Introduction
Sepsis is defined as life-threatening organ
dysfunction caused by a dysregulated host response to infection and
is associated with high mortality and huge social burden (1). Acute kidney injury (AKI) is present in
over 40% of septic patients and increases the mortality up to 70%
(2,3). Mitochondria have important roles in
both physiological and pathophysiological processes, including
calcium homeostasis, cell signaling pathways, transcriptional
regulation, and apoptosis (4).
Previous studies indicated that multiple aspects of mitochondrial
dysfunction contribute to septic AKI, such as the overproduction of
reactive oxygen species (ROS), the depletion of adenosine
triphosphate (ATP), the dissipation of mitochondrial membrane
potential (MMP) and the exacerbation of apoptosis (5–8).
Oxidative stress plays an important role in the
development of mitochondrial dysfunction during septic AKI. When
the antioxidant protection mechanisms and ROS production are
imbalanced, oxidative stress results and leads to the inactivation
of endogenous antioxidant systems, disruption of the electron
transport, uncoupling of mitochondrial oxidative phosphorylation,
and transition of mitochondrial membrane permeability (8–10).
Mitochondria is the primary source of ROS within cells (11). Therefore, mitochondrial oxidative
stress-mediated damage may be the main cause of mitochondrial
dysfunction.
Mitochondria-targeted antioxidants were reported to
block ROS generation within mitochondria as well as preserve
mitochondrial complex respiration, ATP level, and MMP both in
animal and cell models of septic AKI (9,10).
Targeting antioxidants to mitochondria may offer a new therapy in
the future but are not yet available in clinical work. Insulin
therapy is a simple and inexpensive intervention for disturbed
glucose and lipid metabolism during sepsis and has been applied in
critically ill patients (12,13). In
an early experiment, we found insulin infusion increased MMP and
attenuated inflammatory response in liver sections of septic rats
(14). But the effects and
mechanisms of insulin against mitochondrial oxidative stress remain
to be fully elucidated. Under normal conditions, mitochondria
protect from damage by ROS via several interacting antioxidant
systems (15,16).
We hypothesized that insulin could alleviate
mitochondrial oxidative stress and aimed to explore the relations
between insulin therapy and endogenous antioxidant systems as well
as other mechanisms within mitochondria in a rat model of septic
AKI.
Materials and methods
Animals
This study was approved by the Institutional Animal
Care and Use Committee of Southern Medical University (Guangzhou,
China) and conducted in accordance with the Guide for the Care and
Use of Laboratory Animals of the National Institutes of Health
(NIH). Male SD rats (weighing 250 to 300 g) were housed in a 25°C
room temperature, illuminated in a 12 h light/12 h dark cycle with
free access to water and food.
Sepsis model and drug administration
methods
Rats were divided randomly into four groups (n=6 in
each group) as follow: Control group, sham surgery group, cecal
ligation and puncture (CLP) group, and CLP plus insulin group. CLP
and sham surgery were performed according to the method described
by Rittirsch et al (17).
Before operation, all rats were anesthetized with an
intraperitoneal injection of pentobarbital sodium. The rats in the
CLP plus insulin group also received a subcutaneous injection of
insulin glargine (Lantus®; Sanofi-Aventis, Paris,
France) at a dose of 0.5 IU/kg body weight. After surgery, rats
were placed back in cages with free access to water and food, and
the rats of CLP group were resuscitated by injecting prewarmed
normal saline (37°C; 5 ml/100 g body weight) subcutaneously. Rats
were killed at 12 or 24 h after CLP or sham surgery as separate
experiments. At sacrifice, blood specimens were obtained from
abdominal aorta and renal cortices were dissected and stored at
−80°C until analyses.
Serum biochemical assays
The concentrations of blood urea nitrogen (BUN) and
serum creatinine (CRE) were measured using an automatic biochemical
analyzer (Cobas c702; Roche Diagnostics, Mannheim, Germany). The
serum neutrophil gelatinase-associated lipocalin (NGAL) was
estimated by a commercial ELISA kit (EK-Bioscience Biotechnology,
Shanghai, China).
Histology analysis
For histopathological observation, the renal
cortices were fixed, embedded, sectioned, and then subjected to
hematoxylin and eosin (H&E) staining. The images were captured
by a light microscopy (Olympus BX60; Olympus America, Inc.,
Melville, NY, USA).
Measurement of MMP as well as levels
of mitochondrial superoxide dismutase 2 (SOD2) and ROS
Renal mitochondria were isolated using the
Mitochondria Isolation kit (Applygen Technologies Inc., Beijing,
China). MMP was assessed with JC-1 staining and calculated as the
fluorescence ratio of red (JC-1 polymer) to green (JC-1 monomer).
The activity of SOD2 was evaluated using a SOD assay kit-WST
method. Both above diagnosis kits were provided by Beyotime
Institute of Biotechnology (Haimen, China). ROS production within
mitochondria was assessed with
6-chloromethyl-2′,7′-dichlorofluorescein diacetate (CM-H2DCFDA)
staining (Genmed Scientifics Inc., Shanghai, China). Fluorescence
or absorbance was detected by a multifunctional microplate reader
(SpectraMax M5; Molecular Devices, LLC, Sunnyvale, CA, USA).
Tissue biochemical analyses
The renal cortical tissues were homogenized and the
protein concentrations in the supernatant were determined using a
Bradford assay kit. ATP level was detected using a firefly
luciferase-based ATP assay kit. Total nitric oxide (NO) production
was indicated by the concentration of nitrate and nitrite using a
modified Griess reaction method. All above diagnosis kits were
provided by Beyotime Institute of Biotechnology. Chemicals used for
measuring the activity of inducible NO synthase (iNOS) as well as
the contents of total glutathione (GSH) and oxidative GSH (GSSG)
within kidney were obtained from Nanjing Jiancheng Bioengineering
Institute (Nanjing, China). The production of intrarenal uncoupling
protein 2 (UCP2) was estimated by a commercial ELISA kit (Oulu
Biotechnology, Shanghai, China).
Immunohistochemistry
After being deparaffinized and dehydrated, the
embedded kidney sections were treated with 3% hydrogen peroxide to
inactivate endogenous peroxidase. Blocking buffer (10% normal goat
serum) was applied at room temperature for 30 min followed by
sequential application of anti-UCP2 antibody (1:100; Protein Tech
Group, Wuhan, China) and anti-PINK1 (1:100; Bioworld Technology,
Nanjing, China), goat anti-rabbit secondary antibody and
diaminobenzidine tetrahydrochloride substrate (Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA). The images were captured
by a light microscopy (Olympus BX60; Olympus America Inc.).
RT-qPCR
Total RNA was extracted from the renal tissue using
TRIzol reagent and then reverse transcribed and synthesized into
cDNA using RT-PCR kits (Takara Biotechnology Co., Ltd., Dalian,
China). RT-PCR amplification reaction was performed on the
LightCycler 480 (Roche Diagnostics, Laval, QC, Canada). The
quantification cycle (Cq) was obtained from triplicate samples and
averaged. Calculations were based on the ‘ΔΔCq method’ (18) using the equation R
(ratio)=2−ΔΔCq and standardized by the reference gene,
GAPDH. All PCR primers were provided by Generay Biotechnology
(Shanghai, China). SOD1 primer: 5′-TTAGCAGGACAGCAGATGAGT-3′
(forward) and 5′-TCCACGAGAAACAAGATGACT-3′ (reverse). SOD2 primer:
5′-CACTACTACAAAACACCCACC-3′ (forward) and
5′-CTGCTCTAATCAGGACCCACT-3′ (reverse). Glutathione synthetase (GSS)
primer: 5′-CTATGGCACGCTGGTCAAATA-3′ (forward) and
5′-TTCCAAGATCCTGTCCAACAA-3′ (reverse). UCP2 primer:
5′-ACCATTGCACAGAGGAAGG-3′ (forward) and 5′-TCTTGACCACATCAACGGG-3′
(reverse). PINK1 primer: 5′-CCTTCAACAGTTCAGGCGGT-3′ (forward) and
5′-GCCTCGGTGACAGCTAAGTC. PGC1α primer: 5′-GGGGCACATCTGTTCTTCCA-3′
(forward) and 5′-GCTTGACTGGGATGACCGAA. GAPDH primer:
5′-GCCAGCCTCGTCTCATAGACA-3′ (forward) and
5′-AGAGAAGGCAGCCCTGGTAAC-3′ (reverse).
Blood glucose measurement
Capillary blood samples (10 µl) from the tail were
analyzed using a BG meter (Abbott Optimum Xceed; Abbott Diabetes
Care, Inc., Alameda, CA, USA) at 0, 6, 12, 18 and 24 h time point
of the experiment.
Statistical analysis
Data were expressed as mean ± standard deviation.
For multiple comparisons, we used one-way ANOVA, followed by
least-significant difference tests between multiple groups.
Statistical analyses were carried out using SPSS 17.0 software
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Insulin attenuated sepsis-induced
AKI
As shown in Fig. 1A,
indicators of renal injury, including BUN and serum CRE were
dramatically elevated at 24-h timepoint meanwhile serum NGAL were
dramatically elevated both at 12 and 24-h in the CLP group compared
with control or sham surgery group (all P<0.05). Insulin therapy
attenuated these elevations (all P<0.05). Kidney injury was also
examined by H&E staining (Fig.
1B). Renal tissues of the CLP group demonstrated obvious
pathological alternations, such as extensive swelling and
vacuolization of tubular cells. But renal sections of rats treated
with insulin showed lesser pathological changes.
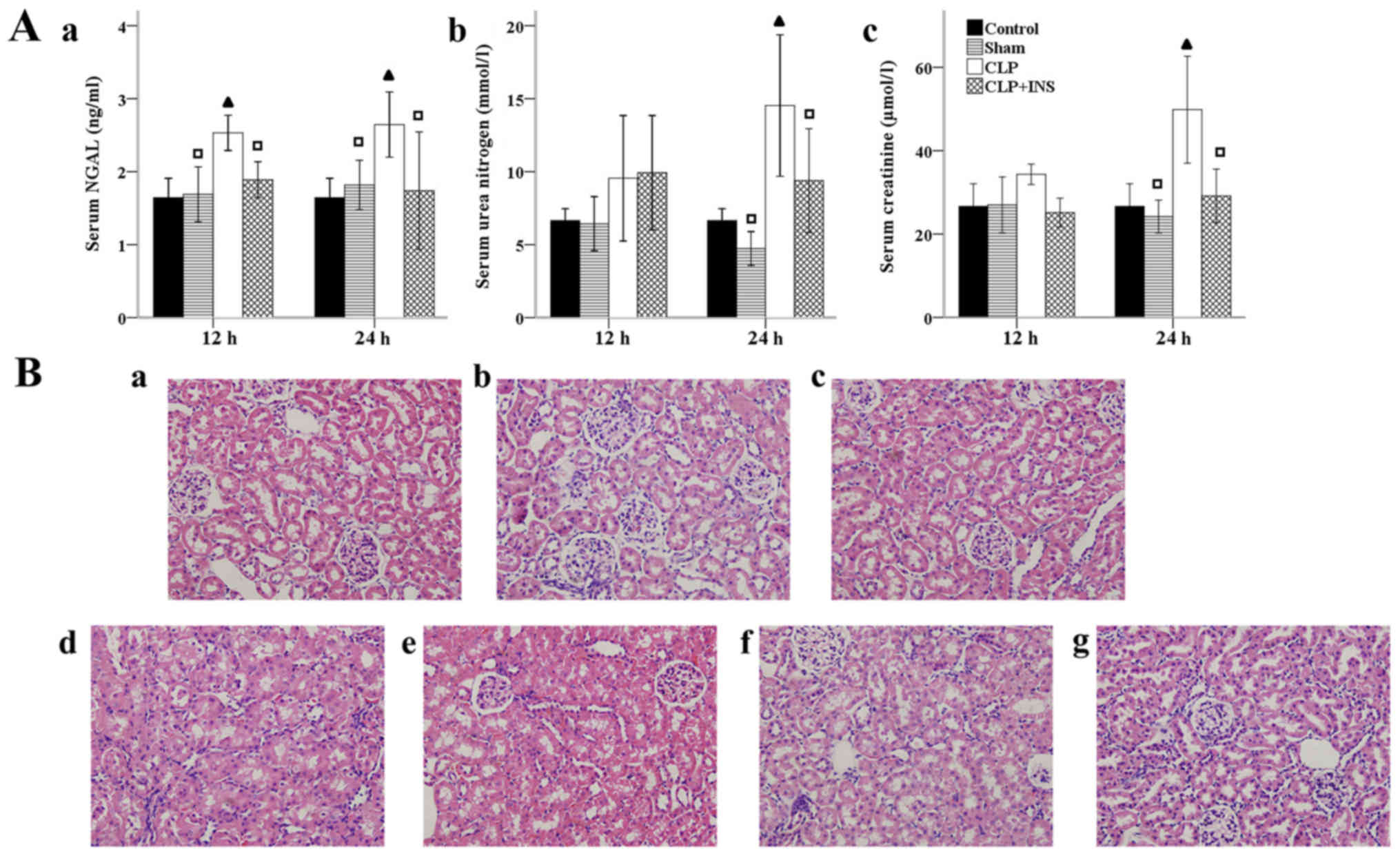 | Figure 1.Effect of insulin (INS) on septic
acute kidney injury. (A) Effect of INS on the levels of (a) serum
neutrophil gelatinase-associated lipocalin (NGAL), (b) urea
nitrogen and (c) creatinine. Results were expressed as mean ± SD.
▲P<0.05 vs. control, □P<0.05 vs. cecal
ligation and puncture (CLP) at same time point. (B) Effect of INS
on sepsis-induced renal pathological changes. Panel a-g (H&E
staining, magnification, ×400) represented control, sham surgery 12
h, sham surgery 24 h, CLP 12 h, CLP 24 h, CLP plus INS 12 h, CLP
plus INS 24 h. |
Insulin preserved renal mitochondrial
function in septic rats
Renal mitochondrial function was analyzed by
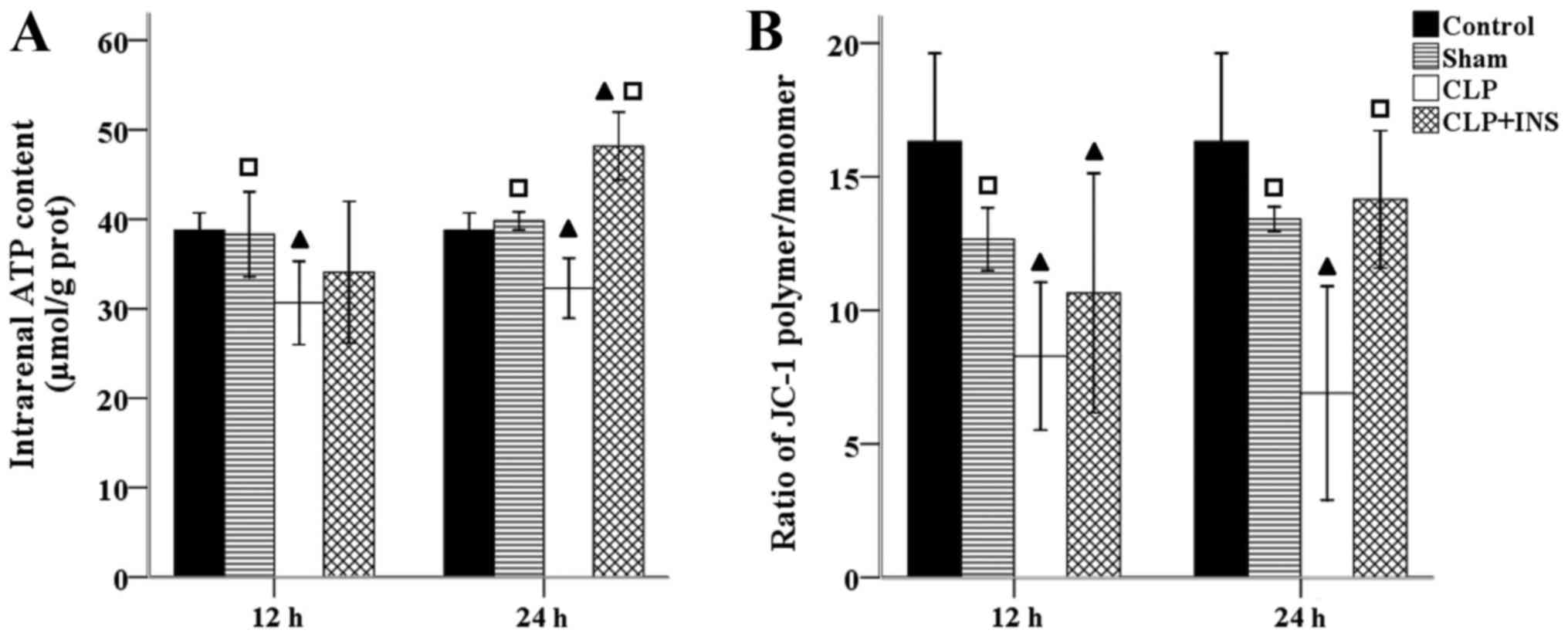
intrarenal ATP level and MMP. Fig. 2
shows that there was a sharp decline in ATP content and MMP in the
CLP group compared with control or sham surgery group (all
P<0.05). However, insulin therapy reversed the depletion of ATP
and dissipation of MMP at 24 h following CLP (both P<0.01).
Insulin ameliorated mitochondrial
oxidative stress in septic rats
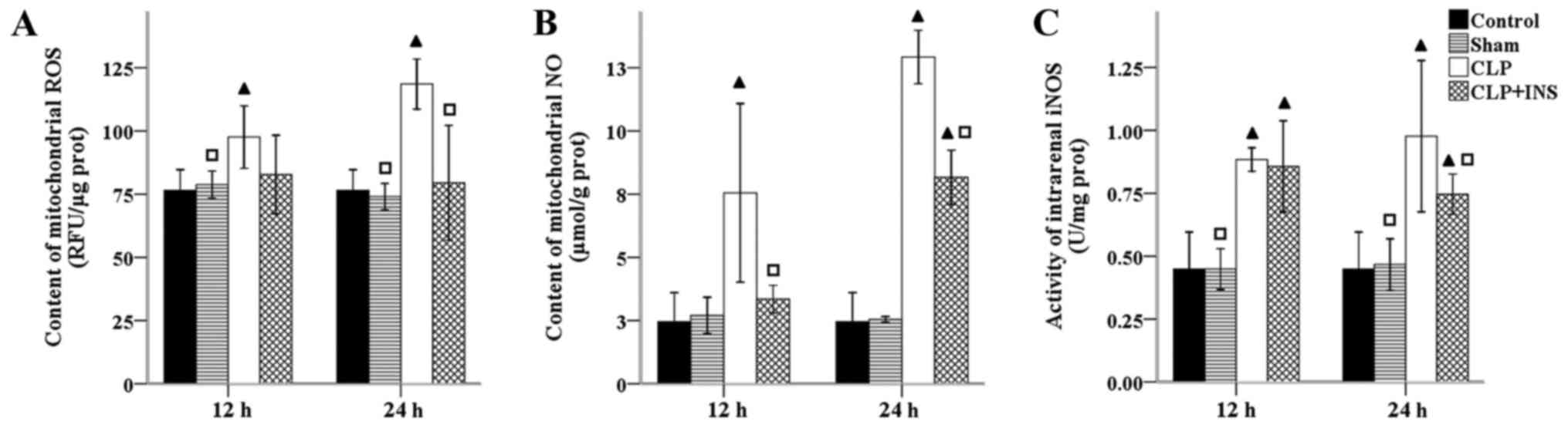
The electron transport chain is the major source of
intracellular ROS and mitochondria are also capable of producing NO
and other reactive nitrogen species (RNS). The overproduction of NO
is normally induced by NOS in sepsis. As shown in Fig. 3, the activity of intrarenal iNOS as
well as production of mitochondrial ROS and NO were dramatically
elevated in the CLP group compared with control or sham surgery
group (all P<0.05). Insulin therapy attenuated these increases
at 24 h following CLP (all P<0.05).
Insulin on expression of mitochondrial
SOD
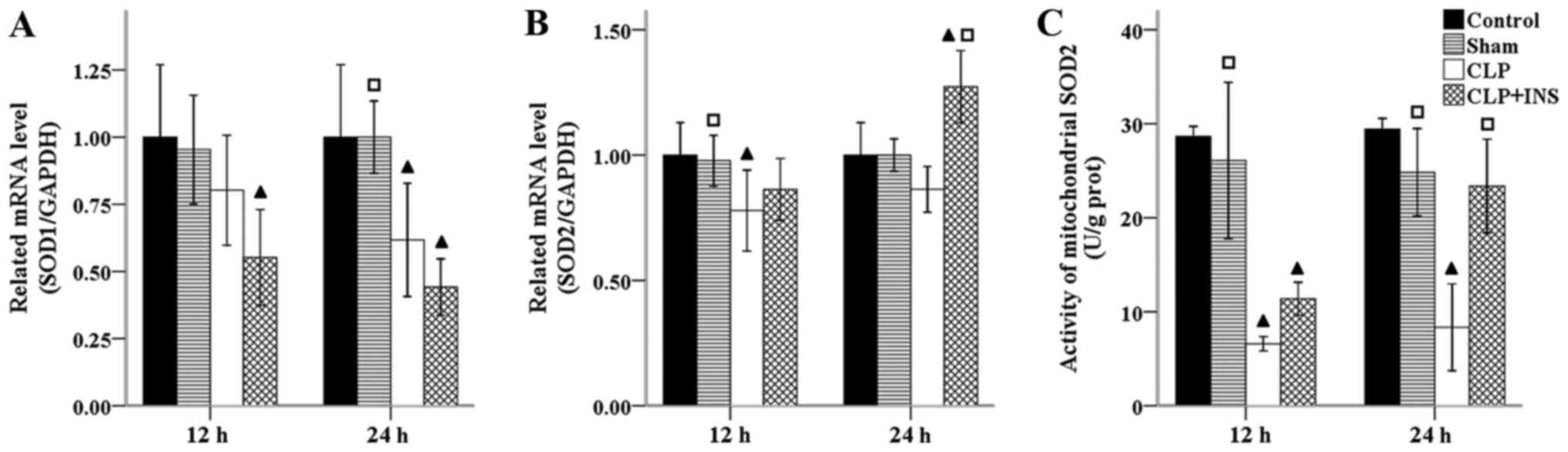
Endogenous antioxidant systems lie the first line of
defense against oxidative stress, since they locate near the site
of mitochondrial ROS formation. Normally, ROS within mitochondria
is converted to hydrogen peroxide (H2O2) by
the action of SOD and then removed by the oxidation of reduced GSH.
As demonstrated in Fig. 4, a
significant decrease in the mRNA level of SOD1 and SOD2 was both
detected in the CLP group compared with control group at 24 and 12
h following CLP respectively (all P<0.05). Insulin therapy
increased the mRNA level of SOD2 but not SOD1 compared with CLP
group at 24-h time point (P<0.001). Concurrently, the activity
of mitochondrial SOD2 was significantly lower in the CLP group than
that of control or sham surgery group (all P<0.001) and
dramatically elevated by insulin treatment at 24 h following CLP
(P<0.001).
Insulin on production of intrarenal
GSH
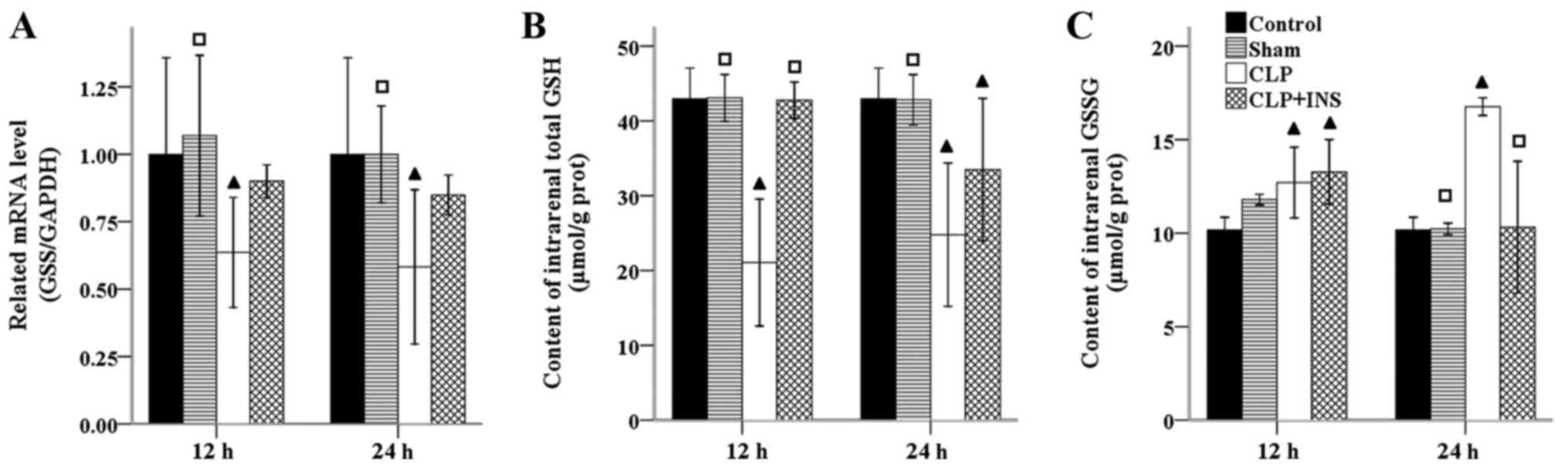
GSH is the most abundant antioxidant within
mitochondria. GSS is the second enzyme in GSH biosynthesis pathway.
As can be seen from Fig. 5, the mRNA
level of GSS was significantly decreased in the CLP group compared
with control or sham surgery group (all P<0.05). However, there
was no significant difference in the mRNA level of GSS between the
CLP group and CLP plus insulin group at the same time point.
Coupled with the transcriptional downregulation of GSS, the content
of total GSH within kidney decreased in the CLP group as compared
with control or sham surgery group (all P<0.001). Although the
content of total GSH in the CLP plus insulin group was higher than
that of CLP group at 12-h time point (P<0.001), but the two
groups had equal level at 24-h. In addition, the GSSH level was
significantly elevated in the CLP group compared with control or
sham surgery group at 24 h following CLP (both P<0.05) and
decreased dramatically in CLP plus insulin group (P<0.05).
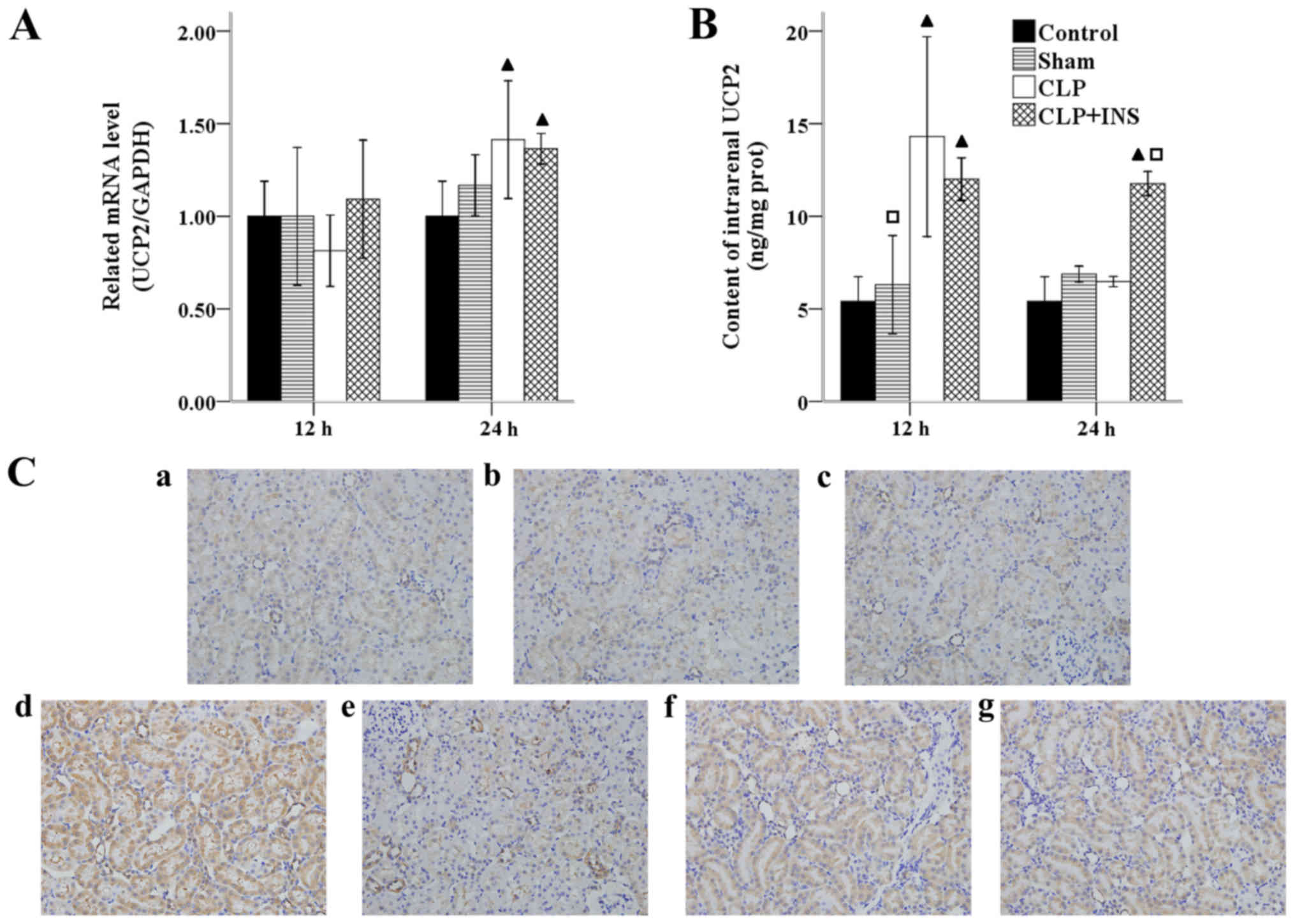
Insulin on expression of UCP2
UCPs, a mitochondrial anion carrier protein, have
been found to decrease ROS emission from mitochondria via induction
a proton leak across the inner membrane. UCP2 is the most wildly
expressed homologue of UCPs. As illustrated in Fig. 6A, the mRNA level of UCP2 increased in
the CLP group and CLP plus insulin group as compared with control
group at 24 h following CLP (both P<0.05). Fig. 6B shows a significant increase of UCP2
level in the CLP group at 12 h as well as in CLP plus insulin group
at 12 and 24 h following CLP compared with control or sham surgery
group (all P<0.01). Moreover, the UCP2 level in the CLP plus
insulin group was higher than that of CLP group at 24 h post CLP
(P<0.01). Similar changes of UCP2 expression were observed by
immunohistochemistry (Fig. 6C).
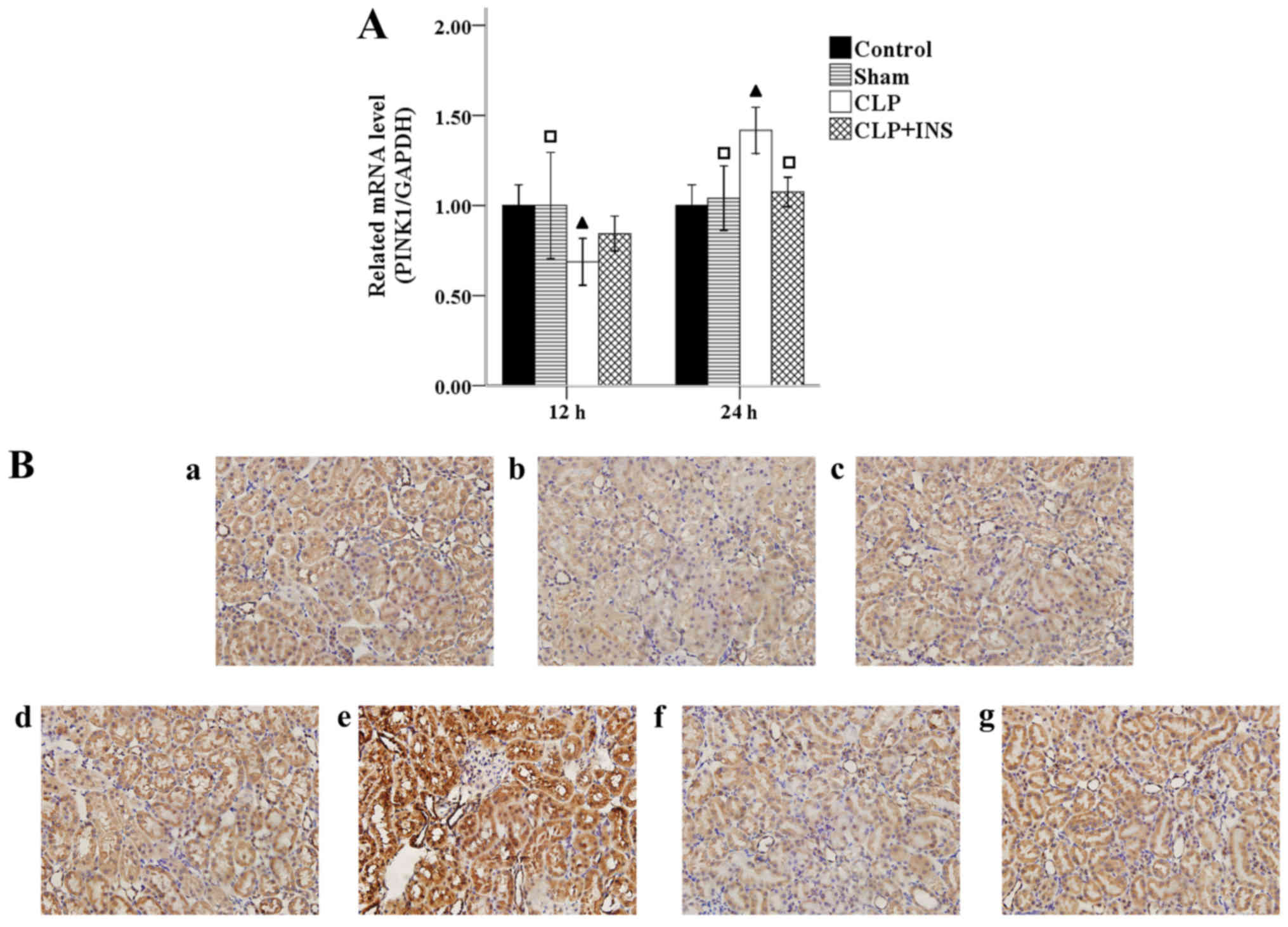
Insulin on expression of PINK1
protein
Accumulation of damaged mitochondria trigger
selective autophagy (mitophagy) to avoid more ROS formation.
Normally, mitophagy is mediated via PINK1-dependent pathway.
Fig. 7A shows that the mRNA level of
PINK1 upregulated in the CLP group compared with control or sham
surgery group at 24 h following CLP (both P<0.05), but CLP plus
insulin group had lower mRNA levels than CLP group at 24-h time
point (P<0.05). Similar changes of PINK1 were demonstrated by
immunohistochemistry (Fig. 7B).
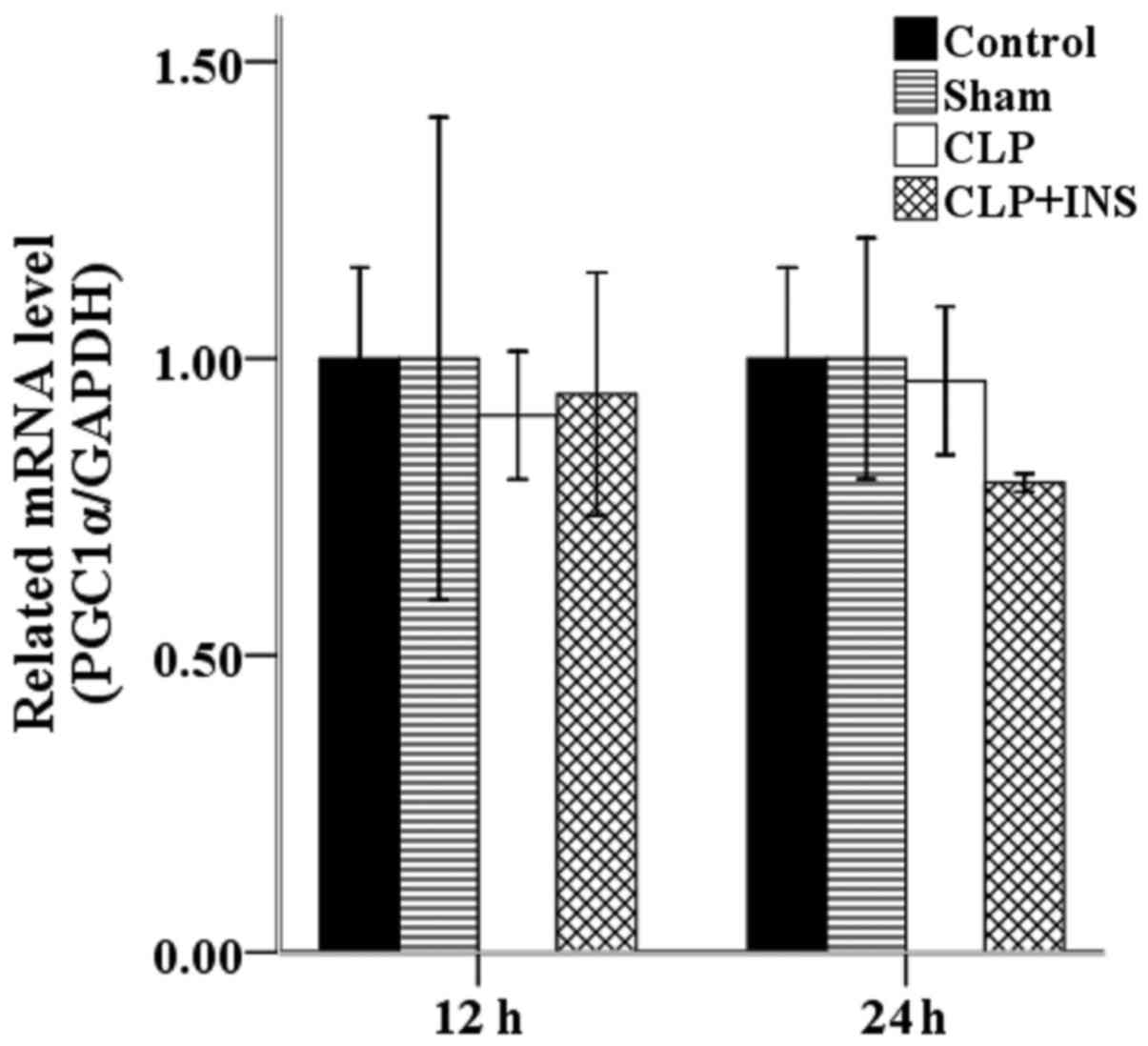
Insulin on the transcription of
PGC1α
Increased mitochondrial mass resulting from
biogenesis can improve endogenous antioxidant system and thereby
attenuate ROS production. PGC1α is a major regulator of
mitochondrial biogenesis. However, as shown in Fig. 8, there was no significant difference
in the mRNA level of PGC1α among the four groups.
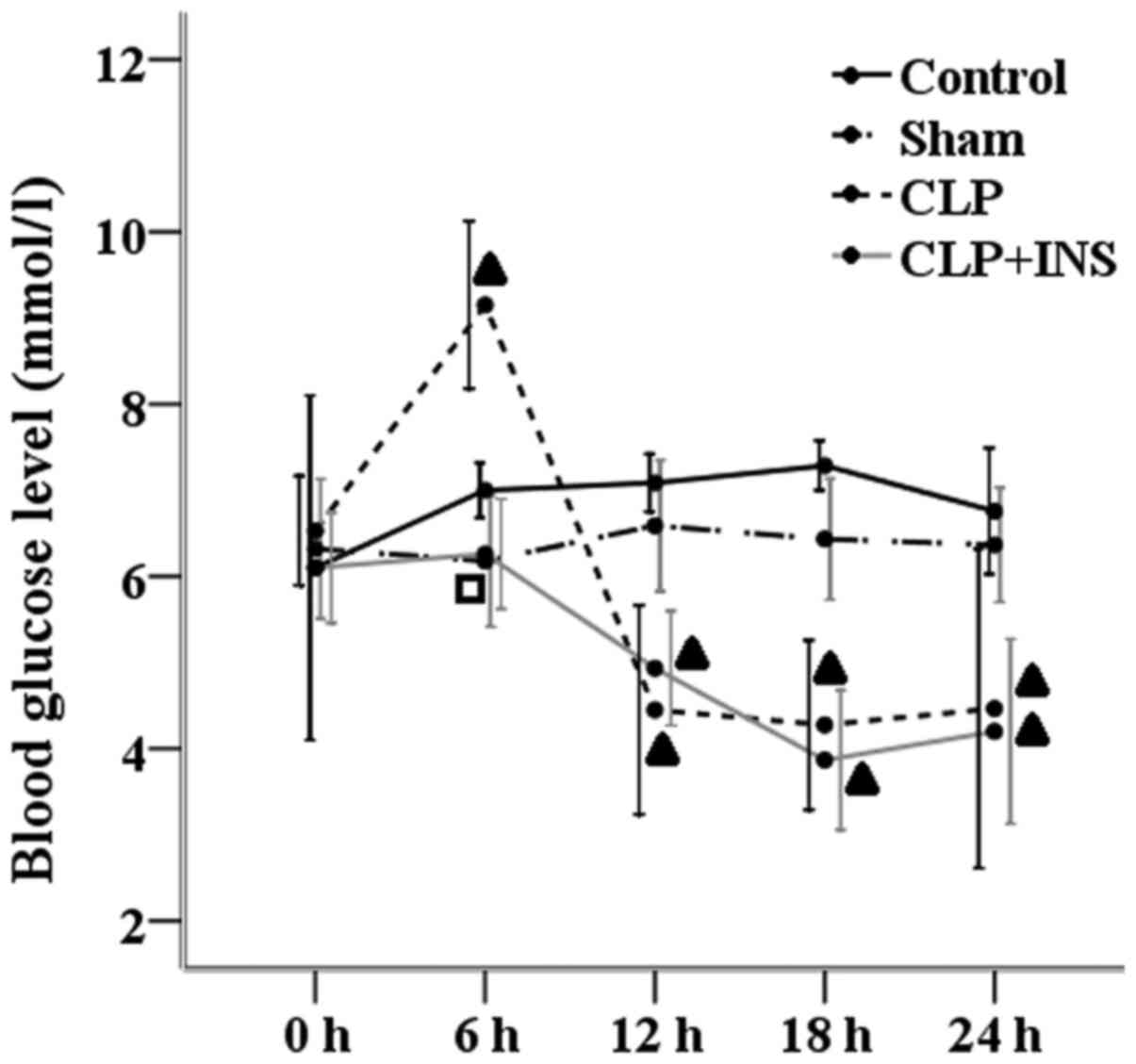
Blood glucose monitoring
As demonstrated in Fig.
9, compared with other three groups, blood glucose levels
increased in the CLP group at 6-h time point. But the CLP group and
CLP plus insulin group had equal blood glucose levels at 0, 12, 18
and 24 h following surgery and both lower than those of the control
group or sham surgery group (P<0.05).
Discussion
Development of organ dysfunction under septic
conditions is now accepted to result from oxidative damage to
mitochondria. The present study demonstrates for the first time
that insulin therapy ameliorates mitochondrial dysfunction via the
suppression of mitochondrial oxidative stress involving
upregulation of SOD2 and UCP2.
AKI is associated with a high morbidity and
mortality in septic patients and lack of sufficient therapy
(2,3). The present study demonstrates that
insulin treatment restored the elevations in BUN, serum CRE and
NGAL following CLP. Concurrently, the histopathological
alternations in renal sections were remarkably reduced in the CLP
plus insulin group as compared with CLP group. The results indicate
that a rat model of sepsis-induced AKI was successfully established
and insulin ameliorated septic AKI.
Mitochondria influence three key processes including
energy homeostasis, autophagy and cell cycle that could potentially
lead to apoptosis (19). Therefore,
mitochondrial dysfunction probably plays a prominent role in the
development of organ dysfunction during sepsis. To our knowledge,
there were no trials directly examined the protective effects of
insulin on mitochondrial dysfunction in septic AKI. The present
study demonstrates that insulin administration reversed the
decreased levels of MMP and ATP in renal sections following
CLP.
In a previous study, the group of Quoilin et
al (8) has already pointed out
the mechanism of ROS-induced ROS formation might be a main cause of
the mitochondrial dysfunction. They revealed that after being
targeted by oxidants, mitochondria became in turn producer of ROS,
thus contributing to aggravate the mitochondrial dysfunction.
Mitochondria is the main source and target of ROS within cells.
Mitochondria-targeted antioxidants such as MnTmPyP and Mito-TEMPO
have been shown to be effective in animal models of septic AKI
(9,10) but still not available in clinical
work. Insulin administration is a simple and inexpensive therapy
for disturbed glucose and lipid metabolism during sepsis and used
widely in ICU (12,13). Our study demonstrates for the first
time that insulin lowered the production of ROS and NO within
mitochondria following CLP; the deceased production of NO was due
to the downregulation of iNOS. ROS cannot cross the mitochondrial
membrane easily and may be more harmful than NO in oxidative damage
to mitochondria (20). Our results
are consistent with the observations that insulin has protective
effects against the inflammatory response and oxidative stress
under septic or other conditions via suppressing ROS generation and
NADPH oxidase, intranuclear nuclear factor-κB and other
inflammatory mediators (21–23).
However, the precise mechanisms of insulin on
mitochondrial oxidative stress remain to be fully elucidated.
Hyperglycemia is common in sepsis and can also induce inflammation,
oxidative stress and apoptosis in sepsis or AKI models (24,25).
Previous studies revealed that insulin therapy exerts the
protective effect against mitochondrial oxidative stress via
lowering blood glucose levels (26).
However, our results demonstrate that blood glucose level do not
differ in the CLP group and CLP plus insulin group except at 6-h
time point, and indicate that insulin has a direct effect on
mitochondrial oxidative stress rather than only an indirect via
glucose modulation. Our results are consistent with the
observations that insulin alleviates the inflammatory response and
oxidative stress in cerebral tissues under septic conditions which
was tested in a rat model (22).
Several antioxidant mechanisms such as endogenous antioxidant
systems, UCPs, mitophagy and mitochondrial biogenesis (15,16)
exist to protect against damage by ROS within mitochondria. There
may be some relationship between insulin therapy and antioxidant
mechanisms within mitochondria.
First of all, we investigated the modulation of
insulin on endogenous antioxidant systems. Our data shows that
insulin therapy upregulated the expression of SOD2 but not SOD1 in
a rat model of septic AKI. In agreement with our findings, Wiryana
reported that intensive insulin therapy can increase the SOD level
in the ICU critically ill patients compared to conventional insulin
therapy (27). ROS is usually
converted into H2O2 by SOD2 or SOD1 located
in the mitochondrial matrix and in the intermembrane space,
respectively (15). SOD2 is
generally thought to play a central role in the maintenance of ROS
levels because it scavenges ROS at the initial step of the radical
chain reaction. We propose that insulin treatment can decrease
overproduction of ROS and consumption of SOD as well as upregulate
the transcription of SOD2 hence leads to the increase of SOD
level.
GSH is the most abundant antioxidant in mitochondria
(28). The main removal of
H2O2 is through the oxidation of reduced
mitochondrial GSH into GSSH. In this study, we show that renal
total GSH content in the CLP plus insulin group was higher than
that of CLP group at 12 h following CLP, but there was no
significant difference between these two groups at 24 h. In a
previous study, the group of Maitra et al has already
demonstrated that rats experienced hyperglycemic phase followed by
euglycemic and hypoglycemic stages during 24 h post CLP (29). Furthermore, Yoneyama and colleagues
revealed that insulin therapy under hyperglycemia could enhance the
GSH content in the liver of septic rats (24). These evidences might explain why
insulin therapy failed to increase the level of GSH at 24 h post
CLP.
UCP2 is highly expressed in kidney and normally
activated directly by ROS to induce proton leak leading to membrane
depolarization and ROS reduction (15). Next, we examined the alteration of
UCP2 during insulin administration post CLP. To our knowledge, our
study reveals for the first time that insulin upregulated UCP2
during septic AKI. Additionally, the UCP2 level in the CLP group
was lower than that of CLP plus insulin group at 24-h time point
which may be due to severe damage of mitochondria in the CLP group.
Concurrently, we previously observed that silencing of UCP2 by
small interfering RNA aggravated mitochondrial dysfunction in
cardiomyocytes under septic conditions (30). Cao et al demonstrated higher
level of ROS and apoptosis in UCP2-knockout mice than those of wild
type (31). On the contrary, insulin
has a bioactive role in the anti-oxidative and anti-apoptotic
effects as well as protection of mitochondria under septic
conditions (21,32,33). All
of these evidences may explain why UCP2 is involved in insulin
ameliorating mitochondrial oxidative stress in sepsis.
In addition, our study reveals that insulin
administration suppressed renal mitophagy indicated by the
downregulation of PINK1 protein during sepsis. Our observation is
consistent with the study of Liu et al revealed that hepatic
mitophagy was suppressed in the presence of insulin resistance and
hyperinsulinemia induced by a high fat diet in mice (34). During various conditions, the
dissipation of potential across mitochondrial inner membrane leads
to the redirection of PINK1 upon import and then trigger the
process of mitophagy (20). We
propose that insulin suppression of mitophagy may be due to the
protection of mitochondrial ultrastructure. Furthermore, mitophagy
requires a fully functional autophagy system, which is under
stringent regulation by signaling pathways particularly mammalian
target of rapamycin (mTOR) (20). As
is well known, class I phosphatidylinositol-3-kinase (PI3K)-AKT
activates mTOR in response to insulin signaling, acting as a
negative regulator of autophagy (35). Thus, insulin itself has the property
of inhibiting autophagy.
Finally, mitochondrial biogenesis was investigated
by the mRNA levels of PGC1α a major regulator of mitochondrial
biogenesis and cellular metabolism. The fall in PGC1α expression
could arise as a consequence of AKI (10). Up to now, there are no trials
directly the modulation of insulin on mitochondrial biogenesis in
sepsis. And our data show that there was no significant difference
among the four group.
In conclusion, this study demonstrates that insulin
ameliorates mitochondrial oxidative stress involving upregulation
of SOD2 and UCP2 in septic AKI. However, SOD pathway is an
energetically costly mechanism to decrease ROS production because
it is reliant on ATP and NADPH which both are decreased in sepsis
(15). Furthermore, our data reveals
that insulin fails to increase GSH synthesis that may impair the
anti-oxidative effects of SOD. On the contrast, UCP2 was reported
to accumulate rapidly in the inner mitochondrial membrane during
mitochondrial reactive oxygen stress in macrophages (36). It seems that upregulation of UCP2
might be a main mechanism of insulin on mitochondrial oxidative
stress under septic conditions.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no.81272070).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
GDC conceived the study, participated in its design,
developed the study and drafted the manuscript. JLZ and YTC made
significant contributions during protocol development and helped
develop the study. JXZ and TW performed the sequencing results
analysis, and coordinated the acquisition and interpretation of
data. QYZ made significant contributions in the study design,
helped interprete the data and revise the manuscript, and was
responsible for the oversight of the present study.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Animal Care and Use Committee of Southern Medical University
(Guangzhou, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Abraham E: New definitions for sepsis and
septic shock: Continuing evolution but with much still to be done.
JAMA. 315:757–759. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bagshaw SM, George C and Bellomo R; ANZICS
Database Management Committee, : Early acute kidney injury and
sepsis: A multicentre evaluation. Crit Care. 12:R472008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schrier RW and Wang W: Acute renal failure
and sepsis. N Engl J Med. 351:159–169. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Galley HF: Bench-to-bedside review:
Targeting antioxidants to mitochondria in sepsis. Crit Care.
14:2302010.PubMed/NCBI
|
|
5
|
Yang RL, Wang XT, Liu DW and Liu SB:
Energy and oxygen metabolism disorder during septic acute kidney
injury. Kidney Blood Press Res. 39:240–251. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pathak E, Macmillan-Crow LA and Mayeux PR:
Role of mitochondrial oxidants in an in vitro model of
sepsis-induced renal injury. J Pharmacol Exp Ther. 340:192–201.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee SY, Lee YS, Choi HM, Ko YS, Lee HY, Jo
SK, Cho WY and Kim HK: Distinct pathophysiologic mechanisms of
septic acute kidney injury: Role of immune suppression and renal
tubular cell apoptosis in murine model of septic acute kidney
injury. Crit Care Med. 40:2997–3006. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Quoilin C, Mouithys-Mickalad A, Lècart S,
Fontaine-Aupart MP and Hoebeke M: Evidence of oxidative stress and
mitochondrial respiratory chain dysfunction in an in vitro model of
sepsis-induced kidney injury. Biochim Biophys Acta. 1837:1790–1800.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Patil NK, Parajuli N, MacMillan-Crow LA
and Mayeux PR: Inactivation of renal mitochondrial respiratory
complexes and manganese superoxide dismutase during sepsis:
Mitochondria-targeted antioxidant mitigates injury. Am J Physiol
Renal Physiol. 306:F734–F743. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Parikh SM, Yang Y, He L, Tang C, Zhan M
and Dong Z: Mitochondrial function and disturbances in the septic
kidney. Semin Nephrol. 35:108–119. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kastl L, Sauer SW, Ruppert T, Beissbarth
T, Becker MS, Suss D, Krammer PH and Gülow K: TNF-α mediates
mitochondrial uncoupling and enhances ROS-dependent cell migration
via NF-κB activation in liver cells. FEBS Lett. 588:175–183. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mesotten D, Swinnen JV, Vanderhoydonc F,
Wouters PJ and Van den Berghe G: Contribution of circulating lipids
to the improved outcome of critical illness by glycemic control
with intensive insulin therapy. J Clin Endocrinol Metab.
89:219–226. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Heuer JG, Sharma GR, Zhang T, Ding C,
Bailey DL, Stephens EJ, Holmes KC, Grubbs RL, Fynboe KA, Chen YF
and Jakubowski JA: Effects of hyperglycemia and insulin therapy on
outcome in a hyperglycemic septic model of critical illness. J
Trauma. 60:865–872. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeng QY, Zhang CM and Qian XH: Protective
effects of continue insulin infusion on liver mitochondrion in the
early stage of septic rats. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi.
25:525–528. 2009.(In Chinese). PubMed/NCBI
|
|
15
|
Mailloux RJ and Harper ME: Uncoupling
proteins and the control of mitochondrial reactive oxygen species
production. Free Radic Biol Med. 51:1106–1115. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mccreath G, Scullion MM, Lowes DA, Webster
NR and Galley HF: Pharmacological activation of endogenous
protective pathways against oxidative stress under conditions of
sepsis. Br J Anaesth. 116:131–139. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rittirsch D, Huber-Lang MS, Flierl MA and
Ward PA: Immunodesign of experimental sepsis by cecal ligation and
puncture. Nat Protoc. 4:31–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gomez H, Ince C, De Backer D, Pickkers P,
Payen D, Hotchkiss J and Kellum JA: A unified theory of
sepsis-induced acute kidney injury: Inflammation, microcirculatory
dysfunction, bioenergetics and the tubular cell adaptation to
injury. Shock. 41:3–11. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Raimundo N: Mitochondrial pathology:
Stress signals from the energy factory. Trends Mol Med. 20:282–292.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dandona P, Ghanim H, Bandyopadhyay A,
Korzeniewski K, Ling Sia C, Dhindsa S and Chaudhuri A: Insulin
suppresses endotoxin-induced oxidative, nitrosative, and
inflammatory stress in humans. Diabetes Care. 33:2416–2423. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Q, Yu W, Shi J, Shen J, Gao T, Zhang
J, Xi F, Li J and Li N: Insulin alleviates the inflammatory
response and oxidative stress injury in cerebral tissues in septic
rats. J Inflamm (Lond). 11:182014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dandona P, Aljada A, Mohanty P, Ghanim H,
Hamouda W, Assian E and Ahmad S: Insulin inhibits intranuclear
nuclear factor kappaB and stimulates IkappaB in mononuclear cells
in obese subjects: Evidence for an anti-inflammatory effect? J Clin
Endocrinol Metab. 86:3257–3265. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yoneyama S, Terashima H, Yamaguchi R,
Tadano S and Ohkohchi N: The manner of the inflammation-boosting
effect caused by acute hyperglycemia secondary to overfeeding and
the effects of insulin therapy in a rat model of sepsis. J Surg
Res. 185:380–387. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Efrati S, Berman S, Hamad RA, Siman-Tov Y,
Chanimov M and Weissgarten J: Hyperglycaemia emerging during
general anaesthesia induces rat acute kidney injury via impaired
microcirculation, augmented apoptosis and inhibited cell
proliferation. Nephrology (Carlton). 17:111–122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Verbruggen SC, Joosten KF, Castillo L and
van Goudoever JB: Insulin therapy in the pediatric intensive care
unit. Clin Nutr. 26:677–690. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wiryana M: The role of intensive insulin
therapy in increasing superoxide dismutase (SOD) and normalizing
hyperglycemia in critically ill patients. Acta Med Indones.
41:59–65. 2009.PubMed/NCBI
|
|
28
|
Galley HF: Oxidative stress and
mitochondrial dysfunction in sepsis. Br J Anaesth. 107:57–64. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maitra SR, Wojnar MM and Lang CH:
Alterations in tissue glucose uptake during the hyperglycemic and
hypoglycemic phases of sepsis. Shock. 13:379–385. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zheng G, Lyu J, Liu S, Huang J, Liu C,
Xiang D, Xie M and Zeng Q: Silencing of uncoupling protein 2 by
small interfering RNA aggravates mitochondrial dysfunction in
cardiomyocytes under septic conditions. Int J Mol Med.
35:1525–1536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cao T, Dong Y, Tang R, Chen J, Zhang CY
and Zen K: Mitochondrial uncoupling protein 2 protects splenocytes
from oxidative stress-induced apoptosis during pathogen activation.
Cell Immunol. 286:39–44. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Leffler M, Hrach T, Stuerzl M, Horch RE,
Herndon DN and Jeschke MG: Insulin attenuates apoptosis and exerts
anti-inflammatory effects in endotoxemic human macrophages. J Surg
Res. 143:398–406. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vanhorebeek I, De Vos R, Mesotten D,
Wouters PJ, De Wolf-Peeters C and Van den Berghe G: Protection of
hepatocyte mitochondrial ultrastructure and function by strict
blood glucose control with insulin in critically ill patients.
Lancet. 365:53–59. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi
J, Liu Z and Cao W: Hepatic autophagy is suppressed in the presence
of insulin resistance and hyperinsulinemia: Inhibition of
FoxO1-dependent expression of key autophagy genes by insulin. J
Biol Chem. 284:31484–31492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Giardina TM, Steer JH, Lo SZ and Joyce DA:
Uncoupling protein-2 accumulates rapidly in the inner mitochondrial
membrane during mitochondrial reactive oxygen stress in
macrophages. Biochim Biophys Acta. 1777:118–129. 2008. View Article : Google Scholar : PubMed/NCBI
|