Introduction
Thyroid cancer is the most prevalent type of
endocrine malignancy and accounts for 1% of cancer cases worldwide.
Based on the degree of differentiation, thyroid cancers are
categorized as differentiated thyroid cancer [DTC, including
papillary thyroid carcinoma (PTC) and follicular thyroid
carcinoma], as well as poorly-differentiated thyroid carcinoma
(PDTC) and anaplastic thyroid cancer (ATC) (1). Most patients with DTC maybe effectively
cured with standard primary treatments and have an excellent
prognosis (2). Although ATC only
accounts for 2% of thyroid cancer cases, it is responsible for more
than half of all thyroid cancer mortalities due to its aggressive
behavior and resistance to conventional therapies (3,4).
Recent molecular pathological studies have indicated
that activation of various tyrosine cascades and inactivation of
the tumor protein (TP) 53 tumor suppressor gene may induce the
progression and dedifferentiation of ATC (5). According to a study on 516 patients
from The Cancer Genome Atlas (TCGA) and Memorial Sloan Kettering
Cancer Center (MSKCC) database, poorly differentiated thyroid
carcinoma (including ATC) had more mutations in TP53, telomerase
reverse transcriptase (TERT) and PI3K than PTC. The signaling
pathways may exert their functions individually or through synergy
with other pathways (6,7). Besides conventional chemotherapy,
multi-kinase-targeted inhibitors are emerging as novel therapeutic
strategies (8). The aim of this
review was to determine the prevalence of the major genetic
alterations and report on the emerging kinase-targeted therapies in
ATC.
PI3K/Akt/mTOR pathway in ATC
The PI3K pathway has a key role in regulating cell
growth, proliferation and survival. This pathway is upregulated in
two ways, firstly by the binding of the p85 subunit of PI3K to the
subunits of activated tyrosine residues present on an activated
growth factor receptor and secondly via direct recognition and
combination of RAS and P110 (9).
mTOR is a regulatory protein of the PI3K/Akt/mTOR pathway and its
activation results in the phosphorylation of 4E-binding protein
(4EBP1) and ribosomal protein S6 (S6k1) both of which regulate the
transcription and translation of critical growth genes (10). Willems et al (11) reported that the activation of mTOR
was higher in lymph node metastases than in primary thyroid
cancers. Phosphorylated (p)-Akt then activates a variety of
downstream factors, including glycogen synthase kinase, Bad, the
forkhead box family of transcription factors, p27 and Mdm2. The
downstream targets of p-Akt have been demonstrated to increase cell
proliferation, motility, protein synthesis and gluconeogenesis, as
well as to inhibit apoptosis (12,13).
p-Akt also affects the nuclear proteins including transcription
factors and nuclear receptors, to form a unique signaling network
(14).
The MSKCC and TCGA contains data for 516 thyroid
cancer patients (84 PDTCs, 33 ATC cases and 399 PTC cases) and
indicated that mutations of PI3K/Akt/mTOR [including mutations of
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α
(PIK3CA) and phosphatase and tens in homolog (PTEN), and of
PIK3C2G, PIK3CG, PIK3C3, PIK3R1, PIK3R2, AKT3, TSC subunit 1
(TSC1), TSC2 and mTOR] were more frequent in PDTCs and ATCs than in
PTCs (Fig. 1A). Analysis of the
association between PI3KCA and overall survival among the 117 PDTC
and ATC cases indicated that patients with mutations of PI3KCA had
an obviously shorter survival time (10.3 vs. 116.69 months;
Fig. 1B and C). Kunstman et
al (15) and Landa et al
(16) performed next-generation
sequencing of thyroid cancer patients and the results revealed that
mutations of PI3K/Akt/mTOR were more frequent in ATCs than in other
types of thyroid cancer.
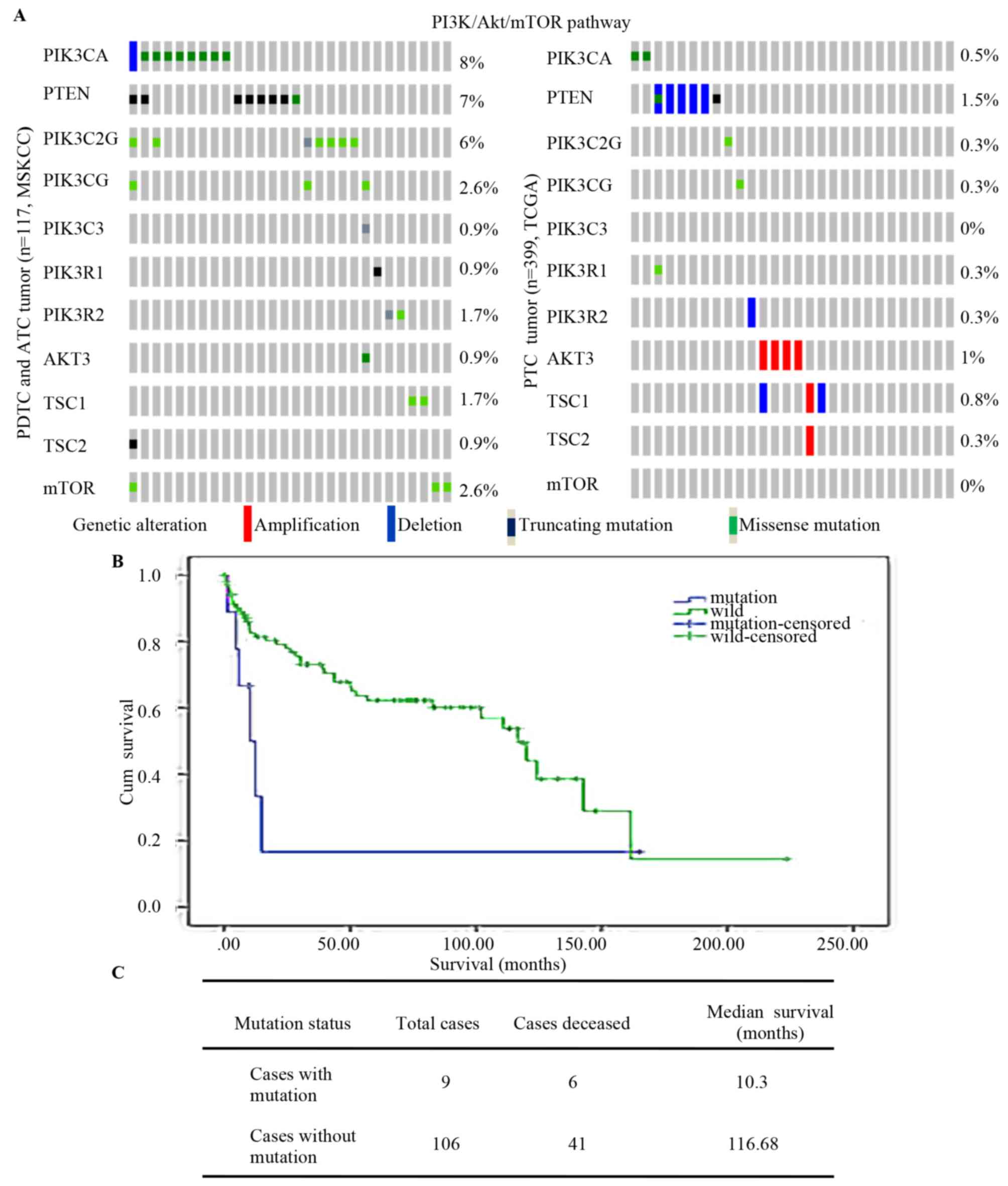 | Figure 1.PI3K/Akt/mTOR pathway mutation in
thyroid cancers. (A) PI3K/AKT/mTOR pathway (includes PIK3CA, PTEN,
PIK3C2G, PIK3CG, PIK3C3, PIK3R1, PIK3R2, AKT3, TSC1, TSC2 and MTOR)
mutation in well-differentiated thyroid tumor types (PTC) and those
with poor differentiation (PDTC and ATC). (B) Kaplan-Meier survival
analysis of 84PDTC and 33 ATC with log-rank P-values indicating
significantly shorter survival in mutation cases (P=0.044). (C)
Overall median survival of 117 PDTC and ATC cases. ATC, anaplastic
thyroid cancer; Cum, cumulative; wild, wild-type; PTEN, phosphatase
and tensin homolog; PIK3CA, phosphatidylinositol-4,5-bisphosphate
3-kinase catalytic subunit α; TCGA, The Cancer Genome Atlas; MSKCC,
Memorial Sloan Kettering Cancer Center; PTC, papillary thyroid
carcinoma; PDTC, poorly differentiated thyroid tumor; TSC1, TSC
subunit 1. |
All of the above suggests that the PI3K/mTOR pathway
may be closely linked to thyroid cancer. Current research focuses
on multiple inhibitors that interfere with different nodes of the
PI3K pathway for use in combination with traditional therapy in
clinical trials (Fig. 2).
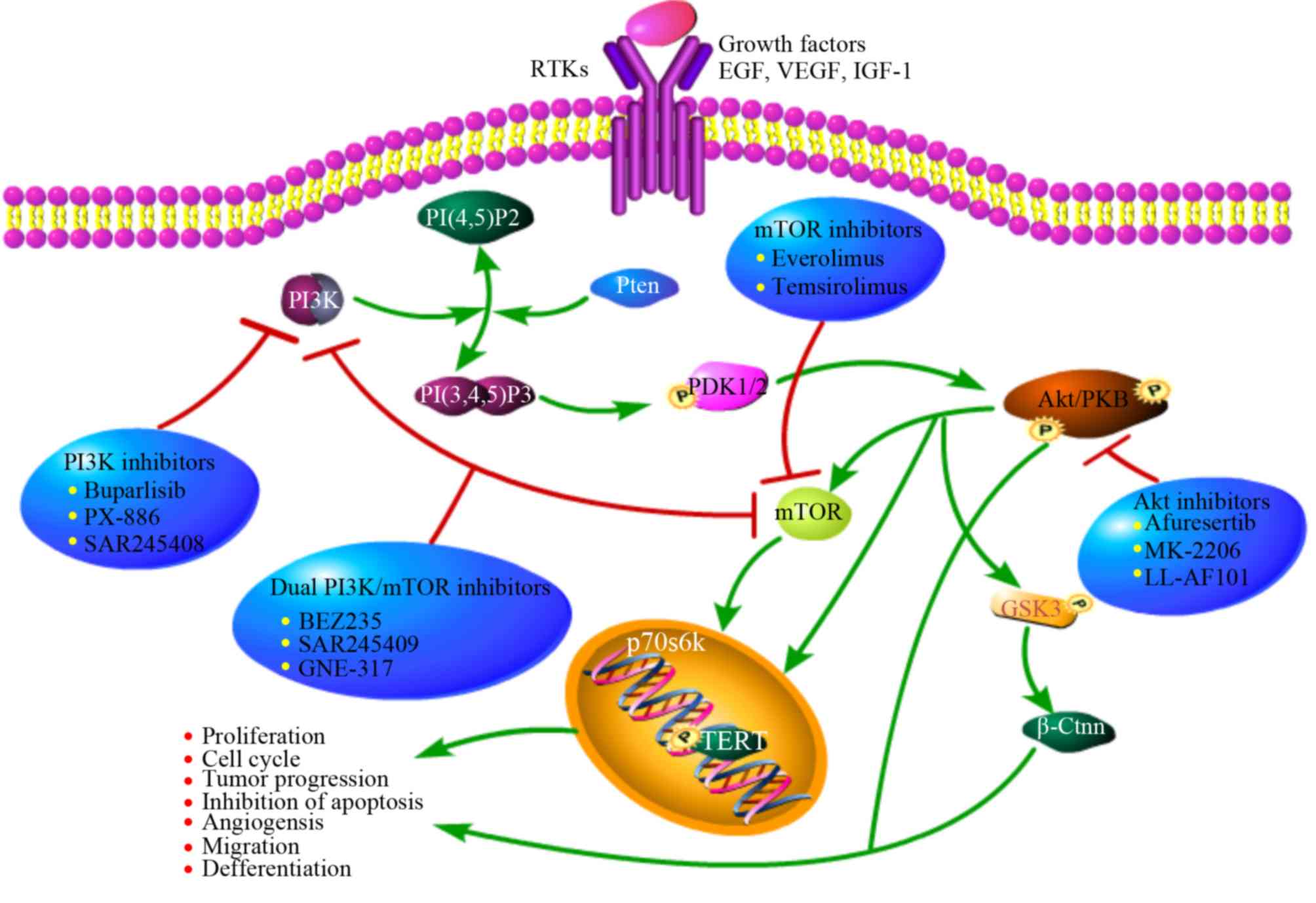 | Figure 2.Overview of PI3K/Akt/mTOR activity
regulation, downstream effectors and target inhibitors. This
pathway is commonly upregulated in tumor cells by upstream
stimulation, activating genetic alterations in PI3K, Akt, mTOR,
loss of function of PTEN or activation via cross-talk pathways.
Specific inhibitors of PI3K, Akt and mTOR may represent a potential
and innovative strategy in tumors with overactivation of this
pathway. PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase;
mTOR, mammalian target of rapamycin; EGF, epidermal growth factor;
VEGF, vascular endothelial growth factor; IGF-1, insulin-like
growth factors-1. |
PI3K inhibitors
NVP-BKM120 (Buparlisib), an inhibitor of pan-class I
PI3K, has been proven to have a role in tumor suppression in a
variety of cell lines and xenograft models, with or without PI3K
alterations (17). NVP-BKM120 has a
bioavailability of >90% and a half-life of 40 h according to a
study on breast cancer (18). It has
been proven that NVP-BKM120 suppresses downstream factors of PI3K,
including downregulation of p-Akt and p-S6R and cancer stem
markers. A previous study by our group suggested that synergistic
action of BKM120 and Prima-1Met effectively suppressed
the proliferation and migration and promoted the differentiation of
thyroid cancer cells in vivo and in vitro (19). Radiation therapy is another important
treatment for thyroid cancer, but low-dose radiation induces the
activation of the PI3K/Akt/mTOR pathway, leading to the occurrence
of epithelial-mesenchymal transition (EMT). EMT is characterized by
reduced expression of E-cadherin and enhanced cell motility, as
well as acquisition of invasive features and metastasis. It has
been reported that NVP-BKM120 efficiently inhibits the activation
of the PI3K/Akt/mTOR pathway, which increases the number of DNA
breaks and promotes the sensitivity to radiotherapy (20,21).
NVP-BKM120 is undergoing phase II/III clinical testing for further
evaluation (22).
mTOR inhibitors
RAD001 (everolimus), a rapamycin analog, is an
allosteric mTORC1 inhibitor. RAD001 has a therapeutic effect in
tumors harboring alterations in the mTOR pathway (23). RAD001 has been tested in a phase II
clinical trial (24) including 28
patients with progressive metastatic or locally advanced
radioactive refractory DTC and 7 patients with ATC. The results
indicated that 17 patients (65%) achieved stable disease (SD) as
the best response, with 15 (58%) exhibiting SD lasting for >24
weeks. Furthermore, treatment with RAD001 increased
progression-free survival in patients with metastatic cancer. As
only 7 patients with ATC were included, it was not possible to draw
any definite conclusions. The results of that study on patients
with ATC were disappointing, with none of the patients benefitting
from treatment. Certain pre-clinical trials evaluating the
anti-tumor activity of everolimus, alone or in combination, are
ongoing, suggesting that mTOR inhibitors including everolimus may
be a promising treatment option for thyroid cancer (25).
Dual PI3K/mTOR inhibitors
BEZ235, a dual PI3K/mTOR inhibitor, reduces PI3K,
mTORC1 and mTORC2 kinase activity, and consistently inactivates
downstream signaling factors via competitive binding to the ATP
binding domain of these enzymes (26,27). For
dual PI3K/mTOR inhibition, BEZ235 has demonstrated a better effect
in terms of therapeutic resistance and synergistic effects. BEZ235
has been effectively evaluated in vitro on several thyroid
cancer cell lines derived from major pathological types, with ATC
exhibiting the greatest sensitivity. BEZ235 generally induces cell
cycle arrest at the G0/G1 phase, and also causes apoptosis in the
most sensitive thyroid cell lines. The experiments also established
that daily treatment with BEZ235 at a dose of 50 mg/kg
significantly reduced the growth of xenograft tumors composed of
8505C ATC cells, without any toxicity observed (27). In addition, BEZ235 was able to
increase the expression of Na+/I− symporter
(NIS) and other thyroid-specific genes, leading to an increase in
or the restoration of the sensitivity to radioactive iodine (RAI)
(28).
AKT inhibitors
Akt is a central and essential point in the PI3K
pathway, and is therefore a promising target for anti-cancer
therapies. Akt inhibitors maybe grouped into various classes,
including ATP-competitive inhibitors (GSK690693 and Afuresertib),
allosteric inhibitors (MK-2206 and PH-316) and irreversible
inhibitors (LL-AF101) (29). MK-2206
is a selective inhibitor of all Akt isoforms, decreasing p-Akt
Thr308 and p-Akt Ser473 levels as well as downstream
phosphorylation. MK-2206 was demonstrated exert dose- and
time-dependent effects on different thyroid cancer cells lines. It
potently inhibited the proliferation of all thyroid cancer cells in
a low-micromolar range (IC50 mostly below or around 0.5
µM). When used synergistically with temsirolimus, MK2206 was able
to completely overcome the side effect of mTOR inhibition of Akt
(30) therefore, MK-2206 is a good
candidate for further investigation as a treatment for ATC.
TP53 in ATC
TP53 lies at the center of a large network of
apoptosis-, invasion-and stem cell-associated genes. The p53
protein is composed of a core DNA-binding domain (DBD) and
regulatory domains, serving as a major barrier against
tumorigenesis (31). Of note,
numerous in vitro and xenograft models have confirmed that
p53 mutation not only abolishes the tumor suppressive function, but
also often acquires new tumorigenic driver activities. This
negative role was termed gain-of-function (GOF). Inactivation of
p53 has been considered a hallmark of advanced thyroid tumors
(32). TP53 mutations have been
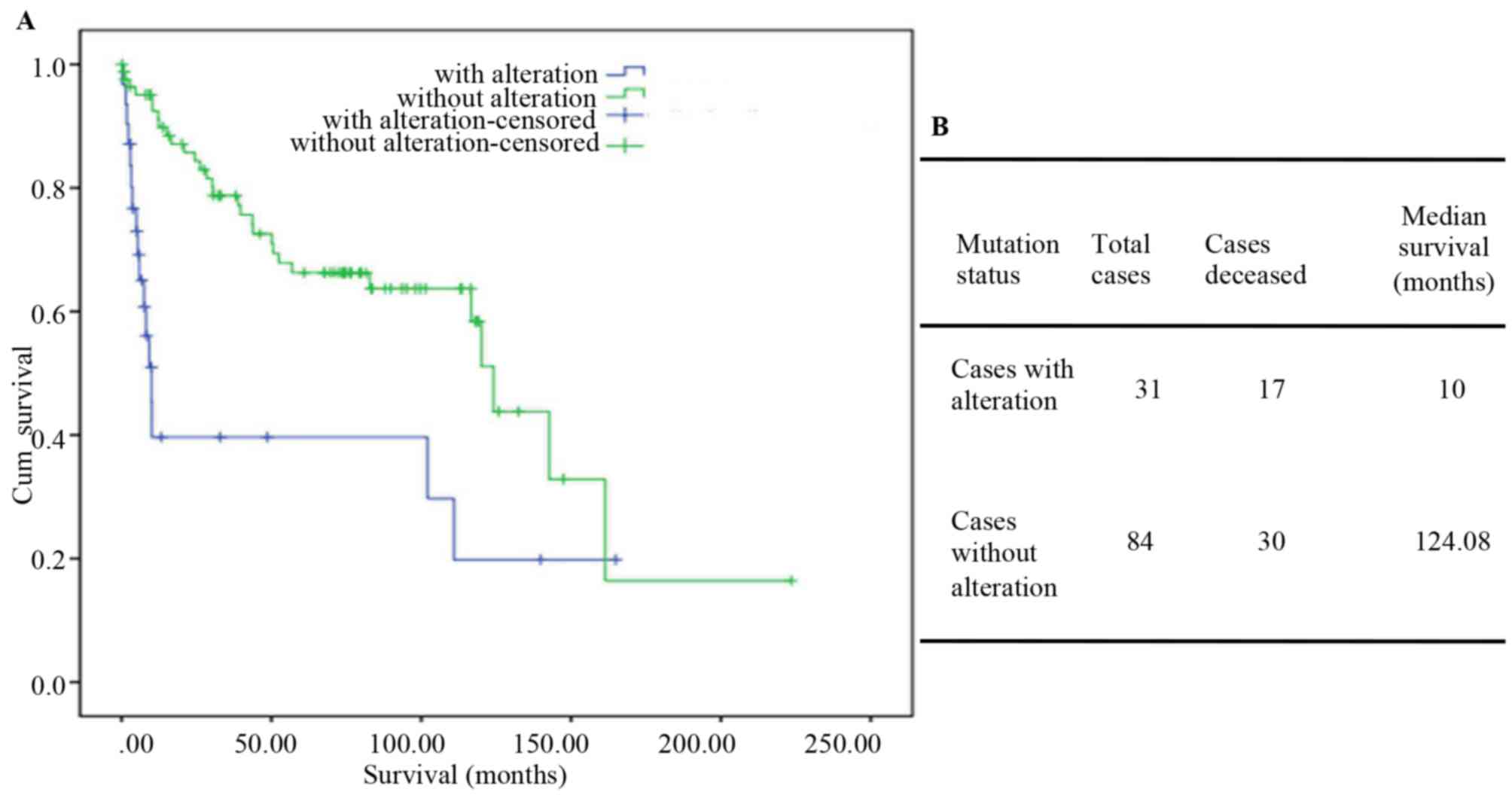
identified in 28.2% of PTCs and in 1% of ATCs and PDTCs. Among the
33 ATC and 84 PDTC patients from MSKCC dataset, the mutation group
had a median survival time of only 10 months (124.08 in the
wild-type TP53 group; Fig. 3A and
B). Mutant p53 enhances signaling through receptors such as
transforming growth factor β receptor, epidermal growth factor
receptor and MET. Unlike wild-type p53, mutant p53 protein has been
verified to escape proteasome-dependent degradation, leading to its
hyper-stabilization in tumors (33).
The degradation resistance of mutant p53 presents a fundamental
problem for therapeutic intervention in tumors with mutant p53.
Restoration of p53 function is essential for
promoting the sensitivity to chemotherapeutic drugs or radiation
therapy. This hypothesis is supported by a study in which
redifferentiation and restoration of cellular responses to
physiological stimuli were achieved after re-expression of
wild-type p53 in ATC (34). Table I summarizes the small molecules that
restore the function of the tumor suppressor gene p53.
 | Table I.Compounds that induce reactivation of
mutant p53. |
Table I.
Compounds that induce reactivation of
mutant p53.
| Type of drug | Drug | Mechanism |
|---|
| Adenovirus gene
therapy | Advexin | Exogenous import
and increase of wide-type p53 expression |
|
| ONYX-015 |
|
|
| CP-31398 | Stabilize the
DNA-binding core domain induce conformational change |
| Compounds that
induce reactivation of mutant p53 | PRIMA-1 | Bind to thiol
groups in the core domain and restore wide-type conformation |
|
|
PRIMA-1Met |
|
|
| RITA | Restore p53
transcriptional activity |
| Compounds that
deplete mutant p53 | 17-AAG | Hsp90 inhibitors,
increase the mutant p53 degradation |
|
| Geldanamycin |
|
|
| LBH589 | Disrupt the
HDAC6/Hsp90/mutant p53 complex |
|
| SAHA |
|
Introduction of wild-type p53
Restoration of wild-type p53 function may be
achieved by introduction of an intact complementary DNA copy of the
p53 gene using a suitable viral vector, in most cases an adenoviral
vector [recombinant adenovirus-p53 (Adp53)]. ONYX-015, the most
prominent and clinically evaluated replication-competent Adp53
vector, has been proven to have a tumor-specific effect, and
proliferates effectively in p53-mutant, but not in p53 wild-type
cells (35). Furthermore,
suppression of p21 via p21-targeting micro-ribonucleic acids may
effectively induce Adp53-mediated apoptosis and autophagy in human
cancer cells (36). Subsequent
studies demonstrated that a higher clinical efficacy was achieved
when ONYX-015 was combined with conventional chemotherapeutic drugs
or radiation. Numerous clinical trials have been performed in
different types of advanced cancers patients (37,38).
However, numerous patients still respond poorly (39). Gendicine (rAd-p53), a
first-generation gene therapeutic, has been licensed for clinical
use for head and neck malignancies in China. In randomized trials
on nasopharyngeal and pancreatic carcinoma, better control was
observed when Gendicine (weekly intra-tumoral injections) was
combined with radiation or chemoradiation therapy (40). However, to date, these vectors have
not been widely used in patients worldwide. The precise mechanism
of action and clinical anti-tumor effect of these vectors requires
further exploration.
Enhancement of the functionality of
endogenous wild-type p53
As mentioned above, the p53 pathway is most likely
also disrupted in a large fraction of wild-type p53-carrying
tumors. Mdm2, a critical negative regulator acting via ubiquitin-
mediated p53 degradation, is frequently overexpressed in wild-type
TP53-carrying tumors, leading to resistance to p53 gene therapy for
cancer (41). Furthermore, mutant
p53 is overexpressed in numerous tumors due to a lack of sufficient
amounts of Mdm2 to trigger p53 degradation. For these reasons, Mdm2
has been an important therapeutic target. Different strategies for
targeting Mdm2 and/or inhibition of p53-Mdm2 binding have been
designed, including Nutlins, reactivation of p53 and induction of
tumor cell apoptosis (RITA) and inhibitor of Hdm2 HLI98ubiquitin
ligase (HLI 373), which have the ability to increase p53 levels and
transcriptional activity (42).
Nutlin-3a stabilizes and activates p53 by blocking p53 binding to
Mdm2, leading to the expression of downstream genes of p53,
including p21, BAX and p53 upregulated modulator of apoptosis
(43). RITA binds to the N-terminus
of p53 and causes a configurable change, resulting in accumulation
of p53 and upregulation of its target genes. RITA induces apoptosis
in wild-type p53-harboring cancer cells, but has little side
effects on normal cells (44). The
availability of these therapies raises hope for the treatment of
wild-type TP53-carrying tumors, with fewer side effects than
traditional chemotherapeutic drugs.
‘Correction’ of mutant p53 protein
As a result of the unfolding of the DBD, mutant p53
loses its role as a tumor suppressor gene and promotes the
development of tumor progression. Therefore, pharmacological
compounds to reactivate mutant p53 through changing it to the
wild-type conformation and activating its transcription have been
developed. These small-molecular drugs, including CP-31398, WR1065,
PRIMA-1, PRIMA-1MET (APR-246), Ellipticine and MIRA-1,
have demonstrated positive effects in cancer, including induction
of massive apoptosis, inhibition of invasion and tumor stem cell
suppression (45).
PRIMA-1MET is able to not only restore the wild-type
conformation of mutant p53, but also that of mutant N-terminal
transactivation domain of TAp63γ (TAp63γ) and N-terminal
transactivation domain of TAp73β (TAp73β) in tumor cells (46). Messina et al (47) successfully validated that
PRIMA-1Met prevented the GOF effect of mutant p53 and
increased the expression of thyroid-specific differentiation
markers, including Tg and NIS, in thyroid cancer cells.
PRIMA-1Metis less effective in wild-type or null p53
thyroid cell lines (BC-PAP and Hth-74 cell viability was
significantly reduced at 1 µM; by contrast, Prima-1 at up to 20 µM
had no effect on TPC-1 and SW-1736 cells). The use of
PRIMA-1Met in combination with irradiation and novel
targeted tyrosine kinase inhibitors is a novel research
hotspot.
Human TERT and B-Raf proto-oncogene,
serine/threonine kinase (BRAF)V600E in advanced thyroid
cancer
Telomerase has a key role in cellular immortality
and tumorigenesis. Its catalytic subunit is TERT. Accumulating
evidence indicates that TERT promoter mutations are associated with
aggressive, metastatic and thyroid stem cell phenotypes (48,49). In
the dataset of TGCA database, ~40% of PDTCs and 73% of ATCs
harbored TERT promoter mutations as compared with 9% of PTCs. In
addition, TERT promoter mutations were rarely detected in normal
parenchyma or in benign lesions (50,51).
BRAF, a member of the RAF family, is a serine-threonine kinase. The
BRAFV600E mutation, a crucial stimulator of the
mitogen-activated protein kinase pathway, is associated with the
radioiodine resistance due to block of NIS
(Na+/I−) symporter expression.
BRAFV600E is more frequently encountered in the
dedifferentiated subtype (52). TERT
promoter mutation is closely combined with BRAFV600E and
RAS mutations. Studies have indicated that the coexistence of
somatic mutations, BRAFV600E and TERT C228T, is strongly
associated with aggressive phenotypes, poor prognosis and
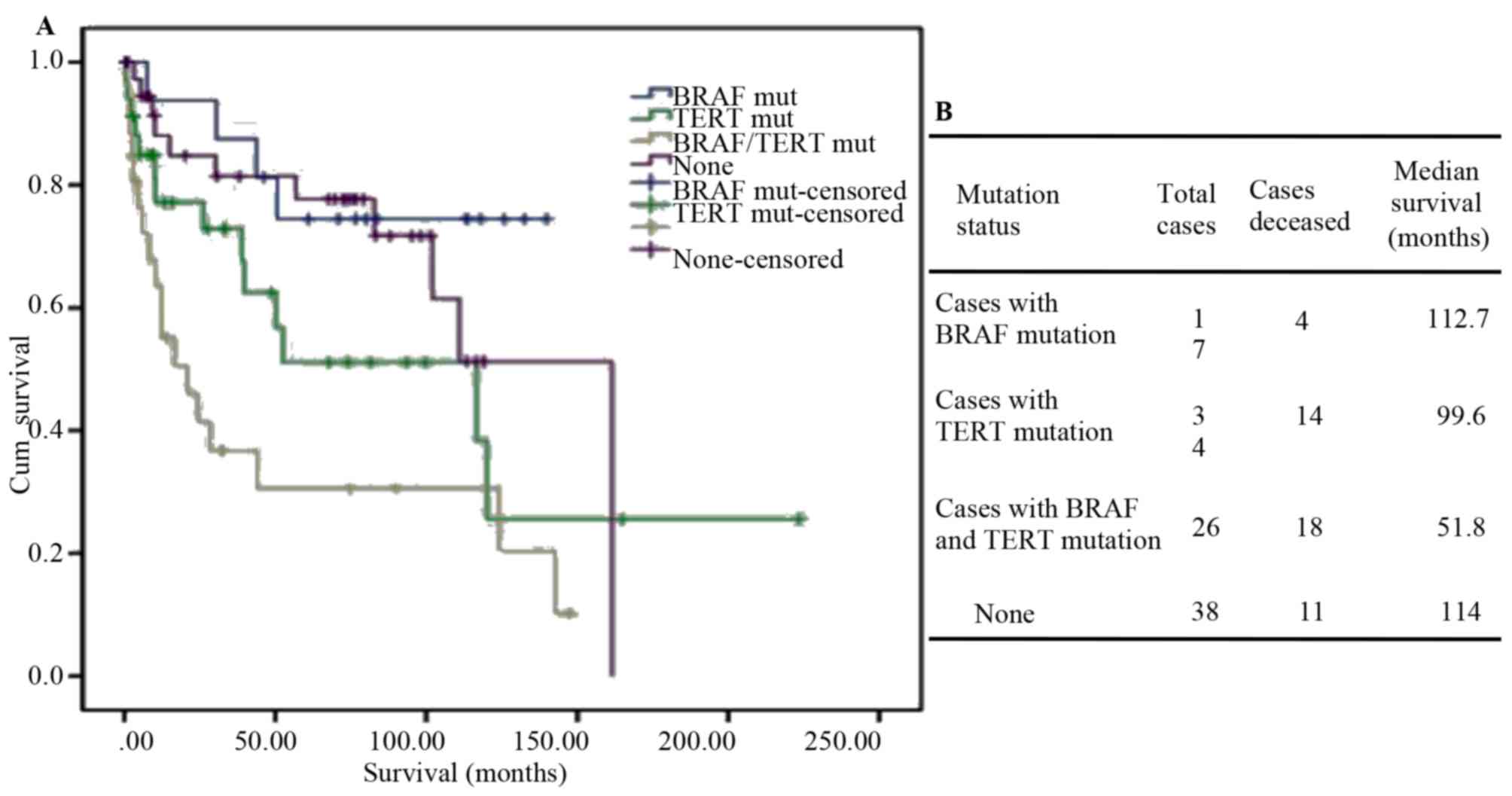
recurrence of ATC (53). Analysis of
the clinical data of the 84 PDTC and 33 ATC cases from MSKCC
revealed that co-existence of BRAF and TERT mutations has a
synergistic effect on aggressiveness in thyroid carcinoma
development. Tumors with both mutations are associated with a high
risk of recurrence and shorter overall survival (Fig. 4A and B). Charles et al
(54) reported that in the mouse
model, PIK3CA was unable to drive thyroid tumorigenesis
independently; however, in mice with both PIK3CA and
BRAFV600E mutations, ATC was observed. Of note, in ATC
cases featuring BRAFV600E in combination with PIK3CA or
TP53 mutations, few, if any, mutations in other known
ATC-associated genes were observed. The mechanism resulting in
progression from DTC to ATC has been a subject of frequent study
with accumulation of mutations in recognized malignancy-associated
genes identified. Therefore, combination of multiple targeted drugs
may achieve an effective breakthrough in the treatment of ATC.
TERT inhibitors and BRAF inhibitors
Vemurafenib (PLX4032) and Dabrafenib (GSK2118436),
highly selective for BRAF V600E-mutant cells, have been
approved by the US Food and Drug Administration for melanoma
therapy (55). The first report of
using vemurafenib to treat metastatic papillary thyroid carcinoma
was in 2013. Vemurafenib appears to have a promising clinical
efficacy in patients with metastatic PTC (56). In a phase II clinical trial, 51
patients (patients with RAI-refractory PTC) were enrolled.
Treatment with BRAF inhibitors for redifferentiation and RAI
reuptake in BRAFV600E-mutant thyroid cancer was also
assessed. The clinical results suggested that the effect was better
at first use (57). Marten and
Gudena (58) reported on an ATC
patient with an early clinical response, demonstrating a decrease
in the subcutaneous metastasis, but follow-up computed tomography
at 2 months after initiation of vemurafenib revealed rapid
progression of the disease with metastases to the central nervous
system and esophagus and progression of pulmonary metastasis. Other
BRAF inhibitors, including PLX-4720 and MLN2480, are also being
tested in the clinic, but have not been evaluated in thyroid
cancer. Clinical data indicate that these inhibitors significantly
improve response rates and overall survival in patients with
BRAFV600E-mutant metastatic melanoma (59,60).
Maggisano et al (61) proved
that inhibition of TERT expression by small interfering RNA
significantly depressed the proliferation and invasion of ATC
cells. BIBR1532, a selective telomerase inhibitor, decreases native
and recombinant human telomerase activity, leading to senescence of
human cancer cells. A study by Bu et al (62) reported that BIBR1532 effectively
decreased the invasion, migration and angiogenesis of PTC cells
in vivo and in vitro. However, at present, studies
assessing the efficacy of BIBR1532 in the treatment of ATC are
rare.
Other agents and approaches
NF-κB allows thyroid cancer cells to acquire
invasive properties and undergo metastasis by upregulating the
expression of matrix metalloproteinases and urokinase-type
plasminogen activator (63).
Triptolide has been used in preclinical studies on ATC, and has
demonstrated an inhibitory effect on angiogenesis and invasion
(64). More clinical research is
required to evaluate the synergistic effect with other
chemotherapeutic agents for thyroid cancer. The Wnt-β-catenin
signaling pathway is associated with cell adhesion and
differentiation (65). Accumulation
of β-catenin may induce nuclear transport and combine with certain
transcription factors, which may activate the transcription of
downstream factors, including c-myc and cyclin D1. The
Wnt-β-catenin pathway may also regulate the level of cyclin D1,
which is associated with lymph node metastases in thyroid cancer
patients. Dickkopf-1, a selective inhibitor of the Wnt-β-catenin
pathway, effectively inhibits the proliferation and migration of
several thyroid cancer cell lines by regulating Wnt-β-catenin and
E-cadherin expression (66). Certain
genes regulate each other and form networks to produce a biological
effect. Further signaling pathways are also associated with the
development and progression of ATC. Sorafenib is a multikinase
inhibitor that targets vascular endothelial growth factor receptor
(VEGFR), platelet-derived growth factor receptor (PDGFR) and RAF.
In the phase II trial of sorafenib, 10 patients with ATC were
enrolled and no objective responses were observed (67). Another two trials assessing sorafenib
in thyroid carcinomas included six ATC patients and similarly no
responses were achieved (68,69).
These results call into question the benefit of this agent in ATC
patients. Another study involved 15 ATC patients treated with
pazopanib (a multikinase inhibitor targeted on VEGFR, PDGFR and
c-kit) in a single-arm, phase II study, with no responses obtained
according to the Response Evaluation Criteria in Solid Tumors
(70). Lenvatinib, a newer,
small-molecule VEGFR inhibitor, was tested in differentiated
thyroid cancer, medullary thyroid carcinoma and ATC in a phase II
study. Only 11 patients with ATC were recruited; however, the
results were encouraging, with three patients exhibiting a partial
response according to the Response Evaluation Criteria in Solid
Tumors, seven with SD and one patient exhibiting progression of
disease (71). Lenvatinib has been
approved in the USA for the treatment of DTC, but it is approved
for all subtypes of thyroid cancer in Japan (67).
Genomic and epigenetic alterations are now being
exploited as molecular targets in thyroid cancer treatment. The
abnormalities of the histones in post-translational modification
have been demonstrated in thyroid cancer development, and they are
regarded as promising molecular targets for the patients that are
resistant to conventional therapies. Compounds/drugs that reverse
the effects of histone modifications by modulating the pattern of
histone acetylation/methylation have been tested in preclinical
models of thyroid cancer to identify their effects on tumor cell
proliferation and/or invasiveness, and their ability to
re-differentiate tumor cells and restore their ability to
accumulate radioactive iodine (72,73).
Conclusion
Precision medical therapy is an exciting technique
for individualized tumor treatment based on the patient's features
and tumor characteristics. ATC is one of the most aggressive human
cancer types and is associated with a low survival rate.
Dysfunction of signaling pathways may increase the proliferation,
dedifferentiation and metastasis of thyroid cancer. Inhibition of
protein tyrosine kinases has been indicated to hold great promise.
These drugs may not only provide a better understanding of tumor
biological characteristics, but also more optimistic outcomes in
the future. Combination of tyrosine kinases inhibitors with
traditional radiotherapy and chemotherapy may represent an improved
treatment strategy.
Acknowledgements
Not applicable.
Funding
The present study was supported by funds from the
National Natural Science Foundation of China (grant nos. 81703904
KZ and 81473452 LZ) and the Education Department of Liaoning
Province, China (‘the Program for Distinguished Professor of
Liaoning Province’).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ and ZL contributed to the design and conception
of the study, and revised it carefully for important intellectual
content. YZ was responsible for acquiring the data by screening the
papers identified on Pubmed and TCGA. RW revised the study
critically for important intellectual content. KZ and ZL were
involved in drafting the study. YZ analyzed and interpreted the
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
O'Neill JP and Shaha AR: Anaplastic
thyroid cancer. Oral Oncol. 49:702–706. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jin S, Borkhuu O, Bao W and Yang YT:
Signaling pathways in thyroid cancer and their therapeutic
implications. J Clin Med Res. 8:284–296. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hsu KT, Yu XM, Audhya AW, Jaume JC, Lloyd
RV, Miyamoto S, Prolla TA and Chen H: Novel approaches in
anaplastic thyroid cancer therapy. Oncologist. 19:1148–1155. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Keutgen XM, Sadowski SM and Kebebew E:
Management of anaplastic thyroid cancer. Gland Surg. 4:44–51.
2015.PubMed/NCBI
|
|
5
|
Saini S, Maker AV, Burman KD and Prabhakar
BS: Genetic aberrations and alterations in signaling cascades
implicated in the pathogenesis of anaplastic thyroid cancer.
Biochim Biophys Acta Rev Cancer. 2018.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guerra A, Di Crescenzo V, Garzi A, Cinelli
M, Carlomagno C, Tonacchera M, Zeppa P and Vitale M: Genetic
mutations in the treatment of anaplastic thyroid cancer: A
systematic review. BMC Surg. 13 (Suppl 2):S442013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu B and Ghossein R: Genomic landscape of
poorly differentiated and anaplastic thyroid carcinoma. Endocr
Pathol. 27:205–212. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perri F, Pezzullo L, Chiofalo MG, Lastoria
S, Di Gennaro F, Scarpati GD and Caponigro F: Targeted therapy: A
new hope for thyroid carcinomas. Crit Rev Oncol Hematol. 94:55–63.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartholomeusz C and Gonzalez-Angulo AM:
Targeting the PI3K signaling pathway in cancer therapy. Expert Opin
Ther Targets. 16:121–130. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Saji M and Ringel MD: The PI3K-Akt-mTOR
pathway in initiation and progression of thyroid tumors. Mol Cell
Endocrinol. 321:20–28. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Willems L, Tamburini J, Chapuis N, Lacombe
C, Mayeux P and Bouscary D: PI3K and mTOR signaling pathways in
cancer: New data on targeted therapies. Curr Oncol Rep. 14:129–138.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cao F, Zhang C, Han W, Gao XJ, Ma J, Hu
YW, Gu X, Ding HZ, Zhu LX and Liu Q: p-Akt as a potential poor
prognostic factor for gastric cancer: A systematic review and
meta-analysis. Oncotarget. 8:59878–59888. 2017.PubMed/NCBI
|
|
13
|
Phyu SM and Smith TAD: Combination
treatment of cancer cells with pan-Akt and pan-mTOR inhibitors:
Effects on cell cycle distribution, p-Akt expression level and
radiolabelled-choline incorporation. Invest New Drugs. 37:424–430.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xing M: Genetic alterations in the
phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer.
Thyroid. 20:697–706. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kunstman JW, Juhlin CC, Goh G, Brown TC,
Stenman A, Healy JM, Rubinstein JC, Choi M, Kiss N, Nelson-Williams
C, et al: Characterization of the mutational landscape of
anaplastic thyroid cancer via whole-exome sequencing. Hum Mol
Genet. 24:2318–2329. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Landa I, Ibrahimpasic T, Boucai L, Sinha
R, Knauf JA, Shah RH, Dogan S, Ricarte-Filho JC, Krishnamoorthy GP,
Xu B, et al: Genomic and transcriptomic hallmarks of poorly
differentiated and anaplastic thyroid cancers. J Clin Invest.
126:1052–1066. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maira SM, Pecchi S, Huang A, Burger M,
Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, et al:
Identification and characterization of NVP-BKM120, an orally
available pan-class I PI3-kinase inhibitor. Mol Cancer Ther.
11:317–328. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sirohi B, Rastogi S and Dawood S:
Buparlisib in breast cancer. Future Oncol. 11:1463–1470. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li Z, Xu X, Li Y, Zou K, Zhang Z, Xu X,
Liao Y, Zhao X, Jiang W, Yu W, et al: Synergistic antitumor effect
of BKM120 with prima-1met via inhibiting PI3K/AKT/mTOR and
CPSF4/hTERT signaling and reactivating mutant P53. Cell Physiol
Biochem. 45:1772–1786. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chang L, Graham PH, Hao J, Ni J, Bucci J,
Cozzi PJ, Kearsley JH and Li Y: Acquisition of
epithelial-mesenchymal transition and cancer stem cell phenotypes
is associated with activation of the PI3K/Akt/mTOR pathway in
prostate cancer radioresistance. Cell Death Dis. 4:e8752013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chang L, Graham PH, Hao J, Ni J, Bucci J,
Cozzi PJ, Kearsley JH and Li Y: PI3K/Akt/mTOR pathway inhibitors
enhance radiosensitivity in radioresistant prostate cancer cells
through inducing apoptosis, reducing autophagy, suppressing NHEJ
and HR repair pathways. Cell Death Dis. 5:e14372014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Massacesi C, Di Tomaso E, Urban P, Germa
C, Quadt C, Trandafir L, Aimone P, Fretault N, Dharan B, Tavorath R
and Hirawat S: PI3K inhibitors as new cancer therapeutics:
Implications for clinical trial design. Onco Targets Ther.
9:203–210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Saran U, Foti M and Dufour JF: Cellular
and molecular effects of the mTOR inhibitor everolimus. Clin Sci
(Lond). 129:895–914. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schneider TC, de Wit D, Links TP, van Erp
NP, van der Hoeven JJ, Gelderblom H, Roozen IC, Bos M, Corver WE,
van Wezel T, et al: Everolimus in patients with advanced
follicular-derived thyroid cancer: Results of a phase II clinical
trial. J Clin Endocrinol Metab. 102:698–707. 2017.PubMed/NCBI
|
|
25
|
Onoda N, Nakamura M, Aomatsu N, Noda S,
Kashiwagi S, Kurata K, Uchino S and Hirakawa K: Significant
cytostatic effect of everolimus on a gefitinib-resistant anaplastic
thyroid cancer cell line harboring PI3KCA gene mutation. Mol Clin
Oncol. 3:522–526. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yi H, Ye X, Long B, Ye T, Zhang L, Yan F,
Yang Y and Li L: Inhibition of the AKT/mTOR pathway augments the
anticancer effects of sorafenib in thyroid cancer. Cancer Biother
Radiopharm. 32:176–183. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin SF, Huang YY, Lin JD, Chou TC, Hsueh C
and Wong RJ: Utility of a PI3K/mTOR inhibitor (NVP-BEZ235) for
thyroid cancer therapy. PLoS One. 7:e467262012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Petrulea MS, Plantinga TS, Smit JW,
Georgescu CE and Netea-Maier RT: PI3K/Akt/mTOR: A promising
therapeutic target for non-medullary thyroid carcinoma. Cancer
Treat Rev. 41:707–713. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nitulescu GM, Margina D, Juzenas P, Peng
Q, Olaru OT, Saloustros E, Fenga C, Spandidos DΑ, Libra M and
Tsatsakis AM: Akt inhibitors in cancer treatment: The long journey
from drug discovery to clinical use (Review). Int J Oncol.
48:869–885. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu R, Liu D, Trink E, Bojdani E, Ning G
and Xing M: The akt-specific inhibitor MK2206 selectively inhibits
thyroid cancer cells harboring mutations that can activate the
PI3K/Akt pathway. J Clin Endocrinol Metab. 96:E577–E585. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pflaum J, Schlosser S and Muller M: P53
family and cellular stress responses in cancer. Front Oncol.
4:2852014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Parrales A and Iwakuma T: Targeting
oncogenic mutant p53 for cancer therapy. Front Oncol. 5:2882015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maslon MM and Hupp TR: Drug discovery and
mutant p53. Trends Cell Biol. 20:542–555. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bullock AN and Fersht AR: Rescuing the
function of mutant p53. Nat Rev Cancer. 1:68–76. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wiman KG: Strategies for therapeutic
targeting of the p53 pathway in cancer. Cell Death Differ.
13:921–926. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Larson C, Oronsky B, Scicinski J, Fanger
GR, Stirn M, Oronsky A and Reid TR: Going viral: A review of
replication-selective oncolytic adenoviruses. Oncotarget.
6:19976–19989. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Galanis E, Okuno SH, Nascimento AG, Lewis
BD, Lee RA, Oliveira AM, Sloan JA, Atherton P, Edmonson JH,
Erlichman C, et al: Phase I–II trial of ONYX-015 in combination
with MAP chemotherapy in patients with advanced sarcomas. Gene
Ther. 12:437–445. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Makower D, Rozenblit A, Kaufman H, Edelman
M, Lane ME, Zwiebel J, Haynes H and Wadler S: Phase II clinical
trial of intralesional administration of the oncolytic adenovirus
ONYX-015 in patients with hepatobiliary tumors with correlative p53
studies. Clin Cancer Res. 9:693–702. 2003.PubMed/NCBI
|
|
39
|
Chen GX, Zhang S, He XH, Liu SY, Ma C and
Zou XP: Clinical utility of recombinant adenoviral human p53 gene
therapy: Current perspectives. Onco Targets Ther. 7:1901–1909.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li J, Pan J, Zhu X, Su Y, Bao L, Qiu S,
Zou C, Cai Y, Wu J and Tham IW: Recombinant adenovirus-p53
(Gendicine) sensitizes a pancreatic carcinoma cell line to
radiation. Chin J Cancer Res. 25:715–721. 2013.PubMed/NCBI
|
|
41
|
Wade M, Li YC and Wahl GM: MDM2, MDMX and
p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 13:83–96.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Oren M, Tal P and Rotter V: Targeting
mutant p53 for cancer therapy. Aging (Albany NY). 8:1159–1160.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Selivanova G: Wild type p53 reactivation:
From lab bench to clinic. FEBS Lett. 588:2628–2638. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Urso L, Calabrese F, Favaretto A, Conte P
and Pasello G: Critical review about MDM2 in cancer: Possible role
in malignant mesothelioma and implications for treatment. Crit Rev
Oncol Hematol. 97:220–230. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Puca R, Nardinocchi L, Porru M, Simon AJ,
Rechavi G, Leonetti C, Givol D and D'Orazi G: Restoring p53 active
conformation by zinc increases the response of mutant p53 tumor
cells to anticancer drugs. Cell Cycle. 10:1679–1689. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bykov VJ and Wiman KG: Mutant p53
reactivation by small molecules makes its way to the clinic. FEBS
Lett. 588:2622–2627. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Messina RL, Sanfilippo M, Vella V, Pandini
G, Vigneri P, Nicolosi ML, Gianì F, Vigneri R and Frasca F:
Reactivation of p53 mutants by prima-1 [corrected] in thyroid
cancer cells. Int J Cancer. 130:2259–2270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu X, Bishop J, Shan Y, Pai S, Liu D,
Murugan AK, Sun H, El-Naggar AK and Xing M: Highly prevalent TERT
promoter mutations in aggressive thyroid cancers. Endocr-Relat
Cancer. 20:603–610. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Melo M, da Rocha AG, Vinagre J, Batista R,
Peixoto J, Tavares C, Celestino R, Almeida A, Salgado C, Eloy C, et
al: TERT promoter mutations are a major indicator of poor outcome
in differentiated thyroid carcinomas. J Clin Endocrinol Metab.
99:E754–E765. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu R and Xing M: TERT promoter mutations
in thyroid cancer. Endocr-Relat Cancer. 23:R143–R155. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jin A, Xu J and Wang Y: The role of TERT
promoter mutations in postoperative and preoperative diagnosis and
prognosis in thyroid cancer. Medicine (Baltimore). 97:e115482018.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dong H, Shen WZ, Yan YJ, Yi JL and Zhang
L: Effects of BRAF(V600E) mutation on Na(+)/I(−) symporter
expression in papillary thyroid carcinoma. J Huazhong Uni Sci
Technolog. Med Sci. 36:77–81. 2016.
|
|
53
|
Shi X, Liu R, Qu S, Zhu G, Bishop J, Liu
X, Sun H, Shan Z, Wang E, Luo Y, et al: Association of TERT
promoter mutation 1,295,228 C>T with BRAF V600E mutation, older
patient age, and distant metastasis in anaplastic thyroid cancer. J
Clin Endocrinol Metab. 100:E632–E637. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Charles RP, Silva J, Iezza G, Phillips WA
and McMahon M: Activating BRAF and PIK3CA mutations cooperate to
promote anaplastic thyroid carcinogenesis. Mol Cancer Res.
12:979–986. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang W: BRAF inhibitors: The current and
the future. Curr Opin Pharmacol. 23:68–73. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kim KB, Cabanillas ME, Lazar AJ, Williams
MD, Sanders DL, Ilagan JL, Nolop K, Lee RJ and Sherman SI: Clinical
responses to vemurafenib in patients with metastatic papillary
thyroid cancer harboring BRAF(V600E) mutation. Thyroid.
23:1277–1283. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Brose MS, Cabanillas ME, Cohen EE, Wirth
LJ, Riehl T, Yue H, Sherman SI and Sherman EJ: Vemurafenib in
patients with BRAF(V600E)-positive metastatic or unresectable
papillary thyroid cancer refractory to radioactive iodine: A
non-randomised, multicentre, open-label, phase 2 trial. Lancet
Oncol. 17:1272–1282. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Marten KA and Gudena VK: Use of
vemurafenib in anaplastic thyroid carcinoma: A case report. Cancer
Biol Ther. 16:1430–1433. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cabanillas ME, Patel A, Danysh BP, Dadu R,
Kopetz S and Falchook G: BRAF inhibitors: Experience in thyroid
cancer and general review of toxicity. Horm Cancer. 6:21–36. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lim AM, Taylor GR, Fellowes A, Cameron L,
Lee B, Hicks RJ, McArthur GA, Angel C, Solomon B and Rischin D:
BRAF Inhibition in BRAFV600E-Positive Anaplastic Thyroid Carcinoma.
J Natl Compr Canc Netw. 14:249–254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Maggisano V, Celano M, Lombardo GE, Lepore
SM, Sponziello M, Rosignolo F, Verrienti A, Baldan F, Puxeddu E,
Durante C, et al: Silencing of hTERT blocks growth and migration of
anaplastic thyroid cancer cells. Mol Cell Endocrinol. 448:34–40.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Bu R, Siraj AK, Divya SP, Kong Y,
Parvathareddy SK, Al-Rasheed M, Al-Obaisi KAS, Victoria IG,
Al-Sobhi SS, Al-Dawish M, et al: Telomerase reverse transcriptase
mutations are independent predictor of disease-free survival in
middle eastern papillary thyroid cancer. Int J Cancer.
142:2028–2039. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Namba H, Saenko V and Yamashita S: Nuclear
factor-kB in thyroid carcinogenesis and progression: A novel
therapeutic target for advanced thyroid cancer. Arq Bras Endocrinol
Metabol. 51:843–851. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhu W, He S, Li Y, Qiu P, Shu M, Ou Y,
Zhou Y, Leng T, Xie J, Zheng X, et al: Anti-angiogenic activity of
triptolide in anaplastic thyroid carcinoma is mediated by targeting
vascular endothelial and tumor cells. Vascul Pharmacol. 52:46–54.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sastre-Perona A and Santisteban P:
Wnt-independent role of β-catenin in thyroid cell proliferation and
differentiation. Mol Endocrinol. 28:681–695. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang L, Shao YY and Ballock RT: Thyroid
hormone interacts with the Wnt/beta-catenin signaling pathway in
the terminal differentiation of growth plate chondrocytes. J Bone
Miner Res. 22:1988–1995. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ito Y, Onoda N, Ito KI, Sugitani I,
Takahashi S, Yamaguchi I, Kabu K and Tsukada K: Sorafenib in
japanese patients with locally advanced or metastatic medullary
thyroid carcinoma and anaplastic thyroid carcinoma. Thyroid.
27:1142–1148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Gupta-Abramson V, Troxel AB, Nellore A,
Puttaswamy K, Redlinger M, Ransone K, Mandel SJ, Flaherty KT,
Loevner LA, O'Dwyer PJ and Brose MS: Phase II trial of sorafenib in
advanced thyroid cancer. J Clin Oncol. 26:4714–4719. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kloos RT, Ringel MD, Knopp MV, Hall NC,
King M, Stevens R, Liang J, Wakely PE Jr, Vasko VV, Saji M, et al:
Phase II trial of sorafenib in metastatic thyroid cancer. J Clin
Oncol. 27:1675–1684. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Bible KC, Suman VJ, Menefee ME, Smallridge
RC, Molina JR, Maples WJ, Karlin NJ, Traynor AM, Kumar P, Goh BC,
et al: A multiinstitutional phase 2 trial of pazopanib monotherapy
in advanced anaplastic thyroid cancer. J Clin Endocrinol Metab.
97:3179–3184. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Takahashi S, Kiyota N, Yamazaki T,
Chayahara N, Nakano K, Inagaki L, Toda K, Enokida T, Minami H,
Imamura Y, et al: A Phase II study of the safety and efficacy of
lenvatinib in patients with advanced thyroid cancer. Future Oncol.
15:717–726. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Sasanakietkul T, Murtha TD, Javid M, Korah
R and Carling T: Epigenetic modifications in poorly differentiated
and anaplastic thyroid cancer. Mol Cell Endocrinol. 469:23–37.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Celano M, Mio C, Sponziello M, Verrienti
A, Bulotta S, Durante C, Damante G and Russo D: Targeting
post-translational histone modifications for the treatment of
non-medullary thyroid cancer. Mol Cell Endocrinol. 469:38–47. 2018.
View Article : Google Scholar : PubMed/NCBI
|