Introduction
Chronic myeloid leukemia (CML) is an acquired
somatic mutation disorder of the hematopoietic stem cells (1) and is the third most common type of
leukemia, constituting ∼15% of all leukemia cases. The natural
course of CML is divided into three phases: chronic phase (CP),
accelerated phase (AP) and blast phase (BP). CML typically begins
with a CP. If left untreated, CML progresses from the CP to the AP
after 3–5 years, followed by a terminal BP of lymphoid or myeloid
phenotype (2). The only possible
cure for CML is allogeneic stem cell transplantation. In the early
CP, the 5-year survival rate following allogeneic transplantation
is 25–70% (3); however, allogeneic
stem cell transplantation is only available to a minority of
individuals. Therefore, the main treatment for CML is chemotherapy
and the conventional chemotherapeutic agents include imatinib
mesylate (IM), interferon (IFN)-α, cytarabine and hydroxyurea. IFN
with or without cytarabine was used as the conventional treatment
for CP leukemia at the end of the 1990s. Currently, imatinib is the
first-line therapy of choice for the CP of CML (4).
CML is characterized by the Philadelphia chromosome,
which encodes the oncogene BCR-ABL. BCR-ABL is a fusion gene
resulting from the reciprocal translocation between BCR and ABL on
chromosomes 22 and 9, respectively (5,6). The
chimeric protein BCR-ABL exhibits an uncontrolled tyrosine kinase
activity and phosphorylates several substrates that activate
multiple signaling pathways, including Ras, signal transducer and
activator of transcription-5 (STAT-5), extracellular
signal-regulated kinase (ERK)/mitogen-activated protein kinase
(MAPK), Janus kinase 2 (JAK-2), phosphatidylinositol-3 kinase
(PI-3K) and nuclear factor (NF)-κB (7,8).
This abnormal signaling leads to the malignant cellular phenotype
of CML, including increased proliferation, inhibition of the
apoptotic response to mutagenic stimuli and reduction of adhesion
to the bone marrow stroma and extracellular matrix (9). IM, an inhibitor of the tyrosine
kinase activity of BCR-ABL, has been successfully used to treat CML
patients in the CP and is considered the first-line therapy for the
CP of CML (10–12). However, IM is less effective in the
AP and BP of CML (13) and certain
patients develop IM resistance (14) due to mutation and amplification of
the BCR-ABL gene. Although inhibitors of farnesyltransferase and
dual Src-family kinase/Abl kinase inhibit the growth of multi-drug
resistant (MDR)-CML cells, side-effects and high cost limit their
clinical application (15).
Therefore, it is imperative to screen novel agents for the AP and
BP of CML.
Fangchinoline is the main chemical constituents of
radix Stephaniae tetrandrae, the dried roots of
Stephaniae tetrandrae S. Moore (Menispermaceae),
which has been shown to possess a wide range of pharmacological
activities, including inhibition of histamine release and
antihypertensive activities (16,17),
anti-inflammatory effects (18–20),
antiplatelet aggregation activities (21), antihyperglycemic actions (22,23),
neuroprotective effects (24) and
antioxidant and radical scavenging activities (25,26).
Previous studies indicated that fangchinoline exhibited significant
antitumor activity in various human cancers, including breast,
prostate and hepatocellular carcinoma. Its antitumor mechanisms
involved in inducing G1/S phase cell cycle arrest include
inhibition of cyclin D1, upregulation of p27, potentiation of
cancer cell apoptosis by upregulating pro-apoptotic B cell lymphoma
(BCL)-2-associated X protein (BAX) and downregulating
anti-apoptotic BCL-2, and initiation of excessive autophagy via
p53/sestrin2/AMP-activated protein kinase (AMPK) signaling
(27–29). Moreover, it has been reported that
fangchinoline reverses the multidrug resistance of antitumor drugs
mediated by P-glycoprotein (P-gp) (30,31).
However, the effect of fangchinoline on CML and the underlying
mechanisms remain unclear.
In the present study, we evaluated the effect of
fangchino-line on the proliferation of K562 cells derived from the
blast crisis of CML and investigated the potential mechanisms
involved. We identified that fangchinoline efficiently inhibits the
growth of K562 cells. Further investigation demonstrated that
fangchinoline induces cell cycle arrest at G0/G1 rather than
apoptosis, which may result from upregulation of cyclin-dependent
kinase (CDK)-N1A and MCL-1, and down-regulation of cyclin D2
(CCND2). These findings suggest the possibility of fangchinoline as
an effective antitumor agent in CML.
Materials and methods
Preparation of fangchinoline
Fangchinoline was kindly provided by Dr H.B. Wang
(School of Life Science and Technology, Tongji University,
Shanghai, China) and was stable when stored at 4°C. It was
dissolved in dimethyl sulf-oxide (DMSO) as a stock solution and
then stored at 4°C. The final DMSO concentration did not exceed
0.1% (v/v), which had no effect on cell growth in any experiment.
Control cells were treated with the same amount of DMSO (0.1%, v/v)
as used in the corresponding experiments.
Cell culture
K562 cells purchased from Cell Bank Type Culture
Collection of Chinese Academy of Sciences (Shanghai, China), were
routinely maintained in RPMI-1640 culture medium (Thermo Fisher
Scientific, Shanghai, China) supplemented with 10% fetal bovine
serum (FBS; HyClone Laboratories Inc., Logan, UT, USA), 100 U/ml
penicillin (Gibco BRL, Grand Island, NY, USA) and 100 μg/ml
streptomycin (Gibco) and grown at 37°C in a humidified atmosphere
of 5% CO2. The study was approved by the ethics
committee of Tongji University, Shanghai, China.
Cell proliferation assay
Cell proliferation was measured by direct counting.
Initially, logarithmically growing cells were seeded into 6-well
culture plates at a density of 2×105 cells/ml and
treated for 24 and 48 h at 0, 1, 3 and 10 μM fangchinoline.
The cells per well were collected and then counted using a
hemocytometer.
Cell viability assay
Cell viability was assessed using a methyl-thiazol
tetrazolium (MTT) assay. Exponentially growing cells were
inoculated into 96-well culture plates with 1×104 cells
per well and treated with a series of concentrations of
fangchinoline (0, 0.1, 0.3, 1, 3 and 10 μM) for 24 and 48 h.
All experiments were conducted parallel with controls (0.1% DMSO).
Then, 20 μl sterile MTT (5 mg/ml, Sigma-Aldrich, St. Louis,
MO, USA) was added to each well. Following further incubation at
37°C for 4 h, the reaction was stopped by adding 150 μl
DMSO. Following agitation on an automated shaker for 10 min,
formazan production was determined by measurement of the
spectrometric absorbance at 490 nm on an enzyme immunoassay
analyzer FlexStation 3™ (Molecular Devices, Sunnyvale, CA, USA).
The percentage of cell proliferation was calculated using the
optical density (OD) as follows: (OD of experimental well - OD of
blank well) / (OD of control well - OD of blank well) x100. The
IC50 values, defined as the concentration of drug that caused 50%
inhibition of absorbance compared with the control cell treated
with DMSO only, were calculated using SPSS 17.0 statistical
software (Aspire Software International, Leesburg, VA, USA).
Cell cycle distribution analysis
Cell cycle distribution was determined by DNA
staining with propidium iodide (PI). Exponentially-growing K562
cells were cultured and treated in 6-well culture plates
(4×105 cells/ml) with 0, 1, 3 and 10 μM
fangchinoline for 24 and 48 h. Cells were then harvested by
centrifugation at 1,200 rpm at 4°C, washed once in
phosphate-buffered saline (PBS) and fixed in 70% cold ethanol
overnight. Cells were centrifuged and resuspended in 0.25 ml PBS
containing 0.2 mg/ml RNase and incubated at 37°C for 1 h. Cells
were added to 2.5 μl 4 mg/ml PI and stored in the dark at
4°C. The cells were analyzed on a flow cytometer (Becton-Dickinson,
San Jose, CA, USA) and the percentage of cells in the different
phases of the cell cycle was analyzed using FlowJo software.
Measurement of apoptosis by flow
cytometry
Apoptotic cells were detected using an
Annexin-V-Fluos staining kit. Cells were seeded in 6-well culture
plates at a density of 4×105 cells/ml, followed by
fangchinoline and DMSO (control) treatment for 24 and 48 h.
Following treatment, cells were collected and washed with PBS.
After centrifugation at 200 x g for 5 min, the cell pellet was
resuspended in 100 μl Annexin-V-Fluos labeling solution (20
μl Annexin-V-Fluos labeling reagent prediluted in 1 ml
incubation buffer and 20 μl PI), incubated for 15 min in the
dark at room temperature and then immediately analyzed with a flow
cytometer (Becton-Dickinson).
RNA extraction and quantitative reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA from each group of cells was extracted
with TRNzol-A+ reagent (Tiangen, Beijing, China).
The first cDNA strand was synthesized using TIANScript RT Kit
(Tiangen) and Oligo(dT) 15 primer from 2 μg total RNA,
according to the manufacturer’s instructions. The primer sequences
for the target genes were as follows: CDKN1A,
5’-CTCATCCCGTGTTCTCCTTT-3′ (forward) and 5′-GTACCACCCAGCGGACAAGT-3′
(reverse); CCND2, 5′-TGGAGCTGCTGTGCCACG-3′ (forward) and
5′-GTGGCCACCATTCTGCGC-3′ (reverse); MCL-1,
5′-GGACATCAAAAACGAAGACG-3′ (forward) and 5′-GCAGCTTTCTTGGTTTATGG-3′
(reverse); BAX, 5′-GATGCGTCCACCAAGAAGCT-3′ (forward) and
5′-CGGCCCCAGTTGAAGTTG-3′ (reverse); β-actin,
5′-GGCTGTATTCCCCTCCATCG-3′ (forward) and
5′-CCAGTTGGTAACAATGCCATGT-3′ (reverse). The PCR amplifications were
performed for 40 cycles of 95°C for 5 sec, 60°C for 20 sec and 72°C
for 10 sec. Real-time quantitative RT-PCR was performed on a
Stratagene Mx3000P system (Stratagene, La Jolla, CA, USA) with
SYBR® Premix Ex Taq Mix (Takara Biotechnology
Co., Ltd., Dalian, China). When cycling was completed, melting
curve analysis was performed to establish the specificity of the
PCR product. Data were collected and stored in Excel format and
analyzed using Mx3000P software version 4.0. The expression level
of cDNA of each candidate gene was internally normalized using
β-actin. The relative quantitative value was expressed using the
2−ΔΔCt method (32),
representing the amount of candidate gene expression with the same
calibrators. Each experiment was performed in duplicates and
repeated three times.
Statistical analysis
The data were analyzed by one-way analysis of
variance using SPSS 17.0 software and P-values were calculated.
Final values are expressed as mean ± standard deviation (SD).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Fangchinoline inhibits K562 cell
proliferation in a dose- and time-dependent manner
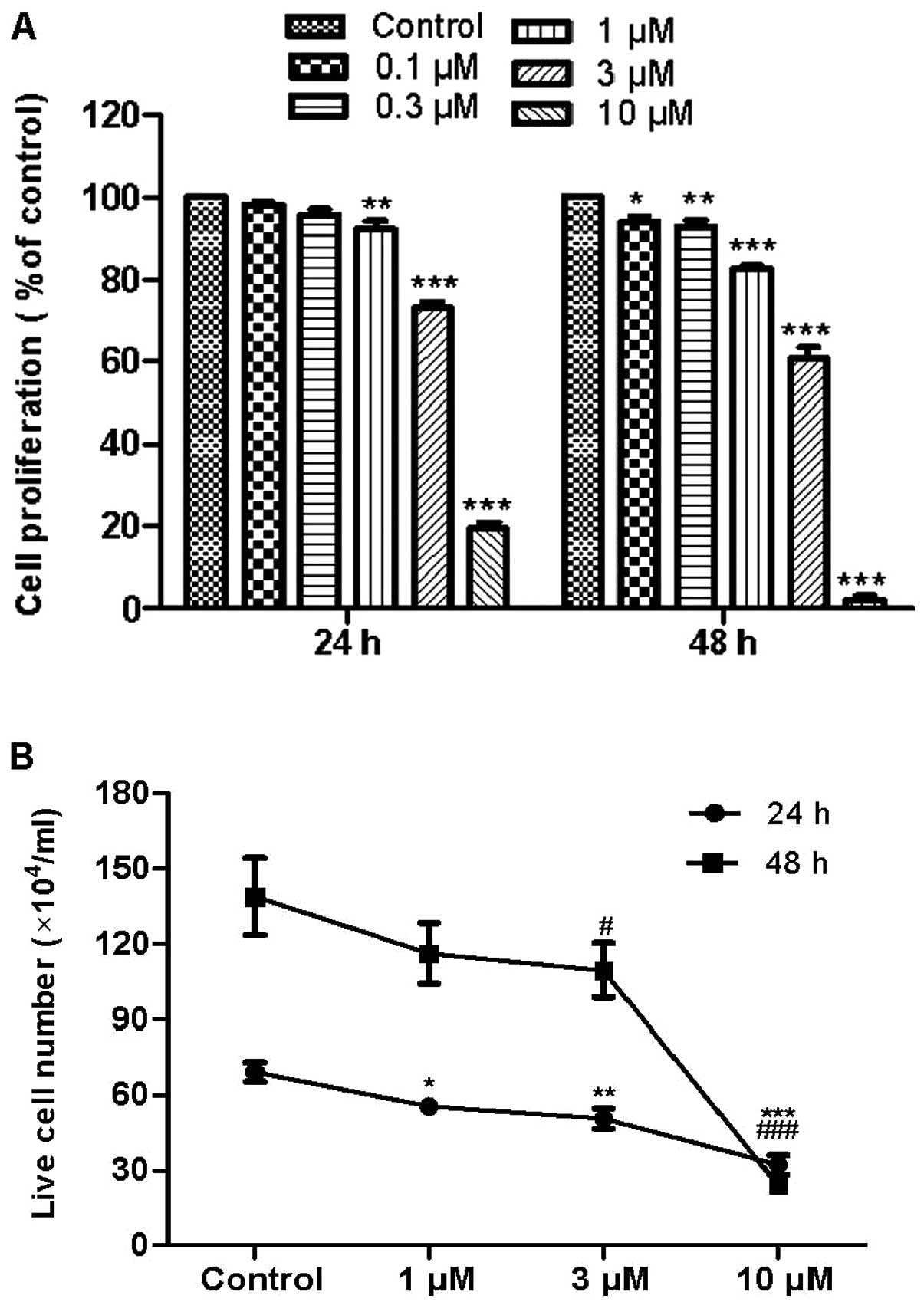
To evaluate the effects of fangchino-line on CML
cell proliferation, exponentially-growing K562 cells were treated
with 0.1, 0.3, 1, 3 and 10 μM fangchinoline. After 24 and 48
h, cell proliferation was determined by the MTT assay. Results
revealed that fangchinoline significantly decreased the percentage
of viable cells as compared with cells without treatment (Fig. 1A). After incubation with 1
μM fangchinoline for 24 and 48 h, cell viability reduced to
92 and 83%, respectively. After incubation with 3 μM
fangchinoline for 24 and 48 h, cell viability reduced to 73 and
61%, respectively. After incubation with 10 μM
fangchino-line for 24 and 48 h, cell viability reduced to 19 and
2%, respectively. The IC50 of fangchinoline treatment for 24 and 48
h was ∼4.82 and 2.65 μM, respectively. To verify the
anti-proliferation effect of fangchinoline, cell proliferation was
also measured by direct counting (Fig.
1B), which is consistent with the results of the MTT assay.
Those results indicated that the anti-proliferation effect of
fangchinoline on K562 cells is in a dose- and time-dependent
manner.
Fangchinoline induces cell cycle arrest
at the G0/G1 phase in K562 cells
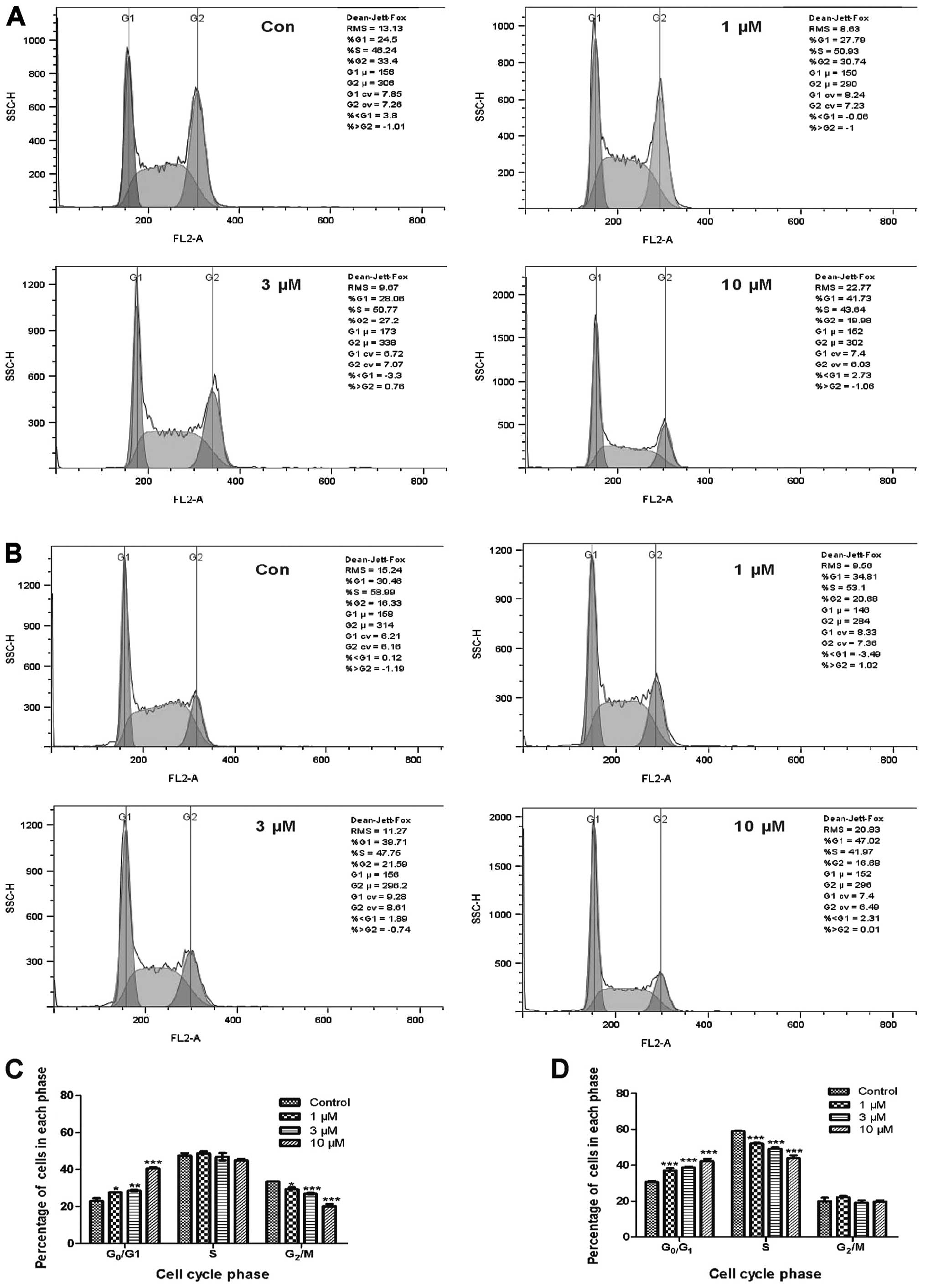
We subsequently analyzed cell cycle distribution in
fangchinoline-exposed cells using flow cytometry following PI
staining. Treatment with fangchinoline for 24 and 48 h resulted in
the accumulation of K562 cells in the G0/G1 phase and a concomitant
depletion of cells in the G2/M or S phases (Fig. 2A–D). After treatment with 1, 3 and
10 μM fangchinoline for 24 h, the proportion of cells in the
G0/G1 phase increased gradually from 23.0±2.05% in the control
group to 27.6±0.29, 28.6±0.86 and 40.6±1.06%, respectively. The
proportion of cells in the G2/M phase was reduced by ∼4, 7 and 13%,
respectively, as compared to the control value (27.1±0.8%; Fig. 2A and C). After treatment with 1, 3
and 10 μM fangchinoline for 48 h, the proportion of cells in
the G0/G1 phase increased gradually from 30.7±0.96 to 37.1±2.1,
38.7±0.89 and 42.3±2.3%, respectively. The proportion of cells in
the S phase was reduced by ∼7, 10 and 15%, respectively, as
compared to the control value (59.0±0.31%; Fig. 2B and D). These results indicate
that fangchinoline induces cell cycle arrest at the G0/G1 phase in
K562 cells.
Effects of fangchinoline on the
expression level of cell cycle-related genes
To explore the mechanism of fangchinoline in
inducing cell cycle arrest at the G0/G1 phase in K562 cells, the
mRNA level of CDKN1A, which inhibits all cyclin-CDK complexes and
CCND2 was investigated using quantitative real-time RT-PCR. The
expression level of CCND2 markedly decreased in 1 μM
fangchinoline-treated cells at 24 h, whereas it significantly
increased in 10 μM fangchinoline-treated cells. By 48 h, the
expression level of CCND2 had markedly declined in each
experimental group (Fig. 3A and
B). After exposure to 10 μM fangchinoline for 24 and 48
h, the mRNA level of CDKN1A markedly increased compared to the
control group. CDKN1A mRNA expression in other experimental groups
tended to increase; however, there was no significant difference
compared with the control group (Fig.
3C and D). These results indicate that cell cycle arrest at the
G0/G1 phase in K562 cells may result from upregulation of CDKN1A
and downregulation of CCND2.
Fangchinoline does not induce apoptosis
in K562 cells
To evaluate whether the anti-proliferation effect of
fangchino-line is required to induce apoptosis, we detected cell
apoptosis with and without fangchinoline treatment using the
Annexin-V-Flous/PI dual-staining assay. As shown in Fig. 4A and B, after treatment with
fangchinoline for 24 and 48 h at various concentrations, the
percentages of apoptotic cells in each group were 0%, which
demonstrated that fangchinoline does not induce apoptosis in K562
cells. To determine the anti-apoptotic mechanism of fangchinoline
in K562 cells, we examined the mRNA levels of BCL-2 family members,
including MCL-1 and BAX. Quantitative real-time RT-PCR analysis
revealed that the mRNA level of MCL-1 significantly increased after
treatment with 10 μM fangchinoline for 24 h. The mRNA level
of BAX slightly increased after treatment with 10 μM
fangchinoline for 48 h; however, the ratio of BAX to MCL-1 mRNA
level was markedly lower than in the control group. Treatment for
24 and 48 h with 1 and 3 μM fangchino-line did not induce
discernible changes in the mRNA level of BAX and MCL-1 (Fig. 4C and D).
Discussion
Previous studies have shown that a number of herbal
extracts and isolated compounds possess antitumor activity.
Tetrandrine is a major compound from radix Stephaniae
tetrandrae. The potent antitumor activity of tetrandrine has
been extensively reported. Fangchinoline is a derivative of
tetrandrine, with structural features similar to tetrandrine.
Several studies have demonstrated that fangchinoline inhibits the
growth of various tumor cells and have determined the mechanisms
involved in inducing G1/S phase cell cycle arrest, potentiating
cancer cell apoptosis and triggering excessive autophagy instead of
inducing apoptosis. However, the effects and the underlying
mechanisms of fangchinoline in human CML cells remain unclear. In
the present study, we observed that fangchinoline exerts a
significant growth inhibition effect in K562 cells and the
inhibition effects are dose- and time-dependent. Further analysis
revealed that fangchinoline treatment triggers cell cycle arrest at
the G0/G1 phase; however, it does not induce apoptosis of K562
cells. These effects are considered a result of the upregulation of
CDKN1A and MCL-1 expression, as well as downregulation of CCND2
expression.
Cell cycle progression is precisely regulated by a
series of cell cycle regulators, including cyclins, CDKS and CDK
inhibitors (CDKIs). Progression through the G1 phase and transition
from G1 into the S phase are regulated by cyclin D, E and their
dependent kinases. D-type cyclins, including CCND1, CCND2 and
CCND3, assemble with CDK4 and CDK6 to form active complexes
(33), The cyclin D-CDK4/6
complexes induce the phosphorylation of the retinoblastoma (Rb)
protein and the release of E2F, which triggers G1 cell cycle
progression (34). Increased
expression of CDKs and cyclins has been observed in the majority of
cancer cells. The deregulation of the cell cycle in the G1 phase
has been implicated in tumor development and proliferation
(35,36). In hematopoietic cells, CCND2 and
CCND3 mediate the G1-S-phase transition and, if overexpressed,
allow for G1-S phase progression under conditions of growth-factor
deprivation. Thus, it is likely that BCR-ABL in K562 cells provides
a mitogenic signal that results in overexpression of CCND2 and
facilitates the G1-S phase transition (37). In our study, we identified that
fangchinoline decreases the mRNA level of CCDN2 and induces G0/G1
growth arrest in K562 cells. These partly account for K562 cell
proliferation inhibition following fangchinoline treatment.
Cyclin/CDK complexes are negatively regulated by two
families of CDK inhibitors. The first class of inhibitors includes
the INK4a proteins. The second family of inhibitors is composed of
Cip/Kip proteins, including p21, p27 and p57 (38). p21 encoded by CDKN1A is a member of
the Cip/Kip family of cyclin-dependent kinase inhibitors known to
be upregulated in response to DNA damage and oxidative stress and
p21 plays an essential role in growth arrest following DNA damage
by binding and inhibiting cyclin/CDK complexes. Expression of p21
in response to DNA damage and other cellular stress is regulated
largely at the transcriptional level by p53-dependent and
-independent mechanisms (39–41).
p21 was originally considered a negative regulator of the cell
cycle and a tumor suppressor. p21 is involved in the regulation of
fundamental cellular programs, including cell proliferation,
differentiation, migration, senescence and apoptosis. It not only
exhibits anti-oncogenic, but also oncogenic properties. The
functions of p21 depend on its intracellular localization. In the
nucleus, it serves as a negative cell cycle regulator and tumor
suppressor, in particular by participating in the launch of a
senescence program. When p21 is localized in the cytoplasm, it acts
as an oncogene by protecting cells against apoptosis (42). In addition, levels of p21 often
determine the cellular response to various drugs. RKO human
colorectal carcinoma cells, which express low levels of p21,
normally undergo apoptosis in response to prostaglandin A2. In
contrast, NIH 3T3 cells and MCF-7 cells express high levels of p21
and undergo G1 arrest in response to prostaglandin A2 (43,44).
In BCR-ABL-transformed hematopoietic cells, BCR-ABL-induced
expression of p21 is localized exclusively in the nucleus. In
BCR-ABL-positive cells, p21 decreases cell proliferation; however,
it does not change the level of spontaneous apoptosis. p21 reduces
IM-and taxol-induced apoptosis in BCR-ABL-positive cells (45). In this study, we demonstrated that
treatment of K562 cells with 10 μM fangchinoline quickly and
significantly increases the mRNA level of CDKN1A. Our results
strongly suggest that p21, as a negative regulator of the cell
cycle, induces cell cycle arrest at the G0/G1 phase in K562 cells
treated with fangchinoline.
Chemotherapy induces apoptosis of tumor cells. In
the Annexin V-Flous/PI dual-staining assay, we did not observe the
occurrence of fangchinoline-induced apoptosis. BCL-2 family members
are critical regulators of apoptosis (46). Proteins of this family are divided
into anti- and pro-apoptotic proteins. Anti-apoptotic BCL-2
proteins, including BCL-2, BCL-XL and MCL-1, prevent the release of
cytochrome c from mitochondria, whereas pro-apoptotic BAX
and BAK participate in the formation of pores in the mitochondria
through which cytochrome c is released (47-50).
MCL-1 has been identified as a BCR/ABL-dependent survival factor in
CML (51) and acts as an
anti-apoptotic factor in various neoplastic cells, including
several leukemia-derived cell lines (52,53).
Upregulation of MCL-1 expression has been implicated in the
chemoresistance of certain malignancies (54). One study demonstrated that MCL-1
inhibits BAX in the absence of MCL-1/BAX interaction and the
anti-apoptotic function of MCL-1 requires the presence of BAX
(55). Hence, we focused on the
expression of MCL-1 and BAX in K562 cells following treatment with
fangchinoline. Our data demonstrated that a high concentration (10
μM) of fangchinoline increases the mRNA level of MCL-1 and
BAX in K562 cells. This may be the main reason why fangchinoline is
unable to induce apoptosis of K562 cells. In addition, there is
accumulating evidence that CDKN1A confers a protective advantage
against apoptosis, which appears to be correlated with a
cytoplasmic translocation of the protein (56–58).
Therefore, fangchinoline-induced upregulation of CDKN1A expression
may also contribute to the survival of K562 cells. However, the
inhibition of apoptotic cell death does not mean that other forms
of cell death occur in K562 cells treated with fangchinoline. The
MTT assay revealed that the growth inhibition rate of K562 cells
reached 98% after treatment for 48 h with 10 μM
fangchinoline. Therefore, we hypothesize that non-apoptotic cell
death occurs in K562 cells treated with 10 μM fangchinoline.
It is necessary to clarify the role of p21 and MCL-1 in
non-apoptotic cell death.
In conclusion, fangchinoline potently increased the
expression of CDKN1A and MCL-1, and decreased the expression of
CCND2. Additionally, it caused cell cycle arrest at the G0/G1 phase
and did not induce apoptosis, resulting in the inhibition of
proliferation in K562 cells. To date, there are no reports on the
adverse reaction of radix Stephaniae tetrandrae. Therefore,
fangchinoline may be a new candidate in the therapeutic strategy of
CML.
Acknowledgements
This study was supported by grants
from the Ministry of Science and Technology (2011CB965100,
2011DFA30480, 2010CB944900, 2010CB945000 and 2011CBA01100), the
National Natural Science Foundation of China (31101061, 31000378,
81170499, 90919028 and 31071306), the Science and Technology
Commission of Shanghai Municipality (11ZR1438500, 11XD1405300) and
the Ministry of Education (IRT1168 and 20110072110039). This study
was also supported by the Chen Guang Project supported by the
Shanghai Municipal Education Commission and Shanghai Education
Development Foundation (12CG19), as well as Fundamental Research
Funds for the Central Universities.
References
|
1
|
Barr RD and Fialkow PJ: Clonal origin of
chronic myelocytic leukemia. N Engl J Med. 289:307–309. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Giles FJ, Cortes JE, Kantarjian HM and
O’Brien SM: Accelerated and blastic phases of chronic myelogenous
leukemia. Hematol Oncol Clin North Am. 18:753–774. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baccarani M, Saglio G, Goldman J, et al:
Evolving concepts in the management of chronic myeloid leukemia.
Recommendations from an expert panel on behalf of the European
LeukemiaNet. Blood. 108:1809–1820. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
von BN and Duyster J: Chronic myelogenous
leukemia: treatment and monitoring. Dtsch Arztebl Int. 107:114–121.
2010.
|
|
5
|
Nowell PC and Hungerford DA: Chromosome
studies on normal and leukemic human leukocytes. J Natl Cancer
Inst. 25:85–109. 1960.PubMed/NCBI
|
|
6
|
Rowley JD: Letter: A new consistent
chromosomal abnormality in chronic myelogenous leukaemia identified
by quinacrine fluorescence and Giemsa staining. Nature.
243:290–293. 1973. View
Article : Google Scholar
|
|
7
|
Barnes DJ and Melo JV: Management of
chronic myeloidleukemia: targets for molecular therapy. Semin
Hematol. 40:34–49. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hamdane M, David-Cordonnier M and
D’Halluin JC: Activation of p65NF-kB protein by p210BCR-ABL in a
myeloid cell line. Oncogene. 15:2267–2275. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deininger MW, Goldman JM and Melo JV: The
molecular biology of chronic myeloid leukemia. Blood. 10:3343–3356.
2000.PubMed/NCBI
|
|
10
|
Druker BJ, Sawyers CL, Kantarjian H, et
al: Activity of a specific inhibitor of the BCR-ABL tyrosine kinase
in the blast crisis of chronic myeloid leukemia and acute
lymphoblastic leukemia with the Philadelphia chromosome. N Engl J
Med. 344:1038–1042. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roskoski RJ: STI-571: an anticancer
protein-tyrosine kinase inhibitor. Biochem Biophys Res Commun.
309:709–717. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Druker BJ: Perspectives on the development
of imatinib and the future of cancer research. Nat Med.
15:1149–1152. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Druker BJ, Talpaz M, Resta DJ, et al:
Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine
kinase in chronic myeloid leukemia. N Engl J Med. 344:1031–1037.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Azam M, Latek RR and Daley GQ: Mechanisms
of autoinhibition and STI-571/imatinib resistance revealed by
mutagenesis of BCR-ABL. Cell. 112:831–843. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Konig H, Holyoake TL and Bhatia R:
Effective and selective inhibition of chronic myeloid leukemia
primitive hematopoietic progenitors by the dual Src/Abl kinase
inhibitor SKI-606. Blood. 111:2329–2338. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakamura K, Tsuchiya S, Sugimoto Y,
Sugimura Y and Yamada Y: Histamine release inhibition activity of
bisbenzylisoquinoline alkaloids. Planta Med. 58:505–508. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim HS, Zhang YH, Oh KW and Ahn HY:
Vasodilating and hypotensive effects of fangchinoline and
tetrandrine on the rat aorta and the stroke-prone spontaneously
hypertensive rat. J Ethnopharmacol. 58:117–123. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hristova M and Istatkova R:
Complement-mediated antiinflammatory effect of
bisbenzylisoquinoline alkaloid fangchinoline. Phytomedicine.
6:357–362. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Choi HS, Kim HS, Min KR, Kim Y, Lim HK,
Chang YK and Chung MW: Anti-inflammatory effects of fangchinoline
and tetrandrine. J Ethnopharmacol. 69:173–179. 2000. View Article : Google Scholar
|
|
20
|
Shen YC, Chou CJ, Chiou WF and Chen CF:
Anti-inflammatory effects of the partially purified extract of
radix Stephaniae tetrandrae: comparative studies of its
active principles tetrandrine and fangchinoline on human
polymorphonuclear leukocyte functions. Mol Pharmacol. 60:1083–1090.
2001.PubMed/NCBI
|
|
21
|
Kim HS, Zhang YH and Yun YP: Effects of
tetrandrine and fangchinoline on experimental thrombosis in mice
and human platelet aggregation. Planta Med. 65:135–138. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsutsumi T, Kobayashi S, Liu YY and
Kontani H: Anti-hyperglycemic effect of fangchinoline isolated from
Stephania tetrandra Radix in streptozotocin-diabetic mice.
Biol Pharm Bull. 26:313–317. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ma W, Nomura M, Takahashi-Nishioka T and
Kobayashi S: Combined effects of fangchinoline from Stephania
tetrandra Radix and formononetin and calycosin from
Astragalus membranaceus Radix on hyperglycemia and
hypoinsulinemia in streptozotocin-diabetic mice. Biol Pharm Bull.
30:2079–2083. 2007.PubMed/NCBI
|
|
24
|
Lin TY, Lu CW, Tien LT, Chuang SH, Wang
YR, Chang WH and Wang SJ: Fangchinoline inhibits glutamate release
from rat cerebral cortex nerve terminals (synaptosomes). Neurochem
Int. 54:506–512. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gülçin I, Elias R, Gepdiremen A, Chea A
and Topal F: Antioxidant activity of bisbenzylisoquinoline
alkaloids from Stephania rotunda: cepharanthine and fangchinoline.
J Enzyme Inhib Med Chem. 25:44–53. 2010.PubMed/NCBI
|
|
26
|
Sekiya N, Hikiami H, Yokoyama K, Kouta K,
Sakakibara I, Shimada Y and Terasawa K: Inhibitory effects of
Stephania tetrandra S. Moore on free radical-induced lysis
of rat red blood cells. Biol Pharm Bull. 28:667–670. 2005.
|
|
27
|
Xing ZB, Yao L, Zhang GQ, Zhang XY, Zhang
YX and Pang D: Fangchinoline inhibits breast adenocarcinoma
proliferation by inducing apoptosis. Chem Pharm Bull (Tokyo).
59:1476–1480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang CD, Huang JG, Gao X, et al:
Fangchinoline induced G1/S arrest by modulating expression of p27,
PCNA and cyclin D in human prostate carcinoma cancer PC3 cells and
tumor xenograft. Biosci Biotechnol Biochem. 74:488–493. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang N, Pan W, Zhu M, Zhang M, Hao X,
Liang G and Feng Y: Fangchinoline induces autophagic cell death via
p53/sestrin2/AMPK signalling in human hepatocellular carcinoma
cells. Br J Pharmacol. 164:731–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He L, Yang J and Hu L: Transmembrane
transport activity of paclitaxel regulated by fangchinoline in
MDR1-mDCK II cells. Zhongguo Zhong Yao Za Zhi. 35:1478–1481.
2010.(In Chinese).
|
|
31
|
He P, Sun H, Jian XX, Chen QH, Chen DL,
Liu GT and Wang FP: Partial synthesis and biological evaluation of
bisbenzylisoquinoline alkaloids derivatives: potential modulators
of multidrug resistance in cancer. J Asian Nat Prod Res.
14:564–576. 2012. View Article : Google Scholar
|
|
32
|
Kenneth JL and Thomas DS: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2–ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sherr CJ: G1 phase progression: Cycling on
cue. Cell. 79:551–555. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Johnson DG and Schneider-Broussard R: Role
of E2F in cell cycle control and cancer. Front Biosci. 3:447–448.
1998.PubMed/NCBI
|
|
35
|
Bischoff JR: Cdk inhibitors in cancer
therapy: What is next? Trends Pharmacol Sci. 29:16–21. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ando K, Ajchenbaum-Cymbalista F and
Griffin JD: Regulation of G1/S transition by cyclins D2 and D3 in
hematopoietic cells. Proc Natl Acad Sci USA. 90:9571–9575. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sherr CJ and Roberts JM: CDK inhibitors:
positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer
Ther. 1:639–649. 2002.PubMed/NCBI
|
|
40
|
Weiss RH: p21Waf1/Cip1 as a therapeutic
target in breast and other cancers. Cancer Cell. 4:425–429. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zu K, Bihani T, Lin A, Park YM, Mori K and
Ip C: Enhanced selenium effect on growth arrest by BiP/GRP78
knockdown in p53-null human prostate cancer cells. Oncogene.
25:546–554. 2006.PubMed/NCBI
|
|
42
|
Romanov VS, Pospelov VA and Pospelova TV:
Cyclin-dependent kinase inhibitor p21 (Waf1): contemporary view on
its role in senescence and oncogenesis. Biochemistry (Mosc).
77:575–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gorospe M, Wang X, Guyton K and Holbrook
N: Protective role of p21 (WAF1/CIP1) against prostaglandin
A2-mediated apoptosis of human colorectal carcinoma cells. Mol Cell
Biol. 16:6654–6660. 1996.PubMed/NCBI
|
|
44
|
Hitomi M, Shu J, Strom D, Harter ML and
Stacey DW: Prostaglandin A2 blocks the activation of G1 phase
cyclin dependent kinase without altering mitogen-activated protein
kinase stimulation. J Biol Chem. 271:9376–9383. 1996. View Article : Google Scholar
|
|
45
|
Forster K, Obermeier A, Mitina O, Simon N,
Warmuth M, Krause G and Hallek M: Role of p21(WAF1/CIP1) as an
attenuator of both proliferative and drug-induced apoptotic signals
in BCR-ABL-transformed hematopoietic cells. Ann Hematol.
87:183–193. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
van Delft MF and Huang DC: How the Bcl-2
family of proteins interact to regulate apoptosis. Cell Res.
16:203–213. 2006.PubMed/NCBI
|
|
47
|
Danial NN and Korsmeyer SJ: Cell death:
critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Labi V, Erlacher M, Kiessling S and
Villunger A: BH3-only proteins in cell death initiation, malignant
disease and anticancer therapy. Cell Death Differ. 13:1325–1338.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Leber B, Lin J and Andrews DW: Embedded
together: the life and death consequences of interaction of the
Bcl-2 family with membranes. Apoptosis. 12:897–911. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wei MC, Zong WX, Cheng EH, et al:
Proapoptotic BAX and BAK: a requisite gateway to mitochondrial
dysfunction and death. Science. 292:727–730. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Aichberger KJ, Mayerhofer M, Krauth MT,
Skvara H, Florian S and Sonneck K: Identification of mcl-1 as a
BCR/ABL-dependent target in chronic myeloid leukemia (CML):
evidence for cooperative antileukemic effects of imatinib and mcl-1
antisense oligonucleotides. Blood. 105:3303–3311. 2005. View Article : Google Scholar
|
|
52
|
Zhou P, Qian L, Kozopas KM and Craig RW:
Mcl-1, a Bcl-2 family member, delays the death of hematopoietic
cells under a variety of apoptosis-inducing conditions. Blood.
89:630–643. 1997.PubMed/NCBI
|
|
53
|
Opferman JT, Iwasaki H, Ong CC, Suh H,
Mizuno S and Akashi K: Obligate role of anti-apoptotic MCL-1 in the
survival of hematopoietic stem cells. Science. 307:1101–1104. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Thallinger C, Wolschek MF, Wacheck V,
Maierhofer H, Gunsberg P and Polterauer P: Mcl-1 antisense therapy
chemosensitizes human melanoma in a SCID mouse xenotransplantation
model. J Invest Dermatol. 120:1081–1086. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Germain M, Milburn J and Duronio V: MCL-1
inhibits BAX in the absence of MCL-1/BAX Interaction. J Biol Chem.
283:6384–6392. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Coqueret O: New roles for p21 and p27
cell-cycle inhibitors: a function for each cell compartment? Trends
Cell Biol. 13:65–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Asada M, Yamada T, Ichijo H, Delia D,
Miyazono K, Fukumuro K and Mizutani S: Apoptosis inhibitory
activity of cytoplasmic p21(Cip1/WAF1) in monocytic
differentiation. EMBO J. 18:1223–1234. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Suzuki A, Tsutomi Y, Akahane K, Araki T
and Miura M: Resistance to Fas-mediated apoptosis: activation of
caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene
family ILP. Oncogene. 17:931–939. 1998. View Article : Google Scholar : PubMed/NCBI
|