Introduction
Ischemic preconditioning (IPC) is a well-established
adaptive response that briefly exposes cardiomyocytes to
iscemia/reperfusion and dramatically improves myocardial tolerance
of ischemic insult (1). During IPC,
adenosine, kappa-opioid receptor, K+ channels and Akt1
have been reported to be the responsible mechanisms (2–6), but
none of them seem to elucidate IPC-induced cardioprotection in a
convincing manner. On the other hand, connexin protein 43 (Cx43)
has been documented to be implicated in preconditioning (7,8) and may
serve as a potential end-effector of IPC-induced cardioprotection
(9). However, it remains unclear
which structure formed by Cx43 protein is involved in the
preconditioning.
Cx43, a member of the connexin family (10), has been found extensively in connexon
proteins of cardiac tissues (11,12). It
consists of four transmembrane regions, two extracellular loops,
and one intracellular loop. The amino- and carboxyl-termini are
located on the cytoplasmic side of the membrane (13,14). The
connexon of two adjacent cardiomyocytes constitutes a gap junction
between the myocardial cells (15).
However, for most of the time, the connexon of cardiomyocytes
remains separate and do not form a gap junction. Thus, hemichannels
are present in the non-junctional regions of individual cells.
These structural domains mediate the intracellular and
extracellular transport of small-molecule substances (16) and cell volume both in extracellular
ischemia and physiological isosmotic situations (17). Cardiomyocytes regulate the
hemichannel permeability by preventing water from entering the
cells and subsequent cell death in a hypotonic extracellular
environment, which is called volume regulation.
During ischemia, metabolic by-products of anaerobic
glycolysis accumulate in the cytoplasmic solute, which creates an
osmotic load in cardiomyocytes (15). During the cell death process,
hemichannels have been documented to serve as potential toxic pores
(18). We suspect that IPC may
provide myocardial protection by blocking the hemichannels, which
in turn mediates the cell volume of cardiomyocytes. Cx43 is present
not only in the inner and outer membranes of mitochondria, but also
in the inner and outer membranes of ventricular myocytes (19,20).
Many studies have investigated the mechanism of IPC with Cx43 in
mitochondria (21) and gap junction
on cell membrane (22,23). However, none of the research
completely explains the IPC-induced cardiomyocyte protection,
suggesting that there may be other mediators or terminal effectors
in the IPC. So this paper focuses on whether Cx43-formed
hemichannels on the cell membrane are involved in IPC and its
mechanism.
The current study employed a lentivirus with
Cx43-silencing shRNA and a hemichannel blocker, octanol or
18a-Glycyrrhizic acid (18a-GA) to block these channels so as to
investigate the mechanism involved in IPC-induced protection. The
study found that both treatments reduced the mortality of
cardiomyocytes.
Materials and methods
Experimental protocol
To investigate the effects of hemichannels on volume
regulation of cardiomyocytes, mouse cardiomyocytes (HL-1; Saiqi
Biological Engineering Company, Shanghai, China), cultured in
vitro under a hypotonic condition (at an osmotic pressure of
150 mOsm/l), were divided into 6 groups: Control,
Hypotonic+dimethylsulphoxide (DMSO) (10 µl; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany); Hypotonic+Octanol or 18a-GA groups,
respectively cultured with hemichannel blockers (octanol and
18a-GA, dissolved in DMSO, both at 100 µmol/l, 10 µl;
Sigma-Aldrich; Merck KGaA); Lentiviral vector, and Cx43-silenced
groups, respectively transfected with a scramble lentiviral vector
(MOI=100) and a lentivirus transfected with Cx43-silencing shRNA
(MOI=100). A normal control group was cultured with an isotonic
solution (at an osmotic pressure of 308 mOsm/l). After the
treatments, cardiomyocytes were collected to assess cell
morphological feature and volumetric (replace with area) changes at
0 min (the moment before the addition of hypotonic solution) and 30
min after the intervention.
Further, we evaluated the effects of
hemichannel-induced volume regulation on cardioprotection conferred
by ischemic preconditioning in vitro. Simulated
ischemia-reperfusion (SIR) was induced by a 7-h anoxia and a
subsequent 6-h reoxygenation and the cardiomyocytes were divided
into 6 groups: Control, SIR+DMSO, SIR+octanol, SIR+18a-GA,
SIR+scramble lentiviral vector, and SIR+Cx43-silenced groups;
Simulated ischemic preconditioning (SIP) was achieved by exposing
the cells to a 1-h anoxia and a 30-min reoxygentaion before a
subsequent 7-h anoxia and 6-h reoxygenation. The cardiomyocytes
received the same treatments as SIR groups and were divided into:
Control, SIP+DMSO, SIP+octanol, SIP+18a-GA, SIP+scramble lentiviral
vector, and SIP+Cx43-silenced groups. A normal control group was
cultured in an isotonic solution (at an osmotic pressure of 308
mOsm/l). The mortality of each group was averaged from 3 samplings
by trypan blue stain assay after the experiment.
Cell culture
Mouse cardiomyocytes were maintained in Dulbecco's
modified Eagle's medium (DMEM; HyClone; GE Healthcare Life
Sciences, Logan, UT, USA) containing 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 1%
penicillin and streptomycin at 37°C and in a moist atmosphere with
5% CO2. Cardiomyocytes in logarithmic growth phase were
seeded in 6-well plates (1×105 cells/well) and cultured
in DMEM for 10–12 h. Three fragments of Cx43-silencing shRNA
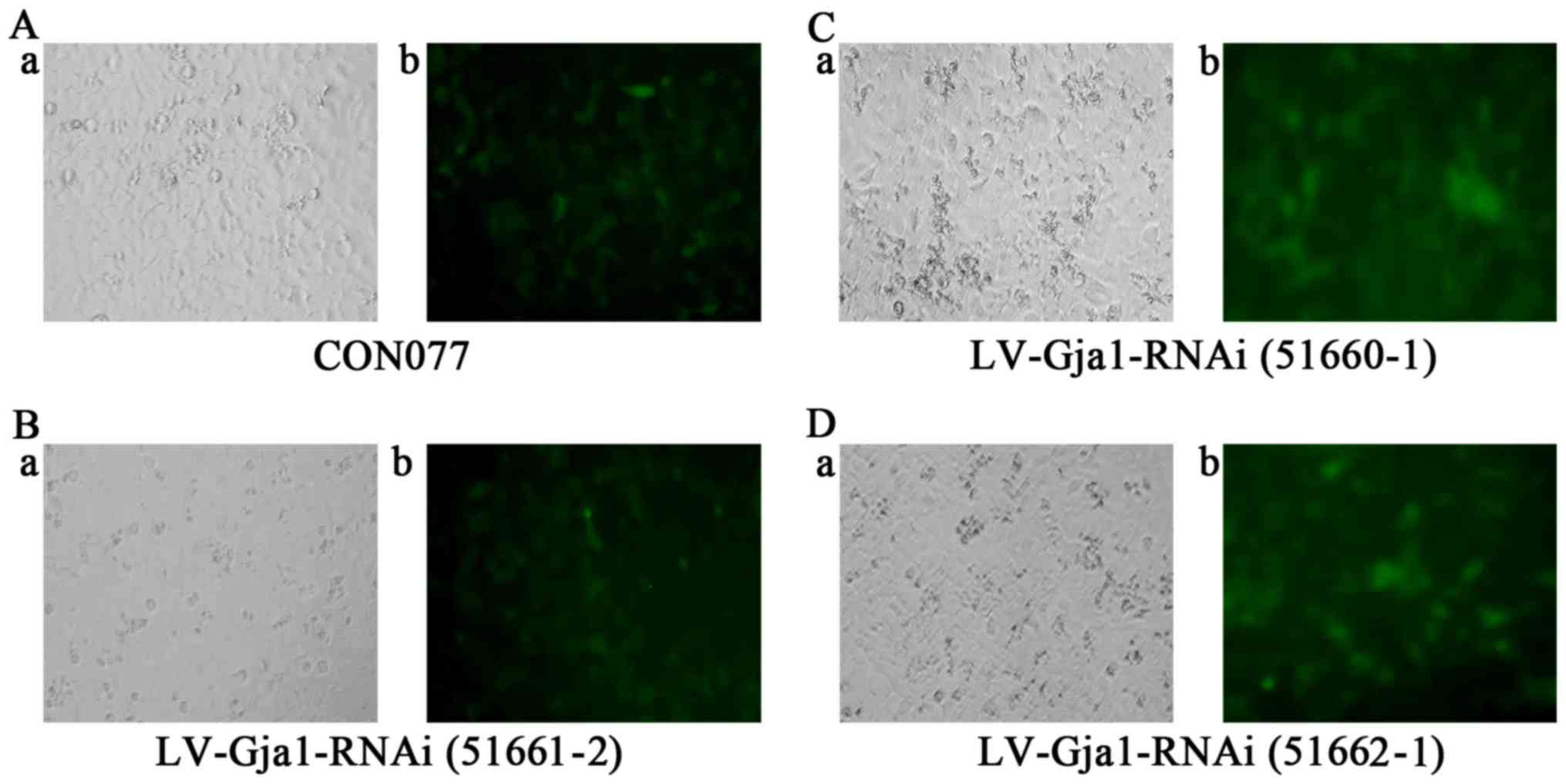
lentiviral vectors [LV-Gja1-RNAi (51660–1), LV-Gja1-RNAi (51661–2) and LV-Gja1-RNAi (51662–1)] and scramble lentiviral vector (CON077;
Gene Company Ltd., Shanghai, China) were then introduced (MOI=100,
for both). The transfection efficiency of the lentiviral vectors
was observed by inverted fluorescence microscopy (Olympus
Corporation, Tokyo, Japan) after puromycin (Sigma-Aldrich; Merck
KGaA) was added to filter successfully-transfected cells after 72
h. Cardiomyocytes with the highest transfection rate in the three
fragments were prepared for subsequent experiments after sampling
checking by western blotting and qPCR.
Cell volumetric regulation
To verify the role of hemichannels in volume
regulation, we simulated the extracellular hypotonic environment
after ischemia-reperfusion. Mouse cardiomyocytes in logarithmic
growth phase were seeded in 12-well plates (5×104
cells/well), including two wells of cardiomyocytes transfected with
scramble lentiviral vectors and two wells with Cx43-silenced
lentivirus (see cell culture, above). Hypotonic solution and
Octanol or 18a-GA were added to corresponding groups simultaneously
under normal ambient conditions. Cell morphological feature and
volumetric (replaced with area) change were observed and
photographed by inverted microscopy within 30 min after the
addition of the solution. The 20 best adherent cells in each group
were chosen to calculate and compare the changes of the areas (at 0
and 30 min after the solution adding) using Image-Pro Plus 6.0.
Mortality of cardiomyocytes
To evaluate the role of hemichannels in IPC, we
simulated the ischemic reperfusion (IR) and IPC conditions with the
cardiomyocytes. Briefly, the cells in the 12-well plates as
previously described were divided into 6 SIR and 6 SIP groups. They
were cultured in hypotonic solution (at an osmotic pressure of 150
mOsm/l), in which the anoxic condition was conducted in a hypoxic
device (containing 95% CO2 and 5% N2, at
37°C) and the reoxygenation condition in a normal cellular
incubator (containing 5% C02, at 37°C). Octanol or
18a-GA was administered into the matching groups just before the
experiment began. A normal control group was cultured in an
isotonic solution (at an osmotic pressure of 308 mOsm/l). The
mortality of each group was averaged from 3 samplings by trypan
blue stain assay after the experimental intervention.
Western blot
To assess the transfection efficiency of the
Cx43-silenced lentivirus, we determined the total protein and
activated (phosphorylated) protein of Cx43 by western blotting.
Normal and transfected cardiomyocytes were lysed in RIPA buffer
containing 1% protease inhibitor and phosphatase inhibitor
(Sigma-Aldrich; Merck KGaA). Protein concentrations of each sample
were determined by Quantitative nucleic acid-protein Analyzer
(Biomate 5; Thermo Fisher Scientific, Inc.). Equal load of proteins
was separated by 10% sodium dodecyl sulphate-polyacrylamide gel
electrophoresis (SDS-PAGE), transferred electrophoretically to
nitrocellulose membranes (Whatman International Ltd., Maidstone,
UK), and then rocked in Tris-buffered saline containing 0.1%
Tween-20 (TBST) and 5% milk at room temperature for 2 h. Membranes
were incubated at 4°C overnight with specific primary antibodies:
rabbit anti-Cx43 (1:1,000), rabbit anti-phospho-Cx43 (1:1,000; Cell
Signaling Technology, Inc., Danvers, MA, USA), and anti-GADPH
antibody (1:2,000; Zhongshan Golden Bridge Biotechnology Co., Ltd.,
Beijing, China). The membranes were further incubated in the
horseradish peroxidase-conjugated secondary antibodies (1:5,000)
(Zhongshan Golden Bridge Biotechnology Co., Ltd.) for 1 h at room
temperature after four rinses in TBST. The blots were visualized
with a chemiluminescent detection kit. The bands were quantified
using Image J software.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR analysis was essential to further confirm
the expression of Cx43. Total ribonucleic acids (RNAs) of normal
and transfected cardiomyocytes were isolated with TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.) and their
concentration and purity were quantified by spectrophotometry with
the software of Quantitative nucleic acid-protein Analyzer. Reverse
transcription and amplification were performed respectively with
the Eastep RT Master Mix and Eastep qPCR Master Mix (Promega
Corporation, Madison, WI, USA) by q PCR in the ABI PRISM 7500
Sequence Detection System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Amplification process was a three-step procedure
of 40 cycles, including denaturation at 95°C for 2 min, annealing
at 95°C for 15 sec, and extension at 60°C for 50 sec followed by a
melting-curve procedure of 1 min at 60°C. β-actin and the target
samples were performed in triplicate. 2−ΔΔCq method was
used for analysis. The primers were listed as follows: Cx43
forward, 5′-TTCATGCTGGTGGTGTCC-3′; reverse,
5′-TTGGCATTCTGGTTGTC-3′; β-actin forward,
5′-AGCGAGCATCCCCCAAAGTT-3′; reverse,
5′-GGGCACGAAGGCTCATCATT-3′.
Statistical analysis
Results were presented as mean ± standard deviation
(SD) and analyzed by Paired Student's t-test or One-way analysis of
variance (ANOVA) followed by a least significant difference (LSD)
for multiple comparisons. A value of P<0.05 indicated the
statistic significance. Data were processed by SPSS 19.0 software
(SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 5.0 software
(GraphPad Software, Inc., La Jolla, CA, USA).
Results
Transfection efficiency of
Cx43-silenced lentivirus
After the Cx43-silenced lentivirus was transfected
into the cardiomyocytes, green fluorescence was observed under an
inverted fluorescence microscope at 72 h after the transfection. A
rough estimate of the photoluminescent ratios of the four
lentiviral vectors was well over 80% (Fig. 1).
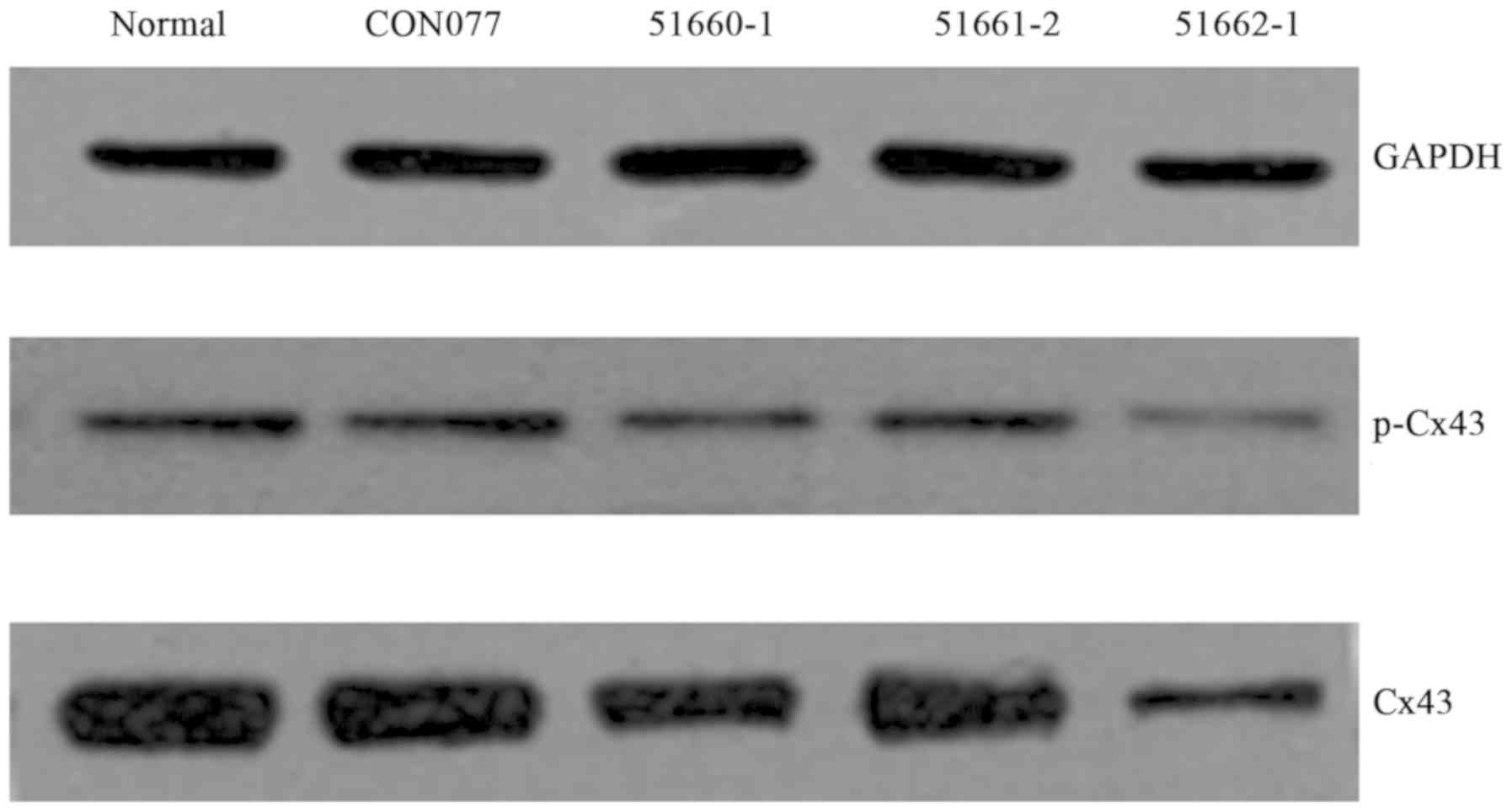
Western blot analysis was repeated 3 times. The
analysis revealed that abundant total protein and phosphorylated
protein of Cx43 were present in normal cardiomyocytes but both
decreased in the cells transfected with Cx43-silenced lentivirus,
especially in the LV-Gja1-RNAi (51662–1) group (Fig.
2). The transfection efficiency was validated again through
RT-qPCR. The RNA expression level coincided with the previous
results (Fig. 3). Compared with that
of the normal cardiomyocytes, the Cx43 expression in cells
transfected with Cx43-silenced lentivirus declined in the following
sequence: Lentiviral vectors LV-Gja1-RNAi (51661–2), LV-Gja1-RNAi (51660–1) and LV-Gja1-RNAi (51662–1) (P<0.05). Therefore, the LV-Gja1-RNAi
(51662–1) was used for subsequent
experiments.
Hemichannels-mediated volume
regulation
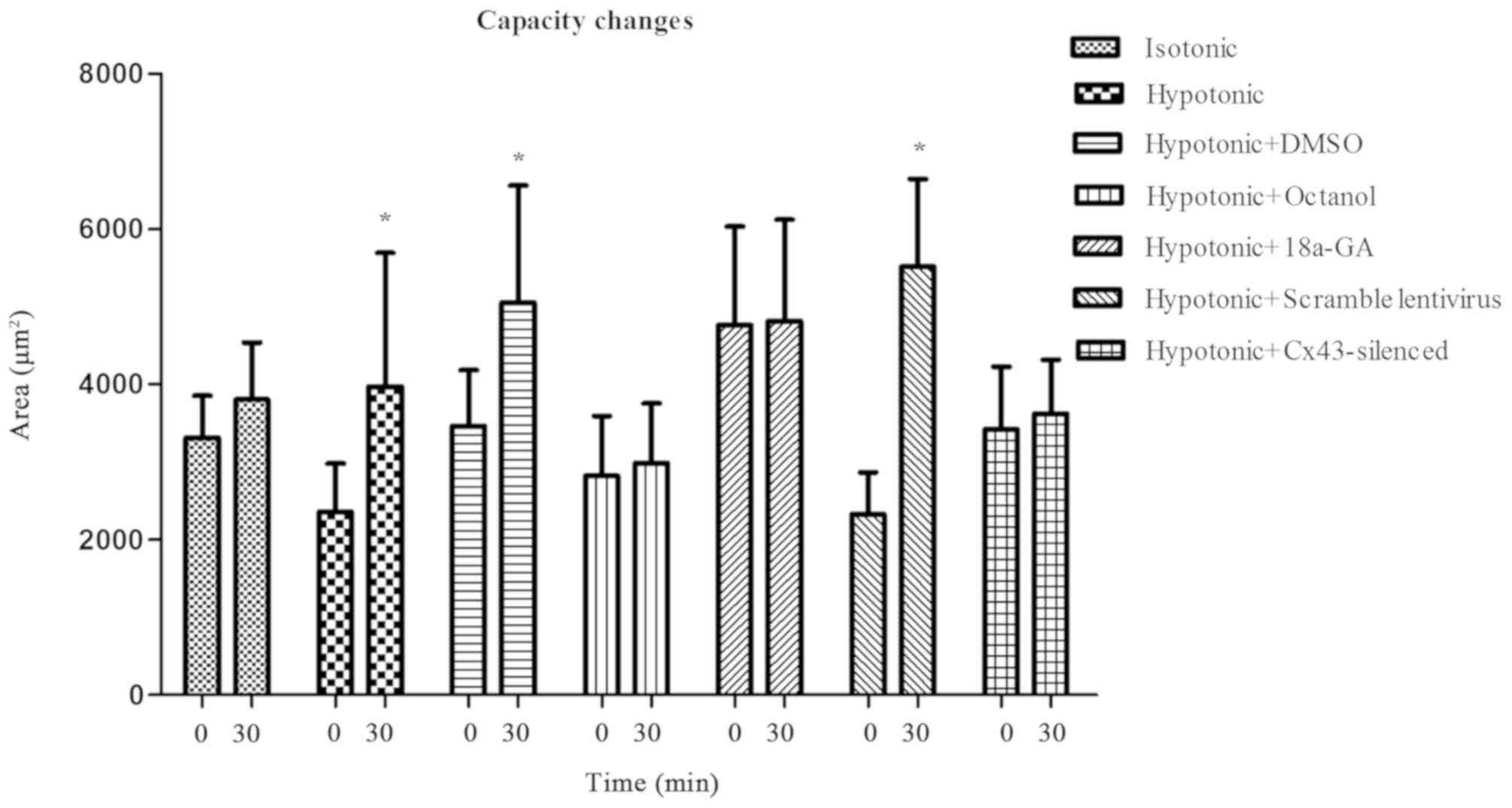
The morphological and volumetric changes of the
cells were determined at 30 min after the addition of hypotonic
solution. Within the 30 min after the solution addition, all cells
remained adherent and showed good viability. When a comparison was
made between the two time points (30 min after the solution
addition vs. 0 min), the cellular area in the hypotonic,
hypotonic+DMSO, hypotonic+scramble lentivirus group augmented
markedly (3,973±1,720 vs. 2,363±619 µm2, 5,050±1,511 vs.
3,464±723 µm2, 5,517±1,128 vs. 2,331±536 µm2,
P<0.05, respectively; on the contrary, the area of the remaining
four groups (Isotonic, hypotonic+octanol, hypotonic+18a-GA,
hypotonic+Cx43-silenced lentivirus) showed no conspicuous changes
(3,804±737 vs. 3,313±543 µm2, 2,990±765 vs. 2,821±773
µm2, 4,817±1,306 vs. 4,762±1,271 µm2,
3,627±688 vs. 3,419±814 µm2, P>0.05, respectively;
Fig. 4). These volumetric changes
indicate that cell edema can be substantially avoided by
obliterating the Cx43-formed hemichannels, demonstrating their
function of accommodating cell volume.
IPC-induced protection by blocking
hemichannels
In both SIR and SIP conditions, the normal control
group reported excellent cell viability and zero cell death but the
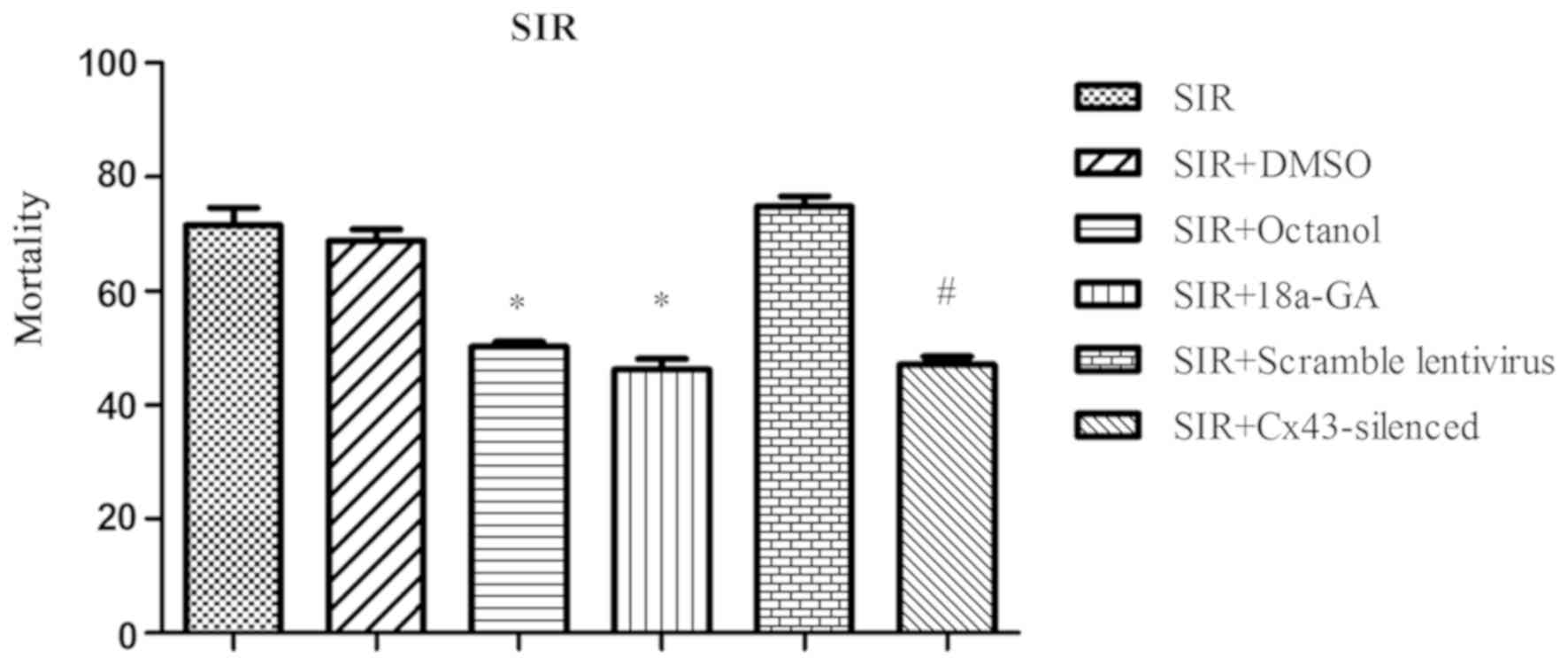
SIR and SIP groups displayed varied mortality rates: the SIR group
reported a mortality of up to 71.50±3.12%; both SIR+octanol and
SIR+18a-GA group showed a much lower death rate when compared with
the SIR+DMSO group (50.19±0.97, 46.20±1.93 vs. 68.83±1.93%,
P<0.05, respectively); similarly, the mortality of the
SIR+Cx43-silenced group decreased noticeably when compared with
that of SIR+scramble lentivirus group (47.17±1.41 vs. 74.78±1.88%,
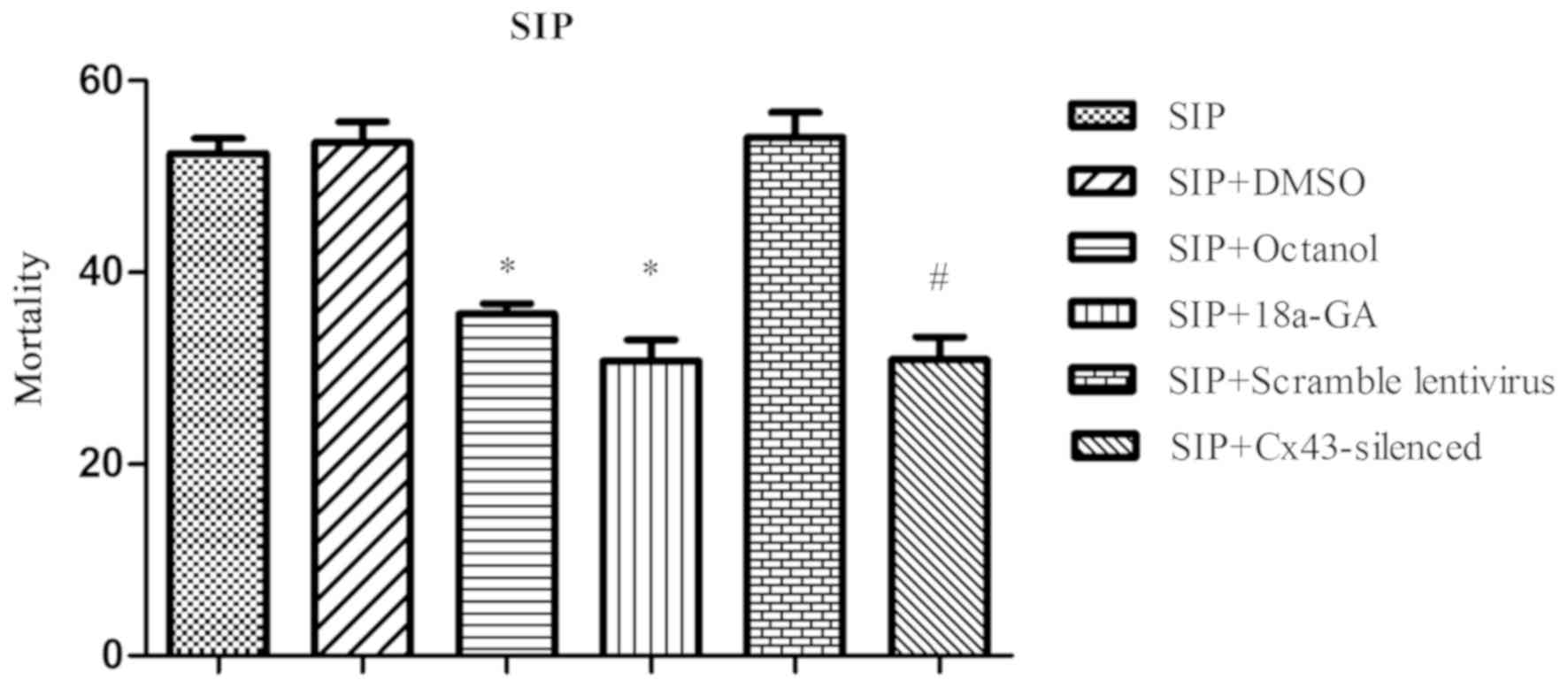
P<0.05; Fig. 5). Likewise, the
mortality of the SIP+octanol and SIP+18a-GA group declined
considerably when compared with that of the SIP+DMSO group
(35.70±1.02, 30.76±2.20 vs. 53.58±2.14)%, P<0.05, respectively);
that of SIP+Cx43-silenced group also decreased distinctly when
compared with the SIP+scramble lentivirus group (30.89±2.37 vs.
54.12±2.55%, P<0.05; Fig. 6).
Of note, the SIP group had a much lower mortality
rate when in comparison with the SIR group, suggesting the
successful modeling and the protective effect of IPC on
cardiomyocytes (52.44±1.53 vs. 71.50±3.12%, P<0.05]. Taken
together, these findings demonstrate that blocking the hemichannels
can further consolidate the IPC-induced cardiomyocyte
protection.
Discussion
The principal aim of the present study was to
investigate the role of the hemichannels of cardiomyocytes in the
protective effect of ischemic preconditioning on the
ischemic/reperfusion injury as a result of capacity regulation. In
this research, we demonstrated that cellular edema was deterred by
blocking hemichannels with blockers or by silencing Cx43 gene,
which apparently enhanced the role of IPC protection. Therefore,
these results suggest that the IPC-induced cardiomyocyte protection
may be mediated by hemichannels.
IPC has been reported as a myocardial protection, in
which after a brief, repeated, nonlethal ischemia/reperfusion, the
myocardial infarction area caused by the subsequent prolonged
ischemia decreased by 75% (1). This
IPC-induced protection is not complicated at all but a decisive
elucidation of its underlying mechanism remains such a challenge
that our understanding up to date is still modest in the available
literature. In the course of ischemia, intracellular osmotic
pressure increases when metabolites accumulate in cardiomyocytes
(24). This increased pressure
creates an osmotic gradient between the intracellular and
extracellular environment and, in turn, leads to cell distension
(25). In cardiomyocytes,
Cx43-formed hemichannels control the intra- and extra-cellular
transfer of water (17). Thus, Cx43
protein is definitely involved in the protective effect against
ischemia (preconditioning) (22).
Our results of SIR and SIP groups showed that the mortalities of
hemichannel-blocked groups decreased noticeably compared with those
of control groups, indicating that the hemichannels participate in
the death caused by ischemia, and that blocking the hemichannel can
reduce the cell mortality. Therefore, the current study confirms
that the involvement of Cx43 in the IPC-induced protection is
achieved by blocking the hemichannels.
Diaz et al speculated that the main
myocardial protective mechanism of IPC is cell volume regulation
(26). Their study focused on cell
volume regulation from the direction of chloride channels, which is
still controversial (27) and awaits
further verification. Naitoh et al supposed that IPC has
distinct effects on interaction of gap junction Cx43 with
PKCepsilon, p38MAPKalpha, and Src during ischemia (28). Miura et al presumed that
PKC-mediated Cx43 phosphorylation contributes to IPC-induced
protection (29). Such studies
ignore the fact that a hemichannel state is prevalent for most of
the cardiomyocytes. The present study probed into the
hemichannel-mediated volume regulation of cardiomyocytes in
hypotonic solution and demonstrated that the cell volume is
regulated by hemichannels, which echoes the findings of our
previous research (30). On the
other hand, Azzam et al claimed that an alleged death factor
after ischemia may spread from cell to cell through gap junctions
formed by Cx43 (31). However, the
hemichannel blocker, heptanol, functioning as gap junction
uncoupling (32), can restrain the
propagation of this factor-the spatial process of cell death
(33–35). Conversely, some research asserted
that cardioprotection of IPC was lost in heterozygous
Cx43-deficient mice (36),
indicating that IPC protection is independent of Cx43-formed gap
junctions or cell-to-cell communication. Unlike those previous
studies (36), which were conducted
in vivo and did not completely knock out the Cx43 gene, the
present study was implemented in vitro, with the Cx43 gene
utterly silenced.
Moreover, the current study found that the mortality
of groups with hemichannels blocked by chemicals and gene deletion
was distinctly lower than that of their corresponding counterparts
in SIR/SIP experiments, which directly addresses the role of volume
regulation in IPC. These findings correspondingly imply that cell
swelling and the loss of cell volume regulation play important
roles in ischemic injury in the myocardium, which is consistent
with the findings of other previous studies (37,38).
Similar research of gap junction ascertained that heptanol can
interfere with gap junction opening (32) and in turn reduces the final
myocardial infarct size during reoxygenation or reperfusion
(7), which lends substantial support
to our conclusion. On the basis of the present findings, we attempt
to propose for the first time that hemichannel-mediated cell volume
regulation is most likely involved in the IPC-induced cardiomyocyte
protection.
However, the inhibition of hemichannel transmission
is not the only mechanism of IPC-induced protection. The mortality
of cardiomyocytes was only partly decreased in SIP groups with
hemichannels blocked by chemicals or by silencing Cx43 gene. Hence,
other mechanisms, other than hemichannel-mediated capacity
regulation, may be involved in the IPC-induced protective effect
and have not been elucidated. An experiment conducted in isolated
cardiomyocytes verified that mitochondrial ROS was engaged in the
IPC protection (39). Another
research showed that during ischemia, IPC confers the
cardioprotection by the gap junction protein Cx43-mediated
signaling pathway of PKCε, p38MAPK, and Src (28). Further studies are still required to
understand other contributory mechanisms involved in the
IPC-induced protection.
There are few methods, other than microscopy, to
accurately determine the capacity of individual cells (40,41). In
this research, the thickness of cardiomyocytes, which were closely
adherent to the plate, was so thin that it was negligible. Thus,
the volume change was replaced with the area when cells were still
active. In the current study, before the addition of hypotonic
solution, the area of cardiomyocytes was calculated as the
baseline. At 30 min after the solution addition, obvious volumetric
change was evident while the cell viability and plate adherence
were maintained. Another technical difficulty in the current study
was to measure the volumetric transformation right at the incidence
of cell death. Nevertheless, the disparity in mortality rates is
sufficient to demonstrate the mechanism of IPC-induced
cardiomyoprotection.
Together, the present study comes up with an
explicit interpretation of how IPC protects myocardium from
ischemia injury. This finding provides a new perspective into
ischemic protection and has a great significance for the protection
of clinical patients with myocardial infarction.
In summary, our results demonstrate that Cx43-formed
hemichannels participate in the volume regulation of cardiomyocytes
and that swelling of cardiomyocytes can be alleviated by blocking
these hemichannels in a hypotonic environment. This
hemichannel-mediated volume regulation contributes to the
IPC-induced cardiomyoprotection.
Acknowledgements
Not applicable.
Funding
This study was primarily supported by the Youth
Foundation of Health Department of Fujian Province, China (grant
no. 2014-ZQN-JC-12), and partially supported by Program for New
Century Excellent Talents in Fujian Province University, China
(grant no. 2015B021), the Youth Foundation of Health Department of
Fujian Province, China (grant no. 2015-ZQN-ZD-12), the Joint Funds
for the Innovation of Science and Technology, Fujian Province
(grant no. 2017Y9007) and the National Natural Science Foundation
of China (grant nos. 81770302 and 81770362).
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JF, YL and WW designed the study. DZ and HL
performed the cell culture. JH, HC and TY participated in the cell
area data analysis. WW analyzed the data and drafted the
manuscript. All authors reviewed the manuscript and approved the
final version.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Murry CE, Jennings RB and Reimer KA:
Preconditioning with ischemia: A delay of lethal cell injury in
ischemic myocardium. Circulation. 74:1124–1136. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Das S, Cordis GA, Maulik N and Das DK:
Pharmacological preconditioning with resveratrol: Role of
CREB-dependent Bcl-2 signaling via adenosine A3 receptor
activation. Am J Physiol Heart Circ Physiol. 288:H328–H335. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Frassdorf J, Weber NC, Obal D, Toma O,
Müllenheim J, Kojda G, Preckel B and Schlack W: Morphine induces
late cardioprotection in rat hearts in vivo: The involvement of
opioid receptors and nuclear transcription factor kappaB. Anesth
Analg. 101:934–941. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu SN, Wu AZ and Sung RJ: Identification
of two types of ATP-sensitive K+ channels in rat ventricular
myocytes. Life Sci. 80:378–387. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kunuthur SP, Mocanu MM, Hemmings BA,
Hausenloy DJ and Yellon DM: The Akt1 isoform is an essential
mediator of ischaemic preconditioning. J Cell Mol Med.
16:1739–1749. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dragasis S, Bassiakou E, Iacovidou N,
Papadimitriou L, Andreas Steen P, Gulati A and Xanthos T: The role
of opioid receptor agonists in ischemic preconditioning. Eur J
Pharmacol. 720:401–408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li G, Whittaker P, Yao M, Kloner RA and
Przyklenk K: The gap junction uncoupler heptanol abrogates infarct
size reduction with preconditioning in mouse hearts. Cardiovasc
Pathol. 11:158–165. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schulz R, Boengler K, Totzeck A, Luo Y,
Garcia-Dorado D and Heusch G: Connexin 43 in ischemic pre- and
postconditioning. Heart Fail Rev. 12:261–266. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schulz R, Cohen MV, Behrends M, Downey JM
and Heusch G: Signal transduction of ischemic preconditioning.
Cardiovasc Res. 52:181–198. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Söhl G and Willecke K: Gap junctions and
the connexin protein family. Cardiovasc Res. 62:228–232. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Smyth JW and Shaw RM: Autoregulation of
connexin43 gap junction formation by internally translated
isoforms. Cell Rep. 5:611–618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang SS, Hong S, Kléber AG, Lee LP and
Shaw RM: A micropatterning approach for imaging dynamic Cx43
trafficking to cell-cell borders. FEBS Lett. 588:1439–1445. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kanter HL, Saffitz JE and Beyer EC:
Molecular cloning of two human cardiac gap junction proteins,
connexin40 and connexin45. J Mol Cell Cardiol. 26:861–868. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lau AF, Hatch-Pigott V and Crow DS:
Evidence that heart connexin43 is a phosphoprotein. J Mol Cell
Cardiol. 23:659–663. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dhein S: Pharmacology of gap junctions in
the cardiovascular system. Cardiovasc Res. 62:287–298. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Veenstra RD, Wang HZ, Beblo DA, Chilton
MG, Harris AL, Beyer EC and Brink PR: Selectivity of
connexin-specific gap junctions does not correlate with channel
conductance. Circ Res. 77:1156–1165. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Quist AP, Rhee SK, Lin H and Lal R:
Physiological role of gap-junctional hemichannels. Extracellular
calcium-dependent isosmotic volume regulation. J Cell Biol.
148:1063–1074. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Decrock E, Vinken M, De Vuyst E, Krysko
DV, D'Herde K, Vanhaecke T, Vandenabeele P, Rogiers V and Leybaert
L: Connexin-related signaling in cell death: To live or let die?
Cell Death Differ. 16:524–536. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boengler K, Dodoni G, Rodriguez-Sinovas A,
Cabestrero A, Ruiz-Meana M, Gres P, Konietzka I, Lopez-Iglesias C,
Garcia-Dorado D, Di Lisa F, et al: Connexin 43 in cardiomyocyte
mitochondria and its increase by ischemic preconditioning.
Cardiovasc Res. 67:234–244. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goubaeva F, Mikami M, Giardina S, Ding B,
Abe J and Yang J: Cardiac mitochondrial connexin 43 regulates
apoptosis. Biochem Biophys Res Commun. 352:97–103. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ruiz-Meana M, Núñez E, Miro-Casas E,
Martínez-Acedo P, Barba I, Rodriguez-Sinovas A, Inserte J,
Fernandez-Sanz C, Hernando V, Vázquez J and Garcia-Dorado D:
Ischemic preconditioning protects cardiomyocyte mitochondria
through mechanisms independent of cytosol. J Mol Cell Cardiol.
68:79–88. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Garcia-Dorado D, Ruiz-Meana M, Padilla F,
Rodriguez-Sinovas A and Mirabet M: Gap junction-mediated
intercellular communication in ischemic preconditioning. Cardiovasc
Res. 55:456–465. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rong B, Xie F, Sun T, Hao L, Lin MJ and
Zhong JQ: Nitric oxide, PKC-ε, and connexin43 are crucial for
ischemic preconditioning-induced chemical gap junction uncoupling.
Oncotarget. 7:69243–69255. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fernández-Jiménez R, García-Prieto J,
Sánchez-González J, Agüero J, López-Martín GJ, Galán-Arriola C,
Molina-Iracheta A, Doohan R, Fuster V and Ibáñez B: Pathophysiology
underlying the bimodal edema phenomenon after myocardial
ischemia/reperfusion. J Am Coll Cardiol. 66:816–828. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Holmes JW, Borg TK and Covell JW:
Structure and mechanics of healing myocardial infarcts. Annu Rev
Biomed Eng. 7:223–253. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Diaz RJ, Armstrong SC, Batthish M, Backx
PH, Ganote CE and Wilson GJ: Enhanced cell volume regulation: A key
protective mechanism of ischemic preconditioning in rabbit
ventricular myocytes. J Mol Cell Cardiol. 35:45–58. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Heusch G, Cohen MV and Downey JM: Ischemic
preconditioning through opening of swelling-activated chloride
channels? Circ Res. 89:E482001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Naitoh K, Yano T, Miura T, Itoh T, Miki T,
Tanno M, Sato T, Hotta H, Terashima Y and Shimamoto K: Roles of
Cx43-associated protein kinases in suppression of gap
junction-mediated chemical coupling by ischemic preconditioning. Am
J Physiol Heart Circ Physiol. 296:H396–H403. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miura T, Ohnuma Y, Kuno A, Tanno M,
Ichikawa Y, Nakamura Y, Yano T, Miki T, Sakamoto J and Shimamoto K:
Protective role of gap junctions in preconditioning against
myocardial infarction. Am J Physiol Heart Circ Physiol.
286:H214–H221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luo Y, Fang J, Fan L, Lin C, Chen Z and
Chen L: Octanol preconditioning alleviates mouse cardiomyocyte
swelling induced by simulated ischemia/reperfusion challenge in
vitro. Nan Fang Yi Ke Da Xue Xue Bao. 32:1419–1422. 2012.(In
Chinese). PubMed/NCBI
|
|
31
|
Azzam EI, de Toledo SM and Little JB:
Direct evidence for the participation of gap junction-mediated
intercellular communication in the transmission of damage signals
from alpha-particle irradiated to nonirradiated cells. Proc Natl
Acad Sci USA. 98:473–478. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garcia-Dorado D, Inserte J, Ruiz-Meana M,
González MA, Solares J, Juliá M, Barrabés JA and Soler-Soler J: Gap
junction uncoupler heptanol prevents cell-to-cell progression of
hypercontracture and limits necrosis during myocardial reperfusion.
Circulation. 96:3579–3586. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Garcia-Dorado D, Rodriguez-Sinovas A and
Ruiz-Meana M: Gap junction-mediated spread of cell injury and death
during myocardial ischemia-reperfusion. Cardiovasc Res. 61:386–401.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen BP, Mao HJ, Fan FY, Bruce IC and Xia
Q: Delayed uncoupling contributes to the protective effect of
heptanol against ischaemia in the rat isolated heart. Clin Exp
Pharmacol Physiol. 32:655–662. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rodriguez-Sinovas A, Garcia-Dorado D,
Ruiz-Meana M and Soler-Soler J: Protective effect of gap junction
uncouplers given during hypoxia against reoxygenation injury in
isolated rat hearts. Am J Physiol Heart Circ Physiol.
290:H648–H656. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schwanke U, Konietzka I, Duschin A, Li X,
Schulz R and Heusch G: No ischemic preconditioning in heterozygous
connexin43-deficient mice. Am J Physiol Heart Circ Physiol.
283:H1740–H1742. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Diaz RJ, Harvey K, Boloorchi A, Hossain T,
Hinek A, Backx PH and Wilson GJ: Enhanced cell volume regulation: A
key mechanism in local and remote ischemic preconditioning. Am J
Physiol Cell Physiol. 306:C1191–C1199. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kloner RA: New observations regarding
post-ischemia/reperfusion myocardial swelling. J Am Coll Cardiol.
65:324–326. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Oldenburg O, Qin Q, Krieg T, Yang XM,
Philipp S, Critz SD, Cohen MV and Downey JM: Bradykinin induces
mitochondrial ROS generation via NO, cGMP, PKG, and mitoKATP
channel opening and leads to cardioprotection. Am J Physiol Heart
Circ Physiol. 286:H468–H476. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Korchev YE, Gorelik J, Lab MJ, Sviderskaya
EV, Johnston CL, Coombes CR, Vodyanoy I and Edwards CR: Cell volume
measurement using scanning ion conductance microscopy. Biophys J.
78:451–457. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Curl CL, Bellair CJ, Harris PJ, Allman BE,
Roberts A, Nugent KA and Delbridge LM: Single cell volume
measurement by quantitative phase microscopy (QPM): A case study of
erythrocyte morphology. Cell Physiol Biochem. 17:193–200. 2006.
View Article : Google Scholar : PubMed/NCBI
|