Introduction
Chronic cerebral hypoperfusion (CCH) plays a key
role in white matter (WM) damage, which contributes to the
progression of dementias such as Alzheimer's disease and vascular
dementia (1–5). The integrity of the brain's WM is
comprised mainly of myelinated axons (6). Thus, the damage of myelinated axons
associated with CCH results in development and progression of
dementia. It is essential to determine early alterations of
myelinated axons and potential mechanisms following CCH, which
provide an experimental basis for the prevention of dementia.
Myelination of axons consist of nodes of Ranvier,
paranodal regions, juxtaparanodal regions, and internodes (7,8). Each
defined region contains the localization of key proteins that are
constituents of either the axolemma or the myelin sheath (9,10).
Contactin-associated protein 2 (Caspr2) localizes to juxtaparanodal
regions in association with a cell adhesion molecule of the
neurexin superfamily and forms a complex with contactin 2 to
generate a membrane scaffold that clusters Kv1 channels (11,12).
Voltage-gated sodium (Na+) channels are concentrated at
nodes of Ranvier, which are separated from each other by
myelin-covered internodes. Nodes of Ranvier are responsible for the
generation and efficient propagation of axon potentials (13,14).
Currently, information on the altered expression of Caspr2 and Nav
following CCH is limited.
In the present study, stenosis of the bilateral
carotid common artery in adult Sprague-Dawley rats was used to
study the nerve conduction, cognitive function and the altered
expression of Caspr2 and Nav in order to reveal its underlying
mechanism, and to further develop therapeutic treatments.
Materials and methods
Animals
Adult male Sprague-Dawley rats (6 months, 200±8.5 g)
were housed at a constant room temperature of 24°C on a 12-h
light-dark cycle, and maintained on an ad libitum diet
(Experimental Animal Center, Third Military Medical University,
China). All the experiments were performed with the approval of the
Third Military Medical University Animal Ethics Committee. Every
effort was made to minimize animal suffering and to keep the number
of animals at a minimum.
Bilateral common carotid artery
stenosis (BCAS) surgery (surgical procedure)
The rats were anaesthetized via intraperitoneal
(i.p.) injection of 3% pentobarbital sodium (45 mg/kg) until their
eyelash reflex disappeared, and were then fixed in the dorsal
position. Under aseptic conditions, a 2-cm incision was made in the
median neck. After separation of the tissue layers, ~1 cm of
bilateral common carotid artery was separated from paratracheal
carotid sheath, ligated using a 1.2-mm diameter needle together
with no. 4 silk thread, and repositioned. The wound was closed, and
the rats were given aspirin as an anticoagulant (30 mg/l) in their
drinking water for three days post-surgery. For rats in the sham
group, the bilateral common carotid artery was exposed but no
ligature was made. The animals were randomly allocated into four
groups of six rats each: Sham, 2, 4 and 12 weeks.
Cerebral blood flow (CBF)
measurement
The animals were anesthetized with sodium
pentobarbital (0.5 mg/kg, i.p.), and the skin overlying the left
skull was reflected. A plastic guide cannula (3 mm outer diameter,
2 mm inner diameter, 4 mm length) was fixed perpendicularly to the
skull at 1 mm posterior and 2.5 mm lateral to bregma using dental
resin. A 2.0-mm straight probe (Probe 418-1 Master probe) was
inserted through the guide cannula, and CBF was recorded after the
surgery using laser-Doppler flowmetry (PF5010LDPM; Perimed AB,
Stockholm, Sweden). CBF values were expressed as a percentage of
the baseline value.
Detection of the transcranial
electrical stimulation motor-evoked potential (MEP)
The transcranial electrical stimulation MEPs was
detected (n=6 at each time point in every group) by Keypoint four
guide evoked potential (Experimental Center, Third Military Medical
University, China). All Sprague-Dawley rats were recorded at a
quiet, electrically shielded room temperature of 24°C. The rats
were anaesthetized via i.p. injection of 3% pentobarbital sodium at
a dose of 45 mg/kg until their eyelash reflex disappeared. EEG was
detected with needle electrodes. The stimulation electrode was
located at 1 mm posterior to the bregma. The recording electrode
was located at 3 mm lateral to the sagittal suture. The recording
electrode was located at the contralateral gastrocnemius. The
monopolar recording on the nasal bone was ground. Electrical
stimulation signals reveal a single square wave electrical pulse
(stimulus intensity 5–12 mA, pulse width 0.2 msec). Each result was
repeated at least twice to obtain a stable waveform. The distance
between stimulating and recording electrodes was measured in all
rats to derive an estimate of the MEP latency.
Morris water maze
Two, four, and 12 weeks after CCH surgery, each rat
(n=6/group) was subjected to a water maze test which consisted of a
large circular pool (diameter, 2 m; height, 1.5 m) filled with
water (25±1°C) that was rendered opaque by the addition of enough
milk to prevent the rats from seeing an underwater platform (5-cm
diameter), located 3.5 cm below the surface. The day before the
start of the training, the rats were acclimatized to water by being
allowed to swim freely in a pool. To measure the acquisition
learning behavior, navigation trials were performed daily (4–8
trials/day) for 5 consecutive days for each rat. The rats were
trained to swim to a hidden platform that was situated below the
surface of the water and was placed at the center of one of the
four quadrants throughout training. During the four trials, each
rat was randomly placed in one of the four starting positions. The
rats were given 2 min to find the platform and to stay on it for 10
sec. If the animals failed to find the platform within the given
time, they were gently guided to the platform and were allowed to
stay on it for 10 sec. The rats were trained to find the hidden
platform in the pool over 10 trials. To evaluate the rat's spatial
retention ability, the space probe trails were carried out on day
6. The platform was removed, and the total distance traveled while
swimming and the swimming distance in the target quadrant for 1 min
were recorded by the tracking system.
Histochemical evaluation of myelinated
axon damage
Two, four, and twelve weeks after CCH surgery, each
rat (n=6/group) was anesthetized and perfused intracardially with
phosphate-buffered saline, followed by 4% paraformaldehyde. The
brains were post-fixed in ice-cold 4% paraformaldehyde for 2 h, and
then cryoprotected in a 30% sucrose solution. Coronal cryostat
sections (20-µm) were mounted onto poly-lysine coated slides.
Similar brain slices were selected for immunohistochemistry
labeling, respectively. For quantitative assessment of myelinated
axons, damaged sections were immunostained using antibodies against
Caspr2. Immunoreactivity was revealed by the primary antibody
against Caspr2 (polyclonal rabbit anti-rat Caspr2 obtained from
Boster Biological Technology Co. Ltd., Wuhan, China). FITC-labeled
anti-rabbit secondary antibody was obtained from Beijing Zhongshan
Golden Bridge Biotechnology Co., Ltd. (Beijing, China). A laser
scanning confocal microscope was used to examine the presence and
distribution of fluorescence in the sections. The Image-Pro Plus
analysis system was used to determine the average optical density
(AOD) value of expression.
Examination of Nav protein levels by
western blot analysis
For total protein extraction, tissues were
transferred to cold lysis buffer containing 0.1 mol/l NaCl, 0.05
mol/l Tris-HCl (pH 7.6), 0.001 mol/l EDTA (pH 8.0), 0.1% Tween-20,
aprotinin (1 µg/ml), and PMSF (100 µg/ml) and homogenized on ice.
The homogenate was centrifuged at 8,500 × g for 10 min, and the
supernatant was stored at 4°C. Equal amounts of protein from each
sample (50 µg) were mixed with 30–35 µl sample buffer and boiled
for 4 min. Samples were separated by electrophoresis on a 10%
SDS-PAGE gel and transferred to nitrocellulose membranes at 100 mA
for 2 h. The membrane was blocked with 5% non-fat dry milk in PBS
containing 0.01% Tween-20 at 4°C overnight. Immunoblots were probed
with specific antibodies: Polyclonal rabbit anti-rat Caspr2
(1:1,000; cat. no. TA326393; Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd.). Blots were washed and incubated with
horseradish peroxidase-conjugated anti-rabbit antibodies (1:2,000;
cat. no. ZDR-5306; Beijing Zhongshan Golden Bridge Biotechnology
Co., Ltd.) for 1 h at 25°C. An endogenous control protein, GAPDH,
was included in each western blot analysis. Then the membranes were
developed using enhanced chemiluminescence reagent. The optical
densities of the specific bands were scanned and measured by image
analysis software (Labworks 4.6, China) and normalized to
GAPDH.
Statistical analysis
Statistical analysis was performed using the SPSS
13.0 software package (Chicago, IL, USA). Values were expressed as
means ± standard deviation (SD). Analysis of variance was used to
determine the significance of differences for multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Changes of CBF in the rats after
surgery
CBF values were recorded for all the animals prior
to and after surgery using laser-Doppler flowmetry. Compared to the
sham group, CBF values in the operation group decreased
significantly after surgery. The mean CBF value in rats undergoing
CCH surgery was reduced significantly to 33.90±5.48% compared to
that of sham animals (P<0.05). There were no marked differences
in CBF in the three stenosis groups.
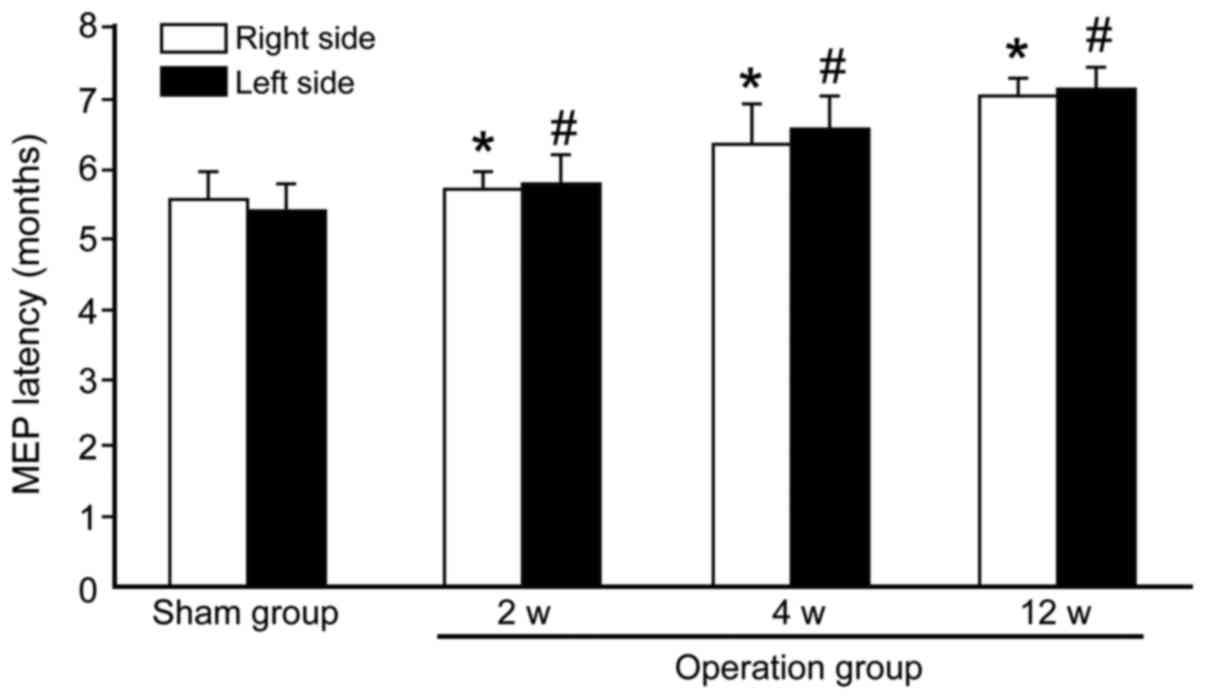
Transcranial electrical stimulation
MEP
After 2, 4 and 12 weeks of surgery, the MEP latency
in the operated groups was significantly prolonged compared to the
values prior to the sham group (Fig.
1). Compared with the sham group (right 5.50±0.41 vs. left
5.38±0.37 msec), the MEP latencies of the operated groups were
significantly longer (right 5.67±0.23 vs. left 5.75±0.27 msec at 2
weeks; right 6.25±0.58 vs. left 6.50±0.44 msec at 4 weeks and right
7.05±0.16 vs. left 7.08±0.29 msec at 12 weeks; P<0.05). The MEP
latencies had no significant change between the left and the right
sides of the rat either in the sham group or the operated groups
(P>0.05).
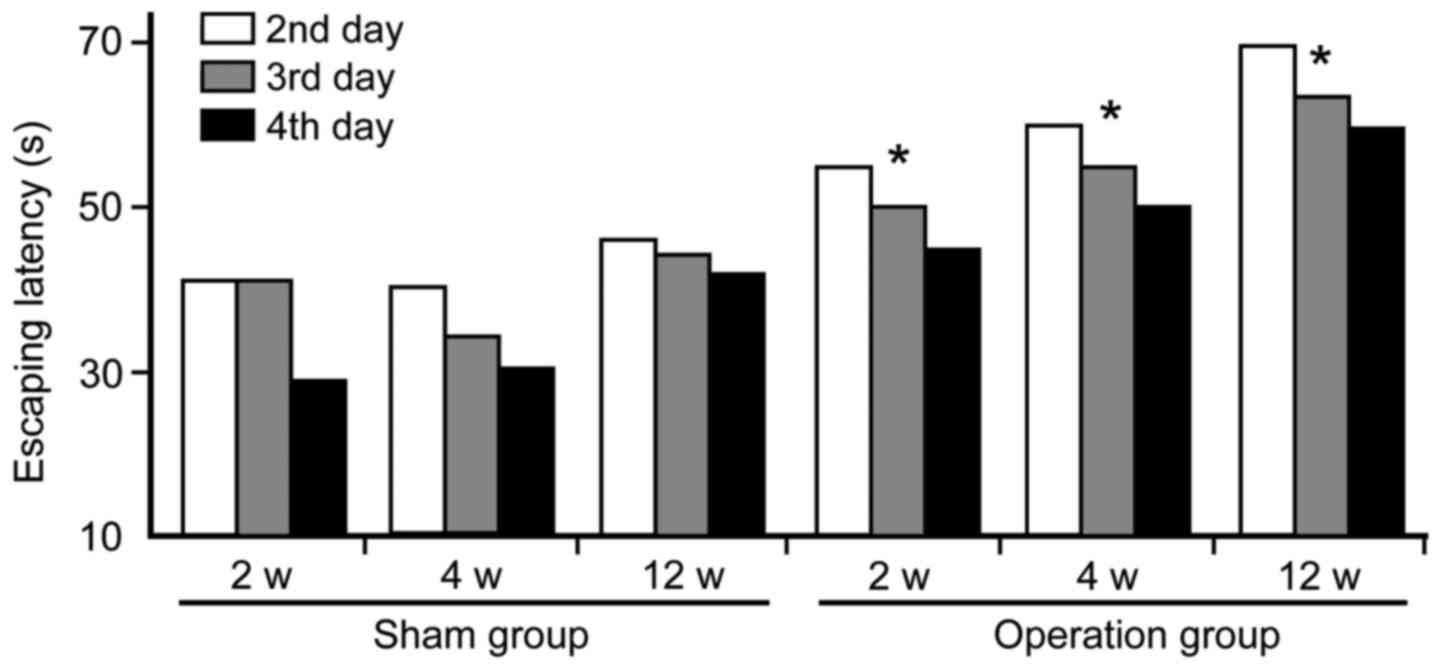
Morris water maze
Morris water maze analyses show that cognitive
functions were markedly damaged at 2, 4 and 12 weeks in the
operated groups following CCH. Compared to the sham group, the
escaping latency of the operation group was significantly prolonged
(Fig. 2). In addition, times of
crossing platform (2.8±0.35 at 2 weeks; 2.1±0.38 at 4 weeks;
1.2±0.50 at 12 weeks) in spatial probing were markedly decreased in
the operation groups as compared with the sham group (3.2±0.77)
(P<0.05). Furthermore, the first time passing hidden platform
(49±3.16 sec at 2 weeks; 65±2.54 sec at 4 weeks; 91.5±3.80 sec at
12 weeks) was prolonged significantly in the operation groups when
compared to the sham group (34±2.94 sec) (P<0.05).
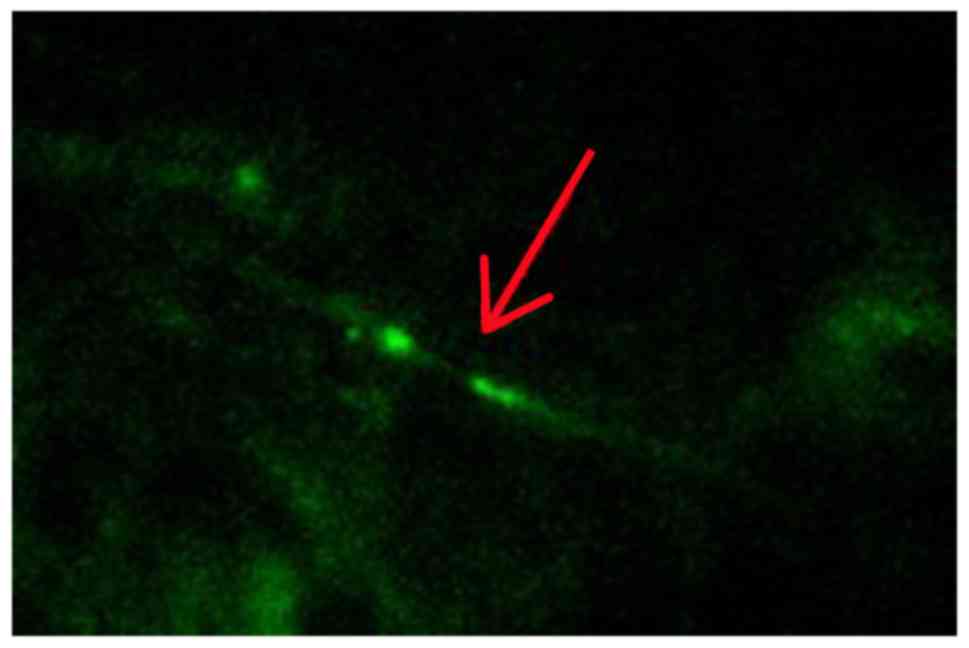
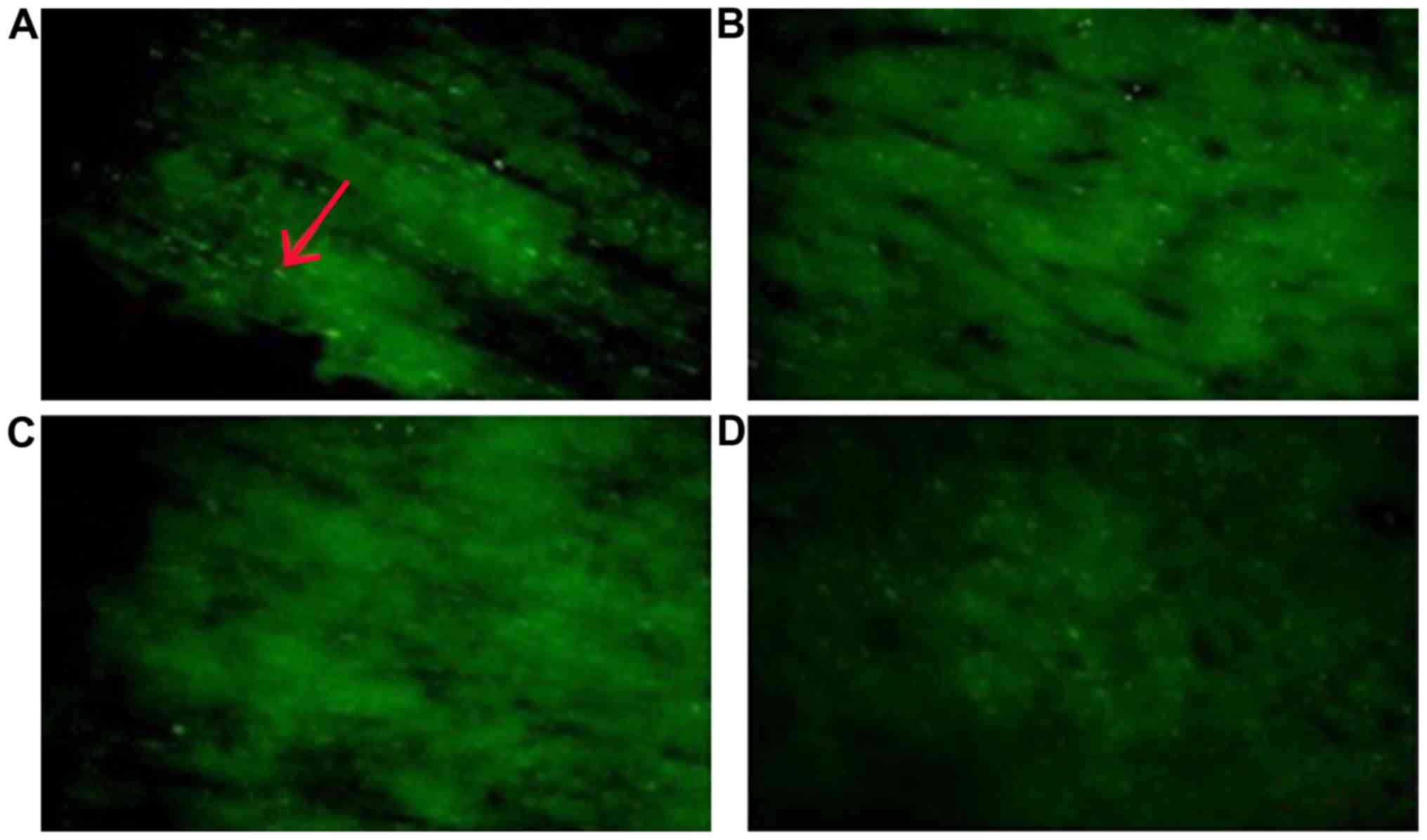
Histochemical evaluation of myelinated
axon damage
Histological analyses revealed that the myelinated
axons were significantly damaged at 2, 4 and 12 weeks in the
operation groups. Compared to the sham group, CCH leads to a large
change in Caspr2 protein expression. Caspr2 protein expression was
detected as fine green particles using immunofluorescence (Fig. 3). As shown in Fig. 4, both the number and intensity of the
particles decreased significantly. Moreover, the intensity of stain
was reduced markedly over time. The distribution pattern of Caspr2
expression also changed markedly. Compared to the sham group, our
results showed the Caspr2 expression was disordered in the
distribution of strip and point in the corpus callosum following
CCH. At 12 weeks after the model preparation, the Caspr2-positive
particles gathered around the area.
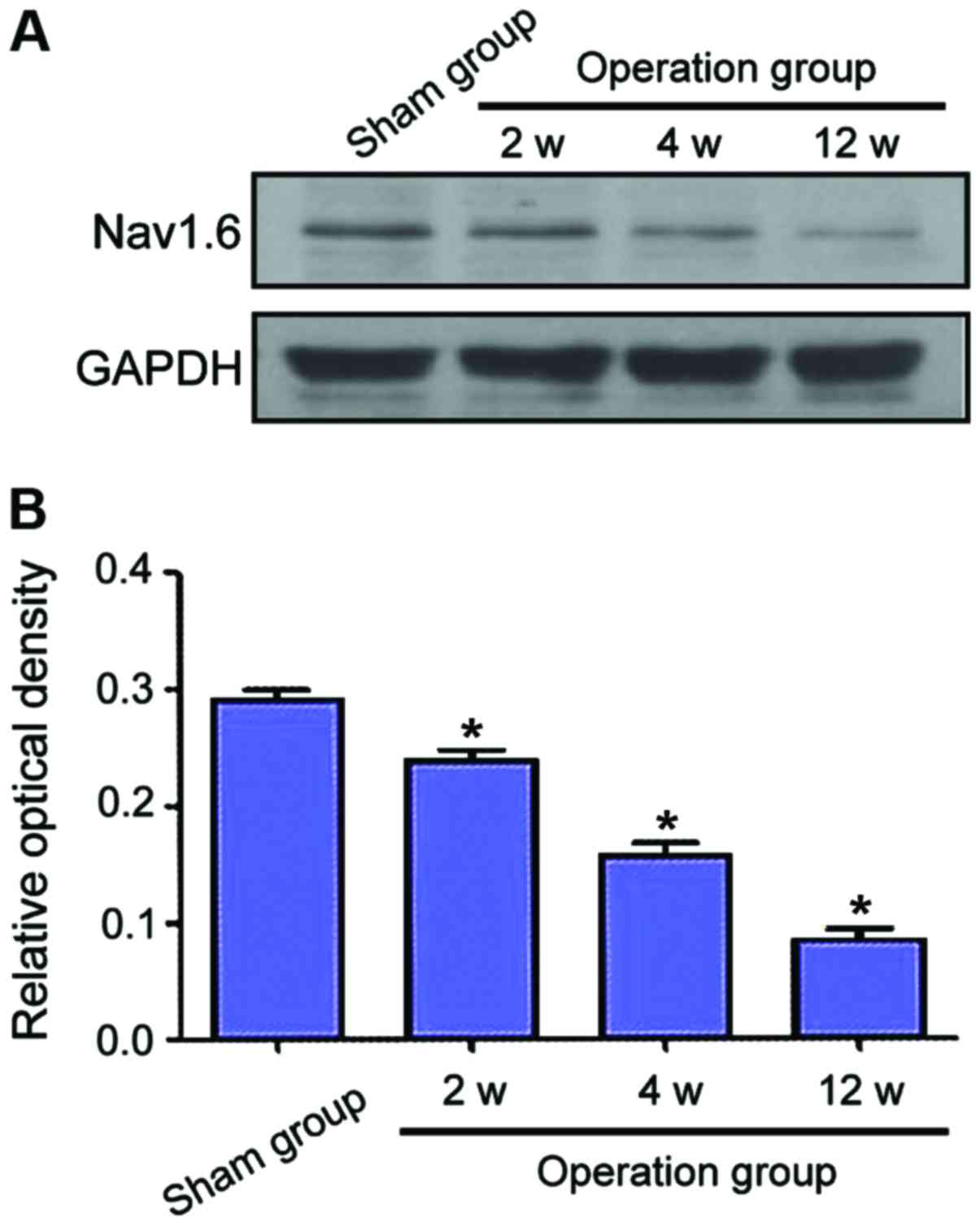
Examination of Nav1.6 protein levels
by western blot analysis
Protein levels of Nav1.6 of each group were
determined by western blot analysis. Compared to the sham group,
Nav1.6 protein levels decreased markedly following CCH (Fig. 5A). In addition, the level of Nav1.6
was reduced over time. As shown in Fig.
5B, the Nav1.6 expression following CCH was significantly
downregulated in the operation groups when compared to the sham
group (P<0.05). Furthermore, there was a marked difference
between the groups at different phases.
Discussion
The present results have shown a progression of the
damage to myelinated axons over time following CCH. In order to
understand the mechanisms and quantify the myelinated axon damage,
we observed the loss of nerve conduction, the damage of cognitive
function and the alterations of Caspr2 and Nav1.6 levels in a rat
model of CCH. The data have demonstrated that Caspr2 levels were
significantly downregulated with abnormal localization. At the same
time, Nav1.6 levels were reduced markedly following CCH.
This study has demonstrated that the nerve
conduction and cognitive function reduced markedly after CCH in
animals, which is consistent with a chronic persistent decrease of
CBF. These results indicate that very early determination of
cognitive function and MEP in patients can provide a promising
strategy for judgment of CCH and prevention of dementia in
clinic.
Recent studies conducted on the correlation of
Caspr2 and Nav expression to damage of myelinated axons have
focused on the potential mechanisms of demyelination disease
(15–17). Some studies indicate that the Caspr2
and Nav expression may be involved in a progressive axonal
denudation and nerve conduction impairment, which contributes to
the demyelination pathology (15–17).
However, studies related to the influence of Caspr2 and Nav
expression on WM damage induced by CCH are limited. In the present
study, alterations in the key proteins of myelinated axons were
observed in a rat model of CCH. The immunohistochemical analysis of
Caspr2 protein revealed that the level and localization changed
significantly. Compared to the sham group, the number and intensity
of the particles decreased significantly over time. Caspr2
expression was found to be disordered in the locations of strip and
point in the corpus callosum following CCH. At 12 weeks after the
operation, the Caspr2 particles gathered around in part of the
area. Thus, the decreasing expression and abnormal locations of the
Caspr2 protein contributed to the myelinated axon damage induced by
CCH. Consistent with the above results, previous studies showed
that Caspr2 expression was downregulated with disruption of normal
localization and diffusion in multiple sclerosis (18,19).
Therefore, CCH results in the alterations of Caspr2 expression,
which contributes to myelinated axon damage. In addition, the
western blot analysis of Nav1.6 protein revealed that the level of
this protein reduced significantly over time, which provided
further support that CCH causes damage to myelinated axons.
Similarly, previous studies showed that the alteration in the
expression of Nav1.6 contributed to the damage to myelinated axons,
which occurred in a variety of neuropathological states including
traumatic brain injury (20,21), spinal injury (22,23),
demyelination disease (24,25) and epilepsy (26,27).
Moreover, it was observed that there was a time-dependent loss of
Nav mRNA and protein in response to CCH as a result of focal
cerebral ischemia (28,29), which corroborates the results of the
present study.
Taken together, this study revealed that there is
progression of Caspr2 and Nav1.6 expression in rat model of CCH
over time. We suggested that the alterations of Caspr2 and Nav1.6
on myelinated axon damage after CCH may be in response to the loss
of nerve conduction and the damage of cognitive function, which
provided molecular basis for the prevention of dementia. However,
the present study revealed that the molecular mechanisms induced by
CCH were limited. There is emerging evidence on distinct protein
architecture alterations after mild cerebral hypoperfusion
(30). Thus, the complexity of the
molecular mechanisms induced by CCH suggests that additional
experiments are required to clarify the WM damage.
In conclusion, our results demonstrate that CCH
results in time-dependent loss of Caspr2 and Nav1.6 protein, which
contributes to the damage of myelinated axons. These results
provide insight into the mechanisms for WM damage in response to
CCH.
References
|
1
|
Akinyemi RO, Mukaetova-Ladinska EB, Attems
J, Ihara M and Kalaria RN: Vascular risk factors and
neurodegeneration in ageing related dementias: Alzheimer's disease
and vascular dementia. Curr Alzheimer Res. 10:642–653. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hainsworth AH, Brittain JF and Khatun H:
Pre-clinical models of human cerebral small vessel disease: utility
for clinical application. J Neurol Sci. 322:237–240. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miki K, Ishibashi S, Sun L, Xu H, Ohashi
W, Kuroiwa T and Mizusawa H: Intensity of chronic cerebral
hypoperfusion determines white/gray matter injury and
cognitive/motor dysfunction in mice. J Neurosci Res. 87:1270–1281.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brown WR, Moody DM, Thore CR, Anstrom JA
and Challa VR: Microvascular changes in the white mater in
dementia. J Neurol Sci. 283:28–31. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fernando MS, Simpson JE, Matthews F,
Brayne C, Lewis CE, Barber R, Kalaria RN, Forster G, Esteves F,
Wharton SB, et al: MRC Cognitive Function and Ageing Neuropathology
Study Group: White matter lesions in an unselected cohort of the
elderly: molecular pathology suggests origin from chronic
hypoperfusion injury. Stroke. 37:1391–1398. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nave K-A: Myelination and support of
axonal integrity by glia. Nature. 468:244–252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Salzer JL: Clustering sodium channels at
the node of Ranvier: close encounters of the axon-glia kind.
Neuron. 18:843–846. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arroyo EJ and Scherer SS: On the molecular
architecture of myelinated fibers. Histochem Cell Biol. 113:1–18.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rios JC, Rubin M, St Martin M, Downey RT,
Einheber S, Rosenbluth J, Levinson SR, Bhat M and Salzer JL:
Paranodal interactions regulate expression of sodium channel
subtypes and provide a diffusion barrier for the node of Ranvier. J
Neurosci. 23:7001–7011. 2003.PubMed/NCBI
|
|
10
|
Susuki K and Rasband MN: Molecular
mechanisms of node of Ranvier formation. Curr Opin Cell Biol.
20:616–623. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peles E and Salzer JL: Molecular domains
of myelinated axons. Curr Opin Neurobiol. 10:558–565. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Girault JA, Oguievetskaia K, Carnaud M,
Denisenko-Nehrbass N and Goutebroze L: Transmembrane scaffolding
proteins in the formation and stability of nodes of Ranvier. Biol
Cell. 95:447–452. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gasser A, Ho TS-Y, Cheng X, Chang K-J,
Waxman SG, Rasband MN and Dib-Hajj SD: An ankyrinG-binding motif is
necessary and sufficient for targeting Nav1.6 sodium channels to
axon initial segments and nodes of Ranvier. J Neurosci.
32:7232–7243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Leterrier C, Brachet A, Dargent B and
Vacher H: Determinants of voltage-gated sodium channel clustering
in neurons. Semin Cell Dev Biol. 22:171–177. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Coman I, Aigrot MS, Seilhean D, Reynolds
R, Girault JA, Zalc B and Lubetzki C: Nodal, paranodal and
juxtaparanodal axonal proteins during demyelination and
remyelination in multiple sclerosis. Brain. 129:3186–3195. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Howell OW, Palser A, Polito A, Melrose S,
Zonta B, Scheiermann C, Vora AJ, Brophy PJ and Reynolds R:
Disruption of neurofascin localization reveals early changes
preceding demyelination and remyelination in multiple sclerosis.
Brain. 129:3173–3185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Howell OW, Rundle JL, Garg A, Komada M,
Brophy PJ and Reynolds R: Activated microglia mediate axoglial
disruption that contributes to axonal injury in multiple sclerosis.
J Neuropathol Exp Neurol. 69:1017–1033. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wolswijk G and Balesar R: Changes in the
expression and localization of the paranodal protein Caspr on axons
in chronic multiple sclerosis. Brain. 126:1638–1649. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zoupi L, Markoullis K, Kleopa KA and
Karagogeos D: Alterations of juxtaparanodal domains in two rodent
models of CNS demyelination. Glia. 61:1236–1249. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mao Q, Jia F, Zhang XH, Qiu YM, Ge JW, Bao
WJ, Luo QZ and Jiang JY: The up-regulation of voltage-gated sodium
channel Nav1.6 expression following fluid percussion traumatic
brain injury in rats. Neurosurgery. 66:1134–1139; discussion 1139.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang JALW, Lin W, Morris T, Banderali U,
Juranka PF and Morris CE: Membrane trauma and Na+ leak
from Nav1.6 channels. Am J Physiol Cell Physiol. 297:C823–C834.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hunanyan AS, Alessi V, Patel S, Pearse DD,
Matthews G and Arvanian VL: Alterations of action potentials and
the localization of Nav1.6 sodium channels in spared axons after
hemisection injury of the spinal cord in adult rats. J
Neurophysiol. 105:1033–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wienecke J, Westerdahl A-C, Hultborn H,
Kiehn O and Ryge J: Global gene expression analysis of rodent motor
neurons following spinal cord injury associates molecular
mechanisms with development of postinjury spasticity. J
Neurophysiol. 103:761–778. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hassen GWFJ, Feliberti J, Kesner L,
Stracher A and Mokhtarian F: Prevention of axonal injury using
calpain inhibitor in chronic progressive experimental autoimmune
encephalomyelitis. Brain Res. 1236:206–215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Black JA, Newcombe J, Trapp BD and Waxman
SG: Sodium channel expression within chronic multiple sclerosis
plaques. J Neuropathol Exp Neurol. 66:828–837. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liao Y, Deprez L, Maljevic S, Pitsch J,
Claes L, Hristova D, Jordanova A, Ala-Mello S, Bellan-Koch A,
Blazevic D, et al: Molecular correlates of age-dependent seizures
in an inherited neonatal-infantile epilepsy. Brain. 133:1403–1414.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yao C, Williams AJ, Cui P, Berti R, Hunter
JC, Tortella FC and Dave JR: Differential pattern of expression of
voltage-gated sodium channel genes following ischemic brain injury
in rats. Neurotox Res. 4:67–75. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blumenfeld H, Lampert A, Klein JP, Mission
J, Chen MC, Rivera M, Dib-Hajj S, Brennan AR, Hains BC and Waxman
SG: Role of hippocampal sodium channel Nav1.6 in kindling
epileptogenesis. Epilepsia. 50:44–55. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yao C, Williams AJ, Hartings JA, Lu XC,
Tortella FC and Dave JR: Down-regulation of the sodium channel
Na(v)1.1 α-subunit following focal ischemic brain injury in rats:
in situ hybridization and immunohistochemical analysis. Life Sci.
77:1116–1129. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reimer MM, McQueen J, Searcy L, Scullion
G, Zonta B, Desmazieres A, Holland PR, Smith J, Gliddon C, Wood ER,
et al: Rapid disruption of axon-glial integrity in response to mild
cerebral hypoperfusion. J Neurosci. 31:18185–18194. 2011.
View Article : Google Scholar : PubMed/NCBI
|