
|
62
|
Kurz S, Thieme R, Amberg R, Groth M,
Jahnke HG, Pieroh P, Horn LC, Kolb M, Huse K, Platzer M, et al:
Correction: The anti-tumorigenic activity of A2M-A lesson from the
naked mole-rat. PLoS One. 13:e01951692018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Birkenmeier G, Müller R, Huse K, Forberg
J, Gläser C, Hedrich H, Nicklisch S and Reichenbach A: Human
alpha2-macroglobulin: Genotype-phenotype relation. Exp Neurol.
184:153–161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Klaus C, Kaemmerer E, Reinartz A,
Schneider U, Plum P, Jeon MK, Hose J, Hartmann F, Schnölzer M,
Wagner N, et al: TP53 status regulates ACSL5-induced expression of
mitochondrial mortalin in enterocytes and colorectal
adenocarcinomas. Cell Tissue Res. 357:267–278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Mashima T, Oh-hara T, Sato S, Mochizuki M,
Sugimoto Y, Yamazaki K, Hamada J, Tada M, Moriuchi T, Ishikawa Y,
et al: p53-defective tumors with a functional apoptosome-mediated
pathway: A new therapeutic target. J Natl Cancer Inst. 97:765–777.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ding S, Tang S, Wang M, Wu D and Guo H:
Acyl-CoA synthetase 5 promotes the growth and invasion of
colorectal cancer cells. Can J Gastroenterol Hepatol.
2017:76157362017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Pitule P, Vycital O, Bruha J, Novak P,
Hosek P, Treska V, Hlavata I, Soucek P, Kralickova M and Liska V:
Differential expression and prognostic role of selected genes in
colorectal cancer patients. Anticancer Res. 33:4855–4865.
2013.PubMed/NCBI
|
|
68
|
Ni R, Shen X, Wu H, Zhu W, Ni J, Huang Z,
Song Y and Gao X: Expression and significance of integrins subunits
in laryngeal squamous cell carcinoma. Lin Chung Er Bi Yan Hou Tou
Jing Wai Ke Za Zhi. 24:686–689. 2010.(In Chinese). PubMed/NCBI
|
|
69
|
Xu Z and Wu R: Alteration in metastasis
potential and gene expression in human lung cancer cell lines by
ITGB8 silencing. Anat Rec (Hoboken). 295:1446–1454. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Huang L, Cai JL, Huang PZ, Kang L, Huang
MJ, Wang L and Wang JP: miR19b-3p promotes the growth and
metastasis of colorectal cancer via directly targeting ITGB8. Am J
Cancer Res. 7:1996–2008. 2017.PubMed/NCBI
|
|
71
|
Tsubouchi H and Roeder GS: The Mnd1
protein forms a complex with hop2 to promote homologous chromosome
pairing and meiotic double-strand break repair. Mol Cell Biol.
22:3078–3088. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yeganeh PN, Richardson C,
Bahrani-Mostafavi Z, Tait DL and Mostafavi MT: Dysregulation of
AKT3 along with a small panel of mRNAs stratifies high-grade serous
ovarian cancer from both normal epithelia and benign tumor tissues.
Genes Cancer. 8:784–798. 2017.PubMed/NCBI
|
|
73
|
Yang Z and Jiang X, Li D, Dong Q, Zhao H
and Jiang X: TCF21 inhibits proliferation and chemoresistance
through the AKT pathway in human gastric cancer. Gene. 682:42–49.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Dai Y, Duan H, Duan C, Zhu H, Zhou R, Pei
H and Shen L: TCF21 functions as a tumor suppressor in colorectal
cancer through inactivation of PI3K/AKT signaling. Onco Targets
Ther. 10:1603–1611. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Shijubo N, Itoh Y and Abe S:
Anti-inflammatory molecule, Clara cell 10 kilodalton protein and
respiratory diseases. Rinsho Byori. 50:370–373. 2002.(In Japanese).
PubMed/NCBI
|
|
76
|
Joyce JA and Fearon DT: T cell exclusion,
immune privilege, and the tumor microenvironment. Science.
348:74–80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kurz S, Thieme R, Amberg R, Groth M,
Jahnke HG, Pieroh P, Horn LC, Kolb M, Huse K, Platzer M, et al: The
anti-tumorigenic activity of A2M-A lesson from the naked mole-rat.
PLoS One. 12:e01895142017. View Article : Google Scholar : PubMed/NCBI
|