Introduction
Hypoxia frequently accompanies such vascular
disorders as atherosclerosis (1),
thrombosis (2) and
ischemia/reperfusion (I/R) injury (3). Myocardial ischemia/reperfusion, in
particular, is a major contributor to cardiomyocyte impairment
(4,5). As the most prevalent disease afflicting
humans (6,7), coronary artery disease is mainly caused
by the hypoxic-ischemic injury to cardiomyocytes (8,9).
Myocardial ischemia and infarction develop when the blood supply to
the myocardium decreases or is discontinued. The mechanism
underlying myocardial ischemia and infarction has yet to be fully
elucidated; however, the oxygen deprivation, i.e. hypoxia, in
myocardial ischemia and infarction can, to varying degrees,
threaten the function and survival of cardiomyocytes (10,11),
although numerous adaptive countermeasures can be induced in the
cardiomyocytes in response to the hypoxic condition (12–14).
Hypoxia-inducible factor 1 (HIF-1) is a
transcription factor that functions as a master regulator of
adaptive responses to reduced O2 environments (10). HIF-1 can improve local
microcirculation, via effects on vascular growth and function, and
regulate O2 utilization, by switching oxidative
metabolism to glycolytic metabolism (15–17).
HIF-1 is composed of HIF-1α and HIF-1β subunits (18). HIF-1α, rather than HIF-1β, is the
O2-regulated subunit during hypoxia (19). Hypoxia and re-oxygenation potently
increase HIF-1 transcriptional activity and HIF-1α protein levels
(20–22). Upregulated HIF-1α activation has been
demonstrated in human hearts under conditions of myocardial
ischemia and infarction (23) and in
patients with coronary artery disease (24,25).
Autophagy is a dynamic catabolic process that
involves the delivery of cellular components to the lysosome for
degradation. Autophagy has been implicated in a wide range of
physiological processes and in the pathogenesis of diverse diseases
(26,27). In addition to its established roles
in the maintenance of homeostasis and adaptation to stress,
autophagy is involved in cell differentiation (28–30).
Autophagy is a closely regulated process that helps to balance the
synthesis, degradation and subsequent recycling of cellular
products; however, little is known about the role of
hypoxia-induced autophagy in hypertension and coronary artery
disease, particularly in cardiomyocytes under hypoxia.
The aim of the present study was to investigate the
autophagy in cardiomyocytes under hypoxia and evaluate the
regulation of HIF-1α in hypoxia-induced autophagy, as well as to
examine the role of autophagy in cell viability.
Materials and methods
Reagents and cell culture
Rapamycin was purchased from Sigma-Aldrich (St.
Louis, MO, USA). The coding sequence of the fusion of
microtubule-associated protein 1 light chain 3 (LC3) with green
fluorescent protein (GFP) was synthesized and cloned into
pcDNA3.1(+) (Invitrogen Life Technologies, Carlsbad, CA, USA) to
construct the LC3-GFP-expressing plasmid. The coding sequence of
HIF-1α was synthesized and cloned into pcDNA3.1(+) to construct the
HIF-1α-pcDNA3.1(+)-expressing plasmid. The HIF-1α small interfering
RNA (siRNA) sequence was designed and the duplexes were produced by
Shanghai GenePharma Co., Ltd. (Shanghai, China).
A rat H9c2 heart cell line was purchased from the
American Type Culture Collection (Manassas, VA, USA), cultured in
Dulbeccos modified Eagles medium (DMEM; Invitrogen Life
Technologies) supplemented with 10% fetal bovine serum (FBS;
Gibco-BRL, Grand Island, NY, USA), 100 U/ml penicillin
(Shijiazhuang Pharmaceutical Group, Shijiazhuang, China) and 100
mg/ml streptomycin (Shijiazhuang Pharmaceutical Group), and grown
in an atmosphere of 5% CO2/95% humidified air at 37°C.
The culture medium was changed every second day.
Quantitative GFP-LC3 analysis and
electron microscopy
Quantitative GFP-LC3 light microscopy autophagy
assays were performed in H9c2 cells following various treatments.
The H9c2 cells grown to 80% confluence were transfected with a
GFP-LC3-expressing plasmid using Lipofectamine® 2000
(Invitrogen Life Technologies). After transfection for 24 h, the
cells were subjected to rapamycin treatment (200 nM)
(Sigma-Aldrich) for another 24 h or were pretreated with hypoxia
for 2 h and analyzed using fluorescence microscopy. In another
experiment, the GFP-LC3-expressing plasmid transfection was
followed by the transfection of the H9c2 cells with
HIF-1α-pcDNA3.1(+), pcDNA3.1(+) or HIF-1α siRNA; 24 h later, the
cells were analyzed using fluorescence microscopy.
RNA isolation and reverse
transcription quantitative polymerase chain reaction analysis
(RT-qPCR)
Total cellular RNA from 2–5×105 cells was
prepared with TRIzol reagent (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), and reverse transcription (RT) was performed
using Moloney Murine Leukemia Virus Reverse Transcriptase (Promega
Corp., Madison, WI, USA). Primer sequences are as follows: are as
following: Beclin 1, F 5-TGAAAATGAGTGTCAGAACT-3 and R
5-CTGTTCACTGTCATCCTCAT-3; autophagy-related gene 5 (Atg5), F
5-TATCAGAGCATGTCACCCTT-3 and R 5-TTCCTGTCTGGCTTGCAGCA-3); Atg7, F
5-GTCCAAGTTCCAGTGGCTGT-3 and R 5-CTCGGGCCCTGCACCTGTGC-3; and
β-actin, F 5-TGTCCACCTTCCAGCAGATGT-3 and R
5-AGCTCAGTAACAGTCCGCCTAGA-3. PCR cycling conditions were as
follows: 42°C for 10 min and 95°C for 20 sec for the reverse
transcription, and 95°C for 10 sec and 60°C for 30 sec for the PCR
reaction, repeated for 35 cycles. For the quantitative analysis of
the mRNA expression of Beclin 1, autophagy-related gene 5 (Atg5)
and Atg7, qPCR was conducted using a LightCycler® 480
system (Roche Diagnostics GmbH, Mannheim, Germany); 1 µg RNA per
sample was converted to cDNA and used for qPCR. Data were
normalized based on β-actin, using the 2−ΔΔCq
method.
Western blot assay
Polyclonal antibodies for β-actin (A2066; 1:1,000)
and LC3-I/LC3-II (L8918, 1:600) were purchased from Sigma-Aldrich.
The Beclin 1 polyclonal antibody was purchased from Santa Cruz
Biotechnology, Inc. (sc-48341; 1:500, Santa Cruz, CA, USA), and the
Atg5 (cat. no. 2630; 1:600) and Atg7 (cat. no. 2631; 1:600)
monoclonal antibodies were purchased from Cell Signaling
Technology, Inc. (Danvers, MA USA). Cell extracts were prepared by
a standard protocol using Cell Lysis Buffer (Cell Signaling
Technology, Inc.), and protein levels were quantified using a
Bio-Rad protein assay (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). Samples were separated using 10% SDS-PAGE electrophoresis and
transferred to a PVDF membranes (EMD Millipore, Billerica, MA,
USA). After blocking with 3% bovine serum albumin (Ameresco, Inc.,
Framingham, MA, USA) overnight at 4°C, the membrane was incubated
overnight again at 4°C with anti-Beclin 1, anti-Atg5, anti-Atg7,
anti-LC3-II or anti-β-actin antibody. Goat anti-mouse IgG or goat
anti-rabbit IgG (Pierce Biotechnology, Inc., Rockford, IL, USA)
secondary antibodies conjugated to horseradish peroxidase and
enhanced chemiluminescence detection systems (GE Healthcare Life
Sciences, Little Chalfont, UK) were used for detection.
Cell viability assay
Cell viability was determined using an MTT assay
(Thermo Fisher Scientific, Inc.). The H9c2 cells were seeded in
96-well plates; after 24 h, the medium was replaced with DMEM
containing 2% FBS. At 24 h after the treatment, the incubation
medium in the test wells was replaced with 50 µl 1X MTT solution,
and the cells were incubated for 2 h at 37°C. Following incubation,
the MTT solution was discarded, and 150 µl dimethylsulfoxide (DMSO)
was added to dissolve the precipitate completely at room
temperature. The optical density was then measured at 570 nm using
a spectrophotometer. The cell viability was expressed as relative
viable cells (%) to control H9c2 cells. All experiments were
performed in three separate experiments.
Statistical analysis
For GFP-LC3 dot number analysis, relative mRNA
expression of Beclin 1, Atg5 and Atg7 to β-actin, and MTT
measurements, data are presented as the mean ± standard error. Data
were analyzed by Student's t-test using GraphPad Prism 5 (GraphPad
Software, Inc., La Jolla, CA, USA), and P<0.05 was considered to
indicate a statistically significant difference.
Results
Hypoxia induces autophagy and the
expression of autophagy-associated molecules in H9c2 cells
H9c2 cells were transfected with GFP-LC3, a
biomarker for autophagy, and exposed to hypoxic conditions for 24
h. LC3, which comprises the two isoforms LC3-I and LC3-II,
typically exhibits diffuse cytosolic distribution. Representative
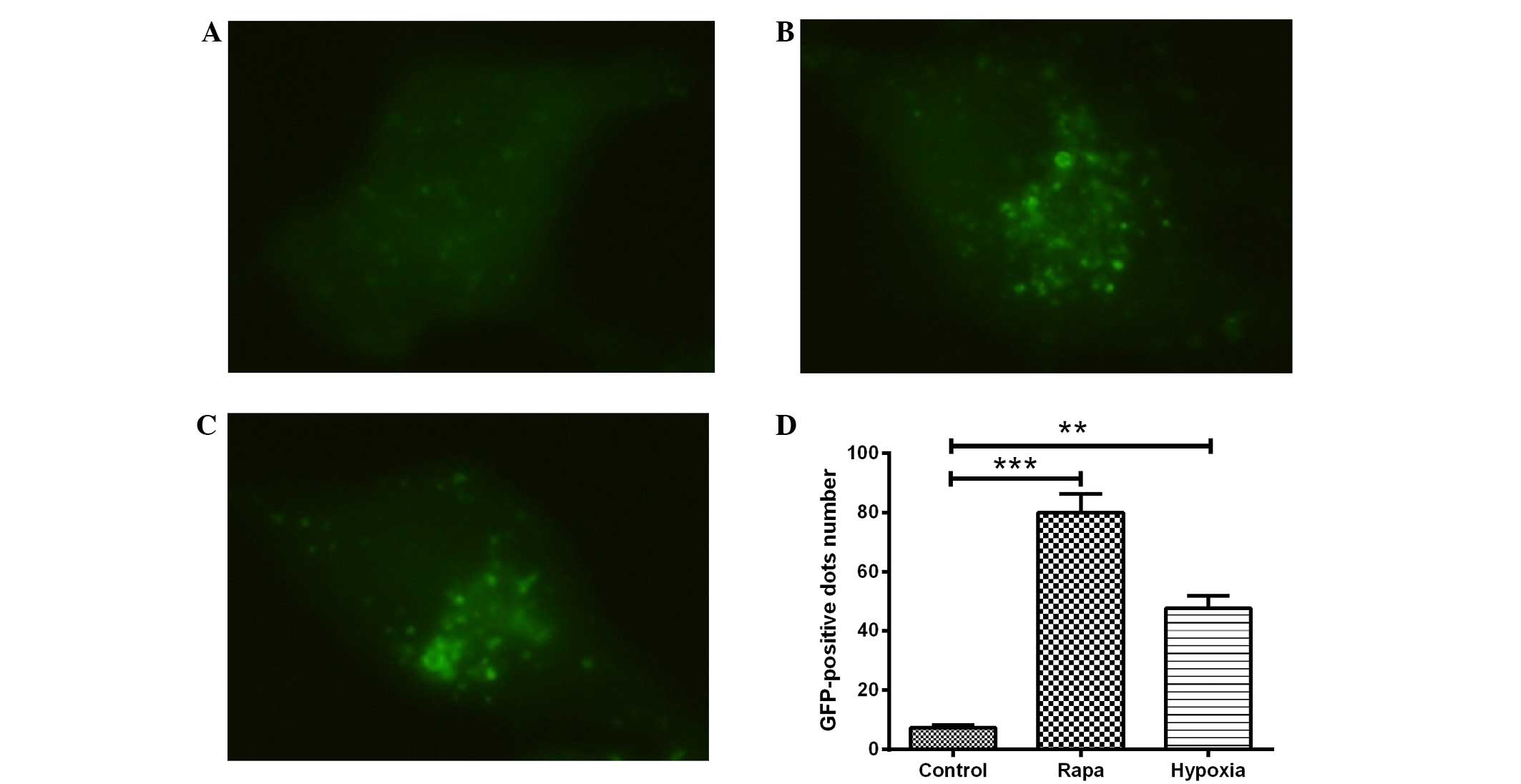
fluorescence images, shown in Fig.
1A–C, indicated that, compared with the DMSO control treatment
(Fig. 1A), treatment with rapamycin
(Fig. 1B) or hypoxia (1%
O2) (Fig. 1C) led to the
redistribution of LC3 to punctuate structures and significantly
increased the number of LC3-GFP-positive vesicles in the H9c2 cells
(P<0.001 or P<0.01; Fig. 1D).
The ultrastructures of the H9c2 cells with hypoxia or with
rapamycin treatment were observed using electron microscopy, and
prominent features of cells with hypoxia or rapamycin treatment
were found in the form of autophagic vacuoles and autolysosomes in
the cytoplasm (data not shown).
When autophagy is activated, the LC3-I protein
localized in the cytoplasm is cleaved, converted into LC3-II and
inserted into autophagosome membranes (31). To detect the expression of LC3-II,
western blotting was performed with the lysates from H9c2 cells
subjected to hypoxia (1% O2) or rapamycin treatment
(Fig. 2A). As part of a type III
phosphoinositide-3 kinase complex, the autophagy gene Beclin 1 is
required for the formation of the autophagic vesicles (32). In addition, ATG products, such as
Atg5 and Atg7, play essential roles in autophagy. As shown in
Fig. 2B and C, the mRNA and protein
expression levels of Atg5, Atg7 and Beclin 1 increased following
hypoxia or rapamycin treatment. Notably, these results indicate
that hypoxia and rapamycin treatment induce autophagy in H9c2
cells.
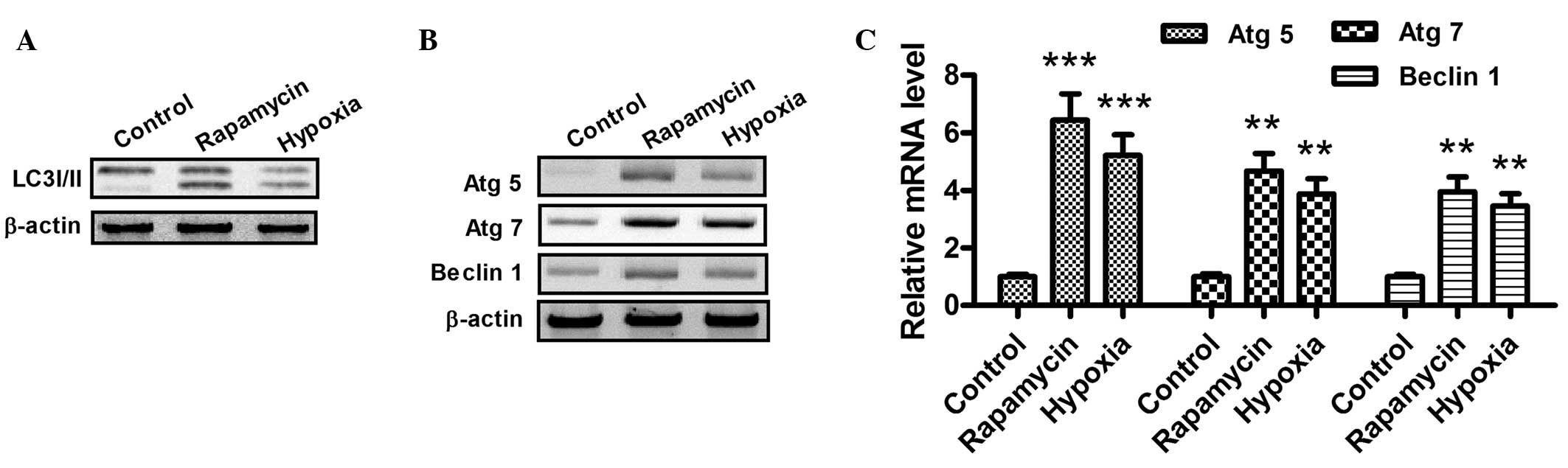 | Figure 2.Hypoxia promotes the conversion of
LC3-I to LC3-II and upregulates the expression of Atg5, Atg7 and
Beclin 1 in cardiocmyocytes. (A and B) Western blot assay of
LC3-I/-II, Atg5, Atg7 and Beclin 1 in H9c2 cells following
rapamycin or hypoxia treatment for 24 h. After the treatment with
dimethylsulfoxide (1:10,000 dilution), 100 nM rapamycin or hypoxia
(1% O2), the cells were lysed and subjected to western
blotting with antibodies against LC3-I/-II, Atg5, Atg7 or Beclin 1.
(C) mRNA levels of Atg5, Atg7 and Beclin 1 in cardiomyocytes, 24 h
after treatment with 100 nM rapamycin or hypoxia (1%
O2). The total cell mRNA was analyzed using a reverse
transcription quantitative polymerase chain reaction. **P<0.01
and ***P<0.001 vs. control. All results were independently
repeated three times. LC3, microtubule-associated protein 1 light
chain 3; Atg, autophagy-related gene. |
Hypoxia-induced autophagy-related
molecules are regulated by HIF-1 in H9c2 cells
To reveal the precise mechanisms underlying the
activation of autophagy in H9c2 cells, the change in HIF-1α
expression was further investigated with a pcDNA3.1 eukaryotic
plasmid and chemically synthesized siRNA. HIF-1, as a key
transcription factor, plays a pivotal role in hypoxia. The HIF-1
overexpression with eukaryotic plasmid or HIF-1 knockdown with
siRNA was induced in the H9c2 cells. As shown in Fig. 3A–D, overexpression of HIF-1
significantly enhanced the occurrence of hypoxia-induced autophagy
(Fig. 3B), whereas the HIF-1
knockdown attenuated the hypoxia-induced formation of autophagic
vacuoles in H9c2 cells (Fig. 3C),
compared with the control H9c2 cells under hypoxia (1%
O2). Furthermore, HIF-1 overexpression markedly
aggravated the transformation of LC3-I to LC3-II (Fig. 3E) and significantly promoted the
expression of Atg5, Atg7 and Beclin 1 (Fig. 3F). By contrast, transfection with
HIF-1 siRNA, which blocked HIF-1 expression, significantly
prevented LC3-II production, as well as the expression of Atg5,
Atg7 and Beclin 1 (Fig. 3E and F).
The results therefore showed that the hypoxia-induced autophagy in
H9c2 cells was dependent on HIF-1α. In combination, the results
suggest that the HIF-1 pathway regulates the activation of
autophagy in H9c2 cells under the hypoxic condition.
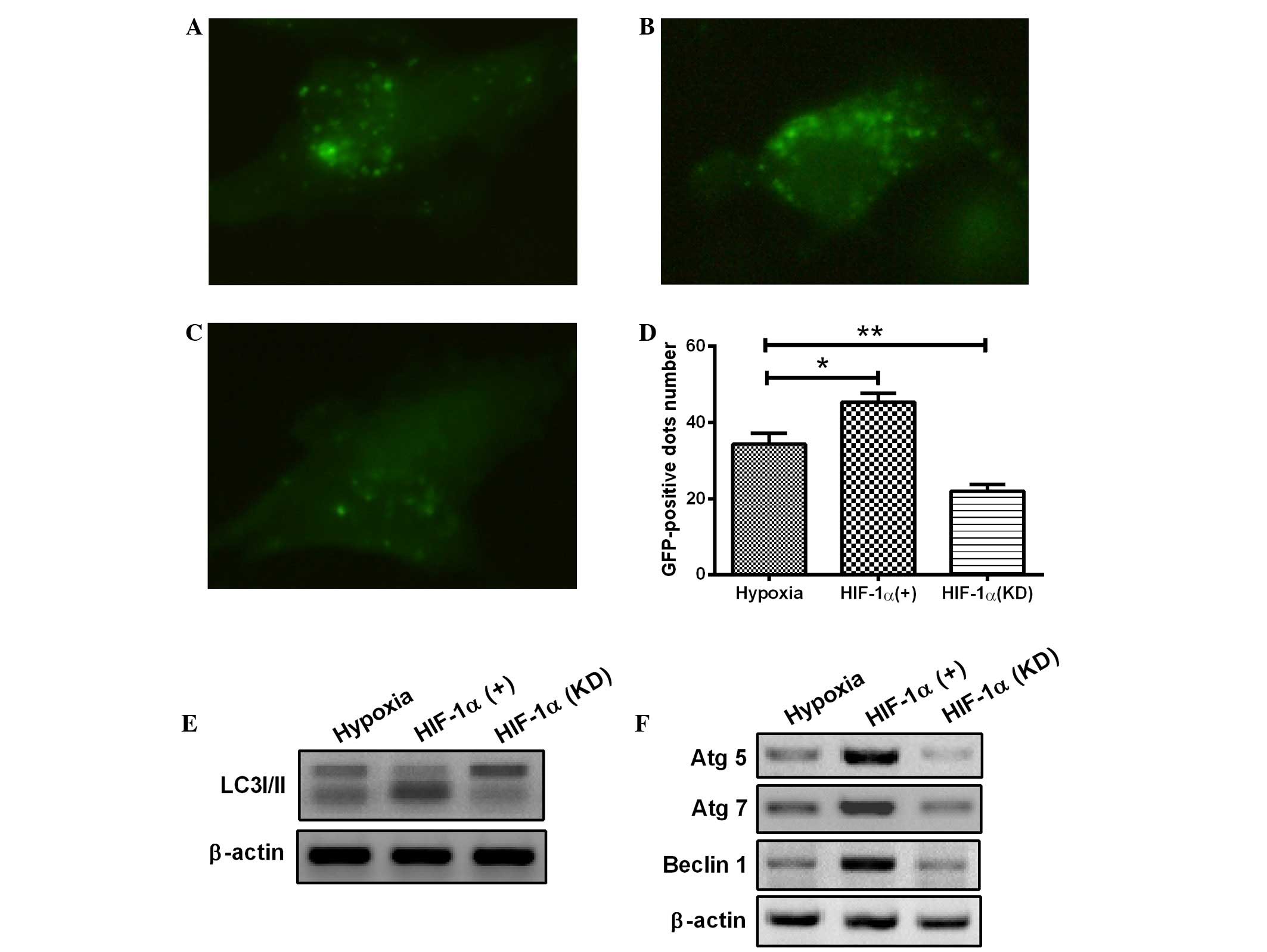 | Figure 3.Hypoxia induces autophagy in
cardiomyocytes through the HIF-1α pathway. (A-D) The cardiomyocytes
were transfected with a plasmid that expressed a GFP-LC3 fusion
protein and (A) pcDNA3.1, (B) pcDNA-HIF-1α or (C) HIF-1α siRNA. At
24 h after transfection, the cells were cultured under hypoxia (1%
O2) for a further 24 h. Following fixation, cells were
immediately visualized using fluorescence microscopy. (D) The
number of punctate GFP-LC3 dots in each cell was counted, and ≥100
cells were included for each group. *P<0.05 and **P<0.01 vs.
hypoxia. (E and F) Following transfection with pcDNA-HIF-1α or
HIF-1α siRNA for 24 h, the cells were lysed and subjected to
western blotting with the antibodies indicated. All results are
averaged for three independent experiments. HIF-1α,
hypoxia-inducible factor 1α; GFP, green fluorescent protein; LC3,
microtubule-associated protein 1 light chain 3; siRNA, small
interfering RNA; Atg, autophagy-related gene. |
HIF-1α-mediated autophagy attenuates
the hypoxia-induced reduction in H9c2 cell viability
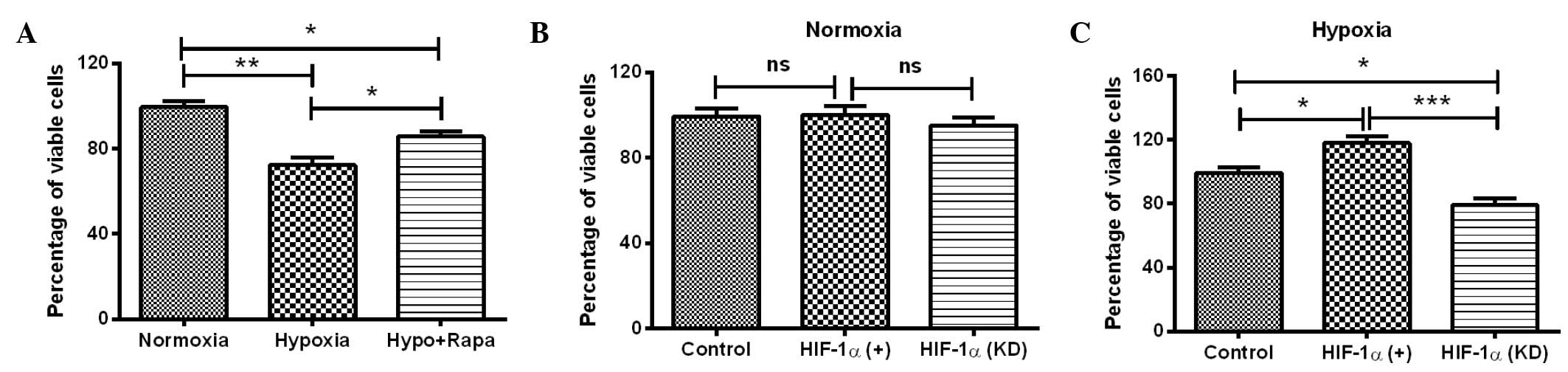
The initial experiments demonstrated that HIF-1
induced significant autophagy and high expression levels of
autophagy-associated molecules in H9c2 cells. To further determine
the effect of HIF-1α-mediated autophagy under hypoxia on cell
viability, the MTT assay was conducted for H9c2 cells under several
conditions: Hypoxia, normoxia, post-rapamycin treatment, HIF-1α
overexpression and HIF-1α knockdown. It was shown that the
viability of H9c2 cells under hypoxia was significantly reduced
compared with the viability of the cells under normoxia (P<0.01;
Fig. 4A). Notably, the reduction in
cell viability was reversed by the autophagy inducer rapamycin
(P<0.05; Fig. 4A). To further
investigate the role of HIF-1α in the hypoxia-induced cell
viability reduction, the viability of H9c2 cells was re-examined
with HIF-1α overexpression or knockdown. As shown in Fig. 4B, manipulation of the HIF-1α level
(overexpression or knockdown) under normoxia had no significant
effect on the cell viability, whereas the upregulation and
downregulation of HIF-1α led to a significantly different effect on
the viability of H9c2 cells under hypoxia, compared with the cells
under hypoxia only. HIF-1α overexpression significantly ameliorated
the reduction in cell viability (P<0.05; Fig. 4C), whereas HIF-1α knockdown enhanced
the hypoxia-induced reduction in cell viability (P<0.05;
Fig. 4C). The difference in cell
viability between the cells with HIF-1α upregulation and HIF-1α
downregulation was particularly significant (P<0.01; Fig. 4C). In combination, these results
suggest that HIF-1α-induced autophagy attenuates the
hypoxia-reduced viability of H9c2 cells.
Discussion
Hypoxia-induced cell death is a major concern in
various clinical settings and plays a critical role in various
physiopathological processes, such as hypoxic/ischemic disease,
organ transplantation, angiogenesis or tumor invasion (33–35). I/R
injury comprises a series of events that may occur together or
separately: Reperfusion arrhythmias, myocardial stunning in
‘reversible mechanical dysfunction’, microvascular damage and cell
death (36–38). Previous studies have demonstrated
that autophagy is a lysosomal degradation process that occurs
within the cell and has crucial physiological cellular functions,
including the degradation of abnormally folded proteins, organelle
turnover and adaptations to such stresses as nutrient depletion,
acidic stress, oxidative stress and I/R injury (39–44).
Furthermore, Macrophage autophagy has been indicated to play a
protective role in advanced atherosclerosis in a mouse model
(45); however, the protective role
of autophagy in cardiomyocytes under hypoxia has not yet been
elucidated.
In the present study, increased autophagic activity
was detected in cardiomyocytes under hypoxia. To the best of our
knowledge, this is the first study to report the effects of hypoxia
on autophagy induction in cardiomyocytes. Several approaches were
adopted to determine the levels of autophagy, including the
analysis of fluorescent LC3-GFP dots and the mRNA and protein
expression levels of autophagy-associated molecules. Cardiomyocytes
were exposed to hypoxia, and an evident increase was found in the
number of autophagic vacuoles and autolysosomes in the cytoplasm,
suggesting enhanced autophagy formation. The occurrence of
autophagy was also confirmed with rapamycin treatment in the H9c2
cell model. The analysis of autophagy-associated molecule levels
confirmed the induction of autophagy by hypoxia or rapamycin: The
expression of LC3-II increased in cells post-hypoxia or -rapamycin
treatment, and the Beclin 1, Atg5 and Atg7 expression increased in
cardiomyocytes post-hypoxia or -rapamycin treatment at both the
mRNA and protein levels. In summary, autophagy could be induced by
hypoxia in cardiomyocytes in vitro.
HIF-1 is a transcription factor that is essential in
the regulation of gene expression to maintain oxygen homeostasis
(46). HIF-1 has been demonstrated
to coordinate adaptive responses to hypoxia at both the cellular
and systemic levels (18,47,48).
Furthermore, HIF-1 has been shown to regulate the expression of
hundreds of target genes involved in angiogenesis, erythropoiesis,
metabolism, autophagy and other adaptive responses to hypoxia
(49). To elucidate the mechanisms
by which cardiomyocytes respond to hypoxia, an HIF-1α
overexpression plasmid and HIF-1α siRNA were used to manipulate the
HIF-1α expression and to investigate the regulatory role of HIF-1α
in hypoxia-induced autophagy. The results demonstrated that the
hypoxia-induced autophagy in cardiomyocytes involved the HIF-1
pathway. A significant increase in autophagy-specific autolysosomes
was observed via the LC3-GFP reporter vector under a fluorescence
microscope in HIF-1α-treated cells. By contrast, HIF-1α knockdown
with siRNA treatment led to considerably less autolysosome
formation in the cardiomyocytes. Furthermore, the cleavage and
recruitment of LC3 to autophagosomes, as well as the expression of
autophagy-associated molecules, were enhanced by HIF-1α
overexpression and attenuated by HIF-1α knockdown. In combination,
these results indicated that the HIF-1 pathway was involved in
hypoxia-induced autophagy in cardiomyocytes.
Hypoxia treatment and HIF-1α overexpression were
both found to induce autophagy; however, their effects on cell
viability differed. Hypoxia appeared to reduce the cell viability,
though accompanied by HIF-1α stimulating, whereas HIF-1α could
significantly inhibit the reduction of cardiomyocyte viability.
However, the overexpression of HIF-1α ameliorated the reduction in
cell viability induced by hypoxia, while the downregulation of
HIF-1α enhanced the hypoxia-induced cell viability reduction. The
autophagy induced by HIF-1 may, therefore, facilitate
cardiomyocytes to overcome the hypoxic injury and increase
survival.
In conclusion, the results of the present study
suggest that hypoxia can induce autophagy in cardiomyocytes through
activation of the HIF-1 pathway. HIF-1α upregulation can increase
autophagy and ameliorate the hypoxia-induced cell viability
reduction. The present study presents a promising foundation for
the development of methods to prevent hypoxic-ischemic injury to
cardiomyocytes.
Acknowledgements
The present study was supported by a grant from the
Inner Mongolia Department of Education Project (no.
NJZY13161(2013-2015).
References
|
1
|
Gautier-Veyret E, Arnaud C, Bäck M, Pépin
JL, Petri MH, Baguet JP, Tamisier R, Lévy P and Stanke-Labesque F:
Intermittent hypoxia-activated cyclooxygenase pathway: Role in
atherosclerosis. Eur Respir J. 42:404–413. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bovill EG and van der Vliet A: Venous
valvular stasis-associated hypoxia and thrombosis: What is the
link? Annu Rev Physiol. 73:527–545. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matsushima S, Kuroda J, Ago T, Zhai P,
Ikeda Y, Oka S, Fong GH, Tian R and Sadoshima J: Broad suppression
of NADPH oxidase activity exacerbates ischemia/reperfusion injury
through inadvertent downregulation of hypoxia-inducible factor-1α
and upregulation of peroxisome proliferator-activated receptor-α.
Circ Res. 112:1135–1149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Talukder MA, Elnakish MT, Yang F,
Nishijima Y, Alhaj MA, Velayutham M, Hassanain HH and Zweier JL:
Cardiomyocyte-specific overexpression of an active form of Rac
predisposes the heart to increased myocardial stunning and
ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol.
304:H294–H302. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Goldhaber JI and Weiss JN: Oxygen free
radicals and cardiac reperfusion abnormalities. Hypertension.
20:118–127. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cheng TO: Coronary heart disease in China.
Hosp Med. 60:4561999.PubMed/NCBI
|
|
7
|
Barth J, Schneider S and von Känel R: Lack
of social support in the etiology and the prognosis of coronary
heart disease: A systematic review and meta-analysis. Psychosom
Med. 72:229–238. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ostádal B: Myocardial ischemic injury and
protection. Exp Clin Cardiol. 9:213–217. 2004.PubMed/NCBI
|
|
9
|
Ost'ádal B, Ost'ádalova I, Skárka L, Kolár
F and Kopecký J: Ischemic injury of the developing heart. Exp Clin
Cardiol. 7:93–98. 2002.PubMed/NCBI
|
|
10
|
Tong W, Xiong F, Li Y and Zhang L: Hypoxia
inhibits cardiomyocyte proliferation in fetal rat hearts via
upregulating TIMP-4. Am J Physiol Regul Integr Comp Physiol.
304:R613–R620. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Botting KJ, McMillen IC, Forbes H,
Nyengaard JR and Morrison JL: Chronic hypoxemia in late gestation
decreases cardiomyocyte number but does not change expression of
hypoxia-responsive genes. J Am Heart Assoc. 3:2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ramjiawan A, Bagchi RA, Blant A, Albak L,
Cavasin MA, Horn TR, McKinsey TA and Czubryt MP: Roles of histone
deacetylation and AMP kinase in regulation of cardiomyocyte PGC-1α
gene expression in hypoxia. Am J Physiol Cell Physiol.
304:C1064–C1072. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Muraguchi T, Kawawa A and Kubota S:
Prohibitin protects against hypoxia-induced H9c2 cardiomyocyte cell
death. Biomed Res. 31:113–122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang W, Peng Y, Wang Y, Zhao X and Yuan Z:
Anti-apoptotic effect of heat shock protein 90 on hypoxia-mediated
cardiomyocyte damage is mediated via the phosphatidylinositol
3-kinase/AKT pathway. Clin Exp Pharmacol Physiol. 36:899–903. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shohet RV and Garcia JA: Keeping the
engine primed: HIF factors as key regulators of cardiac metabolism
and angiogenesis during ischemia. J Mol Med (Berl). 85:1309–1315.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Taylor CT: Mitochondria and cellular
oxygen sensing in the HIF pathway. Biochem J. 409:19–26. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Semenza GL: Hypoxia-inducible factor 1:
Regulator of mitochondrial metabolism and mediator of ischemic
preconditioning. Biochim Biophys Acta. 1813:1263–1268. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl
Acad Sci USA. 92:5510–5514. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Salceda S and Caro J: Hypoxia-inducible
factor 1alpha (HIF-1alpha) protein is rapidly degraded by the
ubiquitin-proteasome system under normoxic conditions. Its
stabilization by hypoxia depends on redox-induced changes. J Biol
Chem. 272:22642–22647. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cai Z, Manalo DJ, Wei G, Rodriguez ER,
Fox-Talbot K, Lu H, Zweier JL and Semenza GL: Hearts from rodents
exposed to intermittent hypoxia or erythropoietin are protected
against ischemia-reperfusion injury. Circulation. 108:79–85. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yuan G, Nanduri J, Bhasker CR, Semenza GL
and Prabhakar NR: Ca2+/calmodulin kinase-dependent
activation of hypoxia inducible factor 1 transcriptional activity
in cells subjected to intermittent hypoxia. J Biol Chem.
280:4321–4328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yuan G, Nanduri J, Khan S, Semenza GL and
Prabhakar NR: Induction of HIF-1alpha expression by intermittent
hypoxia: Involvement of NADPH oxidase, Ca2+ signaling,
prolyl hydroxylases and mTOR. J Cell Physiol. 217:674–685. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee SH, Wolf PL, Escudero R, Deutsch R,
Jamieson SW and Thistlethwaite PA: Early expression of angiogenesis
factors in acute myocardial ischemia and infarction. N Engl J Med.
342:626–633. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hlatky MA, Quertermous T, Boothroyd DB,
Priest JR, Glassford AJ, Myers RM, Fortmann SP, Iribarren C, Tabor
HK, Assimes TL, et al: Polymorphisms in hypoxia inducible factor 1
and the initial clinical presentation of coronary disease. Am Heart
J. 154:1035–1042. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Resar JR, Roguin A, Voner J, Nasir K,
Hennebry TA, Miller JM, Ingersoll R, Kasch LM and Semenza GL:
Hypoxia-inducible factor 1alpha polymorphism and coronary
collaterals in patients with ischemic heart disease. Chest.
128:787–791. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Todde V, Veenhuis M and van der Klei IJ:
Autophagy: Principles and significance in health and disease.
Biochim Biophys Acta. 1792:3–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kundu M, Lindsten T, Yang CY, Wu J, Zhao
F, Zhang J, Selak MA, Ney PA and Thompson CB: Ulk1 plays a critical
role in the autophagic clearance of mitochondria and ribosomes
during reticulocyte maturation. Blood. 112:1493–1502. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pua HH, Guo J, Komatsu M and He YW:
Autophagy is essential for mitochondrial clearance in mature T
lymphocytes. J Immunol. 182:4046–4055. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Srinivas V, Bohensky J and Shapiro IM:
Autophagy: A new phase in the maturation of growth plate
chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells
Tissues Organs. 189:88–92. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wild P, McEwan DG and Dikic I: The LC3
interactome at a glance. J Cell Sci. 127:3–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun Y and Peng ZL: Programmed cell death
and cancer. Postgrad Med J. 85:134–140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rayner BS, Duong TT, Myers SJ and Witting
PK: Protective effect of a synthetic anti-oxidant on neuronal cell
apoptosis resulting from experimental hypoxia re-oxygenation
injury. J Neurochem. 97:211–221. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hartel FV, Holl M, Arshad M, Aslam M,
Gündüz D, Weyand M, Micoogullari M, Abdallah Y, Piper HM and Noll
T: Transient hypoxia induces ERK-dependent anti-apoptotic cell
survival in endothelial cells. Am J Physiol Cell Physiol.
298:C1501–C1509. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bhogal RH, Weston CJ, Curbishley SM, Bhatt
AN, Adams DH and Afford SC: Variable responses of small and large
human hepatocytes to hypoxia and hypoxia/reoxygenation (H-R). Febs
Lett. 585:935–941. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hess ML, Barnhart GR, Crute S, Komwatana
P, Krause S and Greenfield LJ: Mechanical and biochemical effects
of transient myocardial ischemia. J Surg Res. 26:175–184. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jeroudi MO, Hartley CJ and Bolli R:
Myocardial reperfusion injury: Role of oxygen radicals and
potential therapy with antioxidants. Am J Cardiol. 73:2B–7B. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bolli R and Marbán E: Molecular and
cellular mechanisms of myocardial stunning. Physiol Rev.
79:609–634. 1999.PubMed/NCBI
|
|
39
|
Bartolome A, Guillen C and Benito M:
Autophagy plays a protective role in endoplasmic reticulum
stress-mediated pancreatic β cell death. Autophagy. 8:1757–1768.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang T, Qi Y, Liao M, Xu M, Bower KA,
Frank JA, Shen HM, Luo J, Shi X and Chen G: Autophagy is a cell
self-protective mechanism against arsenic-induced cell
transformation. Toxicol Sci. 130:298–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen G, Ke Z, Xu M, Liao M, Wang X, Qi Y,
Zhang T, Frank JA, Bower KA, Shi X and Luo J: Autophagy is a
protective response to ethanol neurotoxicity. Autophagy.
8:1577–1589. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Marino ML, Pellegrini P, Di Lernia G,
Djavaheri-Mergny M, Brnjic S, Zhang X, Hägg M, Linder S, Fais S,
Codogno P and De Milito A: Autophagy is a protective mechanism for
human melanoma cells under acidic stress. J Biol Chem.
287:30664–30676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang D, Ma Y, Li Z, Kang K, Sun X, Pan S,
Wang J, Pan H, Liu L, Liang D and Jiang H: The role of AKT1 and
autophagy in the protective effect of hydrogen sulphide against
hepatic ischemia/reperfusion injury in mice. Autophagy. 8:954–962.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bhogal RH, Weston CJ, Curbishley SM, Adams
DH and Afford SC: Autophagy: A cyto-protective mechanism which
prevents primary human hepatocyte apoptosis during oxidative
stress. Autophagy. 8:545–558. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liao X, Sluimer JC, Wang Y, Subramanian M,
Brown K, Pattison JS, Robbins J, Martinez J and Tabas I: Macrophage
autophagy plays a protective role in advanced atherosclerosis. Cell
Metab. 15:545–553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Semenza GL: HIF-1 and mechanisms of
hypoxia sensing. Curr Opin Cell Biol. 13:167–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Iyer NV, Kotch LE, Agani F, Leung SW,
Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY
and Semenza GL: Cellular and developmental control of O2
homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev.
12:149–162. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun
X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT and
Semenza GL: Impaired physiological responses to chronic hypoxia in
mice partially deficient for hypoxia-inducible factor 1alpha. J
Clin Invest. 103:691–696. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Semenza GL: Regulation of oxygen
homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda).
24:97–106. 2009. View Article : Google Scholar : PubMed/NCBI
|