Introduction
Cervical cancer is the most common type of
gynecological cancer worldwide, accounting for ~8% of all female
malignancies, second only to breast cancer (1,2). Every
year, cervical cancer affects ~500,000 women worldwide, ~250,000 of
which succumb to the disease (3).
Although cervical cancer screening has been popularized globally
(4,5), large numbers of patients with advanced
cervical cancer remain (6). Notably,
hh~85% of novel cases and 80% of fatal cases of cervical cancer
occur in developing countries (7,8). Human
papillomavirus (HPV) is known to be the most common etiological
agent of cervical cancer, and 99% of cervical cancer cases are
attributed to human HPV infection (9,10).
However, HPV infection alone is not sufficient for the malignant
transformation of cervical epithelial cells. Various cofactors and
molecular events are required in order to promote the pathogenic
process of cervical cancer (11,12);
therefore, early detection and treatment of precancerous lesions is
particularly important in order to prevent the progression of
cervical cancer. The molecular pathogenesis of cervical cancer
remains poorly understood. Investigating the molecular mechanisms
underlying the development of cervical cancer, searching for novel
molecular markers for early diagnosis and developing effective
therapeutic targets is urgently required.
E3 ubiquitin ligase isolated by differential display
(EDD) is a human ortholog of the Drosophila melanogaster
hyperplastic discs gene (hyd) (13,14),
which was initially isolated as a progestin-regulated gene in human
T47D breast cancer cells (14,15).
Ubiquitin ligase E3 is able to identify degraded proteins and to
conduct ubiquitin tagging of the substrate. Ubiquitin-mediated
protein degradation is associated with various important protein
signaling pathways, including transcription, cell cycle and DNA
damage (16–20). Previous studies have demonstrated
that EDD participates in the regulation of cyclin levels and cell
cycle progression (21–24), regulates ubiquitination and the
degradation of protein phosphatase (25), and has a role in transcriptional
regulation and the response to DNA damage (15,26–30).
Furthermore, it has previously been demonstrated that EDD is
ectopically overexpressed in certain types of cancer and has an
important role in cancer cell growth, tumorigenesis and drug
resistance (31–36). However the effects of EDD on the
occurrence and progression of cervical cancer and its associated
molecular mechanisms have yet to be fully elucidated.
MicroRNAs (miRNAs or miRs) are a class of non-coding
small RNAs of 20–24 nucleotides in length that regulate gene and
protein expression (37). miRNAs
participate in a diverse range of biological processes including
development, proliferation, differentiation, apoptosis and disease
(38–43). Furthermore, certain miRNAs have been
demonstrated to function as oncogenes or tumor suppressors,
therefore they are directly involved in human cancer, including
liver, lung, breast, colon and brain cancer (44–53). The
miRNA expression profiles of cervical cancer cells and tissues have
previously been analyzed using cDNA cloning (52). The results indicated that aberrant
expression of oncogenic and tumor suppressive miRNAs was required
for cancer cell growth in cervical cancer, and miR-143 and miR-145
were demonstrated to be the tumor suppressive miRNAs. Furthermore,
it was subsequently demonstrated that miR-143 is capable of
inhibiting tumor growth and angiogenesis, inducing cancer cell
apoptosis and cell cycle arrest, increasing chemosensitivity, and
regulating cyclooxygenase stability and expression in colorectal,
lung and pancreatic cancer (44,46,50,54).
These results suggested that miR-143 had an important role in the
carcinogenic process.
The present study aimed to investigate the role of
EDD in the tumorigenicity of cervical cancer, and to further
elucidate the underlying molecular mechanism, in order to improve
the understanding of the pathogenesis of cervical cancer and to aid
in the development of novel therapeutics for the disease.
Materials and methods
Tissue samples and cell lines
Human cervical cancer tissue samples (n=39) and
normal cervical tissue samples (n=13) were obtained from patients
at the Hebei General Hospital (Shijiazhuang, China). The 39
patients with cervical cancer were aged between 27 and 55 years
(average age, 46 years) and had an average weight of 54 kg (weight
range, 48 to 63 kg). All patients in this group had been diagnosed
with stage IA-IVB cervical cancer, according to the FIGO staging
system (55). The 13 normal control
patients were aged between 30 and 55 years and weighed between 48
and 61 kg. The control patients were undergoing a simple
hysterectomy at the Hebei General Hospital due to uterine
leiomyomata. All cancer specimens used in the analyses consisted of
>90% tumor cells, as examined by a gynecologic pathologist.
Cancer specimens from patients with concomitant gynecological
problems were excluded from the study. Informed consent was
obtained from all patients prior to the surgical procedure, and
approval was obtained from the Medical Ethics Committee of the
Hebei General Hospital.
Normal cervical epithelial cells and five cervical
cancer cell lines, including SiHa, HeLa, CaSki, c-41 and c-33A,
were purchased from the American Type Culture Collection (Manassas,
VA, USA). Cell culture was conducted according to methods
previously described by Liu et al (56). Briefly, the cells were cultured in
RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS;
both purchased from Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), 2 mM L-glutamine, 100 µg/ml streptomycin and 100
U/ml penicillin (all obtained from Gibco; Thermo Fisher Scientific,
Inc.). The cells were incubated at 37°C, in an atmosphere
containing 5% CO2.
Cell proliferation, apoptosis and
colony formation assays
SiHa cells (1×104 cells/ml) were seeded
onto 96-well plates, after which cell proliferation and apoptosis
were assessed using the MTT Cell Proliferation/Viability Assay kit
(cat. no. 11465007001; Sigma-Aldrich Chemie Gmbh, Munich, Germany)
and the Annexin-V-FITC Apoptosis Detection kit (cat. no.
APOAF-20TST; Sigma-Aldrich Chemie Gmbh) with a flow cytometer (BD
FACSCalibur™; BD Biosciences, Franklin Lakes, NJ, USA),
respectively, according to the manufacturer's protocols.
Subsequently, SiHa cells (1×104) were suspended in 1.5
ml complete medium [consisting of RPMI 1640 (Invitrogen; Thermo
Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum
(FBS; GE Healthcare Life Sciences, Logan, UT, USA)] containing
0.45% low melting point agarose (Invitrogen; Thermo Fisher
Scientific, Inc.) and then seeded into 35 mm tissue culture plates
containing 1.5 ml complete medium and 0.75% agarose on the bottom
layer, which were then incubated for 10 days at 37°C. The plates
were then stained with 0.005% crystal violet for 30 sec
(Sigma-Aldrich Chemie Gmbh) and the SiHa cell colonies (>0.5 mm
in diameter) were counted under a microscope (Leica DMI3000; Leica
Microsystems GmbH, Wetzlar, Germany), according to a previous study
(57).
Cell transfection
In order to analyze the effects of EDD
overexpression or knockdown, SiHa cells (1×104) were
transfected with adenovirus-expressing Ad-EDD or retrovirus
expressing short hairpin (Sh)-EDD, respectively. Ad-EDD/Ad-negative
control (NC) and Sh-EDD/Sh-NC were designed and synthesized by
Invitrogen (Thermo Fisher Scientific, Inc.). For miR-143 silencing,
SiHa cells (1×104) were transfected with the pLL3.7
lentivirus vector (Addgene, Cambridge, MA, USA) encoding a miR-143
inhibitor sponge. The miR-143 inhibitor sponge was synthesized by
GenePharma (Shanghai, China). The cells were cultured in six-well
plates to 70–80% confluency and transfected using Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.). The cells were
harvested 48 h post-transfection for reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) in
order to determine the transfection efficiency.
Tumor xenograft experiments
Xenograft mice experiments were performed as
previously described (56). Briefly,
SiHa cells (5×106) transfected with Sh-NC, Sh-EDD, Ad-NC
or Ad-EDD were injected subcutaneously into the flanks of female
athymic nude mice (age, 3–4 weeks; n=6/group), purchased from
Shanghai Laboratory Animal Co., Ltd. (Shanghai, China). The mice
were maintained under at 21–22°C under a 12-h light/dark cycle. To
obtain the tumors, the mice were sacrificed by an overdose of
pentobarbital (50 mg/kg; Sigma-Aldrich Chemie Gmbh) 4 weeks
following inoculation.
RT-qPCR analysis
Total RNA (100 ng) was extracted from the harvested
tissue samples and cell lines using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and was purified using
RNase-Free DNase Set (Qiagen, Inc., Valencia, CA, USA), according
to the manufacturer's protocols. Subsequently, 5 ng RNA was reverse
transcribed into cDNA using SuperScript III Reverse Transcriptase
(Invitrogen; Thermo Fisher Scientific, Inc.). qPCR was performed
using TaqMan MicroRNA Assays (Thermo Fisher Scientific, Inc.) on an
ABI 7300 Real-Time PCR System (Applied Biosystems; Thermo Fisher
Scientific, Inc.), according to a previous study (44). The primer sequences were as follows:
EDD forward, 5′-TTAGGCTTTTGGTAAATGGCTGCG-3′ and reverse,
5′-TGAGGGCATAGGCTGGAATCCTTC-3′; miR-143 forward,
5′-AGTGCGTGTCGTGGAGTC-3′ and reverse, 5′-GCCTGAGATGAAGCACTGT-3′;
and β-actin forward, 5′-CATCCTGCGTCTGGACCT-3′ and reverse,
5′-CAGGAGGAGCAATGATCTTG-3′. β-actin was used as the internal
control. EDD, miR-143 and β-actin primers were designed as
previously described by Liu et al (54) and Clancy et al (30). The PCR cycling conditions were as
follows: Pre-heating at 95°C for 5 min, followed by 35 cycles of
denaturation for 30 sec at 95°C, annealing for 1 min at 55°C and
extension for 1 min at 72°C, with a final extension for 5 min at
72°C. Relative gene expression levels were calculated using the
2−ΔΔCq method (58).
Western blotting
Western blotting was performed as previously
described by Zhang et al (44). Briefly, total protein (50 µg) was
extracted from the tissue samples and SiHa cell line using lysis
buffer (20 mM Tris-HCl, pH 7.4, 2 mM EDTA, 25 mM 2-mercaptoethanol,
0.5 mM AEBSF, 0.5% Triton X-100, 2 µg/ml leupeptin and 3 µg/ml
aprotinin) and the protein concentrations were measured using a
Bio-Rad Protein Assay kit (cat. no. 5000002; Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Proteins were separated by 10% SDS-PAGE
and transferred to polyvinylidene difluoride membranes (Merck
Millipore, Darmstadt, Germany). Subsequently, the membranes were
blocked with 10% skimmed milk solution and then incubated overnight
at 4°C with goat anti-EDD (cat. no. sc-9562), rabbit anti-B-cell
lymphoma (Bcl)-2 (cat. no. sc-492), rabbit anti-Bcl-2-associated X
protein (Bax; cat. no. sc-493), goat anti-caspase 3 p11 (cat. no.
sc-1224) and goat anti-GAPDH (cat. no. sc-48166) polyclonal
antibodies (1:2,000; all Santa Cruz Biotechnology, Inc., Dallas,
TX, USA). Following washing with phosphate-buffered saline, the
membranes were incubated with horseradish peroxidase-conjugated
goat anti-rabbit IgG, F(ab')2 (1:3,000; cat. no.
sc-3836) and chicken anti-goat IgG (1:3,000; cat. no. sc-516086;
both Santa Cruz Biotechnology, Inc.) for 45 min at room
temperature, prior to incubation with an enhanced chemiluminescence
substrate (Thermo Fisher Scientific, Inc.). Subsequently, the
membranes were visualized by exposure to ECL film and the band
intensities were quantified using UN-SCAN-IT gel analysis software,
version 5.1 (Silk Scientific, Inc., Orem, UT, USA). GAPDH was used
as a reference gene.
Statistical analysis
All experiments were performed ≥3 times. Data were
presented as the mean ± standard deviation, and were analyzed using
SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA). Statistical
differences between two independent groups were determined using
the unpaired Student's t-test. P<0.05 was considered to indicate
a statistically significant difference.
Results
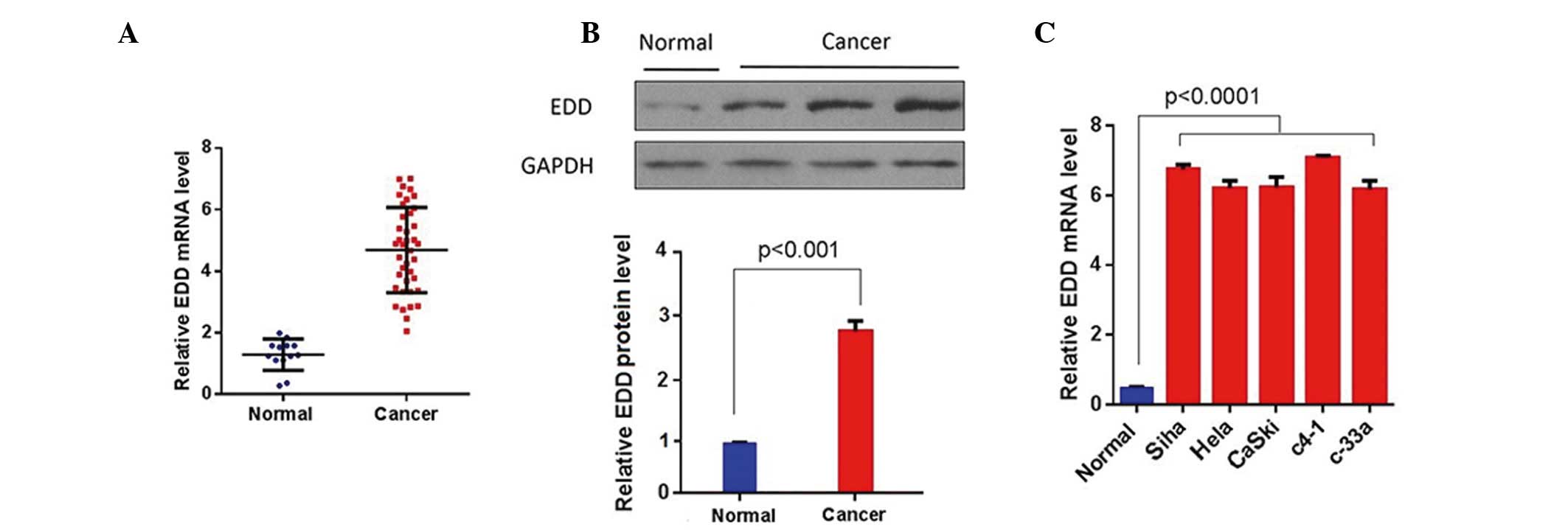
EDD is overexpressed in cervical
cancer tissue samples and cell lines
To investigate the role of EDD in cervical cancer,
the expression levels of EDD were measured in cervical cancer
tissue samples and cell lines using RT-qPCR and western blotting.
EDD was significantly upregulated in the cervical cancer tissue
samples at both the mRNA and protein levels (P<0.01; Fig. 1A and B). Furthermore, the mRNA
expression levels of EDD were significantly increased in the
cervical cancer cell lines, as compared with the normal cervical
epithelial cells (P<0.0001; Fig.
1C). These results suggest that EDD is overexpressed in
cervical cancer tissue samples and cell lines.
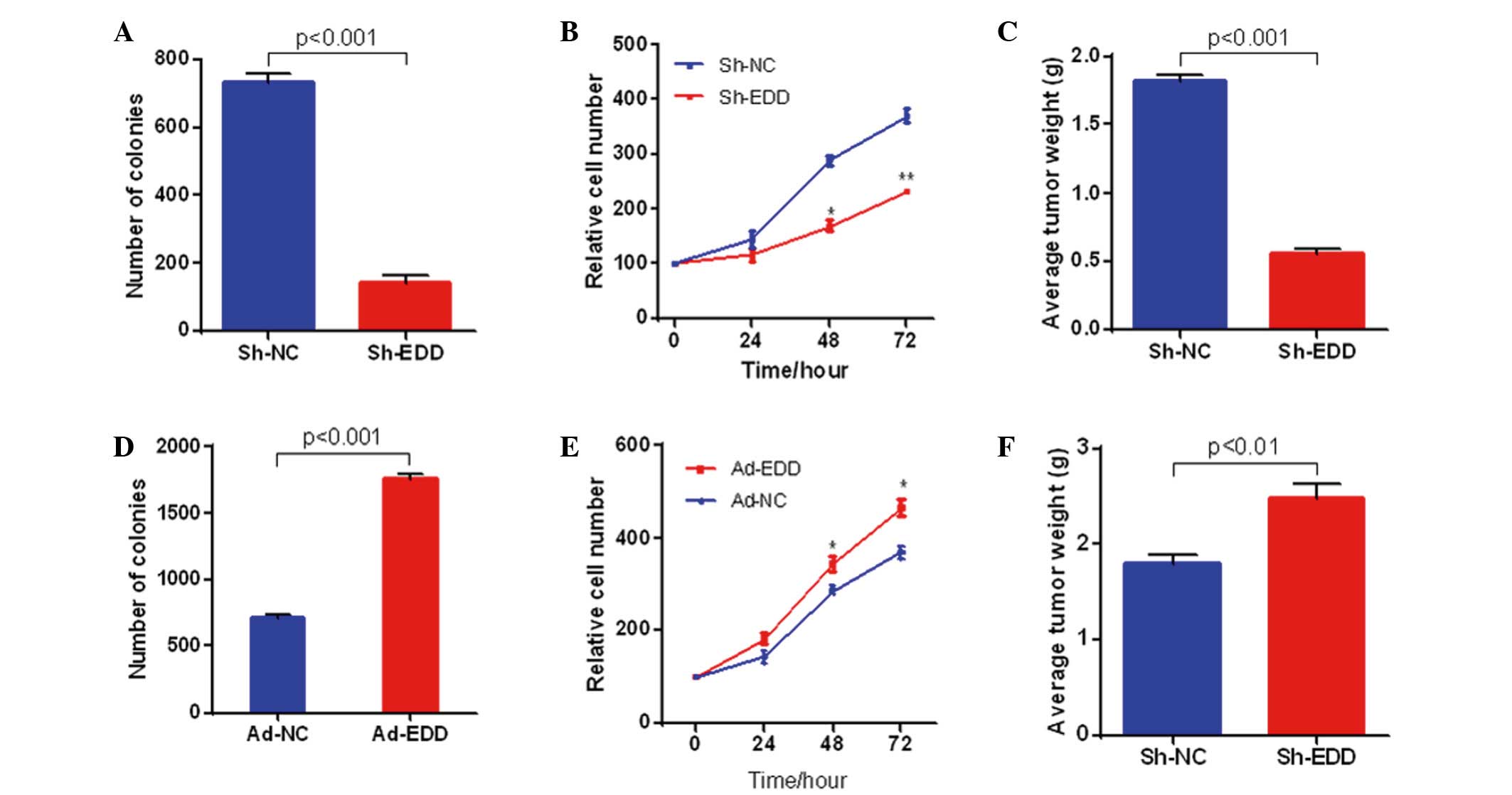
EDD promotes cervical cancer growth in
vitro and in vivo
To further explore the role of abnormal EDD
expression in cervical cancer, EDD expression was knocked down or
upregulated in SiHa cells. EDD knockdown in SiHa cervical cancer
cells significantly inhibited the growth of the cancer cells in
vitro and in vivo. As shown in Fig. 2A and B, colony formation and cell
proliferation were significantly inhibited in the SiHa cells when
EDD was knocked-down in vitro. Furthermore, EDD silencing
significantly suppressed the growth of cervical cancer tumors in
vivo (P<0.001; Fig. 2C). The
opposite results were observed following EDD overexpression. EDD
overexpression significantly increased colony formation, cell
proliferation and tumor growth in cervical cancer (P<0.05;
Fig. 2D-F).
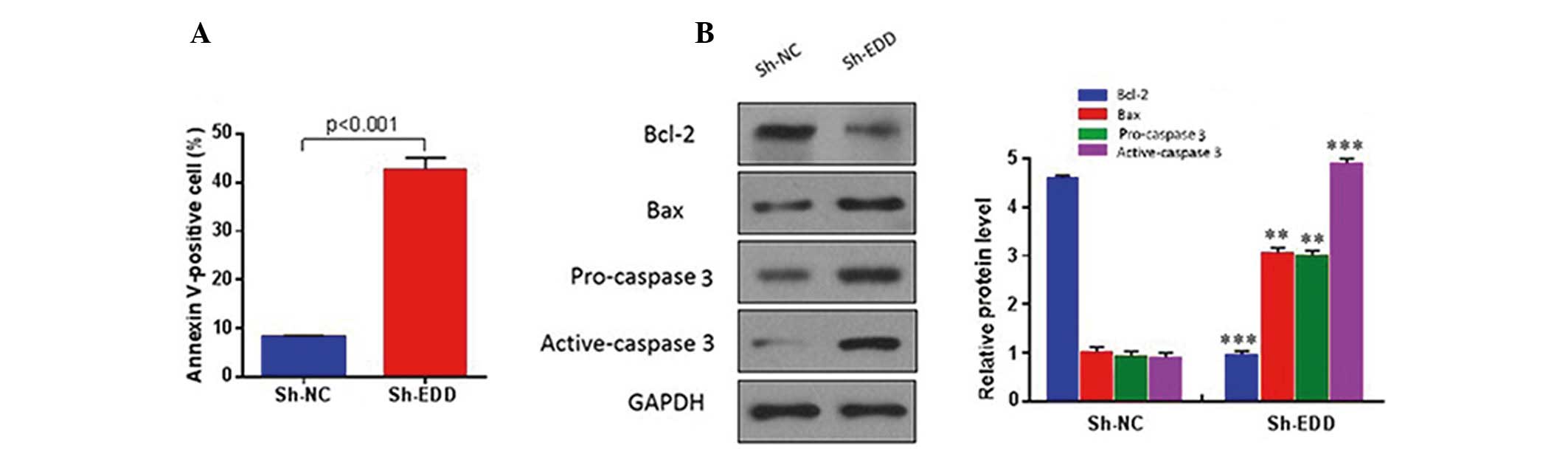
To investigate whether EDD regulates cervical cancer
growth via an apoptotic mechanism, quantitative analysis of
apoptotic cells was performed using fluorescence-activated cell
sorting. The results demonstrated that EDD knockdown significantly
increased the apoptosis of SiHa cells (P<0.001; Fig. 3A), and reduced the protein expression
levels of anti-apoptotic proteins, including Bcl-2, and increased
the protein expression levels of pro-apoptotic proteins, including
Bax and caspase 3 (Fig. 3B). These
results suggest that EDD may have an oncogenic role in cervical
cancer.
EDD regulates the proliferation of
cervical cancer cells via miR-143
As previously demonstrated by Liu et al
(56), miR-143 promotes apoptosis
and inhibits tumor formation in cervical cancer. Therefore, it was
hypothesized that EDD may regulate cervical cancer growth via
miR-143. The expression levels of miR-143 were analyzed in human
cervical cancer tissue samples. The results demonstrated that
miR-143 expression levels were significantly downregulated in
cervical cancer tissue samples, as compared with the normal tissue
samples (P<0.001; Fig. 4A).
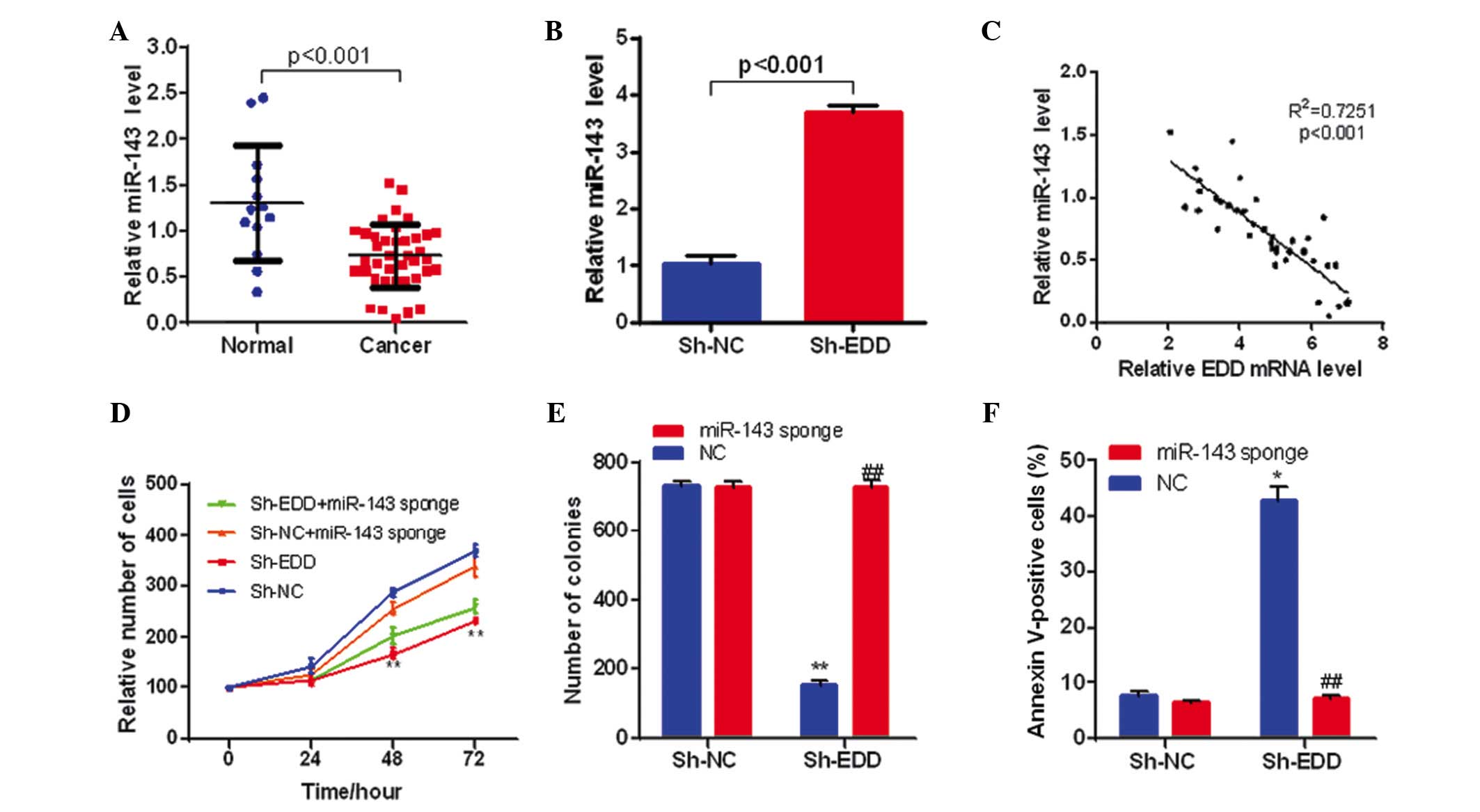 | Figure 4.EDD regulates cervical cancer cell
growth through miR-143. (A) The expression levels of miR-143 are
downregulated in cervical cancer tissue samples (n=13 in normal
group, n=39 in cancer group). (B) EDD knockdown increases miR-143
expression levels in SiHa cells (n=3 in each group). (C) The
expression of miR-143 was negatively correlated with the expression
of EDD in cervical cancer tissue samples (n=39). (D) miR-143
silencing partly reversed the inhibition effect on cell
proliferation caused by EDD knockdown in SiHa cells. **P<0.01,
vs. sh-NC. (E) miR-143 silencing eliminated the effect on colony
formation caused by EDD knockdown in SiHa cells (n=3 in each
group). **P<0.01, vs. sh-NC + NC; ##P<0.01, vs.
sh-EDD + NC. (F) miR-143 silencing prevented the apoptosis caused
by EDD knockdown in SiHa cells (n=3 in each group). *P<0.05, vs.
sh-NC + NC; ##P<0.01, vs. sh-EDD + NC. EDD, E3
ubiquitin ligase isolated by differential display; Sh-NC, short
hairpin negative control; Sh-EDD, short hairpin EDD; miR,
microRNA. |
Following this, the present study investigated
whether the expression levels of miRNA-143 were correlated with
EDD. EDD knockdown significantly increased miR-143 expression
levels in SiHa cells (P<0.001; Fig.
4B), and miR-143 expression was negatively correlated with the
expression levels of EDD in cervical cancer tissue samples
(P<0.001; Fig. 4C), which
suggests that EDD represses miR-143 expression in cervical
cancer.
To further investigate whether miR-143 affected the
function of EDD during the growth of cervical cancer, EDD and
miR-143 were knocked-down and silenced respectively or
simultaneously in SiHa cells. miR-143 sponge was used to eliminate
miR-143 function. miR-143 silencing significantly reversed the
inhibitory growth effect of EDD knockdown in SiHa cells (P<0.01;
Fig. 4D). Furthermore, miR-143
silencing eliminated the effect of EDD knockdown on colony
formation and prevented the apoptosis induced by EDD knockdown in
SiHa cells (P<0.01; Fig. 4E and
F). Therefore, these results demonstrate that EDD may regulate
the proliferation of cervical cancer cells via miR-143.
Discussion
Previous studies have demonstrated that malignancies
are frequently accompanied by the abnormal expression of oncogenes
or tumor suppressor genes (59–65).
EDD, as a human ortholog of the Drosophila melanogaster hyd
gene (14), was shown to be
frequently overexpressed in breast and ovarian cancer, which
suggests that it may have a role in the progression of
gynecological cancer (31,33,36).
Furthermore, Bradley et al (36) demonstrated that EDD downregulation
decreased ovarian cancer cell viability, increased cell apoptosis,
inhibited tumor growth and promoted platinum sensitivity through
mediation of ubiquitin ligase activity. However, the function and
molecular mechanisms of EDD in human cervical cancer have yet to be
elucidated. The results of the present study demonstrated that EDD
expression levels were significantly upregulated in cervical cancer
cell lines and tissues. Functional studies showed that abnormal
expression of EDD impacted cell proliferation and tumor growth in
cervical cancer. Furthermore, EDD knockdown significantly inhibited
colony formation, cell proliferation and tumor growth in
vitro and in vivo via the activation of the apoptosis
signal pathway. These results suggested that EDD may have an
oncogenic role in human cervical cancer. Abnormal expression of EDD
may result in the disorder of ubiquitination and deubiquitination
via ubiquitin ligase E3 and mediate the aberrant expression of
oncogenes or tumor suppressor genes, thus inducing tumor occurrence
and development.
miRNAs are involved in a diverse range of biological
processes (38–43) and previous studies have demonstrated
that certain miRNAs may function as oncogenes or tumor suppressors,
which are directly involved in cancer occurrence and development
(44–53). cDNA cloning demonstrated that miR-143
is a tumor-suppressive miRNA in cervical cancer (52), and Liu et al (56) reported that miR-143 expression was
downregulated in cervical cancer. The results of the present study
demonstrated that miR-143 expression was downregulated in cervical
cancer tissue samples. Furthermore, miR-143 expression levels were
significantly increased following EDD knockdown and were negatively
correlated with the expression of EDD, which suggested that miR-143
may be a functional target of EDD in cervical cancer. Subsequent
functional investigation revealed that miR-143 silencing eliminated
the effect of EDD knockdown on cell proliferation, colony formation
and cell apoptosis in SiHa cells, indicating that miR-143 may be
crucial for the function of EDD in regulating the growth of
cervical cancer cells. These results are concordant with those of a
previous study, which demonstrated that EDD regulates
miRNA-mediated gene silencing and impacts the proliferation of
cancer cells (27). Su et al
(27) identified EDD as a key
mediator for miRNA silencing via genetic screening in mouse
embryonic stem cells. It was demonstrated that E3 ubiquitin ligase
activity was dispensable for EDD function in miRNA silencing
(27). However, the C-terminal
domain of polyadenylate binding protein 1 (PABC) of EDD was
demonstrated to be essential for its silencing function, as EDD
regulated miRNA silencing via its PABC domain and PABC interactors
(27). Furthermore, it has
previously been demonstrated that miR-143 is able to promote
cervical cancer cell apoptosis and inhibit tumor formation by
targeting Bcl-2 (56). These
findings, and the results of the present study, suggest that EDD
may regulate miRNA-143 expression via its PABC domain which, in
turn, impacts carcinogenesis and tumor growth.
In conclusion, the results of the present study
demonstrated that EDD regulates cervical cancer growth in
vivo and in vitro partly via miR-143. Furthermore, EDD
may have an oncogenic role in cervical cancer and may be a
potential target for cervical cancer therapy.
Acknowledgements
The present study was supported by grants from the
Discipline Leaders Project of the Shanghai Municipal Pudong New
Area Health System (grant no. PWRd2012-16) and the Key Discipline
Funding Project of the Shanghai Municipal Pudong New Area Health
Bureau (grant no. PWZx2014-09).
References
|
1
|
World Health Organization: Department of
Reproductive Health and Research and Department of Chronic Diseases
and Health Promotion: Comprehensive cervical cancer control: A
guide to essential practice. World Health Organization. 2006.
|
|
2
|
World Health Organization: GLOBOCAN 2012:
Estimated cancer incidence, mortality and prevalence worldwide in
2012. Lyon, France: International Agency for Research on Cancer.
2014.
|
|
3
|
Anorlu RI: Cervical cancer: The
sub-Saharan African perspective. Reprod Health Matters. 16:41–49.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rositch AF, Silver MI and Gravitt PE:
Cervical cancer screening in older women: New evidence and
knowledge gaps. PLoS Med. 11:e10015862014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Noller K: Intraepithelial neoplasia of the
lower genital tract (cervix, vulva): Etiology, screening,
diagnostic techniques, management. Comprehensive Gynecology (5th).
Mosby Elsevier. (Philadelphia, PA). 2007. View Article : Google Scholar
|
|
6
|
Tewari KS, Sill MW, Long HJ III, Penson
RT, Huang H, Ramondetta LM, Landrum LM, Oaknin A, Reid TJ, Leitao
MM, et al: Improved survival with bevacizumab in advanced cervical
cancer. N Engl J Med. 370:734–743. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dizon DS, Mackay HJ, Thomas GM, Werner TL,
Kohn EC, Hess D, Rose PG and Covens AL: State of the science in
cervical cancer: Where we are today and where we need to go.
Cancer. 120:2282–2288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maguire R, Kotronoulas G, Simpson M and
Paterson C: A systematic review of the supportive care needs of
women living with and beyond cervical cancer. Gynecol Oncol.
136:478–490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schiffman M, Castle PE, Jeronimo J,
Rodriguez AC and Wacholder S: Human papillomavirus and cervical
cancer. Lancet. 370:890–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chaturvedi AK: Beyond cervical cancer:
Burden of other HPV-related cancers among men and women. J Adolesc
Health. 46(Suppl): S20–S26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Burd EM: Human papillomavirus and cervical
cancer. Clin Microbiol Rev. 16:1–17. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dasari S, Wudayagiri R and Valluru L:
Cervical cancer: Biomarkers for diagnosis and treatment. Clin Chim
Acta. 445:7–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tasaki T, Mulder LC, Iwamatsu A, Lee MJ,
Davydov IV, Varshavsky A, Muesing M and Kwon YT: A family of
mammalian E3 ubiquitin ligases that contain the UBR box motif and
recognize N-degrons. Mol Cell Biol. 25:7120–7136. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Callaghan MJ, Russell AJ, Woollatt E,
Sutherland GR, Sutherland RL and Watts CK: Identification of a
human HECT family protein with homology to the Drosophila
tumor suppressor gene hyperplastic discs. Oncogene. 17:3479–3491.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Henderson MJ, Russell AJ, Hird S, Muñoz M,
Clancy JL, Lehrbach GM, Calanni ST, Jans DA, Sutherland RL and
Watts CK: EDD, the human hyperplastic discs protein, has a role in
progesterone receptor coactivation and potential involvement in DNA
damage response. J Biol Chem. 277:26468–26478. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Beaudenon SL, Huacani MR, Wang G,
McDonnell DP and Huibregtse JM: Rsp5 ubiquitin-protein ligase
mediates DNA damage-induced degradation of the large subunit of RNA
polymerase II in Saccharomyces cerevisiae. Mol Cell Biol.
19:6972–6979. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kornitzer D and Ciechanover A: Modes of
regulation of ubiquitin-mediated protein degradation. J Cell
Physiol. 182:1–11. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Connor MK and Seth A: A central role for
the ring finger protein RNF11 in ubiquitin-mediated proteolysis via
interactions with E2s and E3s. Oncogene. 23:2089–2095. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ang XL and Harper Wade J: SCF-mediated
protein degradation and cell cycle control. Oncogene. 24:2860–2870.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vidhyasekaran P: Ubiquitin-proteasome
system-mediated protein degradation in defense signaling. PAMP
Signals in Plant Innate Immunity. Springer. (The Netherlands).
409–430. 2014. View Article : Google Scholar
|
|
21
|
Benavides M, Chow-Tsang LF, Zhang J and
Zhong H: The novel interaction between microspherule protein Msp58
and ubiquitin E3 ligase EDD regulates cell cycle progression.
Biochim Biophys Acta. 1833:21–32. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ling S and Lin WC: EDD inhibits
ATM-mediated phosphorylation of p53. J Biol Chem. 286:14972–14982.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Munoz MA, Saunders DN, Henderson MJ,
Clancy JL, Russell AJ, Lehrbach G, Musgrove EA, Watts CK and
Sutherland RL: The E3 ubiquitin ligase EDD regulates S-phase and
G2/M DNA damage checkpoints. Cell Cycle. 6:3070–3077. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Smits VA: EDD induces cell cycle arrest by
increasing p53 levels. Cell Cycle. 11:715–720. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McDonald WJ, Thomas LN, Koirala S and Too
CK: Progestin-inducible EDD E3 ubiquitin ligase binds to α4
phosphoprotein to regulate ubiquitination and degradation of
protein phosphatase PP2Ac. Mol Cell Endocrinol. 382:254–261. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen HW, Yang CC, Hsieh CL, Liu H, Lee SC
and Tan BC: A functional genomic approach reveals the
transcriptional role of EDD in the expression and function of
angiogenesis regulator ACVRL1. Biochim Biophys Acta.
1829:1309–1319. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Su H, Meng S, Lu Y, Trombly MI, Chen J,
Lin C, Turk A and Wang X: Mammalian hyperplastic discs homolog EDD
regulates miRNA-mediated gene silencing. Mol Cell. 43:97–109. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bethard JR, Zheng H, Roberts L and Eblen
ST: Identification of phosphorylation sites on the E3 ubiquitin
ligase UBR5/EDD. J Proteomics. 75:603–609. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Henderson MJ, Munoz MA, Saunders DN,
Clancy JL, Russell AJ, Williams B, Pappin D, Khanna KK, Jackson SP,
Sutherland RL and Watts CK: EDD mediates DNA damage-induced
activation of CHK2. J Biol Chem. 281:39990–40000. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gwinn D and Sweet-Cordero EA: The
phosphatase PP2A links glutamine to the tumor suppressor p53. Mol
Cell. 50:157–158. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Clancy JL, Henderson MJ, Russell AJ,
Anderson DW, Bova RJ, Campbell IG, Choong DY, Macdonald GA, Mann
GJ, Nolan T, et al: EDD, the human orthologue of the hyperplastic
discs tumour suppressor gene, is amplified and overexpressed in
cancer. Oncogene. 22:5070–5081. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fuja TJ, Lin F, Osann KE and Bryant PJ:
Somatic mutations and altered expression of the candidate tumor
suppressors CSNK1ε, DLG1, and EDD/hHYD in mammary ductal carcinoma.
Cancer Res. 64:942–951. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
O'Brien PM, Davies MJ, Scurry JP, Smith
AN, Barton CA, Henderson MJ, Saunders DN, Gloss BS, Patterson KI,
Clancy JL, et al: The E3 ubiquitin ligase EDD is an adverse
prognostic factor for serous epithelial ovarian cancer and
modulates cisplatin resistance in vitro. Br J Cancer. 98:1085–1093.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kruzelock RP and Short W: Colorectal
cancer therapeutics and the challenges of applied pharmacogenomics.
Curr Probl Cancer. 31:315–366. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoon SY, Lee Y, Kim JH, Chung AS, Joo JH,
Kim CN, Kim NS, Choe IS and Kim JW: Over-expression of human UREB1
in colorectal cancer: HECT domain of human UREB1 inhibits the
activity of tumor suppressor p53 protein. Biochem Biophys Res
Commun. 326:7–17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bradley A, Zheng H, Ziebarth A, Sakati W,
Branham-O'Connor M, Blumer JB, Liu Y, Kistner-Griffin E,
Rodriguez-Aguayo C, Lopez-Berestein G, et al: EDD enhances cell
survival and cisplatin resistance and is a therapeutic target for
epithelial ovarian cancer. Carcinogenesis. 35:1100–1109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bueno MJ, de Pérez Castro I and Malumbres
M: Control of cell proliferation pathways by microRNAs. Cell Cycle.
7:3143–3148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Conrad R, Barrier M and Ford LP: Role of
miRNA and miRNA processing factors in development and disease.
Birth Defects Res C Embryo Today. 78:107–117. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schoolmeesters A, Eklund T, Leake D,
Vermeulen A, Smith Q, Aldred Force S and Fedorov Y: Functional
profiling reveals critical role for miRNA in differentiation of
human mesenchymal stem cells. PLoS One. 4:e56052009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shantikumar S, Caporali A and Emanueli C:
Role of miRNA in diabetes and its cardiovascular complications.
Cardiovasc Res. 93:583–93. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Y, Zhang X, Li H, Yu J and Ren X: The
role of miRNA-29 family in cancer. Eur J Cell Biol. 92:123–128.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Williams AE, Perry MM, Moschos SA,
Larner-Svensson HM and Lindsay MA: Role of miRNA-146a in the
regulation of the innate immune response and cancer. Biochem Soc
Trans. 36:1211–1215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang N, Su Y and Xu L: Targeting PKCε by
miR-143 regulates cell apoptosis in lung cancer. FEBS Lett.
587:3661–3667. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pham H, Rodriguez CE, Donald GW, Hertzer
KM, Jung XS, Chang HH, Moro A, Reber HA, Hines OJ and Eibl G:
miR-143 decreases COX-2 mRNA stability and expression in pancreatic
cancer cells. Biochem Biophys Res Commun. 439:6–11. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang X, Dong Y, Ti H, Zhao J, Wang Y, Li
T and Zhang B: Down-regulation of miR-145 and miR-143 might be
associated with DNA methyltransferase 3B overexpression and worse
prognosis in endometrioid carcinomas. Hum Pathol. 44:2571–2580.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang LQ, Zhang Y, Yan H, Liu KJ and Zhang
S: MicroRNA-373 functions as an oncogene and targets YOD1 gene in
cervical cancer. Biochem Biophys Res Commun. 459:515–520. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Puerta-Gil P, García-Baquero R, Jia AY,
Ocaña S, Alvarez-Múgica M, Alvarez-Ossorio JL, Cordon-Cardo C, Cava
F and Sánchez-Carbayo M: miR-143, miR-222, and miR-452 are useful
as tumor stratification and noninvasive diagnostic biomarkers for
bladder cancer. Am J Pathol. 180:1808–1815. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhu H, Dougherty U, Joseph LJ, Robinson V,
Wu J, Song Z, Mustafi R, Fichera A and Bissonnette M: 1068 EGFR and
c-MYC suppress Mir-143 and Mir-145 in colonic tumorigenesis: Roles
of G1 cell cycle regulators as miRNA targets. Gastroenterol.
136:A164–A165. 2009. View Article : Google Scholar
|
|
51
|
Cho WC: MicroRNAs in cancer - from
research to therapy. Biochim Biophys Acta. 1805:209–217.
2010.PubMed/NCBI
|
|
52
|
Wang X, Tang S, Le SY, Lu R, Rader JS,
Meyers C and Zheng ZM: Aberrant expression of oncogenic and
tumor-suppressive microRNAs in cervical cancer is required for
cancer cell growth. PLoS One. 3:e25572008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tang T, Wong HK, Gu W, Yu MY, To KF, Wang
CC, Wong YF, Cheung TH, Chung TK and Choy KW: MicroRNA-182 plays an
onco-miRNA role in cervical cancer. Gynecol Oncol. 129:199–208.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Qian X, Yu J, Yin Y, He J, Wang L, Li Q,
Zhang LQ, Li CY, Shi ZM, Xu Q, et al: MicroRNA-143 inhibits tumor
growth and angiogenesis and sensitizes chemosensitivity to
oxaliplatin in colorectal cancers. Cell Cycle. 12:1385–1394. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shepherd JH: Cervical and vulva cancer:
changes in FIGO definitions of staging. Br J Obstet Gynaecol.
103:405–406. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liu L, Yu X, Guo X, Tian Z, Su M, Long Y,
Huang C, Zhou F, Liu M, Wu X and Wang X: miR-143 is downregulated
in cervical cancer and promotes apoptosis and inhibits tumor
formation by targeting Bcl-2. Mol Med Rep. 5:753–760.
2012.PubMed/NCBI
|
|
57
|
Hu CE, Liu YC, Zhang HD and Huang GJ: The
RNA-binding protein PCBP2 facilitates gastric carcinoma growth by
targeting miR-34a. Biochem Biophys Res Commun. 448:437–442. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Guo G, Kang Q, Chen Q, Chen Z, Wang J, Tan
L and Chen JL: High expression of long non-coding RNA H19 is
required for efficient tumorigenesis induced by Bcr-Abl oncogene.
FEBS. 588:1780–1786. 2014. View Article : Google Scholar
|
|
60
|
Kennedy AL, Morton JP, Manoharan I, Nelson
DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien
KA, et al: Activation of the PIK3CA/AKT pathway suppresses
senescence induced by an activated RAS oncogene to promote
tumorigenesis. Mol Cell. 42:36–49. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sala A, Bettuzzi S, Pucci S, Chayka O,
Dews M and Thomas-Tikhonenko A: Regulation of CLU gene expression
by oncogenes and epigenetic factors: Implications for
tumorigenesis. Adv Cancer Res. 105:115–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Keyes WM, Pecoraro M, Aranda V,
Vernersson-Lindahl E, Li W, Vogel H, Guo X, Garcia EL, Michurina
TV, Enikolopov G, et al: ΔNp63α is an oncogene that targets
chromatin remodeler Lsh to drive skin stem cell proliferation and
tumorigenesis. Cell Stem Cell. 8:164–176. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Couto SS, Cao M, Duarte PC,
Banach-Petrosky W, Wang S, Romanienko P, Wu H, Cardiff RD,
Abate-Shen C and Cunha GR: Simultaneous haploinsufficiency of Pten
and Trp53 tumor suppressor genes accelerates tumorigenesis in a
mouse model of prostate cancer. Differentiation. 77:103–111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Imanishi Y and Tahara H: Putative
parathyroid tumor suppressor on 1p: Independent molecular
mechanisms of tumorigenesis from 11q allelic loss. Am J Kidney Dis.
38(Suppl 1): S165–S167. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Mercier PL, Bachvarova M, Plante M,
Gregoire J, Renaud MC, Ghani K, Têtu B, Bairati I and Bachvarov D:
Characterization of DOK1, a candidate tumor suppressor gene, in
epithelial ovarian cancer. Mol Oncol. 5:438–453. 2011. View Article : Google Scholar : PubMed/NCBI
|