Introduction
Oral cavity cancer has shown an increased incidence
in recent years in Romania (1) and
also worldwide (2). The most commune
type of oral cavity cancer is squamous cell carcinoma (commonly
referred as ‘Head and Neck Squamous Cell Carcinoma’, HNSCC). The
disease affects several anatomic structures: Oral cavity,
oropharynx, nasopharynx, hypopharynx and larynx (3–5). Most of
HNSCC patients are, unfortunately, diagnosed when the disease has
already progressed to an advanced stage. In this instance the
therapeutic approach is complex and involves the combination of
chemo and radiotherapy either before or after the surgical
procedure based on the protocol established for each patient
(6–8). Thus, the protocols are somewhat
personalized, hower, the development of resistance to conventional
treatments and an increased recurrence rate of primary tumors lead
to a 5-year decline of the survival rate of patients with HNSCC
(9–12). Therefore, additional clinical
approaches of this complex disease are needed. The new therapeutic
strategies have to tackle at least two points, besides increasing
the survival rate, it should reduce the adverse effects of
chemotherapy. The use of natural compounds as adjuvants can be
beneficial by increasing the efficacy of conventional treatments
and very importantly to limit the adverse reactions. The disruption
of normal cellular mechanisms such as proliferation or apoptosis,
is accountable for the development of tumoral processes in
different types of cancer, including HNSCC. Apoptosis, known as
‘programmed cell death’, is a mechanism that provides crucial
control over the cell homeostasis. Apoptosis enables the removal of
cells having DNA mutations, aberrant cellular cycle and that are
prone to malignant transformation (13,14).
Defects observed in the apoptotic pathway associated to cancer have
an important role in conventional oncotherapies (radio- and
chemotherapy) since apoptosis induction needs high dosages of
therapeutic agents. Cisplatin (CisPt), one of the most used
chemotherapeutic agent in HNSCC treatment, has the capacity to
interact with DNA, RNA and different proteins, and by activation of
specific mechanisms, some of them still incompletely known, can
induce apoptosis (15–17).
The elucidation of the mechanisms pertaining to the
apoptosis process is a key step. The restoration of the cellular
mechanisms responsible for tumor cell apoptosis are of upmost
importance in malignant transformation, tumor evasion and
anticancer therapy (18–20). Factors that determine a damaged cell
to go either through apoptosis or to repair the damage and continue
the cell cycle, are still to be discovered. It is well known that
one important apoptosis regulator is the tumor suppressor gene TP53
(TP53). TP53 tumor suppressor has mutations in 40–60% of the HNSCC
cases (21). This is an untimely
event, identified in precancerous lesions and associated with a
poor prognosis. Some studies showed that the rehabilitation of the
TP53 function in an early stage of the disease had no effect, but
can lead to the regression of the tumor in advanced stages
(22,23). This suggests that TP53 tumor
suppressor is not activated in the early phases of the disease, but
can be activated in later phases of tumorigenesis (24,25).
Another important player in the regulation of cell
tumorigenesis-related functions (proliferation, transformation,
differentiation, apoptosis, angiogenesis) is activation of
mitogen-activated protein kinases (MAPKs) pathways such as ERKs
(26–30). There are studies showing that the
activation of the MAPK pathway is correlated with the cancer
prognosis influencing the therapeutic outcome in many types of
cancer (31–33). Correlated once with the intimate
deciphering of molecular pathways that regulate oncogenesis, new
modern and specific therapies able to improve the current
therapeutic methods will be developed. One of the approaches is to
maximize the effectiveness of initial therapy by the use of a
chemotherapeutic drug together with a supporting agent (34). Some studies have been focused on the
discovery of new therapeutic agents obtained from natural compounds
proving anticancer and anti-proliferation effects (35,36). One
of these compounds is curcumin (CRM). CRM is the principal compound
of turmeric extracted from the plant Curcuma longa and has
many diverse properties - anti-inflammatory, anti-bacterial,
anti-fungal, anti-viral and anti-carcinogenic (37). The mechanisms through which CRM
exerts its antitumoral effects are complex and diverse; they appear
to act in the processes of growth and apoptosis and also in
different stages of carcinogenesis (38,39).
Acknowledging all the mentioned issues in the this
type of carcinoma the focus of this study is to investigate how a
natural adjuvant (CRM) supports the apoptotic process induced by a
mono chemical standard agent (CisPt) in an in vitro
experimental model using HNSCC standard cell lines. Moreover, in
our study we investigated the ERK1/2 and/or p53 involvement in
treatment response. The use of adjuvant might have a beneficial
effect decreasing the CisPt doses, therefore reducing the adverse
reactions induced by a chemotherapeutic agent.
Materials and methods
Cell lines culture
The squamous carcinoma cell line PE/CA-PJ49 was from
European Collection of Authenticated Cell Cultures (ECACC cat. no.
0060606). The cell line was obtained from a 57-year old male
patient with tongue carcinoma. The FaDu cell line was obtained from
the American Type Culture Collection (ATCC-HTB-43 cat.). The cell
line was derived from a 56-year-old male patient with pharyngeal
squamous cell carcinoma. Both lines are showing adherent epithelial
type morphology. The cell lines were grown and maintained in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS), 2 mM glutamine, 1% penicillin, and 1%
streptomycin at 37°C in 5% CO2. The sub-confluent
cultures (70–80%) were split 1:4-1:8 (i.e. seeding at 1–3×10,000
cells/cm2) using trypsin-EDTA (0.25% trypsin, 0.03%
EDTA).
The study protocol was approved by the Ethics
Committee of ‘Stefan S. Nicolau’ Virology Institute.
Drugs and treatments
CisPt and CRM (97% purity), were obtained from
Sigma-Aldrich. They were initially dissolved in dimethyl sulfoxide
(DMSO; Sigma-Aldrich) at a concentration of 5 mM. Further, milli-Q
water was used to generate 1 mM stock solutions. The stock
solutions were filtered using a cellulose acetate hydrophilic
filter (0.20 µm) (Sigma-Aldrich). Dilutions used in the
experimental model were done in DMEM to generate the following
concentration ranges: 2–160 µM for CisPt and 5–100 µM for CRM.
Tumor cells were incubated for 6, 24 or 48 h either in the presence
of the drugs (CisPt and/or CRM) or vehicle control (DMSO ≤0.1%).
For inhibition studies of ERK1/2 function, the cells were
pre-incubated for 2 h with 25 µM PD98059 as previously reported
(40). The treated tumor cells were
used to determine cell proliferation, FISH, apoptosis and conserved
as cell pellets at −80°C in order to obtain cell lysates used in
ELISA assays. Non-treated cells were used as controls throughout
the experiments.
Cell viability assay
Tumor cells (1–2×103 cell/well) were
seeded in 96-microwell plates, incubated at 37°C for 24 h to
accomplish full adherence and then treated with different
concentrations of CisPt (2–160 µM) or CRM (5–100 µM). The cell
viability was assessed by the ability of metabolically active cells
to reduce the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) (Sigma-Aldrich) to colored formazan compounds. The
absorbance was measured with an enzyme-linked immunosorbent assay
reader (Dynex plate reader; wavelength 450 nm) (41). The data are presented as the mean
values from at least three different experiments. Untreated cells
served as control having 100% viability. Viability % = (T-B)/(U-B)
×100, (where T, absorbance of treated cells; U, absorbance of
untreated cells, iar B, absorbance of blank).
Cell proliferation assay
CellTiter 96® AQueous One Solution Cell
Proliferation Assay (Promega) was used. The test is based on the
reduction of yellow MTS tetrazolium salt by the viable cells and
generation of colored formazan soluble in the culture medium. The
product was spectrophotometrically quantified by measuring the
absorbance at λ=490 nm (42) using a
Dynex plate reader (DYNEX Technologies-MRS). Results were expressed
as mean values of three determinations ± standard deviation (SD).
Untreated cells served as control and considered to have
proliferation index (PI) equal 1. PI = absorbance of treated
cells/absorbance of untreated cells.
Fluorescence in situ
hybridization
FISH technique was performed after optimization of
the protocol using commercially available probe from Abbott/Vysis
(Vysis) (43,44). According with the manufacturer's
protocol Locus specific identifier LSA TP53/CEP17 FISH Probe Kit
detects the LSI TP53 probe Spectrum Orange target located on
chromosome 17p13.1 and CEP17 (17p11.1-q11.1 Alpha Satellite) probe
Spectrum Green Dual Colour target located on the centromere of
chromosome 17.
Cells and slide preparation: The slides were cleaned
in an ice-cold mixture of 40% methanol and 60% distilled water,
then air dryed and stored at 4°C. Cells were pelleted and suspended
in 0.075 M KCl hypotonic solution for 20 min at 37°C. Tumor cells
were pelleted by centrifugation at 300 × g for 5 min at 4°C and
resuspended in 0.5 ml fixative (3:1 methyl alcohol and glacial
acetic acid). Afterwards the cells were diluted at the appropriate
density and distributed on several locations on the slide. The air
dried slides were incubated at −20°C for 30 min.
FISH probe preparation and hybridization from cell
culture: The slides were denatured for 5 min in 70% formamide/2X
SSC at 73°C. Then the slides were dehydrated by immersion in 70, 80
and 100% cold ethanol solution, 5 min for each step. The slides
were air-dried and 10 µl of the probe LSI TP53/CEP17 was added in
the selected hybridization area. The smears were covered with a
22×22 mm coverslip, sealed and incubated overnight in a humid
chamber at 37°C. Two washes were performed post hybridization,
using washing solutions: 0.4X SSC/0.3% NP-40 at 73°C for 2 min, and
2X SSC/0.1% NP-40 for 2 min. The slides were air-dried and
4′,6-diamidino-2-phenylindole (DAPI II) was added for
counterstaining. The slides were analyzed using a Zeiss Axio Imager
M1 epifluorescence microscope (Zeiss) equipped with filters for
DAPI, SpectrumOrange and SpectrumGreen and a triple filter
(simultaneous DAPI/Orange/Green). Images were acquired at a
magnification of ×1,000 and captured using MetaSystems digital
camera. Images were analyzed using Isis version 5.2, MetaSystems
software for quantitative analysis of samples generated by FISH
technique (Altlussheim). For each sample hybridized signals were
counted in 100 nuclei.
ELISA assay
ELISA assays were used to measure both total and
phosphorylated p53 and ERK1/2proteins in cell lysates. Briefly,
untreated, CisPt and/or CRM treated tumor cells were lysed in PBS
(pH 7.2–7.4) containing 1 mM EDTA, 0.5% Triton X-100, 5 mM NaF, 6 M
urea, 10 µg/ml leupeptin, 10 µg/ml pepstatin, 100 µM PMSF, 3 µg/ml
aprotinin, 2.5 mM sodium pyrophosphate, 1 mM sodium orthovanadate.
The lysate was kept 30 min on ice with stirring every 5 min. The
lysate was centrifuged at 2,000 × g for 5 min at room temperature.
Furthermore, the obtained supernatant was centrifuged at 14,000 × g
for 15 min at 4°C. Protein concentration of the lysates was
measured using Bradford assay. DuoSet_IC Human Total p53 ELISA
[cat. no. DYC1043]; DuoSet_IC Human Phospho-p53 (S15) ELISA [cat.
no. DYC1839]; DuoSet_IC Human/Mouse/Rat Total ERK1/2 [cat. no.
DYC1940] DuoSet_IC Human/Mouse/Rat Phospho- ERK1/2 ELISA [cat. no.
DYC1018B] were purchased from R&D Systems Inc. The protein of
interest was measured using a standard Streptavidin-HRP system
(45). All experiments were
performed in triplicates and sample O.D. was measured at λ=450 nm
using Dynex plate reader.
Analysis of apoptosis
The apoptosis assay was carried out with the Annexin
V-FITC kit using the manufacturer's protocol (BD Pharmingen)
(46). Treated and untreated
1×106 cells/ml were resuspended in cold binding buffer
and staining simultaneously with 5 µl FITC-Annexin V (green
fluorescence) and 5 µl propidium iodide (PI) in the dark at room
temperature for 15 min. Then 400 µl of Annexin V binding buffer was
added and 10,000 cells/sample were acquired using BD Canto II flow
cytometer. The analysis was performed using DIVA 6.2 software in
order to discriminate viable cells (FITC−PI−)
from necrotic cells (FITC+PI+) and early
apoptosis (FITC+PI−) from late apoptosis.
Statistical analysis
Data were analyzed using Student's t-test (paired
type and one tailed distibution). One-way analysis of variation
with P-value of <0.05 was considered statistically
significant.
Results
Culture parameters of the standardized
cell lines used in in vitro experiments
In the present study two human HNSCC cell lines were
used, FaDu derived from pharyngeal squamous carcinoma and
PE/CA-PJ49 obtained from a tongue squamous carcinoma, both cell
lines presenting adherent and epithelial-type morphology. To
achieve the successive passages of cells kept in culture, the
density of cells in the culture plates were carefully supervised in
order to avoid over-population and any sign of aging. At the end of
each experiment, samples from used cells were frozen to create a
batch of cells that can be used anytime to repeat or continue the
experiments. Some studies have shown that the number of cells that
present with polysomy at the level of the 17th chromosome is raised
in oral cavity squamous carcinomas and this may be correlated with
the process of carcinogenesis (47).
The exact role of genetic modifications of TP53 in different stages
of the tumorigenesis process is not completely established, but it
is known that the gene has the ability to induce the restoration of
damaged DNA by activating certain proteins and by stopping the
cellular cycle and induction of apoptosis. Thereby, it has been
tried to identify some possible numeric aberrations of chromosome
17, such as deletion or amplification of TP53 gene in the FaDu and
PE/CA-PJ49 cells. This genetic endeavor was done in order to
discover possible explications between these abnormalities and the
cellular response to therapy. Using the FISH technique, the
intention was to obtain information on the TP53 gene status in the
FaDu and PE/CA-PJ49 tumor cells before starting the experiments.
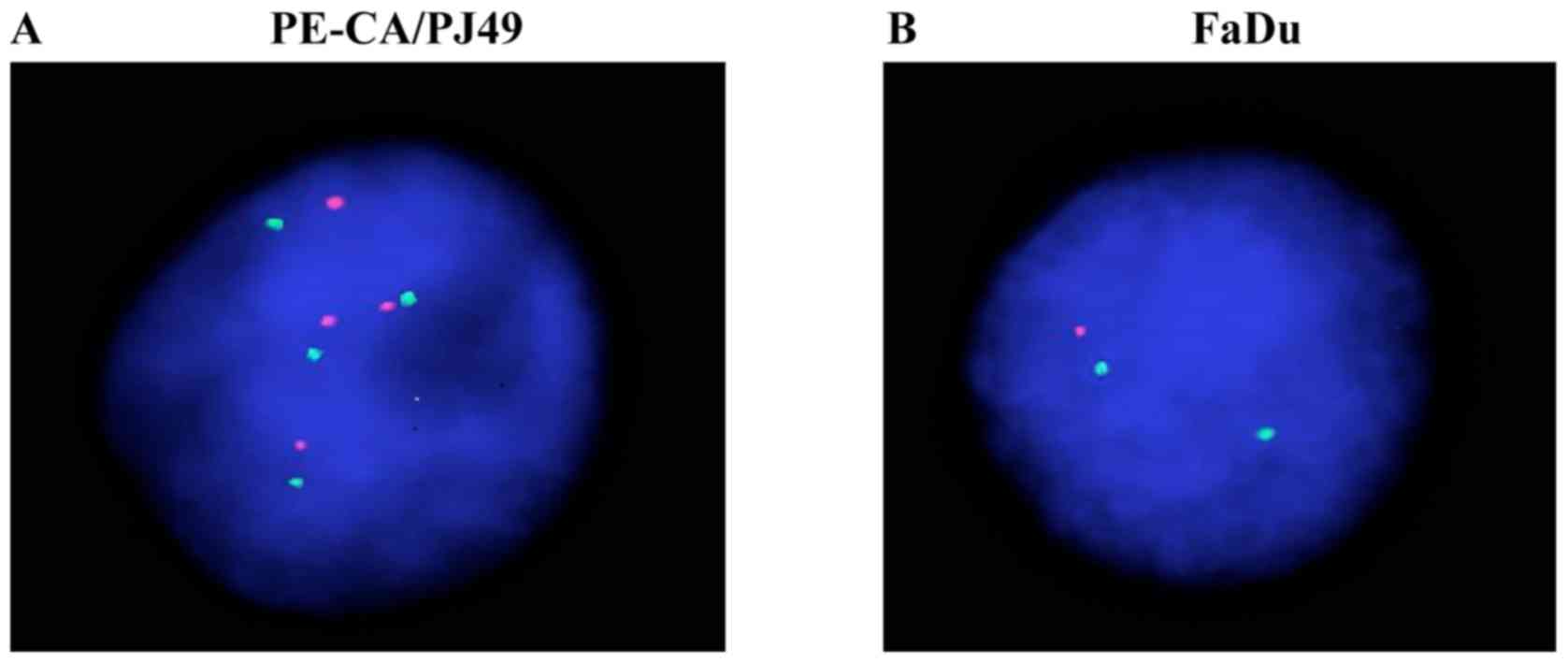
The PE/CA-PJ49 tumor line showed an amplification of the TP53 gene
since all analyzed cells had 4 signals for both TP53 (17p13) (red
dots on Fig. 1A) and 17(D17Z1)
centromere probe (green dots on Fig.
1A). FaDu did not show the amplification of TP53 gene on
chromosome 17. In contrast to this, the cells had only one signal
for TP53 (17p13) (red dot on Fig.
1B) suggesting a deletion of the TP53 gene, without
modification at the level of chromosome 17 which presented only two
signals for the 17(D17Z1) centromere (green dots Fig. 1B).
Effects of cisplatin and/or curcumin
treatment on the cellular viability of HNSCC
Cisplatin is one of the most utilized cytostatic
drugs in the treatment of HNSCC. Unfortunately many patients
develop relapses or cisplatin resistance. A natural compound such
as curcumin might improve and maintain the antitumoral effect of
cisplatin. In order to determine the optimal concentration of the
drug needed to inhibit half of the maximum biological response
(IC50) both tumor cell lines were treated with the
following concentrations 0, 1, 5, 10, 20, 40, 80, 160 µM CisPt or
0, 5, 10, 15, 20, 25, 50, 100 µM CRM. To determine the optimal
treatment time, cells were treated for different time periods (6,
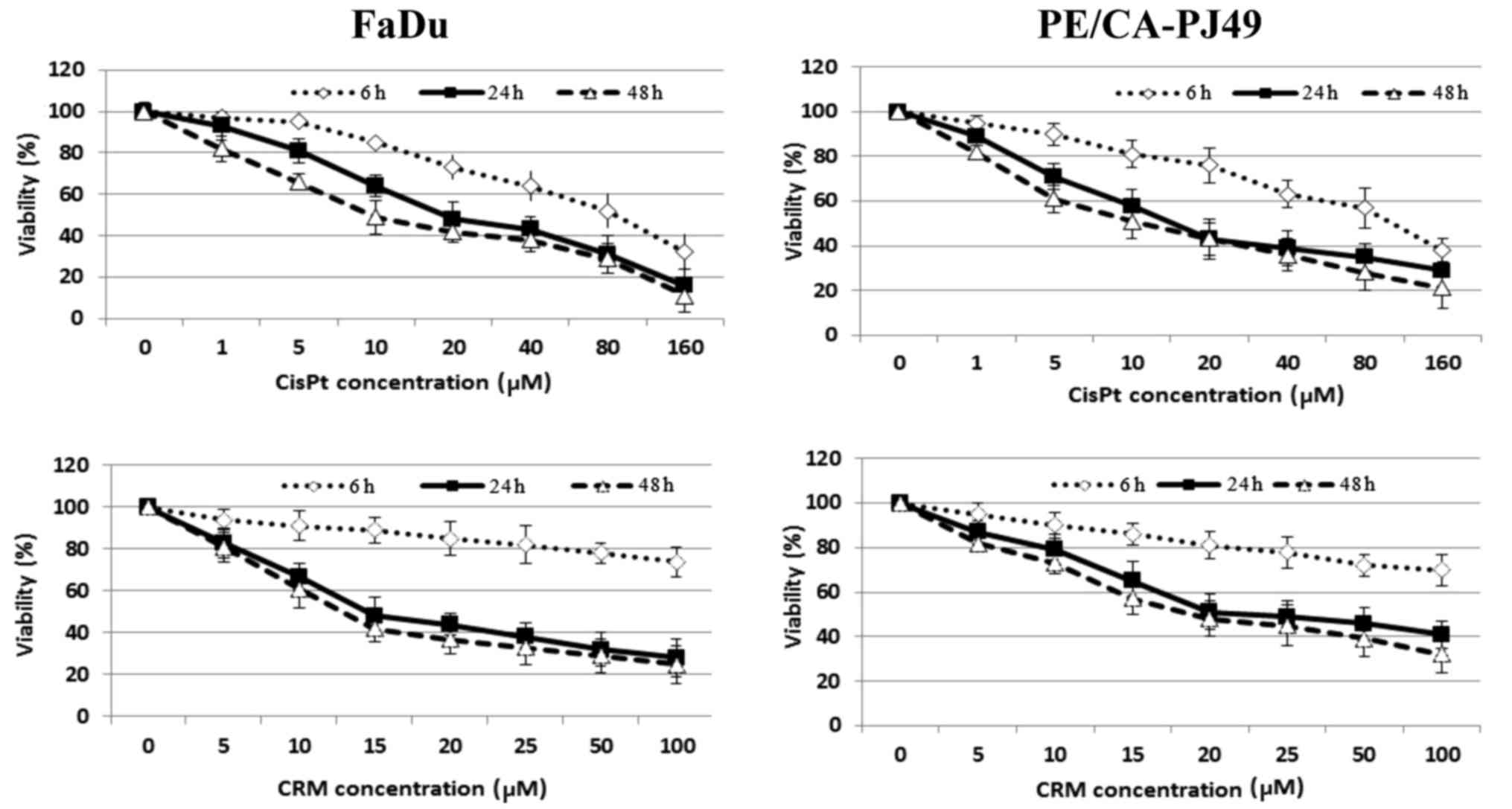
24 or 48 h). The inhibitory effect of CisPt or CRM on the cellular
viability was dose- and time-dependent (Fig. 2). The results showed that 24 h CisPt
treatment had an IC50=11.25 µM for FaDu cells and
IC50=10.55 µM for PE/CA-PJ49 cells. CRM treatment for 24
h reduced the cellular viability with an IC50=13.72 µM
for FaDu cells and IC50=15.20 µM for PE/CA-PJ49 tumor
cells. Based on the obtained results the optimal time of treatment
was 24 h and the optimal concentration was 10 µM for CisPt and 15
µM for CRM (Fig. 2). We tested
concentrations less than 2 µM (data not shown) and they did not
have any effect on the treated FaDu or PE/CA-PJ49 cells.
The effect of the CisPt and/or CRM
treatment on the protein-kinase ERK1/2 expression
MAPKs transmit and amplify the signals involved in
proliferation, as well as in cellular death. Using ELISA assay, we
evaluated the expression and activation of protein-kinase ERK1/2
induced by CisPt and/or CRM treatment of FaDu and PE-CA/PJ49 tumor
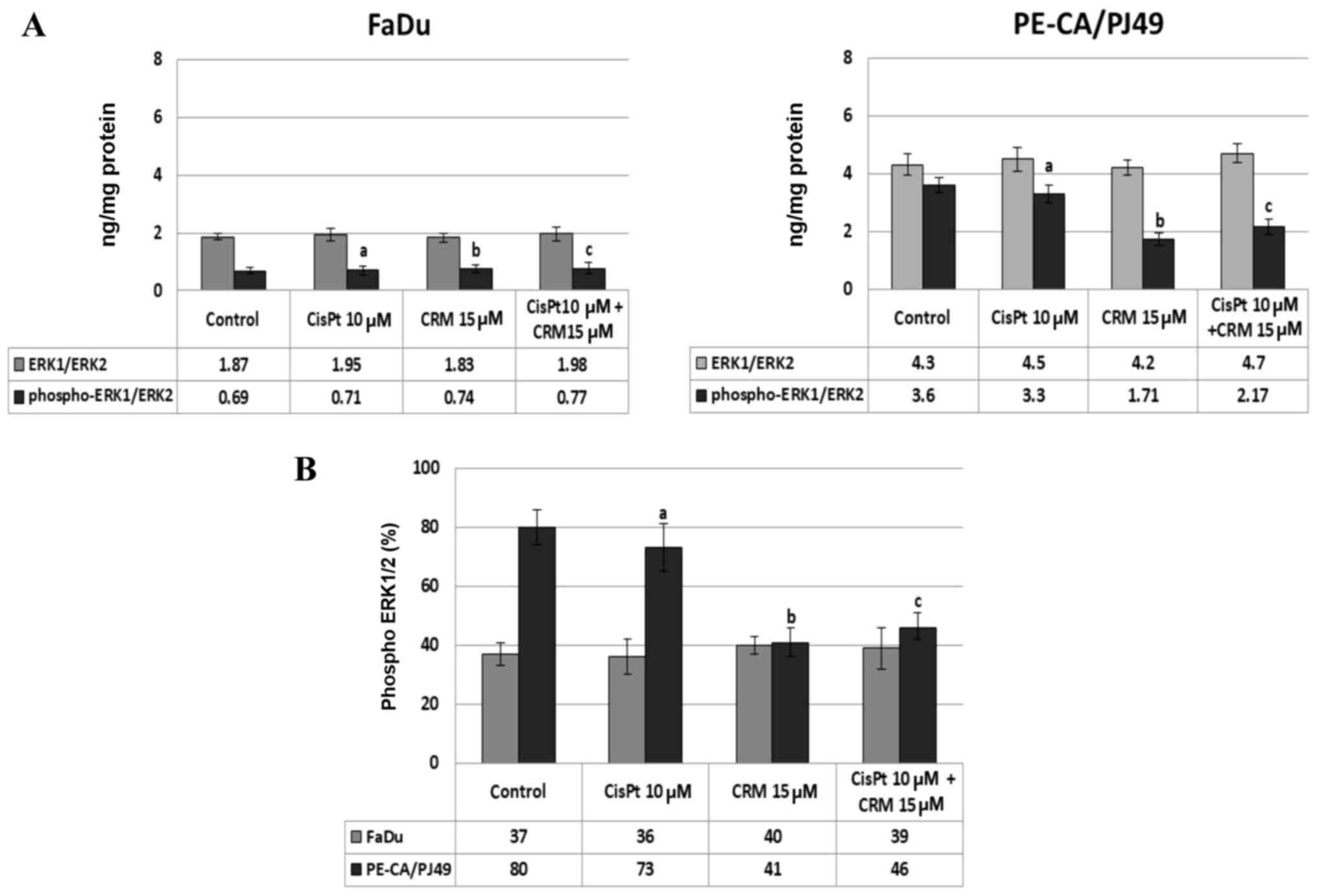
cells. The data show that in the tumor cells treated with CisPt
and/or CRM for different time-points, the ERK1/2 protein-kinase is
highly expressed after 24 h treatment. The untreated PE-CA/PJ49
cells (control) have a statistically significant higher expression
of total ERK1/2 (4.3 ng/mg protein) compared to untreated FaDu
cells (1.87 ng/mg protein) (Fig. 3).
The CisPt and/or CRM treatment applied individually or in
combination did not significantly modify the expression of total
ERK1/2 form in the two analyzed tumor cell lines (Fig. 3A). In order to analyze the effect of
CisPt and/or CRM treatment on the activation of ERK1/2, the level
of ERK1/2 phosphorylation (% phospho-ERK1/ERK2) was quantified. The
results show that FaDu cells responded differently to treatment
compared to PE-CA/PJ49 cells.
In untreated FaDu cells 37% of ERK1/2 protein was
phosphorylated and under the 10 µM CisPt and/or 15 µM CRM treatment
the phosphorylation was not significantly modified compared to the
untreated cells (Fig. 3A and B). In
untreated PE-CA/PJ49 cells 80% of ERK1/2 was phosphorylated
(Fig. 3B), 10 µM CisPt alone
slightly reduced the level of phosphorylation of ERK1/2 to 73% (a;
P=0.03), and 15 µM CRM per se induced a significant inhibition of
ERK1/2 phosphorylation (41%, b; P=0.003) (Fig. 3B).
The combined treatment, CisPt and CRM, led to a
significant reduction (42%) of the ERK1/2 phosphorylation compared
to the control (c; P=0.004) (Fig.
3B). The data show that PE-CA/PJ49 cells having TP53
amplification are more sensitive to CRM treatment. CRM individually
or in combination with CisPt can inhibit phosphorylation of protein
ERK1/2 (Fig. 3B). FaDu having a TP53
deletion and constitutively a lower expression of phospho ERK1/2,
does not respond to any of the treatments (Fig. 3B).
ERK1/2 activation correlates with the
proliferation of tumor cells in HNSCC treated with cisplatin and/or
curcumin
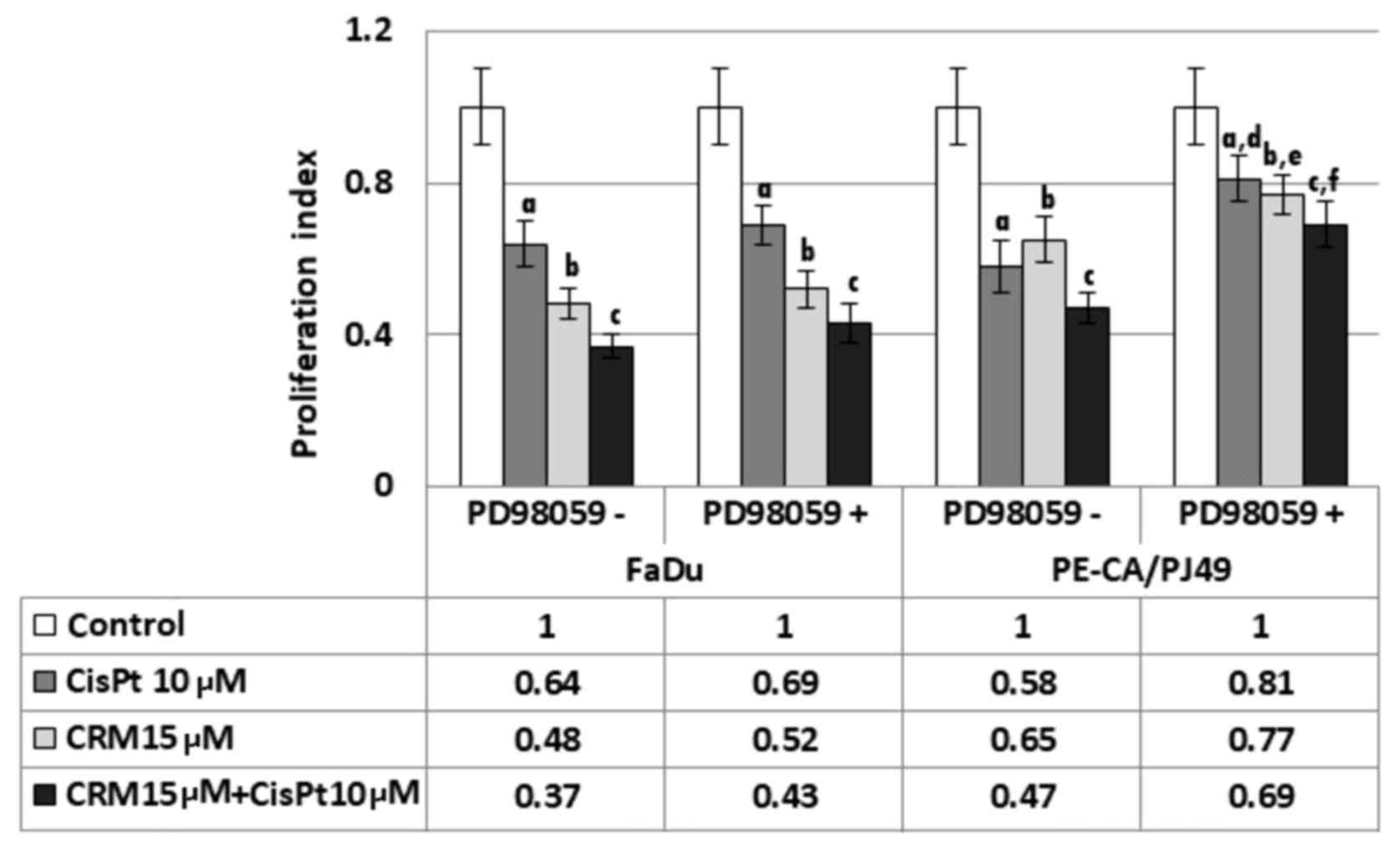
PE/CA-PJ49 and FaDu cells were treated with CisPt
and/or CRM for 6, 24 and 48 h. The cell proliferation index (PI)
was not significantly changed during 6 h treatments. Moreover, PI
for 48 h treatments was not significantly different compared to the
24 h of treatment. CRM treated cells also had a decreased PI (b;
P=0.0004) for FaDu and for PE/CA-PJ49 (b; P=0.0006) compared to the
control (Fig. 4). CisPt treatment
for 24 h determined a significant decrease of the proliferation
process compared to the untreated cells, both in the case of
tumoral cells FaDu (a; P=0.003) and in the cells PE/CA-PJ49 (a;
P=0.0009). CRM treated cells also had a decreased PI (b; P=0.0004)
for FaDu and for PE/CA-PJ49 (b; P=0.0006) compared to the control.
A marked PI inhibition was observed in both cell lines when the
cells were treated simultaneously with CisPt and CRM (Fig. 4). The results are statistically
significant when compared to the control (FaDu cells (c); P=0.0006
and PE/CA-PJ49 (c) P=0.0005) (Fig.
4). Based on the obtained data, CRM had the capacity to
potentiate the effect induced by CisPt treatment on human head and
neck cancer cell lines (Fig. 4).
Since the basal activation status of protein-kinase
ERK1/2 in the analyzed tumor cell lines is different in the
investigated cell lines, we studied the effect of ERK1/2 activation
on the proliferation process as response to the drug treatment.
Thus, the tumor cells were pretreated for 2 h with a specific
ERK1/2 inhibitor, 25 µM PD98059 (concentration determined after
sketching the dose-effect curve) was used for pretreatments, and
then the cells were treated with CisPt and/or CRM for another 24 h.
FaDu cells have a TP53 deletion (Fig.
1) and a constitutively lower expression of phospho ERK1/2
(Fig. 3B). The presence of the
specific inhibitor PD98059 did not influence significantly the
proliferation process compared to control cells (no. PD98059)
(P>0.05) (Fig. 4).
PE-CA/PJ49 cells have TP53 amplification (Fig. 1) and a constitutively high expression
of phospho ERK1/2 (Fig. 3B).
Pretreating with the PD98059 inhibitor of the PE-CA/PJ49 cells
induced an increase of the proliferative activity in the case of
treated cells with CisPt (d; P=0.005) or CRM (e; P=0.007). Similar
effect was obtained in the case of the combined treatment of CRM
and CisPt (f; P=0.003), compared to the cells that were subject to
the same treatment, but in the absence of the PD98059 inhibitor
(Fig. 4). These results show that
the inhibition of the ERK1/2 activity facilitates the restoration
of the proliferative process of the CisPt and/or CRM treated
PE-CA/PJ49 cells. These observations lead to the hypothesis that
proliferation of tumor cells depends on the level of protein-kinase
ERK1/2 activation.
The role of ERK1/2 in the activation
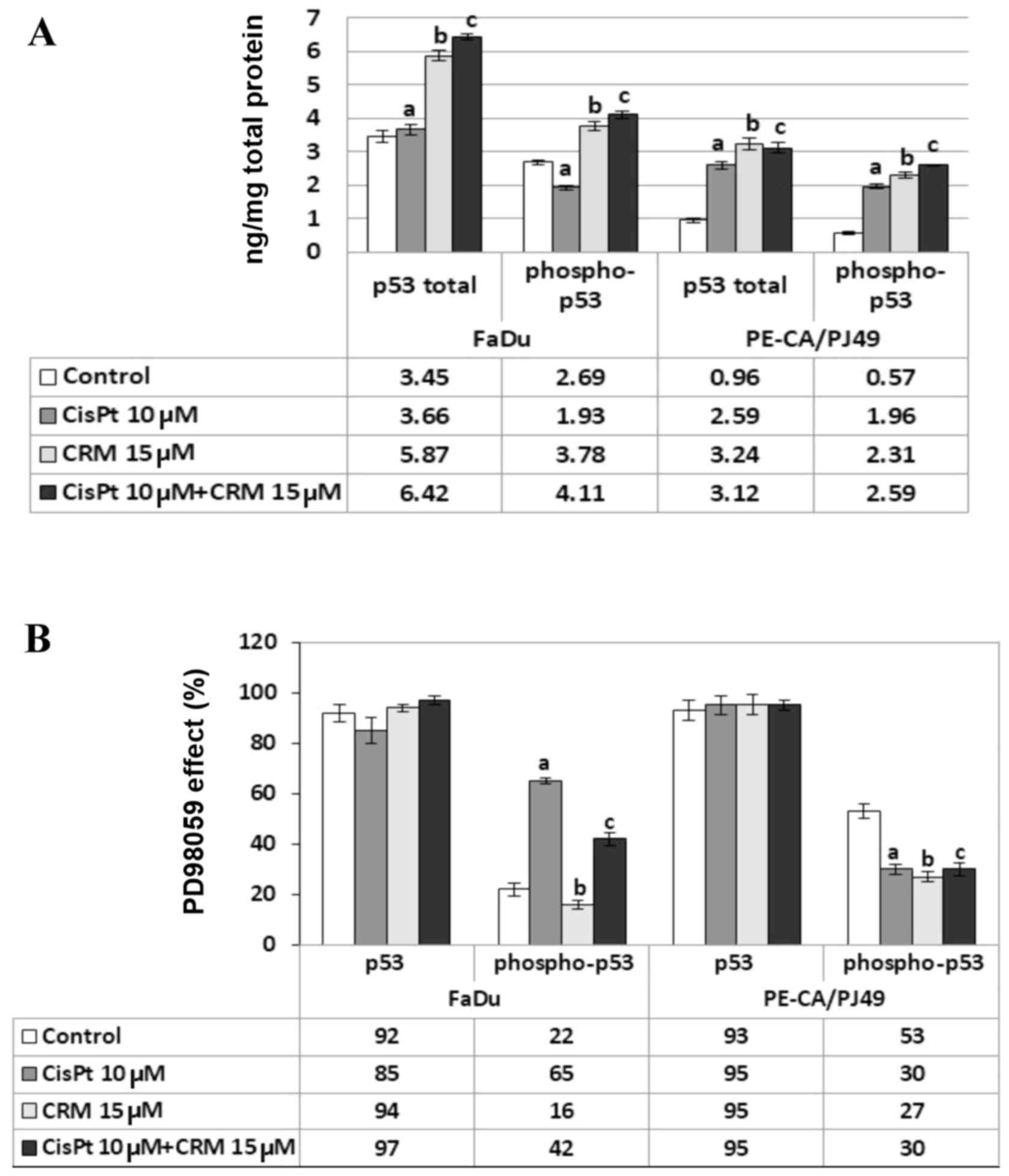
of p53 in HNSCC cells treated with CisPt and/or CRM
FaDu and PE/CA-PJ49 were treated for 24 h as
described, 10 µM CisPt (a; P>0.05) did not affect the expression
of total p53 protein in FaDu cells (Fig.
5A). On the contrary, 15 µM CRM alone (b; P=0.003) or combined
with CisPt (c; P=0.0001) induced a significant increase of total
p53 expression compared to untreated cells (Fig. 5A). In the same cell line, the
expression of phospho-p53 protein was inhibited by CisPt treatment
compared to untreated cells (a; P=0.003) (Fig. 5A). The expression of phospho-p53
showed a significant increase in the presence of either CRM alone
(b; P=0.005) or combined with CisPt (c; P=0.0007) compared to
untreated cells (Fig. 5A).
The analysis of total p53 expression in PE/CA-PJ49
cells showed that either CisPt (a; P=0.00006) or CRM treatment
applied alone (b; P=0.002) induced a significant increase compared
to control cells (Fig. 5A). The same
result was obtained when cells were treated with the combined
treatment (c;P=0.0001) (Fig. 5A).
The p53 phosphorylation in PE/CA-PJ49 cells was amplified by either
CisPt (a; P=0.001) or CRM (b; P=0.0005) treatment compared to
untreated cells. The combined treatment of CisPt and CRM amplified
the expression of phospho-p53 (c; P=0.0002) compared to control,
but the effect of the two agents was not additive (Fig. 5A).
In order to analyze the role of protein kinase
ERK1/2 in the process of p53 phosphorylation, the HNSCC tumor cells
were pretreated with 25 µM PD98059 (specific inhibitor of ERK1/2)
for 2 h. Then, the cells were treated with CisPt and/or CRM for
another 24 h.
As Fig. 5B depicts
the presence of PD98059 inhibitor did not significantly influence
the level of total p53 expression (P>0.05). Untreated FaDu cells
in the presence of PD98059 inhibitor expressed phospho-p53 in 22%,
while in the cells treated with CRM in 16% (b; P=0.03). In the case
of cells treated with CisPt alone the process of phosphorylation
seems to be less affected by the presence of PD98059, the
phosphorylated form of protein p53 being expressed in 65% (a;
P=0.0007). The combined CisPt and CRM treatment in the presence of
ERK1/2 inhibitor led to the decrease of the expression of
phosphorylated p53 to 42% (c; P=0.0004) (Fig. 5B).
In the PE/CA-PJ49 untreated cells, the presence of
PD98059 reduced the phospho-p53 expression to 53% (Fig. 5B). The presence of the specific
ERK1/2 inhibitor significantly affected the level of p53
phosphorylation induced by either CisPt (30%) (a; P=0.007) or CRM
(27%) (b; P=0.005) treatment. The combined CisPt and CRM treatment
in the presence of PD98059 kept the phosphorylated form of protein
p53 to 30% (c; P=0.007) since the effect of the two agents was not
additive (Fig. 5B).
ERK1/2 in the modulation of HNSCC
apoptosis induced by CisPt and/or CRM treatment
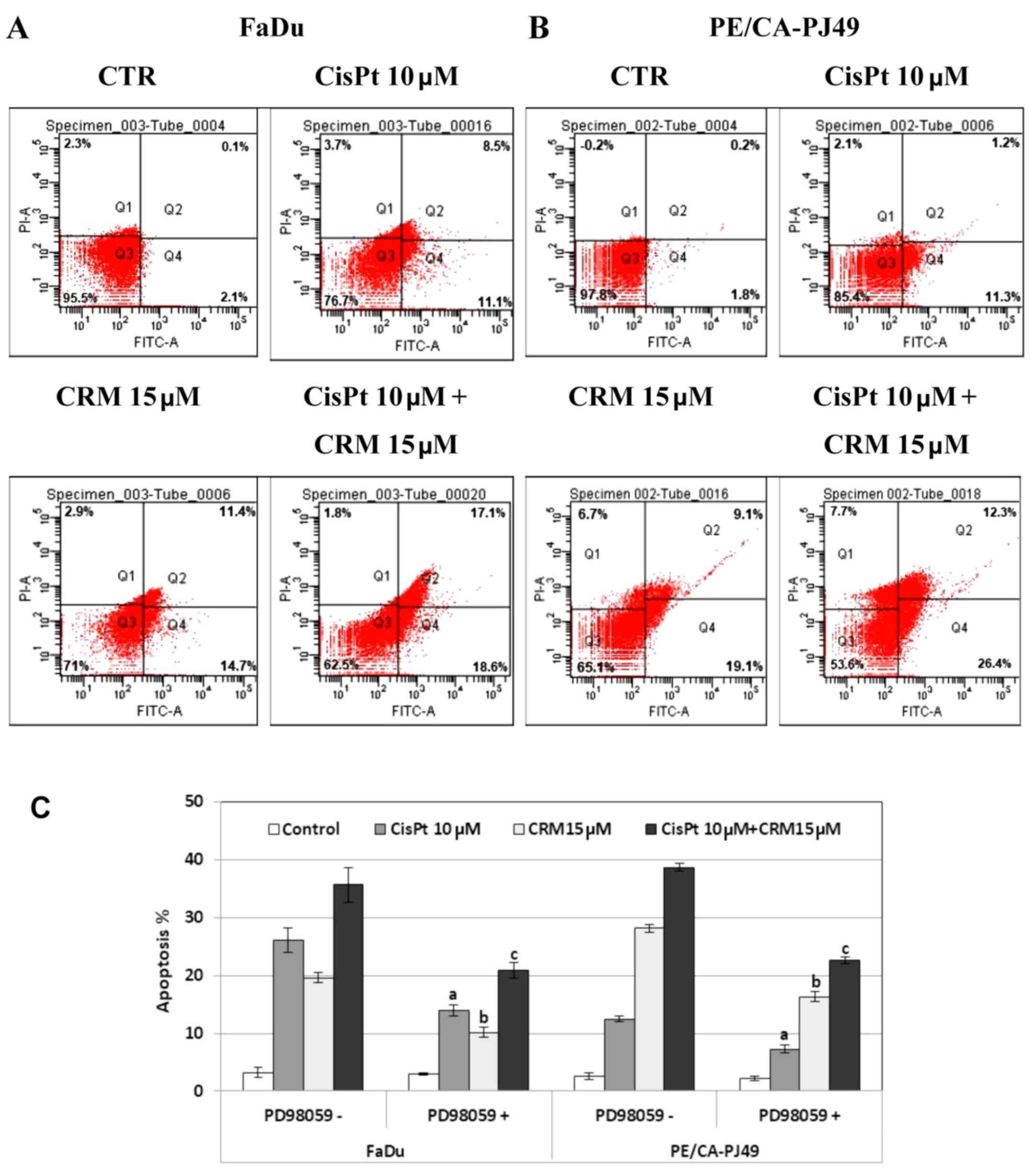
FaDu and PE/CA-PJ49 were treated for 24 h with 10 µM
CisPt, 15 µM CRM, combined treatment and compared to control cells
(untreated). CisPt treated FaDu cells had 9 times enhancement of
the apoptotic events (19.6%) compared to untreated cells (2.2%).
CRM treated FaDu cells had a 12 times higher apoptosis (26.1%) than
untreated cells. The combined treatment of CisPt and CRM induced
35.7% apoptosis. This indicates a 16 times higher apoptosis than in
the untreated cells. Moreover, apoptosis was almost twice higher
(1.8×) than the value obtained for CisPt treatment alone. This
indicates that CRM amplified the apoptotic process and supported
the tumoricidal effect of CisPt (Fig.
6A).
CisPt treatment of PE/CA-PJ49 cells induced a 6×
higher apoptosis (12.5%) compared to untreated cells (2.0%). In the
same manner, apoptosis induced by CRM alone (28.8%) or combined
treatment of CisPt and CRM (38.7%) was 14× and respectively 19×
higher compared to untreated cells. CRM significantly amplified the
effect induced by CisPt treatment on the PE/CA-PJ49 apoptotic
process since apoptosis was 3× higher than the value obtained for
CisPt treatment (Fig. 6B).
Our data support the CRM apoptosis inducer
capabilities considering that CRM had the capacity to induce
apoptosis in both tumor lines. Also, CRM potentiated the effect
induced by CisPt treatment.
In order to analyze the role of ERK1/2 protein
kinase in the modulation of the apoptotic process, HNSCC tumor
cells were pretreated for 2 h with 25 µM ERK1/2 specific inhibitor,
PD98059. Then the cells were treated for 24 h with CisPt and/or
CRM, and subjected to flow cytometric analysis of the apoptotic
process. As shown in Fig. 6C, the
PD98059 inhibition of ERK1/2 protein kinase led to a decrease of
the apoptotic cell percentage in both cell lines compared to
untreated cells. The decreased was statistically significant for
all treatments as follows: FaDu cells (a) CisPt treated P=0.007;
(b) CRM treated P=4.4E-05; (c) combined treatment; P=0.004;
PE/CA-PJ49 cells (a) CisPt treated P=0.006; (b) CRM treated
P=5.1E-05; (c) combined treatment P=0.0003. These results
demonstrate the involvement of ERK1/2 protein kinase in the
apoptotic mechanisms induced by CisPt and/or CRM on the analyzed
tumor cells.
Discussion
The essential step during carcinogenesis of oral
squamous cell carcinomas is the acquisition of genetic instability
that occurs at the nucleotide or the chromosome level (48). The genotypic abnormalities such as
polysomy of chromosome 17 may be associated with the development of
an early recurrence or second primary tumors and can influence the
therapy response (49). In this
study the effect of CisPt and/or CRM treatment on two HNSCC tumor
cell lines (FaDu and PE/CA-PJ49) were analyzed. One of the most
frequent genetic abnormalities associated with HNSCC affects p53
onco-supressor gene. Abnormalities in the p53 gene cause an
inefficient checkpoint system for the repair and destruction of
mutant cells. The cell lines were analyzed using FISH analysis to
detect the possible alterations of chromosome 17 involving the TP53
gene. PE/CA-PJ49 cell line had an amplification of the TP53 gene
associated with polysomy in chromosome 17. FaDu tumor line
presented a deletion in gene TP53, without chromosome 17
modifications. This data can explain the different response of each
cell line to CisPt and/or CRM treatment.
Genetic modifications, as well as p53 protein
expression alterations could influence the activation of several
intracellular signaling pathways, such as ERK1/2 and therefore it
could contribute to the modulation of the response to therapy. In
many studies, the ERK signaling pathway is associated with
proliferative (50) and cellular
differentiation processes (51) on
one hand, and on the other hand, there are studies showing that
this pathway has a role in the apoptotic process (52–54).
This suggests its importance in the modulation of the response to
antitumor therapy (55). The
activation status of ERK1/2 in HNSCC cell lines was evaluated to
establish if the cellular response to CisPt and/or CRM treatment is
influenced by the level of ERK1/2 activation. Expression of the
total and phosphorylated ERK1/2 protein was quantified in treated
and untreated cells. The data show that the total and
phosphorylated ERK1/2 protein expression is much higher in
untreated PE/CA-PJ49 than in FaDu cells. FaDu cell line treated
with CisPt and/or CRM did not show a significant change of ERK1/2
phosphorylation. A significant decrease of phospho ERK1/2 was
observed in PE/CA-PJ49 cells treated with CisPt and CRM. The
constitutively expressed activated ERK1/2 protein-kinase was
different in the two tumor cell lines. This was reflected in
downstream events such as the cell proliferation process. The
inhibition of ERK1/2 activity using PD98059 did not significantly
affect the proliferation of the cells (e.g. FaDu) having a low
expression of the protein. Cells with a constitutively high
expression of ERK1/2 protein-kinase (PE/CA-PJ49) responded to the
presence of the inhibitor by reversing the proliferative process.
This suggests the existence of an ERK1/2 protein-kinase activation
threshold which modulates the proliferative process of tumor
cells.
How p53 responds to the drug treatment of tumor
cells having a different expression pattern of ERK1/2 was
evaluated. In untreated cells, the expression of total and
phosphorylated p53 was higher in FaDu compared to PE/CA-PJ49 cells
(Fig. 5A). This difference can be
associated with the amplification of p53 gene in PE/CA-PJ49 cells
and the p53 deletion in tumor cells FaDu (Fig. 1). CRM treatment induced an increase
of the total p53 protein expression associated with an increase of
the phosphorylation process in both cell lines. The response of
FaDu cells to CisPt treatment led to a decrease of the
phosphorylation process without affecting the expression of total
p53 protein, while CisP induced a significant increase of p53
phosphorylation in PE/CA-PJ49 tumor cells (Fig. 5A and B).
The response of FaDu CRM treated cells in the
presence of ERK1/2 inhibitor PD98059 suggest that the ERK1/2 might
be involved in the p53 phosphorylation. The response of FaDu CisPt
treated cells to the presence of the same inhibitor suggests that
p53 upstream events might follow a different pathway than ERK1/2.
Regardless of the treatment of PE/CA-PJ49 cells, phosphorylation of
p53 involved ERK1/2 activation. Moreover, the results support the
involvement of ERK1/2 and phospho p53 in CisPt and/or CRM induced
apoptosis. Many studies support the antitumor and pro-apoptotic
effect of CRM (56–58). It is not well known if the effect of
CRM is due to the existence of a link between the activation level
of ERK1/2 and the apoptotic process. The results of our study show
that CRM increased the apoptotic process in both tumor cell lines.
CRM acts as an apoptosis-inducer factor and more importantly
potentiates the effect induced by CisPt treatment (Fig. 6).
Conclusions and future directions. The results
showed that the tumor cell line FaDu presented a deletion of the
TP53 gene, while cell line PE-CA/PJ49 presented polysomy. Both
modifications are associated with cell proliferation and response
to therapy. The use of an adjuvant (CRM) can increase the
efficiency of chemotherapy (CisPt) effect by modulating cell
activation processes such as ERK1/2 phosphorylation. The presence
of the adjuvant can decrease the required dose of drug, therefore
reducing the chemotherapeutic adverse reactions of the agent.
The data show that the ERK1/2 way of action either
on cell proliferation or apoptosis depends on the type of cell
characteristics and therapeutic agents. In order to achieve an
efficient personalized therapy, more investigations are necessary
for a better functional interpretation of the intracellular
signaling pathways.
In conclusion, evaluating the level of ERK1/2
activation in tumor cells can be a useful tool for an
individualized treatment plan, but in order to achieve this goal
extended investigations on patient tumor specimens are needed.
Acknowledgements
Not applicable.
Funding
This study was supported by Grants
PN-III-P1-1.2-PCCDI-2017-0341/2018,
PN-III-P1-1.2-PCCDI-2017-0782/2018, PN 19.29.01.01, and by Ministry
of Research and Innovation in Romania, under Program 1 - The
Improvement of the National System of Research and Development,
Subprogram 1.2 - Institutional Excellence - Projects of Excellence
Funding in RDI, contract no. 7PFE/16.10.2018.
Availability of data and materials
The data sets used and/or analysed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
MB and MTN, designed the research, data acquisition,
analysis and interpretation of data, and wrote the manuscript. MB,
MM and GGPM performed the experiments, data acquisition, analysis
and interpretation of data, statistical analysis and manuscript
drafting. VR, GI, LIB, RH, NR and CC contributed to data collection
and interpretation, statistical analysis, critical revision of the
manuscript for important intellectual content. All the authors read
and approved the final manuscript, and had equal contribution.
Ethics approval and consent to
participate
The study protocol was approved by the Ethics
Committee of ‘Stefan S. Nicolau’ Virology Institute.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CisPt
|
cisplatin
|
|
CRM
|
curcumin
|
|
ERK1/2
|
extracellular signal-regulated
kinase
|
|
FISH
|
fluorescence in situ
hibridization
|
|
PD98059
|
(2′-amino-3′-methoxyflavone)
|
|
MTT
|
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
|
|
DMSO
|
dimethyl sulfoxide
|
|
EDTA
|
etylenediaminetetraacetic acid
|
|
PBS
|
phosphate-buffered saline
|
|
FaDu
|
human pharynx squamous cell
carcinoma
|
|
PE/CA-PJ49
|
human tongue squamous cell
carcinoma
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
|
TP53
|
the tumor suppressor gene TP53
|
|
DAPI II
|
4′,6-Diamidino-2-phenylindole
|
|
SSC buffer
|
the saline-sodium citrate buffer
|
|
NP-40
|
detergent solution
|
|
FITC
|
fluorescein isothiocyanate
|
|
PI
|
propidium iodide
|
|
PMSF
|
phenylmethylsulfonyl fluoride
|
|
ELISA
|
enzyme-linked immunosorbent assay.
|
References
|
1
|
Ursu RG, Danciu M, Spiridon IA, Ridder R,
Rehm S, Maffini F, McKay-Chopin S, Carreira C, Lucas E, Costan VV,
et al: Role of mucosal high-risk human papillomavirus types in head
and neck cancers in Romania. PLoS One. 13:e01996632018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Forman D, Bray F, Brewster DH, Gombe
Mbalawa C, Kohler B, Piñeros M, Steliarova-Foucher E, Swaminathan R
and Ferlay J: Cancer Incidence in Five Continents VolX.
International Agency for Research on Cancer; Lyon: 2014
|
|
3
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kumar M, Nanavati R, Modi TG and Dobariya
C: Oral cancer: Etiology and risk factors: A review. J Cancer Res
Ther. 12:458–463. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vigneswaran N and Williams MD:
Epidemiologic trends in head and neck cancer and aids in diagnosis.
Oral Maxillofac Surg Clin North Am. 26:123–141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vermorken JB, Remenar E, van Herpen C,
Gorlia T, Mesia R, Degardin M, Stewart JS, Jelic S, Betka J, Preiss
JH, et al EORTC 24971/TAX 323 Study Group, : Cisplatin,
fluorouracil, and docetaxel in unresectable head and neck cancer. N
Engl J Med. 357:1695–1704. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jimenez L, Jayakar SK, Ow TJ and Segall
JE: Mechanisms of invasion in head and neck cancer. Arch Pathol Lab
Med. 139:1334–1348. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Voiculescu VM, Caruntu C, Solomon I, Lupu
M, Ilie MA, Boda D, Constantin C and Neagu M: Squamous cell
carcinoma: Biomarkers and potential therapeutic targetsHuman Skin
Cancers - Pathways, Mechanisms, Targets and Treatments. Blumenberg
M: IntechOpen; London: pp. 135–159. 2018
|
|
9
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhai TT, van Dijk LV, Huang BT, Lin ZX,
Ribeiro CO, Brouwer CL, Oosting SF, Halmos GB, Witjes MJH,
Langendijk JA, et al: Improving the prediction of overall survival
for head and neck cancer patients using image biomarkers in
combination with clinical parameters. Radiother Oncol. 124:256–262.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lupu M, Caruntu A, Caruntu C, Boda D,
Moraru L, Voiculescu V and Bastian A: Non-invasive imaging of
actinic cheilitis and squamous cell carcinoma of the lip. Mol Clin
Oncol. 8:640–646. 2018.PubMed/NCBI
|
|
12
|
Boda D, Docea AO, Calina D, Ilie MA,
Caruntu C, Zurac S, Neagu M, Constantin C, Branisteanu DE,
Voiculescu V, et al: Human papilloma virus: Apprehending the link
with carcinogenesis and unveiling new research avenues (Review).
Int J Oncol. 52:637–655. 2018.PubMed/NCBI
|
|
13
|
Hassan M, Watari H, AbuAlmaaty A, Ohba Y
and Sakuragi N: Apoptosis and molecular targeting therapy in
cancer. BioMed Res Int. 2014:1508452014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Neagu M, Constantin C, Popescu ID, Zipeto
D, Tzanakakis G, Nikitovic D, Fenga C, Stratakis CA, Spandidos DA
and Tsatsakis AM: Inflammation and metabolism in cancer cell -
mitochondria key player. Front Oncol. 9:3482019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cagnol S and Chambard JC: ERK and cell
death: Mechanisms of ERK-induced cell death - apoptosis, autophagy
and senescence. FEBS J. 277:2–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang J, Zhang J, Shi C, Liu L and Wei Y:
Survival, recurrence and toxicity of HNSCC in comparison of a
radiotherapy combination with cisplatin versus cetuximab: A
meta-analysis. BMC Cancer. 16:6892016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Neagu M, Caruntu C, Constantin C, Boda D,
Zurac S, Spandidos DA and Tsatsakis AM: Chemically induced skin
carcinogenesis: Updates in experimental models (Review). Oncol Rep.
35:2516–2528. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kass JI, Moskowitz HS and Grandis JR:
Oncogenomics/Proteomics of Head and Neck Cancers. Head and Neck
CancerMultimodality Management. 2nd. Springer International
Publishing; New York, NY: pp. 101–114. 2016
|
|
19
|
Chen L and Tweddle DA: p53, SKP2, DKK3 as
MYCN target genes and their potential therapeutic significance.
Cancer Molecular Targets and Therapeutic. Front Oncol. 2:1732012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boda D: Cellomics as integrative omics for
cancer. Curr Proteomics. 10:237–245. 2013. View Article : Google Scholar
|
|
21
|
Powell E, Piwnica-Worms D and
Piwnica-Worms H: Contribution of p53 to metastasis. Cancer Discov.
4:405–414. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Samatar AA and Poulikakos PI: Targeting
RAS-ERK signalling in cancer: Promises and challenges. Nat Rev Drug
Discov. 13:928–942. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Solomon I, Voiculescu VM, Caruntu C, Lupu
M, Popa A, Ilie MA, Albulescu R, Caruntu A, Tanase C, Constantin C,
et al: Neuroendocrine factors and head and neck squamous cell
carcinoma: An affair to remember. Dis Markers. 2018:97878312018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang SH, Sharrocks AD and Whitmarsh AJ:
MAP kinase signalling cascades and transcriptional regulation.
Gene. 513:1–13. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lupu M, Caruntu A, Caruntu C, Papagheorghe
LM, Ilie MA, Voiculescu V, Boda D, Constantin C, Tanase C, Sifaki
M, et al: Neuroendocrine factors: The missing link in non melanoma
skin cancer (Review). Oncol Rep. 38:1327–1340. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Burotto M, Chiou VL, Lee JM and Kohn EC:
The MAPK pathway across different malignancies: A new perspective.
Cancer. 120:3446–3456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Urosevic J, Nebreda AR and Gomis RR: MAPK
signaling control of colon cancer metastasis. Cell Cycle.
13:2641–2642. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tanase CP, Neagu M and Albulescu R: Key
signaling molecules in pituitary tumors. Expert Rev Mol Diagn.
9:859–877. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang M and Huang CZ: Mitogen-activated
protein kinase signaling pathway and invasion and metastasis of
gastric cancer. World J Gastroenterol. 21:11673–11679. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peng Q, Deng Z, Pan H, Gu L, Liu O and
Tang Z: Mitogen-activated protein kinase signaling pathway in oral
cancer. Oncol Lett. 15:1379–1388. 2018.PubMed/NCBI
|
|
31
|
Kumari R, Chouhan S, Singh S, Chhipa RR,
Ajay AK and Bhat MK: Constitutively activated ERK sensitizes cancer
cells to doxorubicin: Involvement of p53-EGFR-ERK pathway. J
Biosci. 42:31–41. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang J, Luo L, Wang D, Guo B, Li J, Yang Z
and Tang D: Combination adjuvant chemotherapy with targeted drugs
for treatment of colorectal cancer: A network meta-analysis. J Cell
Biochem. 119:1521–1537. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bailon-Moscoso N, Cevallos-Solorzano G,
Romero-Benavides JC and Orellana MI: Natural compounds as
modulators of cell cycle arrest: Application for anticancer
chemotherapies. Curr Genomics. 18:106–131. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Seca AM and Pinto DC: Plant secondary
metabolites as anticancer agents: Successes in clinical trials and
therapeutic application. Int J Mol Sci. 19:E2632018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shanmugam MK, Rane G, Kanchi MM, Arfuso F,
Chinnathambi A, Zayed ME, Alharbi SA, Tan BK, Kumar AP and Sethi G:
The multifaceted role of curcumin in cancer prevention and
treatment. Molecules. 20:2728–2769. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Miao Y, Zhao S, Gao Y, Wang R, Wu Q, Wu H
and Luo T: Curcumin pretreatment attenuates inflammation and
mitochondrial dysfunction in experimental stroke: The possible role
of Sirt1 signaling. Brain Res Bull. 121:9–15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shi D, Xu Y, Du X, Chen X, Zhang X, Lou J,
Li M and Zhuo J: Co-treatment of THP-1 cells with naringenin and
curcumin induces cell cycle arrest and apoptosis via numerous
pathways. Mol Med Rep. 12:8223–8228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Patel PB, Thakkar VR and Patel JS:
Cellular effect of curcumin and citral combination on breast cancer
cells: Induction of apoptosis and cell cycle arrest. J Breast
Cancer. 18:225–234. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Neagu M, Constantin C, Tanase C and Boda
D: Patented biomarker panels in early detection of cancer. Recent
Pat Biomark. 1:10–24. 2011. View Article : Google Scholar
|
|
40
|
Chen XY, Cai HZ, Wang XY, Chen QY, Yang H,
Chen YJ and Tang YP: Application of the ERK signaling pathway
inhibitor PD98059 in long-term in vivo experiments. Genet Mol Res.
14:18325–33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Barltrop JA, Owen TC, Cory AH and Cory JG:
5-(3-carboxymethoxyphenyl)-2-(4,5-dimenthylthiazoly)-3-(4-sulfophenyl)tetrazolium,
inner salt (MTS) and related analogs of
3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT)
reducing to purple water-soluble formazans as cell-viability
indicators. Bioorg Med Chem Lett. 1:611–614. 1991. View Article : Google Scholar
|
|
42
|
Petrică-Matei GG, Iordache F, Hainăroşie R
and Bostan M: Characterization of the tumor cells from human head
and neck cancer. Rom J Morphol Embryol. 57 (Suppl 2):791–799.
2016.PubMed/NCBI
|
|
43
|
Hirshberg A, Yarom N, Amariglio N, Yahalom
R, Adam I, Stanchescu R, Ben-Dov I, Taicher S, Rechavi G and
Trakhtenbrot L: Detection of non-diploid cells in premalignant and
malignant oral lesions using combined morphological and FISH
analysis - a new method for early detection of suspicious oral
lesions. Cancer Lett. 253:282–290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Salido M, Tusquets I, Corominas JM, Suarez
M, Espinet B, Corzo C, Bellet M, Fabregat X, Serrano S and Solé F:
Genetic alterations of chromosome 17 in human breast carcinoma
studied by fluorescence in situ hybridization and molecular DNA
techniques. Breast Cancer Res. 7:267–273. 2005. View Article : Google Scholar
|
|
45
|
Sunahori K, Nagpal K, Hedrich CM, Mizui M,
Fitzgerald LM and Tsokos GC: The catalytic subunit of protein
phosphatase 2A (PP2Ac) promotes DNA hypomethylation by suppressing
the phosphorylated mitogen-activated protein kinase/extracellular
signal-regulated kinase (ERK) kinase (MEK)/phosphorylated ERK/DNMT1
protein pathway in T-cells from controls and systemic lupus
erythematosus patients. J Biol Chem. 288:21936–21944. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bundscherer A, Malsy M, Lange R, Hofmann
P, Metterlein T, Graf BM and Gruber M: Cell harvesting method
influences results of apoptosis analysis by Annexin V staining.
Anticancer Res. 33:3201–3204. 2013.PubMed/NCBI
|
|
47
|
Zedan W, Mourad MI, El-Aziz SMA, Salamaa
NM and Shalaby AA: Cytogenetic significance of chromosome 17
aberrations and P53 gene mutations as prognostic markers in oral
squamous cell carcinoma. Diagn Pathol. 10:22015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jin C, Jin Y, Wennerberg J, Akervall J,
Dictor M and Mertens F: Karyotypic heterogeneity and clonal
evolution in squamous cell carcinomas of the head and neck. Cancer
Genet Cytogenet. 132:85–96. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Papavasileiou D, Tosios K, Christopoulos
P, Goutas N and Vlachodimitropoulos D: Her-2 immunohistochemical
expression in oral squamous cell carcinomas is associated with
polysomy of chromosome 17, not Her-2 amplification. Head Neck
Pathol. 3:263–270. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang L, Liu T, Nishioka M, Aguirre RL, Win
SS and Okada N: Activation of ERK1/2 and cyclin D1 expression in
oral tongue squamous cell carcinomas: Relationship between
clinicopathological appearances and cell proliferation. Oral Oncol.
42:625–631. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chambard JC, Lefloch R, Pouysségur J and
Lenormand P: ERK implication in cell cycle regulation. Biochim
Biophys Acta. 1773:1299–1310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Aguzzi A, Maggioni D, Nicolini G, Tredici
G, Gaini RM and Garavello W: MAP kinase modulation in squamous cell
carcinoma of the oral cavity. Anticancer Res. 29:303–308.
2009.PubMed/NCBI
|
|
53
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Engelbrecht AM, Gebhardt S and Louw L: Ex
vivo study of MAPK profiles correlated with parameters of apoptosis
during cervical carcinogenesis. Cancer Lett. 235:93–99. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Squatrito M, Brennan CW, Helmy K, Huse JT,
Petrini JH and Holland EC: Loss of ATM/Chk2/p53 pathway components
accelerates tumor development and contributes to radiation
resistance in gliomas. Cancer Cell. 18:619–629. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hasima N and Aggarwal BB: Cancer-linked
targets modulated by curcumin. Int J Biochem Mol Biol. 3:328–351.
2012.PubMed/NCBI
|
|
57
|
Baharuddin P, Satar N, Fakiruddin KS,
Zakaria N, Lim MN, Yusoff NM, Zakaria Z and Yahaya BH: Curcumin
improves the efficacy of cisplatin by targeting cancer stem-like
cells through p21 and cyclin D1-mediated tumour cell inhibition in
non-small cell lung cancer cell lines. Oncol Rep. 35:13–25. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Borges GÁ, Rêgo DF, Assad DX, Coletta RD,
De Luca Canto G and Guerra EN: In vivo and in vitro effects of
curcumin on head and neck carcinoma: A systematic review. J Oral
Pathol Med. 46:3–20. 2017. View Article : Google Scholar : PubMed/NCBI
|