Introduction
Breast cancer is the most common cancer among women
worldwide and ranks first and second in cancer mortality rates
among women in undeveloped and developed regions, respectively.
According to GLOBOCAN 2012 estimates, 1.67 million women were
diagnosed with breast cancer in 2012, representing an increase in
breast cancer incidence of >20% since 2008 (1). Although it is considered a cancer with
a relatively good prognosis if diagnosed and treated in a timely
manner, the mortality rates from breast cancer remain high,
particularly in developing countries, likely because it is often
diagnosed at advanced stages (2,3). The
majority of breast cancer-related mortalities are due to the
development of distant metastasis, for which no effective
treatments exist (4,5). Although numerous chemotherapeutic
agents are available for the treatment of cancer metastases, no
improvement in the median duration of survival has been observed,
and the molecular events underlying the progression to metastasis
are not completely understood (6).
Apoptosis, also known as programmed cell death, is a
process in living organisms that is necessary for the maintenance
of proper development and the elimination of cell damage or excess.
Apoptosis is characterized by distinct biochemical and
morphological changes, including DNA fragmentation, plasma membrane
blebbing and loss of cell volume. There are two major pathways by
which apoptotic cell death can be induced: The intrinsic (or
mitochondrial) pathway and the extrinsic (or death receptor)
pathway. The intrinsic pathway regulates the activity of proteins
of the survivin and B-cell lymphoma 2 (BCL-2) families. The latter
family includes myeloid cell leukemia-1 (MCL-1), which plays an
integral role in cell survival and apoptosis (7), and BCL-2-associated X protein (BAX), a
pro-apoptotic protein that induces the release of cytochrome
c from mitochondria to the cytosol, where it binds to
apoptotic peptidase activating factor 1 and facilitates the
formation of the apoptosome, leading to the activation of caspase-9
and eventual cell death. The extrinsic pathway is activated by
specific ligands that engage death receptors. This process involves
Fas, which binds to and activates the caspase-8 protein (8,9). Thus,
caspases are central regulators of the apoptotic process, and are
involved in the two major apoptosis pathways (10).
Metformin, an oral biguanide drug, has been used
widely to treat type 2 diabetes and pre-diabetic conditions for
>40 years due to its good tolerability profile and low cost. In
addition to its anti-diabetic effect, epidemiological studies and
basic research have suggested that metformin may reduce the risk of
cancer in diabetic patients (11–15).
Moreover, a number of clinical studies have shown that the survival
rate of cancer patients is improved by treatment with metformin
(16–19). These results suggest that metformin
might potentially be used as an anticancer drug for different types
of cancer. However, the effects and possible mechanisms of action
of metformin in the proliferation and apoptosis of breast cancer
cells have not been explored in depth. The present study was
designed to address this deficiency by investigating the cytotoxic
mechanism of metformin in MDA-MB-231 and MDA-MB-435 human breast
cancer cells.
Materials and methods
Reagents and antibodies
Metformin and propidium iodide (PI) were purchased
from Sigma-Aldrich. (St. Louis, MO, USA). The fluorescent dyes JC-1
and dihydroethidium (DHE) were purchased from Nanjing KeyGen
Biotech Co., Ltd. (Nanjing, China). Rabbit anti-MCL-1 (ab32087;
1:500) and anti-BCL-2 (ab32124; 1:1,000) monoclonal antibodies were
obtained from Abcam (Cambridge, UK). Rabbit anti-BAX (5023;
1:1,000) monoclonal antibody was purchased from Cell Signaling
Technology, Inc. (Beverly, MA, USA), and rabbit anti-β-actin
polyclonal antibody (sc-130657; 1:1,000) was obtained from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA). Horseradish peroxidase
(HRP)-conjugated goat anti-mouse IgG (BL001A; 1:5,000) and goat
anti-rabbit IgG (BL003A; 1:5,000) were purchased from Biosharp
(Suzhou, China).
Cell lines and cell culture
MDA-MB-231 and MDA-MB-435 breast cancer cells were
purchased from Shanghai Cell Bank (Shanghai, China). The cells were
inoculated in fresh Dulbecco's modified Eagle's medium containing
10% fetal calf serum (both Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), 100 U/ml penicillin and 100 mg/l streptomycin
(both Sigma-Aldrich). Cultures were maintained at 37°C in a 5%
CO2 humidified atmosphere.
Cell viability assay
Breast cancer cells were cultured in a 96-well plate
for 24 h at an initial density of 1×105 cells/well,
prior to treatment with various concentrations (1.25, 2.5, 5, 10
and 20 mM) of metformin for 24, 48 or 72 h at 37°C. Subsequently,
20 µl 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) solution (5.0 mg/l; Sigma-Aldrich) was added to each well and
the cells were incubated for a further 4 h. The medium was then
removed by gentle aspiration and 150 µl dimethyl sulfoxide
(Sigma-Aldrich) was added to each well to dissolve the resulting
crystals. Absorbance was read at 490 nm using a microplate reader
(Synergy HT; BioTek Instruments, Inc., Winooski, VT, USA). Cell
viability was assessed by comparison with control cells treated
with vehicle alone.
Colony formation
Colony-forming rates of the tumor cells were
determined using a colony formation assay. The breast cancer cells
were seeded at 500 cells/well in 6-well plates and incubated for 24
h at 37°C. The medium was removed and the cells were then treated
with various concentrations of metformin (0, 0.1, 0.5 and 2.0 mM)
using standard cell culture conditions under a 5% CO2
humidified atmosphere at 37°C. After 8 days, the dishes were washed
twice with phosphate-buffered saline (PBS), fixed with
paraformaldehyde (Sigma-Aldrich) at −20°C for 10 min, and then
stained with crystal violet (Thermo Fisher Scientific, Inc.).
Cellular adenosine triphosphate (ATP)
levels
Cellular ATP levels were determined by a
luciferase-based assay with an ATP Bioluminescence Assay kit (Merck
Millipore, Darmstadt, Germany), according to the manufacturer's
protocol. Briefly, breast cancer cells (2×106) were
seeded in each well of a 24-well plate and allowed to reach the
exponential growth phase prior to being treated with various
concentrations of metformin (10, 20 and 40 mM) for 24 h at 37°C.
After 5 h, the cells were harvested and centrifuged at 10,000 × g
for 5 min at 4°C. Supernatants (100 µl) were mixed with 100 µl ATP
detection working solution in a white 96-well plate. Measurements
were obtained using a luminometer (GloMax® 96 Microplate
Luminometer; Promega Corporation, Sunnyvale, CA, USA) at an
emission maximum of ~560 nm for 300 sec.
Mitochondrial membrane potential
(∆ψm)
The ∆ψm was assessed using the JC-1 Apoptosis
Detection kit (Beyotime Institute of Biotechnology, Jiangsu,
China). Briefly, cells were plated in a 12-well plate at a density
of 2×106 cells/well and treated with metformin (5 mM)
for 24 h at 37°C. Cells were washed once with PBS and incubated at
37°C for 30 min in medium containing 0.5 ml JC-1. Then, the
supernatant was removed and cells were rinsed twice with JC-1
staining buffer. Fluorescence images were observed within 30 min
using a fluorescent microscope (Eclipse Ti-U; Nikon Corporation,
Tokyo, Japan). A decline in the ratio of red to green fluorescence
indicated a loss of ∆ψm.
Reactive oxygen species (ROS)
levels
The determination of ROS levels was based on the
oxidation of DHE. Breast cancer cells were seeded at a density of
2×105 cells/well in a six-well plate for 24 h and
incubated with 20 mM metformin for 1, 3 and 6 h. The cells were
then treated with DHE (5 mM, Beyotime Institute of Biotechnology)
for 20 min at 37°C in the dark. The cells were then washed twice
and harvested in PBS. The fluorescence of DHE was detected with a
flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) with
excitation at 488 nm and emission at 530 nm.
PI staining and caspase inhibition
assays
Apoptosis of the breast cancer cells was determined
using a PI apoptosis detection kit (Nanjing KeyGen Biotech Co.,
Ltd.) according to the manufacturer's protocol. Briefly, breast
cancer cells (2×106) were plated in each well of a
24-well plate and allowed to reach the exponential growth phase
before being treated with various concentrations of metformin (5,
10 and 20 mM) for 24 h. The cells were then harvested and collected
by centrifugation at 1,500 × g for 10 min at 4°C, and resuspended
in 200 µl ice-cold binding buffer. The cell suspensions were
incubated with 5 µl PI for 10 min at room temperature in the dark,
and the percentage of apoptotic cells was measured by flow
cytometry (Accuri™ C6; BD Biosciences). For caspase inhibitor
assays, the cells were pretreated with a pan-caspase inhibitor
(z-VAD-FMK; Sigma-Aldrich, Shanghai, China; 20 µM) for 2 h and then
treated with various concentrations of metformin (2.5, 5, 10 and 20
mM) for an additional 24 h. The extent of apoptosis was then
determined using the MTT assay.
Western blot analysis
MDA-MB-231 cells treated with metformin (5 mM) were
collected at various time points (0, 6, 16 and 24 h), washed twice
with ice-cold PBS, and incubated in radioimmunoprecipitation assay
protein lysis buffer (Beyotime Institute of Biotechnology) for 30
min at 4°C. The lysates were centrifuged at 13,000 × g for 10 min
at 4°C. The concentrations of total lysate protein were detected by
a standard Bradford assay (Bio-Rad Laboratories, Inc., San Diego,
CA, USA), resolved by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (Beyotime Institute of Biotechnology), and
transferred to nitrocellulose membranes (Bio-Rad Laboratories,
Inc.). The nitrocellulose membranes were blocked with 5% non-fat
milk at room temperature for 1 h and then incubated overnight at
4°C with rabbit anti-BCL-2 (1:1,000), anti-MCL-1 (1:500), anti-BAX
(1:1,000) and anti-β-actin (1:1,000) primary antibodies. After
washing the membranes three times for 10 min each with
Tris-buffered saline containing Tween-20, they were incubated with
HRP-conjugated secondary antibodies (1:5,000 dilution). Proteins
were visualized using an Enhanced Chemiluminescence-Plus kit
(Nanjing KeyGen Biotech Co., Ltd.).
Statistical analysis
All experiments were performed at least in
triplicate. Data are expressed as the means ± standard error of the
mean (SEM). Statistical analyses were performed using one-way
analysis of variance with SPSS software version 20.0 (IBM SPSS;
Armonk, NY, USA). Differences were considered statistically
significant at P<0.05.
Results
Metformin inhibits the viability and
colony formation of breast cancer cells
MDA-MB-231 and MDA-MB-435 cells were treated with
metformin at different concentrations (1.25, 2.5, 5, 10 or 20
mmol/l) for 24, 48 or 72 h. MTT and colony formation assays were
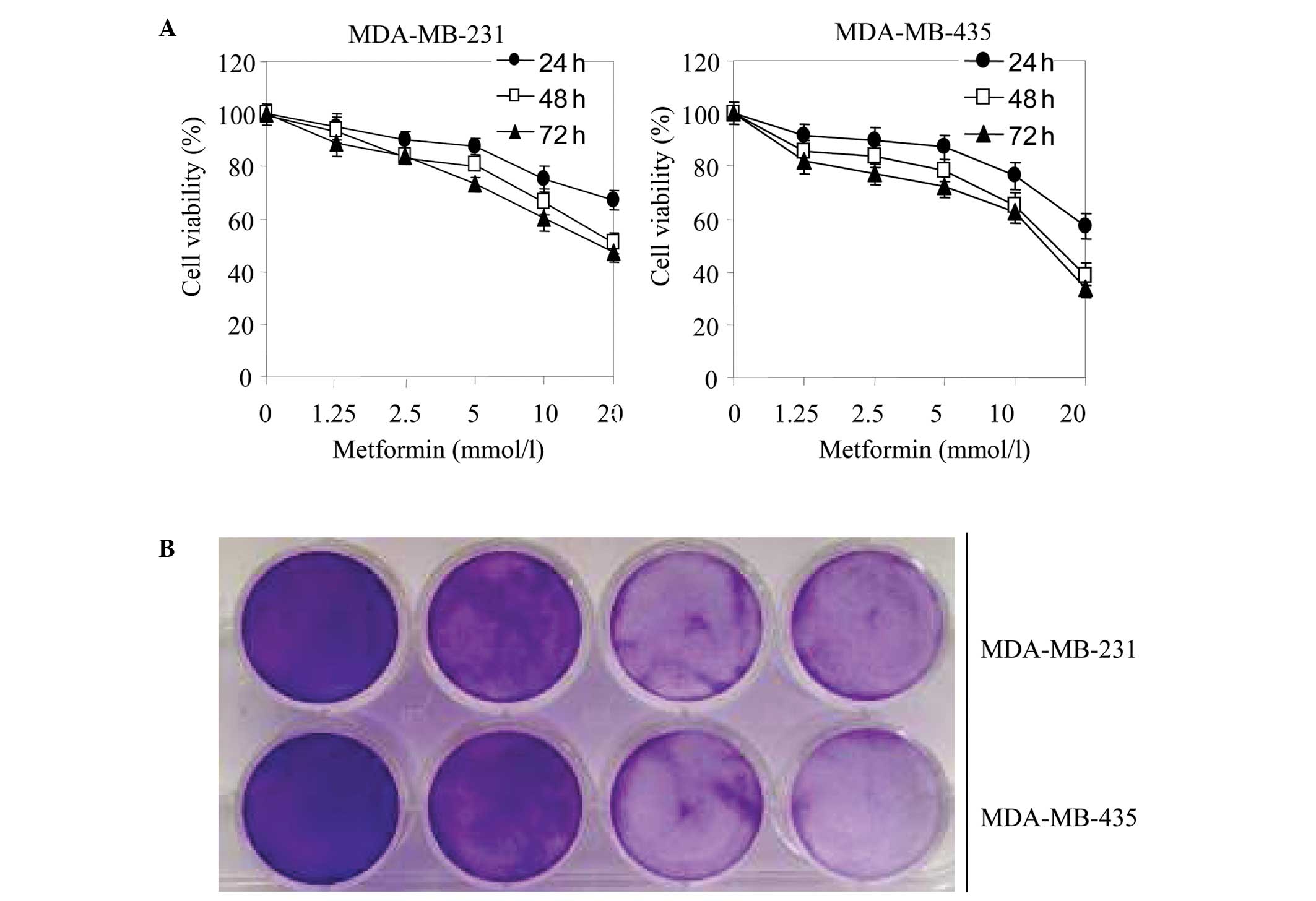
then conducted to assess cell proliferation. As shown in Fig. 1A, metformin significantly decreased
cell viability in a concentration- and time-dependent manner. In
addition, metformin inhibited cell colony formation in a
concentration-dependent manner (Fig.
1B).
Metformin decreases ∆ψm and the
production of ATP
The ∆ψm and its loss are near-universal hallmarks
of, and critical steps for, subsequent cell death (20). In addition, ∆ψm and the generation of
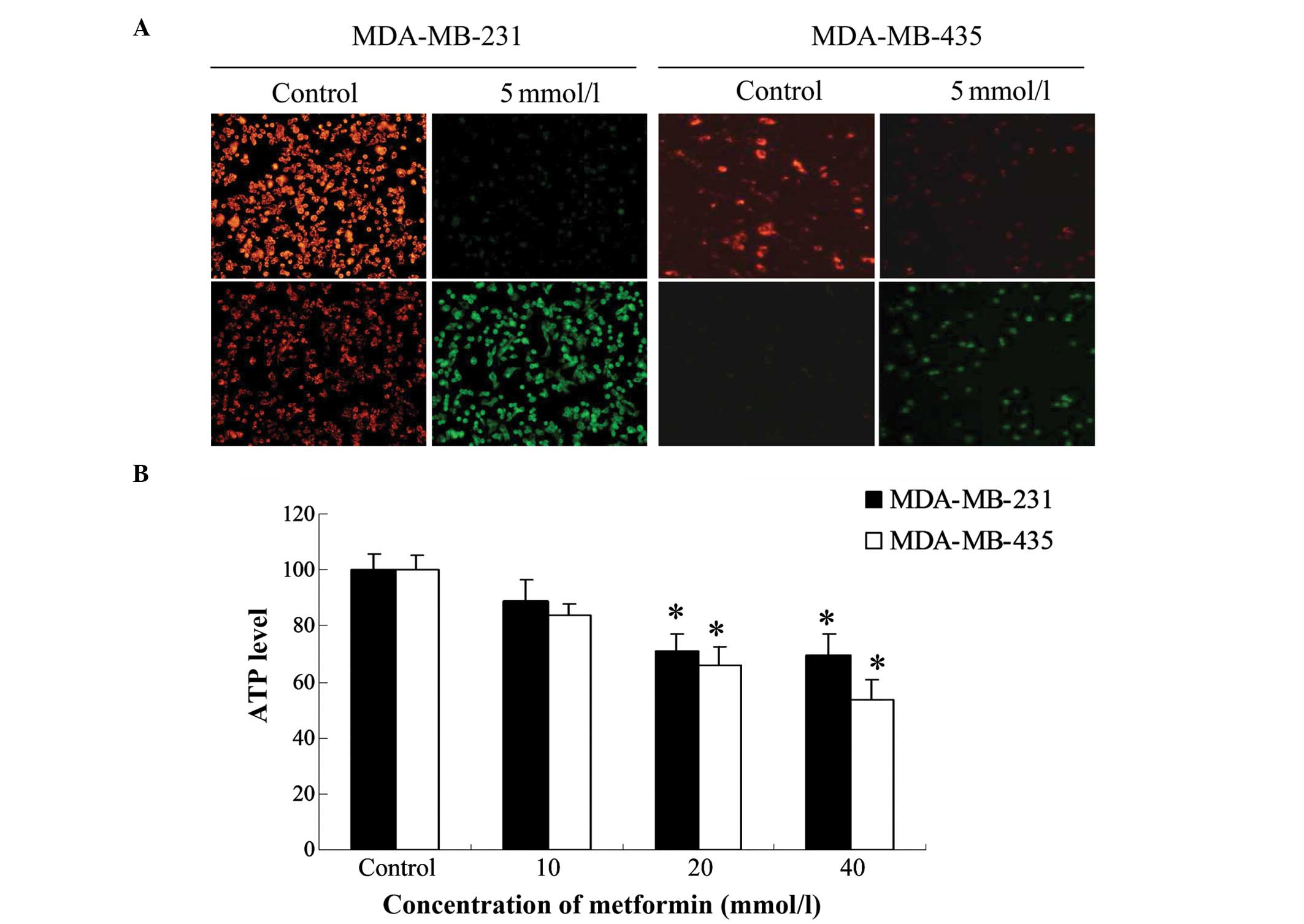
ATP reflect mitochondrial function. Therefore, the effect of a 24-h
treatment with metformin (5 mM) on ∆ψm was investigated in
MDA-MB-231 and MDA-MB-435 cells. The ∆ψm was reduced in
metformin-treated cells as shown by the significant reduction in
red fluorescence and increase in green fluorescence generated by
JC-1 (Fig. 2A). The shift from red
to green fluorescence was more notable in MDA-MB-231 cells than in
MDA-MB-435 cells.
To study the effect of metformin on cellular ATP
production, the two breast cancer cell lines were treated with
different concentrations of metformin (10, 20 and 40 mM) for 24 h.
The ATP levels of metformin-treated MDA-MB-231 and MDA-MB-435 cells
were reduced compared with those in the respective control group.
The lowest level was observed at a metformin concentration of 40 mM
in MDA-MB-435 cells (Fig. 2B). In
MDA-MB-231 cells, the lowest ATP level was observed following
treatment with 20 mM metformin, and was not further reduced when
the concentration of metformin was increased to 40 mM.
Metformin increases ROS
production
As ROS generation is a common after-effect of a
reduction in ∆ψm (21,22), changes in the levels of ROS were
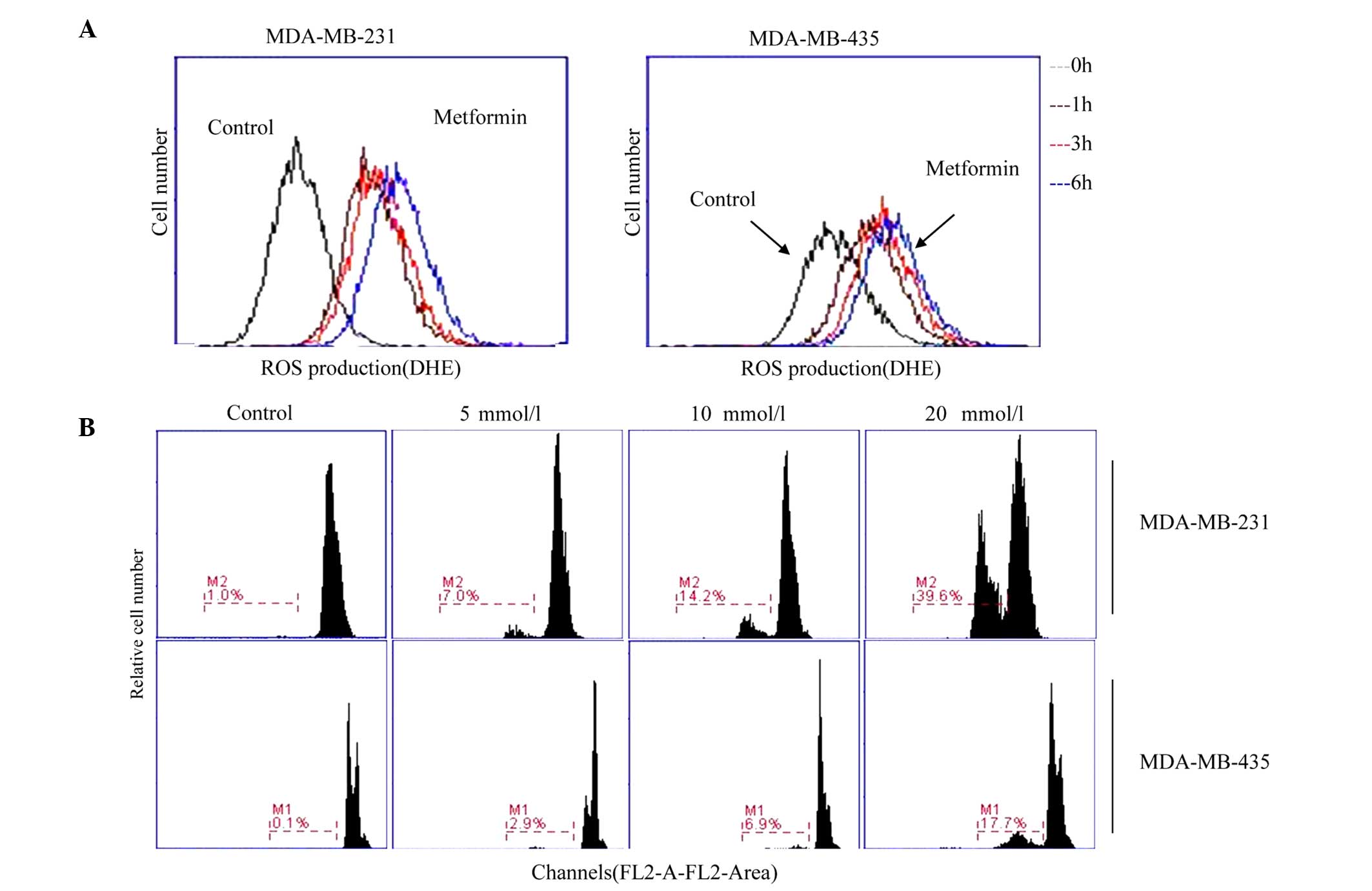
measured by DHE fluorescence. MDA-MB-231 and MDA-MB-435 cells
treated with metformin (20 mM) for various durations (1, 3 and 6 h)
showed an increase in green fluorescence in a time-dependent manner
(Fig. 3A). Flow cytometric analyses
demonstrated that 20 mM metformin caused an increase in ROS
production, as compared with the control cells. The results of flow
cytometry were consistent with the fluorescence images, suggesting
that metformin caused an accumulation of ROS in breast cancer
cells.
Metformin-induced apoptosis is a
caspase-dependent process
The type of cell death induced in breast cancer
cells treated with metformin was determined by PI staining.
Following incubation with metformin for 24 h, the extent of
apoptosis in MDA-MB-231 cells markedly increased from 7.0% with 5
mM metformin to 39.6% with 20 mM metformin. MDA-MB-435 cells were
less sensitive to metformin, as the percentage of cells undergoing
apoptosis only increased from 2.9 to 17.7% (Fig. 3B).
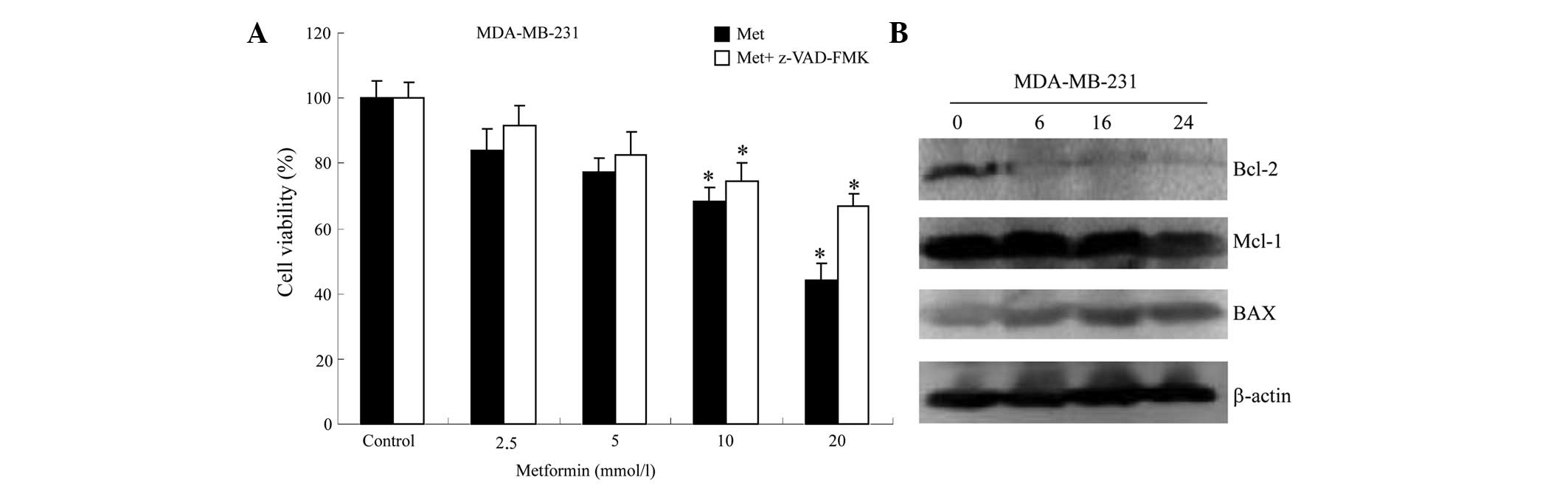
To confirm whether the metformin-induced cell death
observed in MDA-MB-231 cells was a result of apoptosis, the
pan-caspase inhibitor z-VAD-FMK was used to examine the role of
caspases in the process. The results of the MTT assay revealed that
the viability of MDA-MB-231 cells was significantly attenuated by
z-VAD-FMK, confirming that the process is caspase-dependent
(Fig. 4A).
Metformin changes the expression of
apoptosis-associated proteins
To confirm whether metformin-induced apoptosis
activates the mitochondrial pathway, MDA-MB-231 cells were
incubated in the absence or presence of metformin and then
harvested for western blot analyses. Since the mitochondrial
pathway appeared to be involved in the induction of intrinsic
apoptosis, the levels of anti- and pro-apoptotic proteins that
dysregulate the mitochondrial balance were measured. Incubation of
cells with metformin time-dependently upregulated the levels of the
pro-apoptotic protein BAX and downregulated the levels of the
anti-apoptotic proteins BCL-2 and MCL-1 (Fig. 4B). This indicates that
metformin-induced apoptosis is a mitochondria-mediated process.
Discussion
Recent studies have shown that metformin exerts
antitumor effects in vivo and in vitro (23–25). In
addition, metformin has been demonstrated to exert anticancer
activity against hepatocellular carcinoma through inhibition of the
mechanistic target of rapamycin (mTOR) translational pathway in an
AMP-activated protein kinase (AMPK)-independent manner. This leads
to G1 arrest in the cell cycle and subsequent cell apoptosis
through the mitochondria-dependent pathway (26). Nonetheless, the mechanisms underlying
these actions remain unknown in breast cancer.
Apoptosis suppresses cell proliferation (27), and the BCL-2 family of proteins plays
an important role in the response to apoptosis of different cell
types, including myocardial, endothelial and cancer cells (28–31).
Furthermore, these proteins are primarily responsible for
initiating apoptosis through an intrinsic (mitochondrial) pathway,
where signals directly received by the cells can initiate a cascade
of events leading to a commitment to cell death (32,33).
BAX, a pro-apoptotic member of the BCL-2 protein family, which
normally localizes to the cytosolic compartment, translocates to
and oligomerizes on the outer mitochondrial membrane to form
channels through the membrane. The relative ratio of pro-apoptotic
proteins, such as BAX, to anti-apoptotic proteins, such as BCL-2,
determines cell survival or death. Thus, a high ratio of BAX/BCL-2
is associated with greater vulnerability to apoptotic activation
(34).
Metformin is a partial inhibitor of complex 1 of the
mitochondrial electron transport chain (35,36),
causing an abnormal flow of electrons to oxygen and leading to the
accumulation of ROS within the mitochondrial matrix. ROS are
considered to be involved in the pathogenesis of various diseases,
including cancer and inflammation. ROS are also necessary for tumor
cell proliferation, secretion, differentiation and defense.
However, high levels of ROS can induce tumor cell apoptosis and
senescence (37). The mitochondrial
pathway is important for the induction of apoptosis by
chemotherapeutic agents. Moreover, ROS induce the collapse of the
∆ψm, thereby triggering a series of mitochondria-associated events,
including apoptosis (38).
The potency of metformin against cancer cells has
been linked to the generation of ROS (39–41).
Consistent with these previous reports, the results of the present
study indicated that metformin increased the level of ROS in
MDA-MB-231 and MDA-MB-435 cells. A reduction of ∆ψm also induces
apoptosis by releasing pro-apoptotic factors such as cytochrome
c from the mitochondrial inner space to the cytosol.
Cytosolic cytochrome c participates in the activation of
caspase-8 and caspase-9. This, in turn, activates the executioner
caspase-3 to induce cell apoptosis (42). In the present study, metformin
treatment significantly increased the ROS level, decreased ∆ψm, and
activated caspase family enzymes in MDA-MB-231 and MDA-MB-435
cells. In addition, metformin increased the expression of the
pro-apoptotic protein BAX and decreased levels of the
anti-apoptotic proteins BCL-2 and MCL-1 in MDA-MB-231 cells,
leading to an increased ratio of BAX/BCL-2. This result concurs
with other reports showing that a high BAX/BCL-2 ratio is
associated with cytochrome c release and a reduction in ∆ψm
(43).
In conclusion, the present study demonstrated that
metformin treatment effectively reduced cell viability and induced
apoptotic cell death in human breast cancer cells. Metformin also
induced activation of the caspase-dependent pathway, accumulation
of ROS, and a reduction in ΔΨm and ATP production in human breast
cancer cells. These results suggest that metformin-induced
apoptosis is mediated by the accumulation of ROS via the
mitochondria-mediated apoptotic pathway. This study provides
further support for the development of metformin as a novel
therapeutic agent for the treatment of human breast cancer.
Acknowledgements
The present study was supported by a grant from the
Natural Science Foundation of China (grant no. 81272739).
References
|
1
|
Carter D: New global survey shows an
increasing cancer burden. Am J Nurs. 114:172014. View Article : Google Scholar
|
|
2
|
Taha FM, Zeeneldin AA, Helal AM, Gaber AA,
Sallam YA, Ramadan H and Moneer MM: Prognostic value of serum
vascular endothelial growth factor in Egyptian females with
metastatic triple negative breast cancer. Clin Biochem.
42:1420–1426. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Coelho BA, Belo AV, Andrade SP, Amorim WC,
Uemura G and da Silva Filho AL: N-acetylglucosaminidase,
myeloperoxidase and vascular endothelial growth factor serum levels
in breast cancer patients. Biomed Pharmacother. 68:185–189. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Breidenbach M, Rein DT, Schöndorf T, Khan
KN, Herrmann I, Schmidt T, Reynolds PN, Vlodavsky I, Haviv YS and
Curiel DT: A new targeting approach for breast cancer gene therapy
using the heparanase promoter. Cancer Lett. 240:114–122. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gnant M, Balic M, Petru E, Raunik W,
Singer CF, Steger GG, Watzke IM and Brodowicz T: Treatment of bone
metastases in patients with advanced breast cancer. Breast Care
(Basel). 7:92–98. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Coleman RE: Clinical features of
metastatic bone disease and skeletal morbidity. Clin Cancer Res.
12:6243s–6249s. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sano M, Hayashi E, Murakami H, Kishimoto
H, Fukuzawa R and Nemoto N: Mcl-1, an anti-apoptotic Bcl-2 family
member, essentially overlaps with insulin-producing cells in
nesidioblastosis. Virchows Arch. 452:469–470. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arnoult D, Parone P, Martinou JC,
Antonsson B, Estaquier J and Ameisen JC: Mitochondrial release of
apoptosis-inducing factor occurs downstream of cytochrome c release
in response to several proapoptotic stimuli. J Cell Biol.
159:923–929. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Debatin KM: Apoptosis pathways in cancer
and cancer therapy. Cancer Immunol Immunother. 53:153–159. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Algire C, Amrein L, Zakikhani M, Panasci L
and Pollak M: Metformin blocks the stimulative effect of a
high-energy diet on colon carcinoma growth in vivo and is
associated with reduced expression of fatty acid synthase. Endocr
Relat Cancer. 17:351–360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang ZJ, Zheng ZJ, Kan H, Song Y, Cui W,
Zhao G and Kip KE: Reduced risk of colorectal cancer with metformin
therapy in patients with type 2 diabetes: A meta-analysis. Diabetes
Care. 34:2323–2328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang ZJ, Zheng ZJ, Shi R, Su Q, Jiang Q
and Kip KE: Metformin for liver cancer prevention in patients with
type 2 diabetes: A systematic review and meta-analysis. J Clin
Endocrinol Metab. 97:2347–2353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Emami Riedmaier A, Fisel P, Nies AT,
Schaeffeler E and Schwab M: Metformin and cancer: From the old
medicine cabinet to pharmacological pitfalls and prospects. Trends
Pharmacol Sci. 34:126–135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nair V, Pathi S, Jutooru I, Sreevalsan S,
Basha R, Abdelrahim M, Samudio I and Safe S: Metformin inhibits
pancreatic cancer cell and tumor growth and downregulates SP
transcription factors. Carcinogenesis. 34:2870–2879. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bayraktar S, Hernadez-Aya LF, Lei X,
Meric-Bernstam F, Litton JK, Hsu L, Hortobagyi GN and
Gonzalez-Angulo AM: Effect of metformin on survival outcomes in
diabetic patients with triple receptor-negative breast cancer.
Cancer. 118:1202–1211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
He X, Esteva FJ, Ensor J, Hortobagyi GN,
Lee MH and Yeung SC: Metformin and thiazolidinediones are
associated with improved breast cancer-specific survival of
diabetic women with HER2+ breast cancer. Ann Oncol. 23:1771–1780.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hou G, Zhang S, Zhang X, Wang P, Hao X and
Zhang J: Clinical pathological characteristics and prognostic
analysis of 1,013 breast cancer patients with diabetes. Breast
Cancer Res Treat. 137:807–816. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lega IC, Austin PC, Gruneir A, Goodwin PJ,
Rochon PA and Lipscombe LL: Association between metformin therapy
and mortality after breast cancer: A population-based study.
Diabetes Care. 36:3018–3026. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shirakata Y and Koike K: Hepatitis B virus
X protein induces cell death by causing loss of mitochondrial
membrane potential. J Biol Chem. 278:22071–22078. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zamzami N, Marchetti P, Castedo M,
Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B and
Kroemer G: Sequential reduction of mitochondrial transmembrane
potential and generation of reactive oxygen species in early
programmed cell death. J Exp Med. 182:367–377. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gottlieb E, Vander Heiden MG and Thompson
CB: Bcl-x (L) prevents the initial decrease in mitochondrial
membrane potential and subsequent reactive oxygen species
production during tumor necrosis factor alpha-induced apoptosis.
Mol Cell Biol. 20:5680–5689. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bao B, Wang Z, Ali S, Ahmad A, Azmi AS,
Sarkar SH, Banerjee S, Kong D, Li Y, Thakur S and Sarkar FH:
Metformin inhibits cell proliferation, migration and invasion by
attenuating CSC function mediated by deregulating miRNAs in
pancreatic cancer cells. Cancer Prev Res (Phila). 5:355–364. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen TW, Liang YN, Feng D, Tao LY, Qi K,
Zhang HY, Wang HX, Lin QS and Kong H: Metformin inhibits
proliferation and promotes apoptosis of HER-2 positive breast
cancer cells by downregulating HSP90. J BUON. 18:51–56.
2013.PubMed/NCBI
|
|
25
|
Erices R, Bravo ML, Gonzalez P, Oliva B,
Racordon D, Garrido M, Ibañez C, Kato S, Brañes J, Pizarro J, et
al: Metformin at concentrations corresponding to the treatment of
diabetes, potentiates the cytotoxic effects of carboplatin in
cultures of ovarian cancer cells. Reprod Sci. 20:1433–1446. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiong Y, Lu QJ, Zhao J and Wu GY:
Metformin inhibits growth of hepatocellular carcinoma cells by
inducing apoptosis via mitochondrion-mediated pathway. Asian Pac J
Cancer Prev. 13:3275–3279. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Circu ML and Aw TY: Glutathione and
modulation of cell apoptosis. Biochim Biophys Acta. 1823:1767–1777.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Smith MA and Schnellman RG: Calpains,
mitochondria and apoptosis. Cardiovasc Res. 96:32–37. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Andersen JL and Kornbluth S: The tangled
circuitry of metabolism and apoptosis. Mol Cell. 49:399–410. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Petsophonsakul P, Pompimon W and
Banjerdpongchai R: Apoptosis induction in human leukemic
promyelocytic HL-60 and monocytic U937 cell lines by goniothalamin.
Asian Pac J Cancer Prev. 14:2885–2889. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cory S and Adams JM: The Bcl2 family:
Regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Green DR and Kroemer G: The
pathophysiology of mitochondrial cell death. Science. 305:626–629.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rondelet B, Dewachter C, Kerbaul F, Kang
X, Fesler P, Brimioulle S, Naeije R and Dewachter L: Prolonged
overcirculation-induced pulmonary arterial hypertension as a cause
of right ventricular failure. Eur Heart J. 33:1017–1026. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
El-Mir MY, Nogueira V, Fontaine E, Avéret
N, Rigoulet M and Leverve X: Dimethylbiguanide inhibits cell
respiration via an indirect effect targeted on the respiratory
chain complex 1. J Biol Chem. 275:223–228. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Owen MR, Doran E and Halestrap AP:
Evidence that metformin exerts its anti-diabetic effects through
inhibition of complex 1 of the mitochondrial respiratory chain.
Biochem J. 348:607–614. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Behrend L, Henderson G and Zwacka RM:
Reactive oxygen species in oncogenic transformation. Biochem Soc
Trans. 31:1441–1444. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aldea MD, Petrushev B, Soritau Q,
Tomuleasa CI, Berindan-Neagoe I, Filip AG, Chereches G, Cenariu M,
Craciun L, Tatomir C, et al: Metformin plus sorafenib high impacts
temozolomide resistant glioblastoma stem-like cells. J Buon.
19:502–511. 2014.PubMed/NCBI
|
|
40
|
Haugrud AB, Zhuang Y, Coppock JD and
Miskimins WK: Dichloroacetate enhance apoptotic cell death via
oxidative damage and attenuates lactate production in
metformin-treated breast cancer cells. Breast Cancer Res Treat.
147:539–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim EH, Kim MS, Cho CK, Jung WG, Jeong YK
and Jeong JH: Low and high linear energy transfer radiation
sensitization of HCC cells by metformin. J Radiat Res. 55:432–442.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang X: The expanding role of mitochondria
in apoptosis. Genes Dev. 15:2922–2933. 2001.PubMed/NCBI
|
|
43
|
Ibrahim MY, Hashim NM, Mohan S, Abdulla
MA, Kamalidehghan B, Ghaderian M, Dehghan F, Ali LZ, Arbab IA,
Yahayu M, et al: α-Mangostin from Cratoxylum arborescens
demonstrates apoptogenesis in MCF-7 with regulation of NF-κB and
Hsp70 protein modulation in vitro and tumor reduction in vivo. Drug
Des Devel Ther. 8:1629–1647. 2014. View Article : Google Scholar : PubMed/NCBI
|