Introduction
Kawasaki disease (KD) is an acute and systemic
autoimmune vasculitis with unknown etiology, primarily occurring in
pediatric patients aged <5 years (1,2). It is
well known as the leading cause of acquired heart disease in
children in developed countries (3).
Although the etiology remains elusive, KD contributes to systemic
inflammation and has a clear preference for coronary arteries.
The histological manifestations of KD-associated
coronary arteritis are inflammatory cell infiltration with
breakdown of the extracellular matrix, particularly of the elastic
tissue in the vascular media, resulting in coronary artery aneurysm
(CAA) formation (4). Mortality in KD
is usually caused by ischemic cardiomyopathy (5).
Limited knowledge of the etiologic factors and cell
molecular pathology of vasculitis has hampered the discovery of
more effective KD treatments or therapies (6–8). At
present, single-dose intravenous γ globulin (IVIG) is used to
effectively reduce the prevalence of CAAs and is the preferred
treatment for preventing coronary lesions in pediatric patients
with KD (9), but an estimated 10–20%
of patients are not sensitive to this treatment which results in
poor prognosis (3). The optimum
treatment for IVIG non-responders remains inconclusive, and drugs
for secondary or ‘rescue’ treatment vary by center. Thus, it is
important to explore the pathogenesis of KD and identify
alternative treatments.
Soluble epoxide hydrolase inhibitors (sEHi) protect
the cardiovascular system in multiple ways (10–12),
e.g., by inhibiting the deactivation of epoxyeicosatrienoic acids
(EETs) and at times by sEH-mediated effects on
inflammation-associated diols (13,14).
EETs are essential for maintaining the normal function of an
organism and may cause significant vasodilation, alleviate
inflammation, inhibit the migration of vascular smooth muscle cells
and platelet aggregation, promote fibrinolysis and reduce adhesion
factor expression (15–17). Furthermore, it has been reported that
EETs are involved in vascular repair by inducing angiogenesis
(18). Inhibition of sEH increases
the positive roles of EETs in atherosclerosis, hypertension,
myocardial hypertrophy, ischemic heart disease, diabetes-associated
heart failure and metabolic syndrome in vivo (19–24).
However, it remains elusive whether sEHi have any
therapeutic effect in KD. Therefore, the present study aimed to
determine whether the sEHi 12-(3-adamantan-1-yl-ureido)-dodecanoic
acid (AUDA) promotes the vascular repair of human coronary arterial
endothelial cells (HCAECs) and reduces inflammation in the coronary
artery in a KD mouse model induced by Lactobacillus casei
cell wall extract (LCWE). The present study further sought to
reveal the role of the EET/peroxisome proliferator activated
receptor γ (PPARγ) pathway in the effect of AUDA on HCAECs and the
mouse model of KD. The results suggest a potential role of AUDA in
promoting the vascular repair of HCAECs and in alleviating the
inflammatory response in KD.
Materials and methods
Cell culture and treatments
HCAECs were obtained from the Wuhan Culture
Collection and maintained in endothelial culture medium (ECM) with
5% fetal bovine serum (FBS), 1% penicillin/streptomycin solution
and 1% endothelial cell growth supplement (all from ScienCell
Research Laboratories, Inc., San Diego, CA, USA) at 37°C in 5%
CO2 in air. HCAECs were treated with different
concentrations of AUDA (0, 1, 10, 50 or 100 µmol/l) for 24 h.
To further investigate the role of the PPARγ pathway
in the role of AUDA in HCAECs, the PPARγ antagonist GW9662 was
used. HCAECs were cultured with GW9662 (5 µmol/l) for 30 min,
followed by the addition of 100 µmol/l AUDA.
Cell migration assay
For migration assays, 24-well Transwell plates with
8-µm pore size and 6.5 mm-diameter polycarbonate filters (Costar;
Corning Incorporated, Corning, NY, USA) were used. HCAECs (100 µl)
were resuspended in serum-free ECM at a density of 1×105
cells/ml and 100 µl was seeded onto the upper chamber, while ECM
supplemented with 5% FBS was added to the lower chamber. Following
24-h culture, the migrated cells were fixed with 4%
paraformaldehyde for 20 min, washed with PBS and stained with 100
µl 0.1% crystal violet for 30 min. Quantitative analysis of
migrated cells was performed. Cells in 10 randomly selected fields
per well were observed and counted under a phase-contrast
microscope (magnification, ×100; Olympus BH2; Olympus, Tokyo,
Japan). Experiments were performed in triplicate.
Cell adhesion assay
At 90% confluence, HCAECs were seeded into 96-well
culture plates coated in fibronectin (BD Biosciences, San Jose, CA,
USA) at density of 1×104 cells/well and cultured for 1 h
at 37°C. Following incubation, non-adherent cells were washed with
PBS three times, followed by fixation with 4% paraformaldehyde for
20 min, and staining with 100 µl 0.1% crystal violet for 30 min.
Adherent cells in 10 randomly selected fields per well were
observed and counted under a phase-contrast microscope
(magnification, ×100; Olympus BH2; Olympus). Experiments were
performed in triplicate.
Capillary-like tube formation
assay
Matrigel® (BD Biosciences) was thawed on
ice overnight and once thawed, 50 µl was added to each well of a
96-well plate and incubated for 1 h at 37°C to solidify. HCAECs
were seeded into 96-well plates pre-coated with
Matrigel® at a density of 1×104 cells/well
and incubated at 37°C for 6 h. Images of tube formation were
captured using an inverted light microscope (magnification, ×100;
Olympus BH2; Olympus). Segment lengths were measured using ImageJ
software (version 1.44p; National Institutes of Health, Bethesda,
MD, USA) in 5 randomly selected fields per well and the average
segment length per field was calculated.
Cell proliferation assay
Cell counting kit-8 (CCK-8; Beyotime Institute of
Biotechnology, Haimen, China) was used to determine the
proliferation of HCAECs following the manufacturer's protocol. The
CCK-8 assay utilizes the yellow formazan dye produced following the
reduction of tetrazolium salt WST-8 by the mitochondria of live
cells to determine cell activity.
In brief, 2×103 cells in 100 µl medium
were added to each well of a 96-well plate and cultured overnight.
Cells were subsequently treated with various concentrations (0, 1,
10, 50 or 100 µmol/l) of AUDA for 24 h. Following incubation, 10 µl
CCK-8 solution was added and cells were incubated at 37°C for a
further 4 h. The optical density was measured at 490 mm using a
microplate reader (Bio-Rad Laboratories, Hercules, CA, USA).
LCWE
LCWE was prepared as previously described (25). The concentration of LCWE (in PBS) was
measured by analyzing the rhamnose content determined via a
phenol-sulfuric acid colorimetric assay.
Mice
In total 20, male (wild-type) C57BL/6 mice (age, 4–6
weeks; weight, 18–20 g) were obtained from the Animal Centre of
Shandong Medical University (Shandong, China). All mice were
maintained under specific pathogen-free conditions (20–26°C, 40–70%
humidity, 12-h light/dark cycle with access to full-valence
granular rat feedstuff and sterile water ad libitum). Mice
were randomly divided into four groups: PBS, LCWE, LCWE+AUDA and
LCWE+AUDA+GW9662. In each group, mice were injected
intraperitoneally with 0.5 ml PBS alone; PBS supplemented with 0.5
mg LCWE; PBS supplemented with 0.5 mg LCWE and 10 mg/kg AUDA
(Cayman Chemical, Wuhan, China); or PBS supplemented with 0.5 mg
LCWE, 10 mg/kg AUDA and 10 mg/kg GW9662 (MedchemExpress, Shanghai,
China), respectively. Following 14-day induction, mice were
sacrificed.
ELISA
The supernatant of HCAECs and total protein of
murine hearts were used for the detection of matrix
metallopeptidase (MMP)-9, interleukin (IL)-1β and tumor necrosis
factor (TNF)-α by means of ELISA. Lysis buffer (Lichen, Shanghai,
China) was used for the homogenization of murine hearts, and total
protein was extracted following the manufacturer's instructions.
Protein levels of MMP-9, IL-1β and TNF-α were examined using ELISA
kits (MMP-9, cat. no. TY02784B; IL-1β, cat. no., lc-005; TNF-α,
cat. no. lc-007; all Yingxin Laboratory Equipment Co., Ltd.,
Shanghai, China).
Statistical analysis
Prism 5.0 (GraphPad Software, Inc., La Jolla, CA,
USA) was used for data analysis. Data are expressed as the mean ±
standard error. The significance of differences among several
groups was determined using one-way analysis of variance with
Bonferroni correction. P<0.05 was considered to indicate a
statistically significant difference.
Results
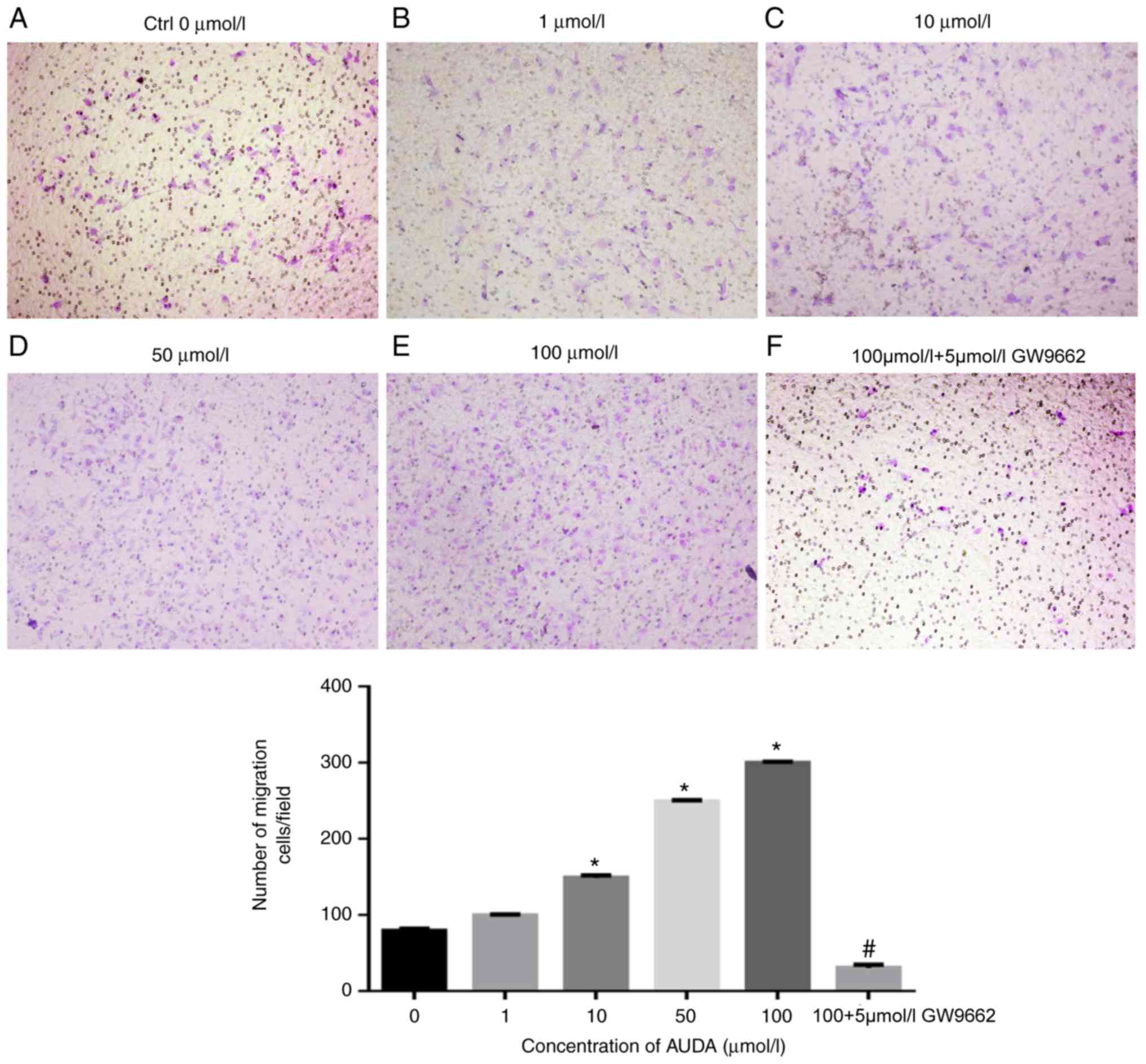
Effect of AUDA on the migration of
HCAECs
As presented in Fig.
1, AUDA augmented the migratory ability of HCAECs in a
dose-dependent manner. Compared with that in the control group (0
µmol/l AUDA), treatment with 10, 50 and 100 µmol/l AUDA resulted in
a significant increase in HCAEC migration (P<0.05). To further
investigate the effect of AUDA on HCAECs, the PPARγ antagonist
GW9662 was used to examine the PPARγ signaling pathway. Following
pre-treatment with GW9662 (5 µmol/l), the AUDA-induced increase in
HCAEC migration was significantly suppressed (P<0.05).
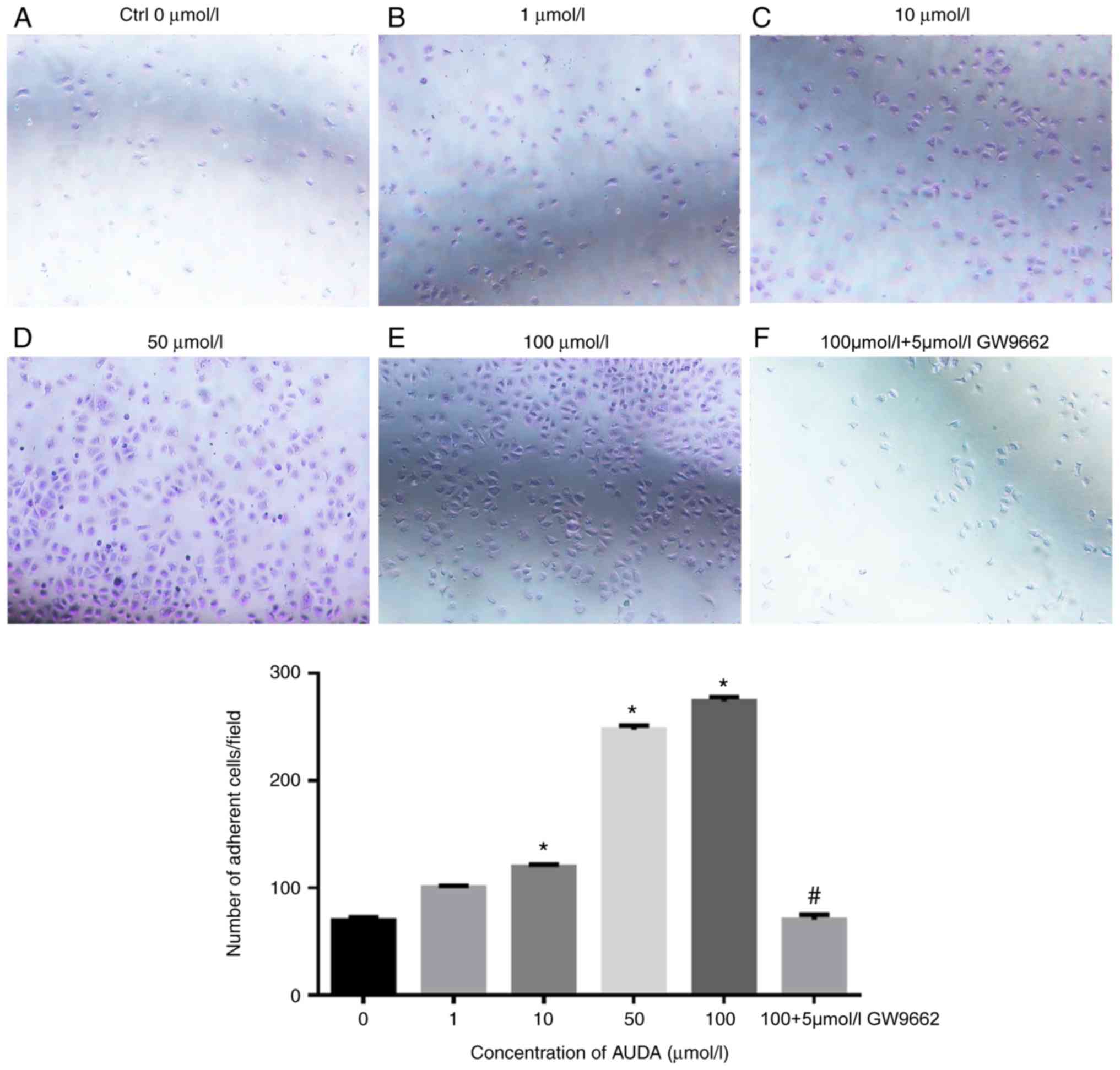
Effect of AUDA on the adhesion of
HCAECs
Next, the effect of AUDA on the adhesion of HCAECs
was evaluated, revealing that AUDA increased cell adhesion in a
dose-dependent manner. Compared with that in the control group (0
µmol/l AUDA), 10, 50 and 100 µmol/l AUDA significantly increased
the adhesion ability of HCAECs (P<0.05). However, cell adhesion
was severely impaired by pre-treatment with GW9662 followed by AUDA
(P<0.05; Fig. 2).
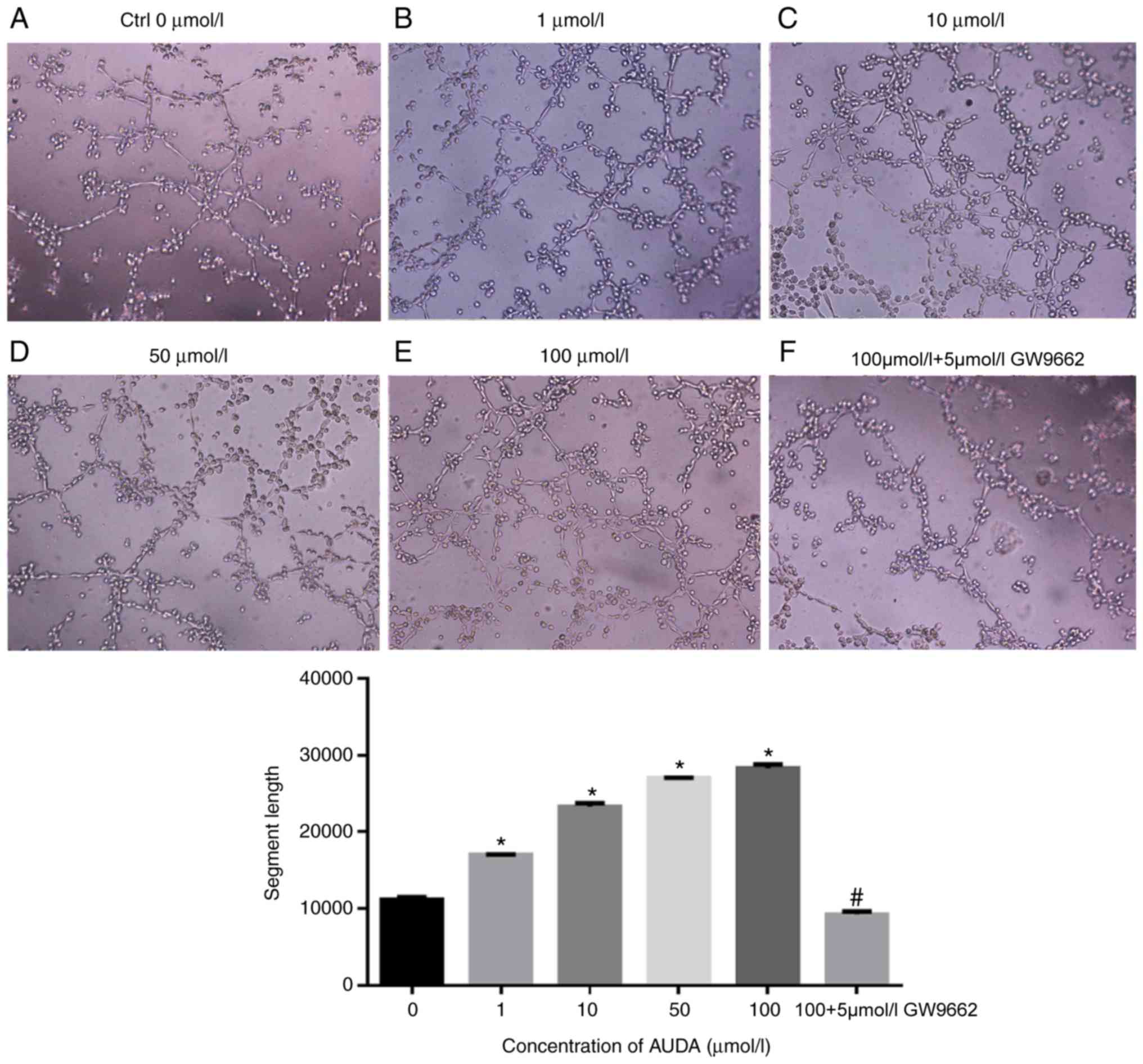
Effect of AUDA on in vitro
angiogenesis in HCAECs
The effect of AUDA on capillary tube formation was
then evaluated as a measure of in vitro angiogenesis in
HCAECs. AUDA increased the capillary tube formation of HCAECs in
vitro in a dose-dependent manner (Fig. 3). In comparison with that observed in
the control group (0 µmol/l AUDA), 1, 10, 50 and 100 µmol/l AUDA
markedly promoted the capillary tube formation ability of HCAECs
in vitro (P<0.05), while pre-treatment with GW9662
followed by AUDA significantly blocked tube formation (P<0.05;
Fig. 3).
Effect of AUDA on the proliferation of
HCAECs
To further explore the role of AUDA in the
proliferation of HCAECs, HCAECs were treated with different
concentrations of AUDA (1, 10, 50 and 100 µmol/l) and the cell
proliferation was determined using the CCK-8 assay. The results
presented in Fig. 4A indicate that
AUDA increased the proliferation of HCAECs in a dose-dependent
manner. Compared with that in the control group (0 µmol/l AUDA), 1,
10, 50 and 100 µmol/l AUDA significantly increased the
proliferation of HCAECs (P<0.05). However, the cell
proliferation was severely attenuated by pre-treatment with GW9662
(P<0.05; Fig. 4A).
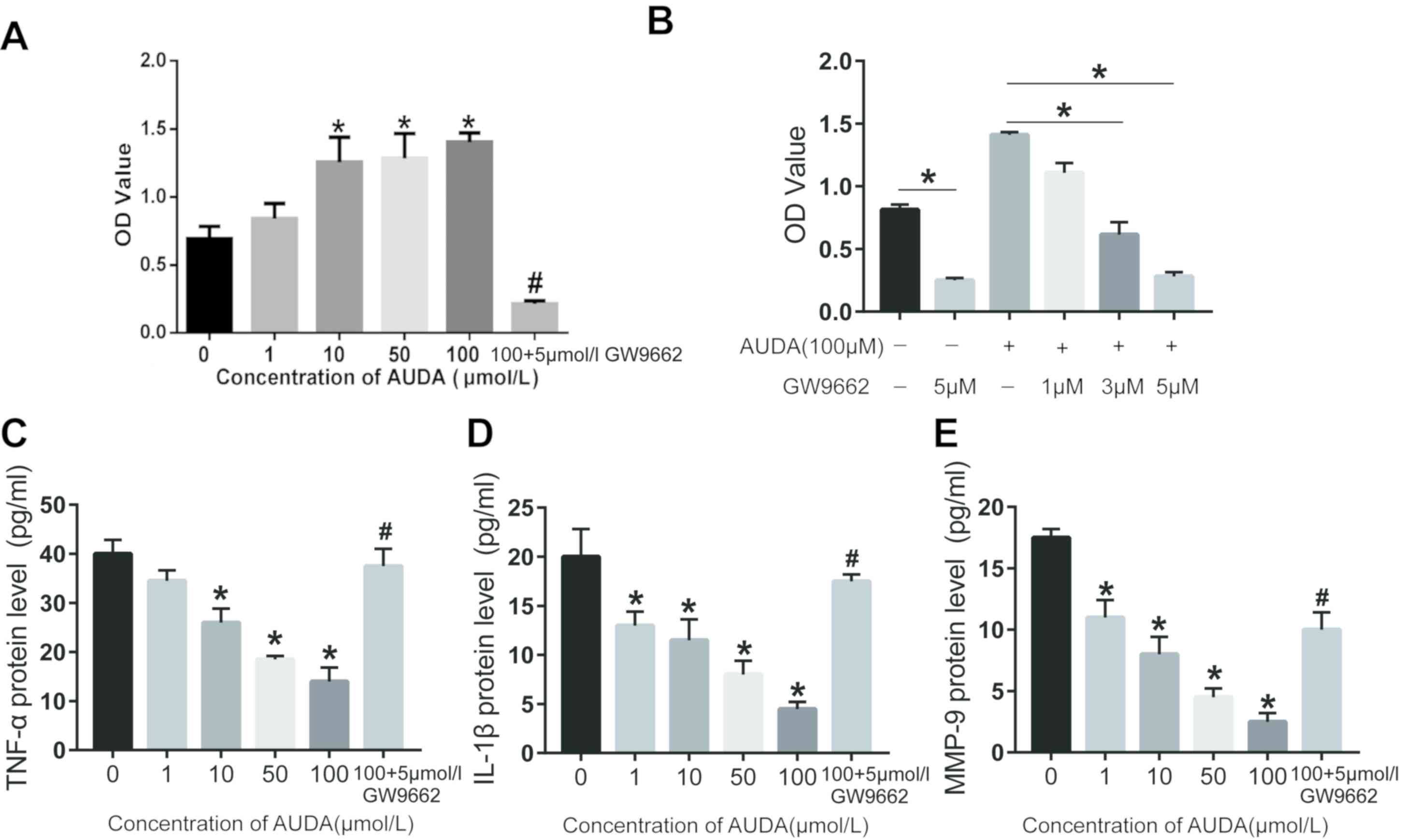 | Figure 4.Effect of AUDA on HCAEC
proliferation. Cell proliferation was determined using a Cell
Counting Kit-8 assay and OD values at 490 nm are presented. (A)
Proliferation of HCAECs treated with 0, 10, 50 or 100 µmol/l AUDA
with or without 5 µmol/l GW9662. (B) Proliferation of HCAECs
treated with 100 µmol/l AUDA combined with different concentrations
(0, 1, 3 or 5 µM) of GW9662. Expression levels of (C) TNF-α, (D)
IL-1β and (E) MMP-9 were detected by ELISA. Values are expressed as
the mean ± standard error of the mean (n=3/group). *P<0.05 vs.
Ctrl (0 µmol/l); #P<0.05 vs. 100 µmol/l AUDA. OD,
optical density; AUDA, 12-(3-adamantan-1-yl-ureido)-dodecanoic
acid, an inhibitor of soluble epoxide hydrolase; HCAECs, human
coronary arterial endothelial cells; MMP, matrix metallopeptidase;
IL, interleukin; TNF, tumor necrosis factor. |
Effects of AUDA on the PPARγ signaling
pathway of HCAECs
To further confirm the effect of AUDA on the PPARγ
signaling pathway, the proliferation of HCAECs treated with 100
µmol/l AUDA combined with different concentrations of GW9662 was
detected. As presented in Fig. 4B,
the cell proliferation was attenuated by pre-treatment with GW9662
in a dose-dependent manner. The expression of the inflammatory
factors TNF-α, IL-1β and MMP-9 in HCAECs was then determined using
ELISA. The results proved that AUDA inhibited the expression of
TNF-α, IL-1β and MMP-9 in a dose-dependent manner (Fig. 4C-E). In comparison to the control
group (0 µmol/l AUDA), 10, 50 and 100 µmol/l AUDA significantly
inhibited the expression of inflammatory factors in HCAECs
(P<0.05). However, the inhibitory effect of AUDA on the
inflammatory factors was abrogated by pre-treatment with GW9662
followed by AUDA (P<0.05; Fig.
4C-E).
Inflammation in mouse cardiac
tissues
To elucidate the potential role of AUDA in
vivo, a KD mouse model was induced by injection of LCWE. Next,
the inflammatory responses in the heart tissues of different groups
of mice, as determined by examination of the protein expression of
the inflammatory factors TNF-α, IL-1β and MMP-9, were assessed by
ELISA at 14 days after drug injection. As presented in Fig. 5, the relative protein expression of
TNF-α, IL-1β and MMP-9 was significantly higher in the hearts of
mice injected with LCWE than in those injected with PBS alone
(P<0.05). In the AUDA+LCWE injection group, TNF-α, MMP-9 and
IL-1β expression levels were lower than those in the LCWE group
(P<0.05), while pre-treatment with GW9662 abrogated the effect
of AUDA (P<0.05).
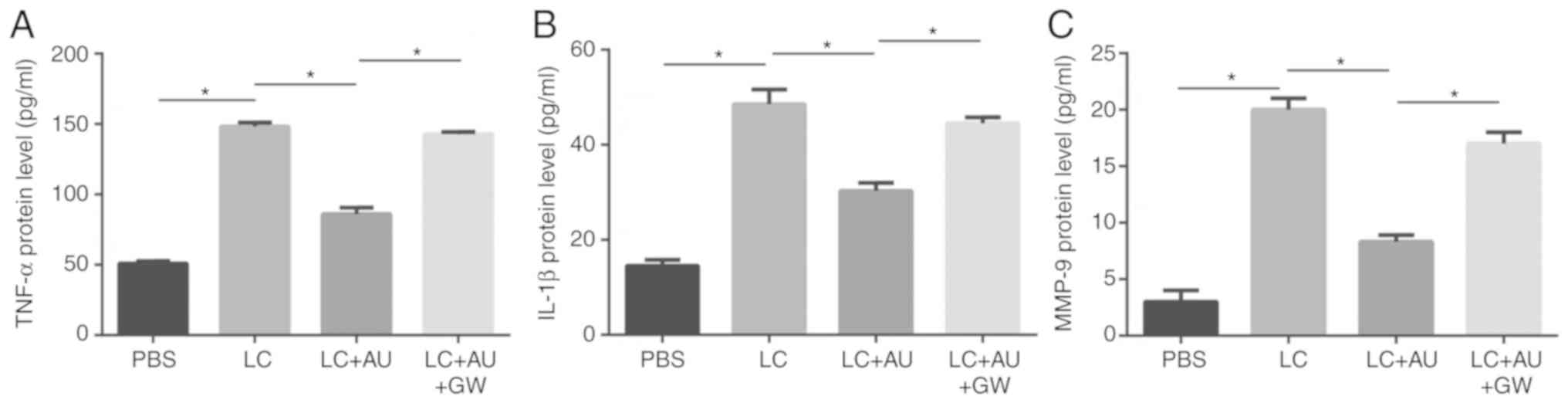 | Figure 5.Quantitative analysis of TNF-α, IL-1β
and MMP-9 protein expression. (A) TNF-α, (B) IL-1β and (C) MMP-9
protein expression levels were detected by ELISA in the heart
tissue samples of mice in different groups. Values are expressed as
the mean ± standard error of the mean (n=3/group). *P<0.05.
Groups: PBS, HCAECs treated with PBS; LC, HCAECs treated with LCWE;
LC+AU, HCAECs treated with LCWE and AUDA; LC+AU+GW, HCAECs treated
with LCWE, AUDA and GW9662. MMP, matrix metallopeptidase; IL,
interleukin; TNF, tumor necrosis factor; AUDA,
12-(3-adamantan-1-yl-ureido)-dodecanoic acid, an inhibitor of
soluble epoxide hydrolase; LCWE, Lactobacillus casei cell
wall extract. |
Discussion
KD is a systemic vasculitis of unknown origin,
frequently occurring in pediatric patients. Coronary artery
lesions, particularly giant CAAs and severe myocarditis, are
potentially fatal complications of KD (26); therefore, the development of
effective therapies is important for improving the outcome for KD
patients. The results of the present study demonstrated the utility
of the sEHi AUDA in improving the vascular repair of HCAECs and
reduce inflammatory reactions in the coronary artery in KD. The
present results also indicated that these effects are dependent on
PPARγ.
KD causes systemic inflammation and has a clear
preference for coronary arteries (27). Coronary arteritis in KD with
characteristics of inflammatory cell infiltration and the
destruction of elastic tissue in the vascular media results in the
occurrence of CAAs (4). EETs have
multiple biological functions in normal and pathophysiological
processes, exerting anti-inflammatory (28,29),
anti-fibrotic (30), anti-apoptotic
(31) and anti-oxidant (32) effects. Furthermore, EETs are known to
have anti-inflammatory effects on blood vessels (33). It is therefore suggested that EETs
have an immunomodulatory effect with potential clinical
applications in chronic inflammatory diseases.
However, EETs are rapidly degraded to
dihydroxyeicosatrienoic acids with low activity in vivo
(13). There is evidence that the
suppression of sEH increases EET levels, which represents a
possible strategy for improving the biological activity of EETs
(34). In the present study, the
efficacy of the sEHi AUDA in modulating the inflammatory response
was investigated.
To further study the anti-inflammatory role of AUDA
in KD, a mouse model of KD was employed, and the protein expression
levels of IL-1β, TNF-α and MMP-9 were measured following AUDA
treatment. There is evidence that the expression of TNF-α, IL-1β
and MMP-9 is associated with the degree of inflammatory infiltrates
in the coronary artery walls of LCWE-injected mice (35). Circulating TNF-α contributes to the
pathogenesis of coronary artery inflammation as a pivotal
pro-inflammatory cytokine, and its expression is significantly
upregulated during the KD-associated inflammatory response in a
mouse model (36). There is also
evidence that the expression of TNF-α in the coronary artery
results in the upregulation of MMP-9 in vascular smooth muscle
cells, as well as localized electrolytic activity and the matrix
destruction of surrounding coronary arteries (37,38). In
addition, it has been indicated that the serum levels of IL-1β are
markedly increased in KD patients in comparison to those in
age-matched healthy controls (39).
In view of this, the protein expression levels of TNF-α, IL-1β and
MMP-9 were examined in the hearts of KD model mice treated with
AUDA. The results demonstrated that the protein expression levels
were reduced in the mouse model of KD following AUDA treatment,
which suggests that AUDA may reduce heart inflammation, and thereby
serve a potential role in the therapeutic treatment of KD.
Certain subgroups of KD have a risk of myocardial
ischemia from coronary artery thrombosis and stenosis (3). A previous study indicated that EETs
promote the vascular repair of endothelial cells via potent
pro-mitogenic, pro-migratory and pro-angiogenic effects (40–42). In
the present study, it was hypothesized that EETs accelerate the
recovery of coronary arteries by promoting the proliferation,
migration, adhesion and tube formation ability of HCAECs. To verify
this hypothesis, the role of the sEHi AUDA on cell proliferation,
migration, adhesion and in vitro angiogenesis of HCAECs was
examined. The results of the current study demonstrated that in
HCAECs, AUDA promoted cell adhesion, migration, proliferation and
tube formation in a dose-dependent manner. AUDA promoted the
migration and adhesion of HCAECs. These results suggest that AUDA
may promote cell migration and adhesion in HCAECs through
interaction with cell surface receptors, leading to cytoskeletal
rearrangement, which can serve as a scaffold for cascades of signal
transducing molecules (43).
However, this hypothesis requires further verification. Taken
together, these results suggest that AUDA may be involved in
promoting coronary artery recovery. PPARγ activation has been
reported to involve the anti-inflammatory functions of EETs
(44). PPARs belong to the nuclear
receptor superfamily and act as ligand-activated transcription
factors. PPARs include three subtypes: PPARa, PPARb/δ and PPARγ.
PPARγ is overexpressed in the skeletal muscle, liver, vascular
wall, kidney and heart. PPAR activators have an anti-inflammatory
role in a variety of cells through suppressing the levels of
pro-inflammatory genes. These results suggest that PPARs have a
regulatory role in inflammation and have potential therapeutic
applications for chronic inflammatory diseases (45). In addition, a previous study has
indicated that EETs act as ligands and endogenous activators for
PPARγ (46). Thus, it was speculated
that the EET/PPARγ pathway may be responsible for the function of
AUDA on HCAECs. The present results indicated that AUDA enhanced
HCAEC adhesion and migration in a dose-dependent manner and, this
AUDA-induced effect was eliminated following treatment with GW9662,
a PPARγ ligand antagonist. These results suggest that AUDA may be
involved in promoting metastasis and adhesion by regulating the
PPARγ pathway. Furthermore, in the KD mouse model, GW9662 markedly
enhanced the protein levels of TNF-α, IL-1β and MMP-9. Taken
together, the results of the present study suggest that AUDA may
exert its angiogenic and anti-inflammatory effects via the
EET/PPARγ pathway. In brief, the present study suggests that EETs
may act in a PPARγ-dependent manner.
In conclusion, the present study investigated the
role of AUDA in HCAECs and a mouse model of KD. The results
demonstrated that treatment with AUDA reduced the protein
expression levels of TNF-α, MMP-9 and IL-1β in the KD mouse model
and that the vascular repair by HCAECs was markedly increased.
These results suggest that AUDA positively modulates the vascular
repair function of HCAECs in vitro and alleviates
inflammation in heart tissue, demonstrating AUDA as a potential
therapeutic treatment of KD.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation (grant no. 30900730), the
Technology Development Plan of Shandong Province (grant no.
2014GSF118066), the Shandong Province Natural Science Foundation
(grant nos. Y2008C44 and Z2008C14) and the Shandong Province
Foundation for Excellent Young and Midlife Scholars (grant no.
2005BS02003).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ND designed the experiments and prepared the
manuscript. QK, DL, ZC and MW analyzed and interpreted the data. CZ
revised the manuscript and provided technical assistance and
advice. All authors read and approved the final manuscript.
Ethical approval and consent to
participate
All animal experiments were performed in accordance
with the protocol approved by the Animal Care Committee of Shandong
University (Ji'nan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Greco A, De Virgilio A, Rizzo MI,
Tombolini M, Gallo A, Fusconi M, Ruoppolo G, Pagliuca G,
Martellucci S and de Vincentiis M: Kawasaki disease: An evolving
paradigm. Autoimmun Rev. 14:703–709. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fimbres AM and Shulman ST: Kawasaki
disease. Pediatr Rev. 29:308–316. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McCrindle BW, Rowley AH, Newburger JW,
Burns JC, Bolger AF, Gewitz M, Baker AL, Jackson MA, Takahashi M,
Shah PB, et al: Diagnosis, treatment, and long-term management of
kawasaki disease: A scientific statement for health professionals
from the american heart association. Circulation. 135:e927–e999.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kato H, Sugimura T, Akagi T, Sato N,
Hashino K, Maeno Y, Kazue T, Eto G and Yamakawa R: Long-term
consequences of Kawasaki disease. A 10-to 21-year follow-up study
of 594 patients. Circulation. 94:1379–1385. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burns JC, Shike H, Gordon JB, Malhotra A,
Schoenwetter M and Kawasaki T: Sequelae of Kawasaki disease in
adolescents and young adults. J Am Coll Cardiol. 28:253–257. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rowley AH, Baker SC, Orenstein JM and
Shulman ST: Searching for the cause of Kawasaki disease-cytoplasmic
inclusion bodies provide new insight. Nat Rev Microbiol. 6:394–401.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rowley AH, Baker SC, Shulman ST, Garcia
FL, Fox LM, Kos IM, Crawford SE, Russo PA, Hammadeh R, Takahashi K
and Orenstein JM: RNA-containing cytoplasmic inclusion bodies in
ciliated bronchial epithelium months to years after acute Kawasaki
disease. PLoS One. 3:e15822008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rowley AH, Shulman ST, Garcia FL,
Guzman-Cottrill JA, Miura M, Lee HL and Baker SC: Cloning the
arterial IgA antibody response during acute Kawasaki disease. J
Immunol. 175:8386–8391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Newburger JW, Takahashi M, Beiser AS,
Burns JC, Bastian J, Chung KJ, Colan SD, Duffy CE, Fulton DR, Glode
MP, et al: A single intravenous infusion of gamma globulin as
compared with four infusions in the treatment of acute Kawasaki
syndrome. N Engl J Med. 324:1633–1639. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ingraham RH, Gless RD and Lo HY: Soluble
epoxide hydrolase inhibitors and their potential for treatment of
multiple pathologic conditions. Curr Med Chem. 18:587–603. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Simpkins AN, Rudic RD, Roy S, Tsai HJ,
Hammock BD and Imig JD: Soluble epoxide hydrolase inhibition
modulates vascular remodeling. Am J Physiol Heart Circ Physiol.
298:H795–H806. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang LN, Vincelette J, Cheng Y, Mehra U,
Chen D, Anandan SK, Gless R, Webb HK and Wang YX: Inhibition of
soluble epoxide hydrolase attenuated atherosclerosis, abdominal
aortic aneurysm formation, and dyslipidemia. Arterioscler Thromb
Vasc Biol. 29:1265–1270. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Spector AA, Fang X, Snyder GD and
Weintraub NL: Epoxyeicosatrienoic acids (EETs): Metabolism and
biochemical function. Prog Lipid Res. 43:55–90. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeldin DC, Kobayashi J, Falck JR, Winder
BS, Hammock BD, Snapper JR and Capdevila JH: Regio- and
enantiofacial selectivity of epoxyeicosatrienoic acid hydration by
cytosolic epoxide hydrolase. J Biol Chem. 268:6402–6407.
1993.PubMed/NCBI
|
|
15
|
Spector AA and Norris AW: Action of
epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell
Physiol. 292:C996–C1012. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Newman JW, Morisseau C and Hammock BD:
Epoxide hydrolases: Their roles and interactions with lipid
metabolism. Prog Lipid Res. 44:1–51. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Miller AW, Dimitropoulou C, Han G, White
RE, Busija DW and Carrier GO: Epoxyeicosatrienoic acid-induced
relaxation is impaired in insulin resistance. Am J Physiol Heart
Circ Physiol. 281:H1524–H1531. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bellien J, Joannides R, Richard V and
Thuillez C: Modulation of cytochrome-derived epoxyeicosatrienoic
acids pathway: A promising pharmacological approach to prevent
endothelial dysfunction in cardiovascular diseases? Pharmacol Ther.
131:1–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chiamvimonvat N, Ho CM, Tsai HJ and
Hammock BD: The soluble epoxide hydrolase as a pharmaceutical
target for hypertension. J Cardiovasc Pharmacol. 50:225–237. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oguro A, Fujita N and Imaoka S: Regulation
of soluble epoxide hydrolase (sEH) in mice with diabetes: High
glucose suppresses sEH expression. Drug Metab Pharmacokinet.
24:438–445. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chaudhary KR, Abukhashim M, Hwang SH,
Hammock BD and Seubert JM: Inhibition of soluble epoxide hydrolase
by trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid
is protective against ischemia-reperfusion injury. J Cardiovasc
Pharmacol. 55:67–73. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qiu H, Li N, Liu JY, Harris TR, Hammock BD
and Chiamvimonvat N: Soluble epoxide hydrolase inhibitors and heart
failure. Cardiovasc Ther. 29:99–111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai
HJ, Kim IH, Tuteja D, Mateo RK, Singapuri A, et al: Prevention and
reversal of cardiac hypertrophy by soluble epoxide hydrolase
inhibitors. Proc Natl Acad Sci USA. 103:18733–18738. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Imig JD and Hammock BD: Soluble epoxide
hydrolase as a therapeutic target for cardiovascular diseases. Nat
Rev Drug Discov. 8:794–805. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lehman TJ, Walker SM, Mahnovski V and
McCurdy D: Coronary arteritis in mice following the systemic
injection of group B Lactobacillus casei cell walls in
aqueous suspension. Arthritis Rheum. 28:652–659. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Noguchi S, Saito J, Kudo T, Hashiba E and
Hirota K: Safety and efficacy of plasma exchange therapy for
Kawasaki disease in children in intensive care unit: Case series.
JA Clin Rep. 4:252018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee Y, Schulte DJ, Shimada K, Chen S,
Crother TR, Chiba N, Fishbein MC, Lehman TJ and Arditi M:
Interleukin-1β is crucial for the induction of coronary artery
inflammation in a mouse model of Kawasaki disease. Circulation.
125:1542–1550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Node K, Huo Y, Ruan X, Yang B, Spiecker M,
Ley K, Zeldin DC and Liao JK: Anti-inflammatory properties of
cytochrome P450 epoxygenase-derived eicosanoids. Science.
285:1276–1279. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cai Z, Zhao G, Yan J, Liu W, Feng W, Ma B,
Yang L, Wang JA, Tu L and Wang DW: CYP2J2 overexpression increases
EETs and protects against angiotensin II-induced abdominal aortic
aneurysm in mice. J Lipid Res. 54:1448–1456. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao G, Tu L, Li X, Yang S, Chen C, Xu X,
Wang P and Wang DW: Delivery of AAV2-CYP2J2 protects remnant kidney
in the 5/6-nephrectomized rat via inhibition of apoptosis and
fibrosis. Hum Gene Ther. 23:688–699. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao G, Wang J, Xu X, Jing Y, Tu L, Li X,
Chen C, Cianflone K, Wang P, Dackor RT, et al: Epoxyeicosatrienoic
acids protect rat hearts against tumor necrosis factor-α-induced
injury. J Lipid Res. 53:456–466. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen W, Yang S, Ping W, Fu X, Xu Q and
Wang J: CYP2J2 and EETs protect against lung ischemia/reperfusion
injury via anti-inflammatory effects in vivo and in vitro. Cell
Physiol Biochem. 35:2043–2054. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Spector AA: Arachidonic acid cytochrome
P450 epoxygenase pathway. J Lipid Res. 50 (Suppl):S52–S56. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Larsen BT, Gutterman DD and Hatoum OA:
Emerging role of epoxyeicosatrienoic acids in coronary vascular
function. Eur J Clin Invest. 36:293–300. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gao F, Si F, Feng S, Yi Q and Liu R:
Resistin enhances inflammatory cytokine production in coronary
artery tissues by activating the NF-κB signaling. Biomed Res Int.
2016:32964372016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hui-Yuen JS, Duong TT and Yeung RS:
TNF-alpha is necessary for induction of coronary artery
inflammation and aneurysm formation in an animal model of Kawasaki
disease. J Immunol. 176:6294–6301. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lau AC, Duong TT, Ito S and Yeung RS:
Matrix metalloproteinase 9 activity leads to elastin breakdown in
an animal model of Kawasaki disease. Arthritis Rheum. 58:854–863.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lau AC, Duong TT, Ito S, Wilson GJ and
Yeung RS: Inhibition of matrix metalloproteinase-9 activity
improves coronary outcome in an animal model of Kawasaki disease.
Clin Exp Immunol. 157:300–309. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Maury CP, Salo E and Pelkonen P:
Circulating interleukin-1 beta in patients with Kawasaki disease. N
Engl J Med. 319:1670–1671. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Michaelis UR, Fisslthaler B,
Barbosa-Sicard E, Falck JR, Fleming I and Busse R: Cytochrome P450
epoxygenases 2C8 and 2C9 are implicated in hypoxia-induced
endothelial cell migration and angiogenesis. J Cell Sci.
118:5489–5498. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cheranov SY, Karpurapu M, Wang D, Zhang B,
Venema RC and Rao GN: An essential role for SRC-activated STAT-3 in
14,15-EET-induced VEGF expression and angiogenesis. Blood.
111:5581–5591. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Potente M, Michaelis UR, Fisslthaler B,
Busse R and Fleming I: Cytochrome P450 2C9-induced endothelial cell
proliferation involves induction of mitogen-activated protein (MAP)
kinase phosphatase-1, inhibition of the c-Jun N-terminal kinase,
and up-regulation of cyclin D1. J Biol Chem. 277:15671–15676. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kireeva ML, Mo FE, Yang GP and Lau LF:
Cyr61, a product of a growth factor-inducible immediate-early gene,
promotes cell proliferation, migration, and adhesion. Mol Cell
Biol. 16:1326–1334. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Deng Y, Theken KN and Lee CR: Cytochrome
P450 epoxygenases, soluble epoxide hydrolase, and the regulation of
cardiovascular inflammation. J Mol Cell Cardiol. 48:331–341. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Delerive P, Fruchart JC and Staels B:
Peroxisome proliferator-activated receptors in inflammation
control. J Endocrinol. 169:453–459. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang
X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD and Shyy JY:
The antiinflammatory effect of laminar flow: The role of PPARgamma,
epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl
Acad Sci USA. 102:16747–16752. 2005. View Article : Google Scholar : PubMed/NCBI
|