Introduction
Caries bacteria are a major cause of dental pulpal
inflammation and infection (1), and
the dental pulp is surrounded by a rigid physical barrier that
resists pathogenic challenges (2).
The integrity of the barrier undergoes inevitable and irreversible
changes due to adverse conditions such as aging, and the caries
bacteria and their products penetrate into the pulp tissue if the
integrity of the barrier is damaged (2). Gram-negative anaerobic bacteria become
dominant in the microflora as carious infection progresses to the
pulp-dentin interface (3).
Lipopolysaccharide (LPS), the major cell wall component of
gram-negative bacteria, is widely used as a pathogen-associated
molecule to mimic clinical pathological conditions (4,5). Studies
have increasingly demonstrated that cell death is caused by
apoptosis, necrosis or pyroptosis, which can be induced by
inflammation (6,7). These findings suggest that bacterial
infections may trigger an inflammatory response that can lead to
cell death.
Pyroptosis, often referred to as inflammatory cell
death, induces the release of inflammatory cytokines interleukin
(IL) 1β and IL-18 by activating inflammatory complexes composed of
caspase-1 (8). NOD-like receptor
protein 3 (NLRP3) inflammasome-mediated caspase-1 activation and
the subsequent secretion of pro-inflammatory cytokines including
IL-1β and IL-18 are markers for pyroptosis (9). NLRP3 inflammasome has been reported to
be expressed in dental pulp tissue (10). However, whether the NLRP3
inflammasome/caspase-1/IL-1β/IL-18 signaling pathway in dental pulp
cells is activated during dental pulp inflammation has not yet been
investigated to the best of the authors' knowledge. The NF-κB
family of inducible transcription factors serve an essential role
in different aspects of immune responses (11). A previous study has demonstrated that
the activation of NF-κB may promote the transcriptional expression
of inflammatory factors (12). The
major NF-κB inhibitor (IκB) family member that regulates the
classical NF-κB pathway is IκBα, which is characterized by its
dynamic changes and signal-induced NF-κB activation (11). Thus, targeting the NF-κB pathway may
be an effective therapy for dental pulpal inflammation.
As a highly conserved catabolic pathway, autophagy
is involved in multiple pathological conditions and in numerous
physiological processes including cell death (13) and differentiation (14), neurodegeneration (15), immunity (16) and organ development (17,18).
Previous studies have also revealed that autophagy participates in
odontoblast aging (19) and tooth
development (20). Autophagy is
usually accompanied by changes in microtubule-associated protein 1
light chain 3 (LC3) α and p62 (21,22).
Autophagy exerts protective roles in the early stage of inflamed
odontoblasts, whereas the over-induction of autophagy leads to
odontoblast cell death (23). LPS
can stimulate autophagy in endothelial cells, cardiomyocytes and
macrophages (24–26); however, the effects of autophagy on
LPS-induced cytotoxicity and whether autophagy in dental pulp cells
could be activated by LPS have yet to be investigated.
The present study hypothesized that LPS induced
NLRP3/caspase-1-dependent pyroptosis in dental pulp cells, which
may be inhibited by autophagy and the NF-κB signaling pathway.
Therefore, the aims of the present study were to: i) Investigate
whether pyroptosis in dental pulp cells was activated in response
to LPS; and ii) elucidate the effects of autophagy during the
process and determine if the NF-κB signal pathway was involved.
Materials and methods
Cell isolation, culture and
identification
As described previously (27), human dental pulp cells were isolated
from intact, caries-free supernumerary teeth that had been freshly
extracted from six healthy children aged between 7 and 10 years
old. The study protocol was approved by the Ethical Committee of
Nanjing Medical University (approval no. NM20180510). Written
informed consent was obtained from the children's parents or legal
representatives. The teeth were maintained in phosphate buffer
saline (PBS) and split open following extraction. Under sterile
conditions, the dental pulp tissues were removed and minced using a
surgical knife, transferred to a centrifuge tube and centrifuged at
300 × g for 5 min at room temperature. The supernatant was removed
and the sample was incubated at 37°C for 45 min with type I
collagen and centrifuged again at 300 × g for 5 min at room
temperature. Following removal of the supernatant, the sample was
washed three times with α-MEM medium (Gibco; Thermo Fisher
Scientific, Inc.) containing 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.) and centrifuged at 300 × g for 5
min at room temperature. The obtained cells were suspended in a-MEM
medium containing 20% FBS, seeded in cell culture bottles and
incubated at 37°C with 5% CO2.
Dental pulp cells at the logarithmic growth phase at
the third passage were washed with PBS twice and digested with PBS
containing 0.25% trypsin for single-cell suspension preparation.
The cell suspension (100 µl) was added to an Eppendorf tube
(3×109 cells/tube) and incubated with rat-anti-human
monoclonal antibodies CD29-PE (cat. no. bs-0486R-PE; 1:100),
CD105-PE (cat. no. bs-0579R-PE; 1:100), CD146-PE (cat. no.
bs-1618R-PE; 1:100), CD34-PE (cat. no. bs-0646R-PE; 1:100), CD45-PE
(cat. no. bs-0522R-PE; 1:100), CD90-PE (cat. no. bs-0778R-PE;
1:100; all from Bioss Biotechnology Co., Ltd.) and rat-anti-human
STRO-1 (cat. no. ab214086; 1:500; Abcam) at 4°C for 30 min,
followed by incubation for 15 min in the dark, washing with PBS,
centrifugation at 300 × g for 5 min at room temperature, suspension
and fixation with PBS containing 1% paraformaldehyde. Background
markers were identified using homotype controlled monoclonal
antibodies. Flow cytometry (Becton, Dickinson and Company) and
FlowJo software version 7.6.2 (FlowJo LLC) were used to analyze the
cells. According to the results, the cells with the best purity,
defined as the highest positive cell surface antigen rate, were
selected for subsequent experiments.
Pure dental pulp cells at passages 3 to 4 were used
for drug treatment. Briefly, different concentration of LPS (0, 10,
100, 200, 300, 500 and 1,000 µg/l), rapamycin (2 µM),
3-methyladenine (3-MA; 10 mM) and BAY11-7082 (2.5 µM; all from
Sigma-Aldrich; Merck KGaA) were used to pretreat the dental pulp
cells for 72 h.
Western blot analysis
Western blot analysis was conducted as previously
described (28). The total protein
was extracted from human dental pulp cells (2×106) using
RIPA lysis buffer containing Halt Protease and Phosphatase
Inhibitor Cocktail (Pierce; Thermo Fisher Scientific, Inc.).
Bicinchoninic acid protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.) was applied to determine the protein
concentration. Proteins (30 µg/lane) were separated by SDS-PAGE
(10% gel) and transferred to a PVDF membrane (Bio-Rad Laboratories,
Inc.). The membrane was blocked with 5% skim milk in PBS-Tween-20
for 2 h. Membranes were incubated primary antibodies overnight at
4°C, and with secondary antibodies for 2 h at room temperature. In
addition, to detect the protein expression of IL-1β, cell culture
media were collected and the proteins in the media were
precipitated using trichloroacetic acid protein precipitation
method as previously described (29). The band signal was developed using an
Enhanced Chemiluminescence kit (Beyotime Institute of
Biotechnology) and exposed to X-ray film. The relative quantity of
proteins was determined by Image J (version 1.47; National
Institutes of Health) and normalized to loading controls. The
antibodies used were as follows: Anti-β-tubulin (50 kDa; rabbit;
1:1,000; cat. no. ab6046; Abcam), anti-IL-1β (17 kDa; rabbit;
1:1,000; cat. no. 12703; Cell Signaling Technology, Inc.),
anti-pro-IL-1β (17 kDa; rabbit; 1:1,000; cat. no. 83186; Cell
Signaling Technology, Inc.), anti-caspase-1 (45 kDa; rabbit;
1:1,000; cat. no. ab74279; Abcam), anti-pro-caspase-1 (20/22 kDa;
rabbit; 1:1,000; cat. no. 4199; Cell Signaling Technology, Inc.),
anti-phosphorylated (p-)NF-κB (65 kDa; rabbit; 1:1,000; cat. no.
3039; Cell Signaling Technology, Inc.), anti-NF-κB (65 kDa; rabbit;
1:1,000; cat. no. 8242; Cell Signaling Technology, Inc.), anti-IκBα
(36 kDa; rabbit; 1:1,000; cat. no. ab32518; Abcam), anti-p62 (62
kDa; mouse; 1:1,000; cat. no. ab56416; Abcam) and anti-LC3-II/LC3-I
(~16 and ~18 kDa; rabbit; 1:1,000; cat. no. ab51520; Abcam), and
the secondary antibodies were horseradish peroxidase-conjugated
goat anti-mouse/rabbit IgG (1:2,000; sc-516102/ sc-2357; Santa Cruz
Biotechnology, Inc.).
ELISA
The cultured supernatant IL-18 content was
determined by ELISA using a human IL-18 kit (cat. no. SEA064Hu;
USCN Life Sciences, Inc.). The assay was performed according to the
manufacturer's instructions and the results were calculated
relative to standard curves prepared for IL-18.
Cell viability
Cell viability was determined by sulforhodamine B
(SRB) assay (cat. no. 230162, Sigma-Aldrich; Merck KGaA). Briefly,
the dental pulp cells were seeded in 96-well plates at a density
1×106 cells/ml and cultured to ~70–80% confluence. Next,
the cells were fixed with 50% trichloroacetic acid at 4°C for 1 h
and stained by 0.4% SRB solution for 30 min at room temperature.
SRB was measured as the absorbance at 565 nm using a Bio-Rad
microplate reader (Bio-Rad Laboratories, Inc.).
Statistical analysis
Data are presented as the mean ± SEM. GraphPad Prism
7 (GraphPad Software, Inc.) was used for statistical analysis.
Statistical comparisons between two and among multiple groups were
performed using Student's t-test and one-way analysis of variance
followed by Dunnett's test, respectively. P<0.05 was considered
to indicate a statistically significant difference.
Results
Pyroptotic cell death is induced by
LPS in cultured dental pulp cells
Human dental pulp cells were examined by flow
cytometry, and the results demonstrated that these cells were
positive for CD29, CD90, CD105, CD146 and STRO-1, but negative for
the cell surface antigens CD34 and CD45 (Fig. 1A). These surface antigens were
consistent with those expressed on dental pulp cells in a previous
study (30), indicating that human
dental pulp cells were successfully cultured and isolated in the
current study. The effect of LPS stimulation on dental pulp cells
were investigated by SRB assay after treating the cells with
different concentrations of LPS for 72 h; the results demonstrated
that LPS reduced cell viability in a concentration-dependent manner
(Fig. 1B). When the cells were
treated with 300 µg/l LPS, cell viability significantly decreased
(P<0.01); thus 300 µg/l LPS was selected for subsequent
experiments. A significant decrease in dental pulp cell viability
was observed in the LPS group at 72 h compared with the control
group (Fig. 1C). The secretion
levels of the pyroptosis markers IL-1β and IL-18 and caspase-1
activation were assessed by western blotting and ELISA following
LPS treatment for 24, 48 and 72 h. Western blot assay results
demonstrated that LPS upregulated the protein levels of
extracellular IL-1β (Fig. 1D) and
caspase-1 (Fig. 1E) in a
time-dependent manner without affecting the accumulation of
pro-IL-1β (Fig. 1F) and
pro-caspase-1 (Fig. 1G) in the
dental pulp cells (Fig. 1H).
Similarly, ELISA assay data demonstrated that LPS increased the
extracellular content of IL-18 at 48 and 72 h (Fig. 1I). These results demonstrated that
LPS increased the extracellular secretion of IL-1β and IL-18 and
caspase-1 activation, suggesting that LPS may induce pyroptotic
cell death in cultured dental pulp cells. Based on these results,
72 h was selected as the treatment duration for the following
experiments.
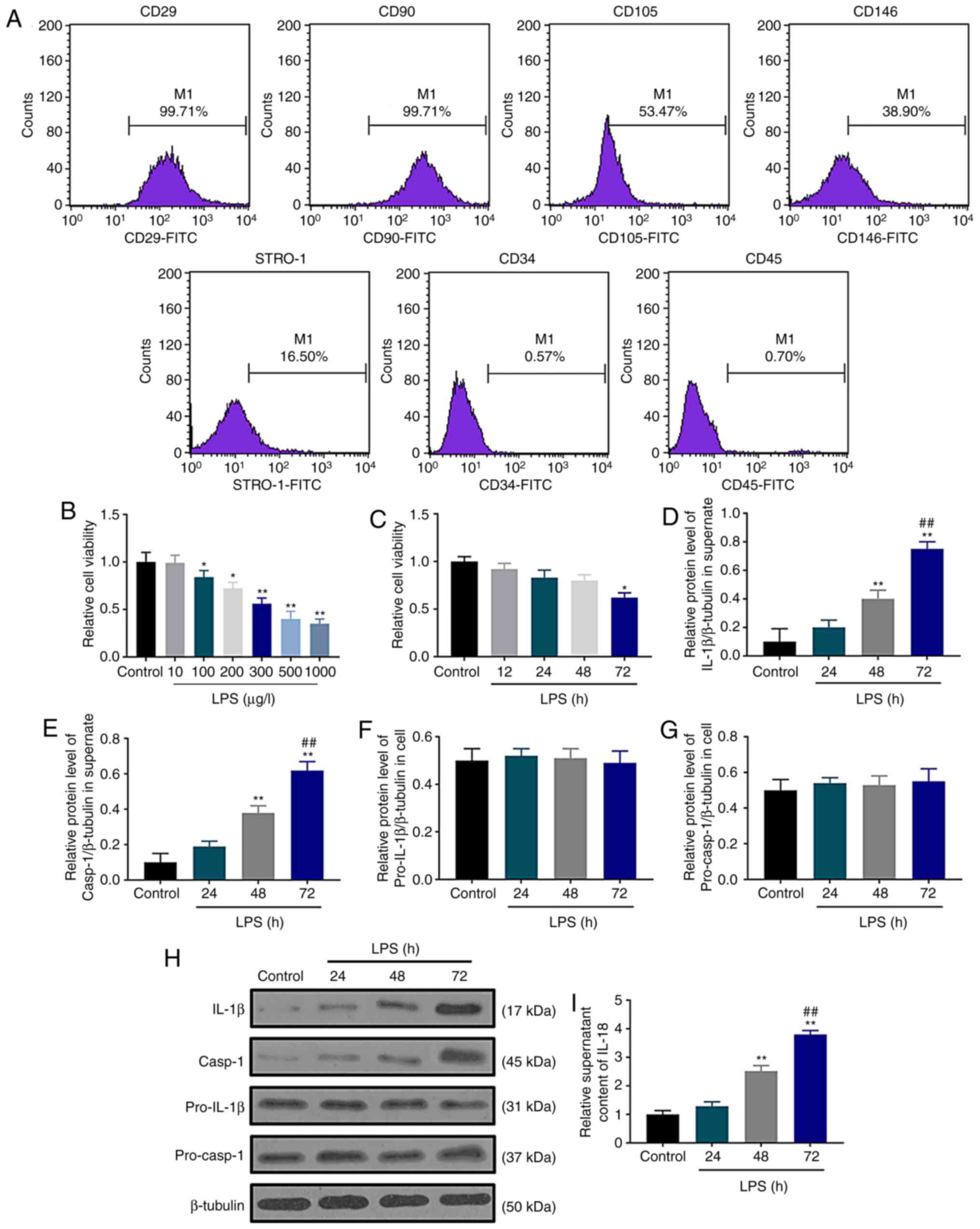 | Figure 1.Pyroptotic cell death is induced by
LPS in cultured dental pulp cells. (A) Human dental pulp cell
surface marker expression was examined using flow cytometry. (B)
Effects of different concentrations of LPS (0, 10, 100, 200, 300,
500 and 1,000 µg/l) on human dental pulp cell viability. (C) Human
dental pulp cell viability was inhibited by LPS as determined by
sulforhodamine B assay. (D and E) The protein levels of (D) IL-1β
and (E) caspase-1 were normalized to that of β-tubulin. (F and G)
The protein levels of (F) (pro)-IL-1β and (G) (pro)-caspase-1 were
normalized to that of β-tubulin. (H) Western blot assay was used to
detect the contents of IL-1β, caspase-1, pro-IL-1β and
pro-caspase-1. (I) IL-18 contents were determined by ELISA.
*P<0.05 and **P<0.01 vs. control; ##P<0.01 vs.
48 h. LPS, lipopolysaccharide; IL, interleukin. |
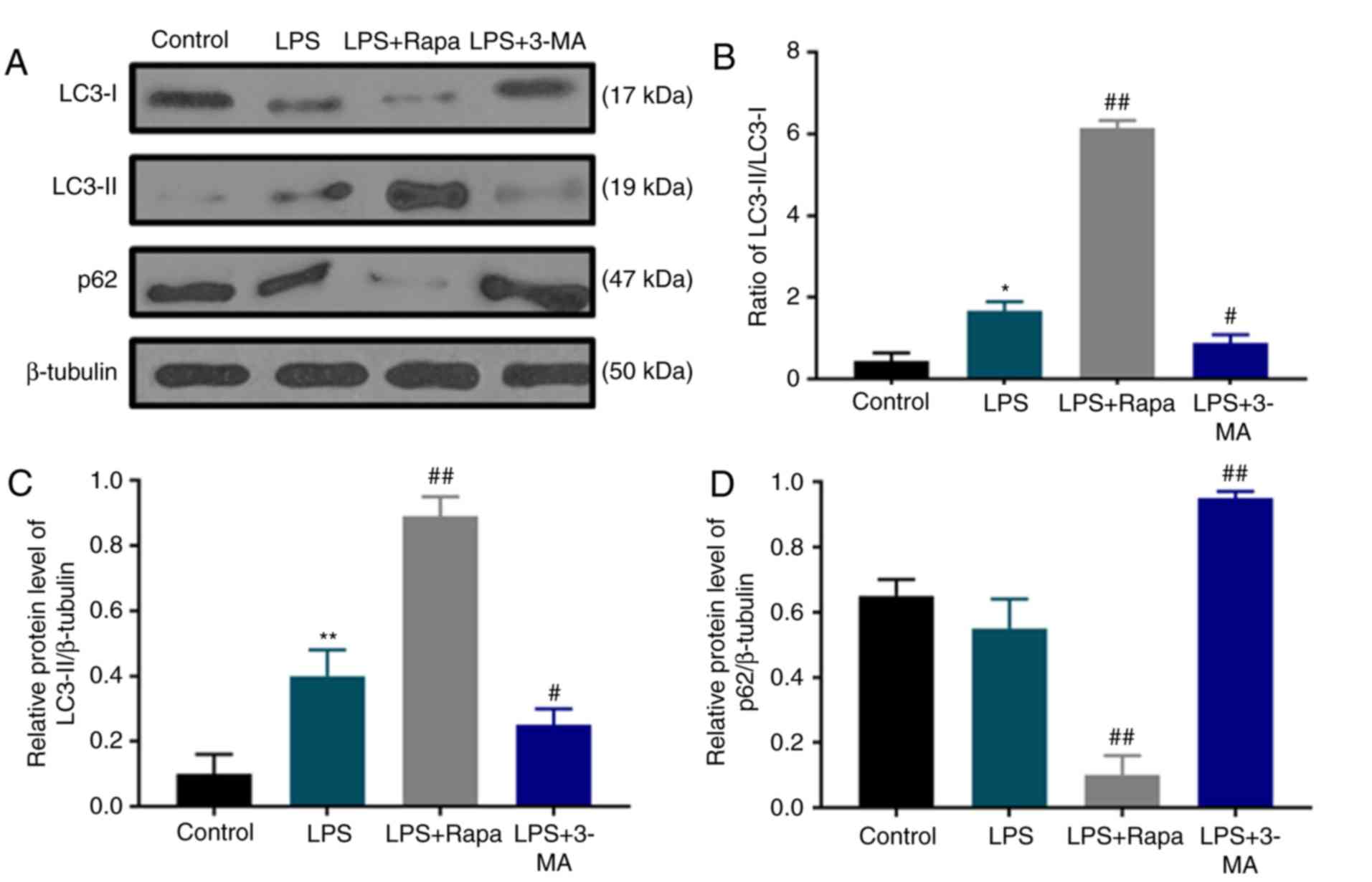
Autophagy is activated by LPS
To study the effect of LPS stimulation on dental
pulp cell autophagy and whether chemical modulators of autophagy
may regulate the autophagy of dental pulp cells under LPS
treatment, the protein levels of autophagy markers LC3 and p62 were
determined by western blotting (Fig.
2A). The results revealed that under LPS stimulation, the ratio
of LC3-II/LC3-I protein levels was increased with notable
upregulation of LC3-II compared with the control group (Fig. 2B and C); however, p62 accumulation
was slightly decreased (Fig. 2A and
D). In addition, the LC3-II/LC3-I ratio was increased and
LC3-II content and p62 were further upregulated by rapamycin
treatment, whereas 3-MA treatment produced opposite effects to
rapamycin (Fig. 2), indicating that
rapamycin induced autophagy, whereas 3-MA inhibited autophagy in
dental pulp cells under LPS treatment.
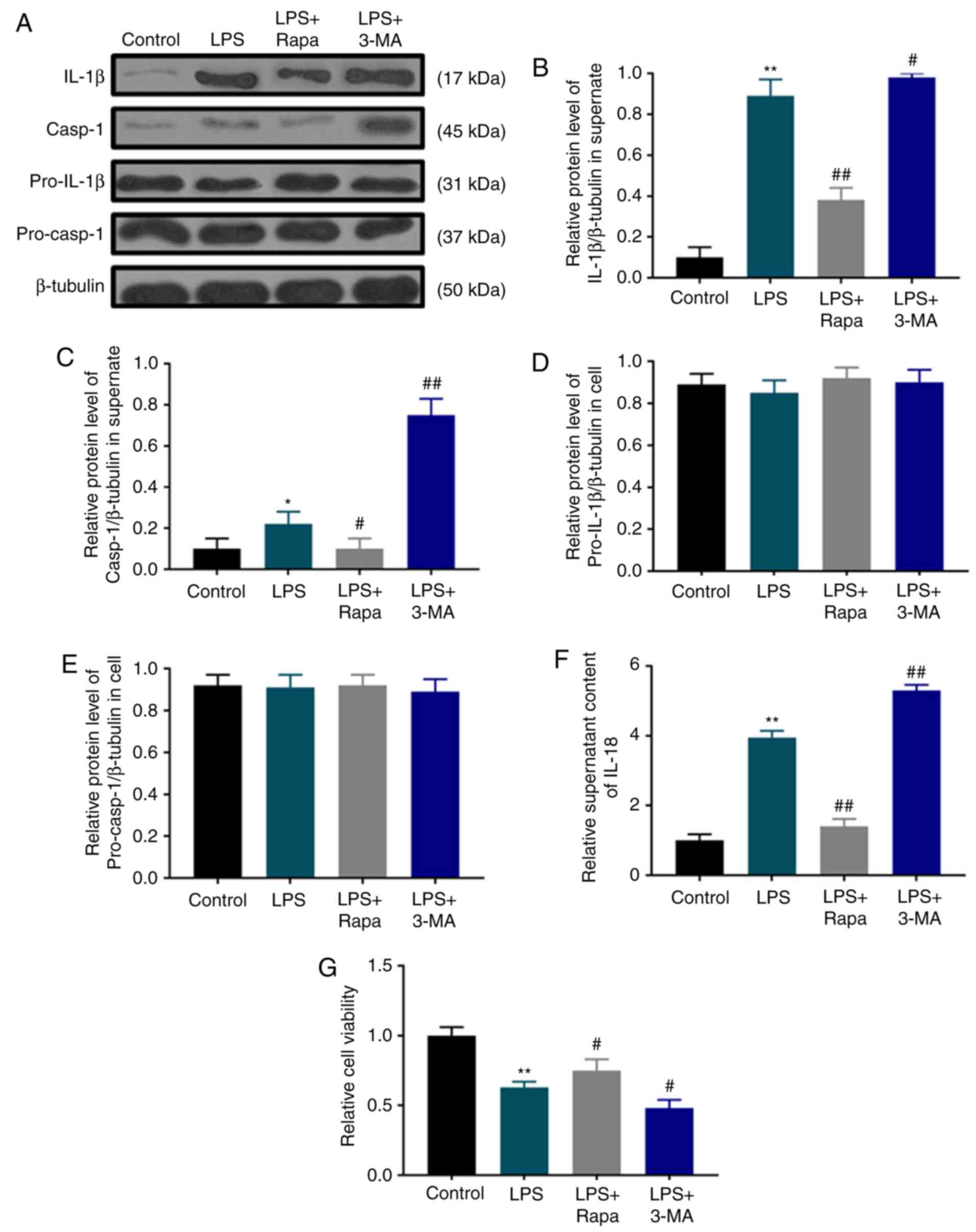
LPS-induced pyroptotic cell death is
suppressed by autophagy
Autophagy is activated by LPS; however, its role in
pyroptotic cell death is unclear. Therefore, the effects of
rapamycin and 3-MA on pyroptotic cell death and the viability of
dental pulp cells were investigated under LPS treatment. Pyroptotic
characteristics such as IL-1β (Fig. 3A
and B) and IL-18 secretion (Fig.
3F), as well as caspase-1 activation (Fig. 3A and C) in the LPS group were
significantly inhibited by rapamycin, but enhanced by 3-MA without
affecting the levels of pro-IL-1β (Fig.
3A and D) and pro-caspase-1 (Fig. 3A
and E). Consistently, SRB assay results demonstrated that
rapamycin treatment significantly attenuated the LPS-induced
decrease in dental pulp cell viability, whereas 3-MA pretreatment
exerted an opposite effect (Fig.
3G).
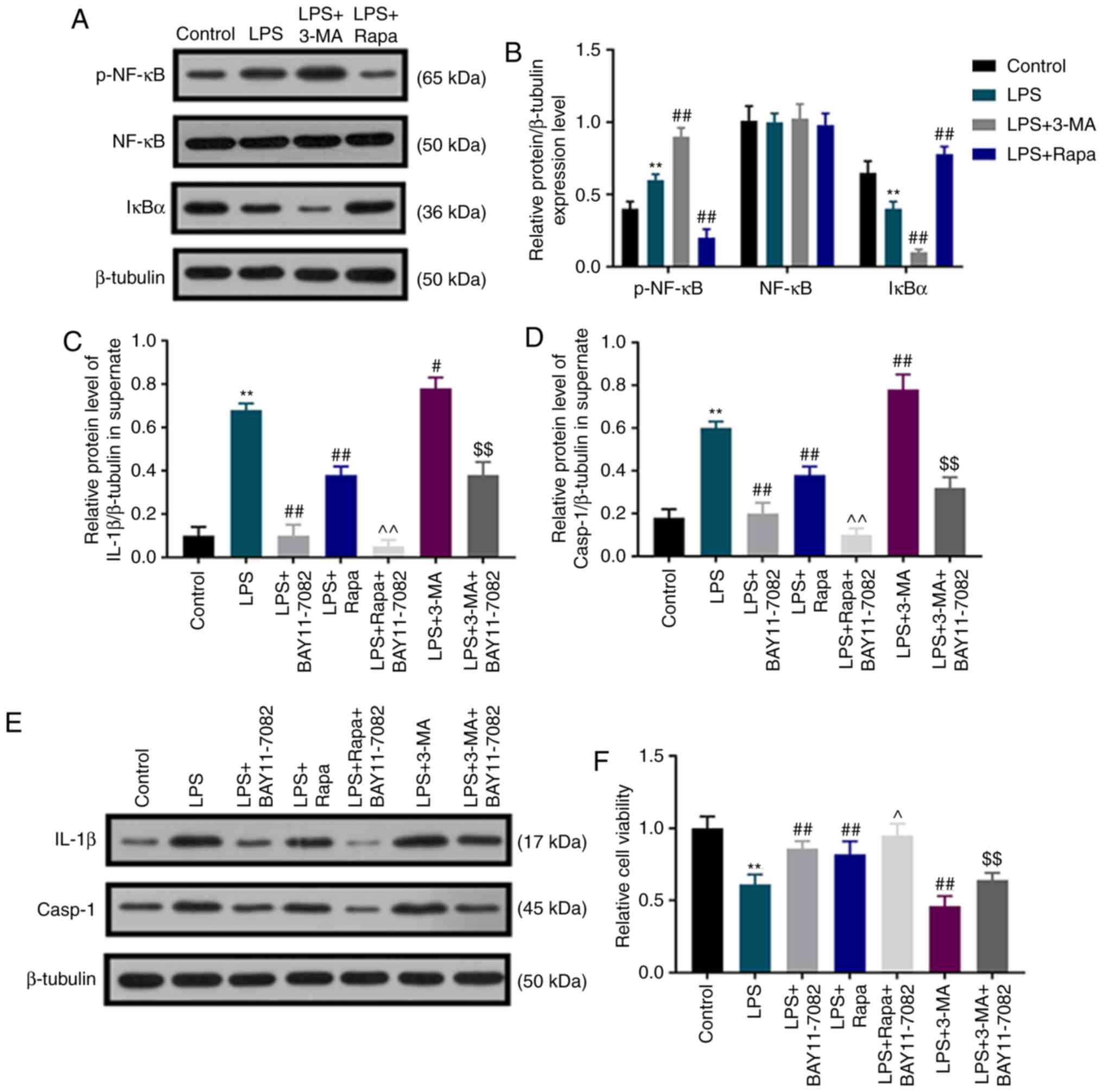
Autophagy inhibits LPS-induced
pyroptotic death of dental pulp cells by regulating the NF-κB
signaling pathway
To detect whether autophagy inhibited LPS-induced
pyroptotic dental pulp cell death by regulating the NF-κB signaling
pathway, the levels of p-NF-κB, NF-κB and IκBα were determined by
western blot assay, and the NF-κB signaling pathway inhibitor
BAY11-7082 was used. Under LPS stimulation, the protein level of
p-NF-κB was significantly upregulated, whereas that of IκBα was
downregulated in dental pulp cells compared with that in the
control group, and this effect was further enhanced by 3-MA, but
weakened by rapamycin (Fig. 4A and
B). No significant difference was observed in the expression of
NF-κB among all groups. Under BAY11-7082 and LPS treatment, there
was an evident decrease in the elevated IL-1β and caspase-1
activation induced by LPS. Of note, rapamycin inhibited the
elevation of IL-1β and caspase-1 activation induced by LPS, and
these inhibitory effects were further enhanced by BAY11-7082
(Fig. 4C-E). 3-MA promoted
LPS-induced IL-1β elevation and caspase-1 activation, which was
partially reversed by BAY11-7082 (Fig.
4C-E). In addition, the results demonstrated that BAY11-7082
partially reversed the decreased dental pulp cell viability induced
by LPS; BAY11-7082 further increased the promoting effects of
rapamycin on dental pulp cell viability, but attenuated the
inhibitory effect of 3-MA on cell viability (Fig. 4F).
Discussion
The present study investigated the role of
pyroptotic cell death in inflamed dental pulp cells. The results
demonstrated that LPS induced pyroptotic cell death in cultured
dental pulp cells; this was inhibited by autophagy induction. In
addition, autophagy was increased to a certain extent under LPS
treatment, suggesting that the activation of autophagy may be a
self-help measure in inflamed dental pulp cells, and the activation
was enhanced by rapamycin-induced autophagy. Thus, it was
hypothesized that rapamycin may be regarded as a potential drug
candidate for inducing autophagy, which is likely to inhibit
LPS-induced pyroptotic cell death in cultured dental pulp cells.
The LPS-activated p-NF-κB/IκBα signaling pathway was inhibited by
rapamycin-induced autophagy, whereas 3-MA-inhibited autophagy
produced effects opposite to those of LPS.
Dental pulp cells are the primary targets for
inflammatory agents, and the mechanism underlying dental pulp cell
fate determination in an inflammatory microenvironment needs to be
studied to protect dental pulp cells. To determine whether
LPS-stimulated dental pulp cells induced inflammation, it was
identified that pyroptosis-induced pro-inflammatory cytokines IL-1β
and caspsase1 (18,31,32)
increased significantly as LPS treatment time increased, suggesting
that LPS-stimulated dental pulp cells could mimic the pyroptotic
death of dental pulp cells; these were therefore used as a model
for exploring the mechanism of pyroptotic dental cell death.
Apoptosis-associated speck like protein (ASC) and NLRP3 are
involved in the activation of caspase-1 (33,34).
Jiang et al (10) have
reported that the NLRP3/caspase-1 pathway exhibits a biological
role in the innate immune response mounted by human dental pulp
fibroblasts. In the present study, LPS activated caspase-1 in
dental pulp cells, which is associated with the formation of NLRP3
inflammatory corpuscles (3). Further
activation of the inflammasome induces pyroptosis (35). However, in present study, the
expression of NLRP3 and ASC were not examined; this is a limitation
and requires further study.
Several studies have determined the expression
levels of autophagy molecules in aging human odontoblast and dental
pulp cells (36–39). It has been reported that autophagy
induction serves a protective role against hypoxic stress in human
dental pulp cells (40). Increased
levels of autophagy molecules including ATG5, LC3-II and Beclin-1
have been identified in adult human dental pulp, especially in aged
pulp cells (41). Under LPS
stimulation, autophagy-related molecules are differentially
expressed in adult pulp tissue and aged human dental pulp cells
(39). In the current study, the
ratio of LC3-II/LC3-I was increased following LPS treatment.
Autophagy agonist rapamycin further increased the ratio of
LC3-II/LC3-I, whereas the inhibition of autophagy by 3-MA reversed
these effects. The results also demonstrated that rapamycin
inhibited the elevation of IL-1β, caspase-1 and IL-18 following LPS
stimulation, whereas 3-MA generated opposite effects to those of
LPS. These results demonstrated that autophagy was activated in
LPS-treated dental pulp cells and that targeting autophagy may be
an effective therapy for dental pulpal inflammation.
NF-κB is an important transcription factor that
regulates inflammation and is a part of an essential signaling
pathway involved in the LPS-induced expression of cytokines
(42). Previous studies have
demonstrated that autophagy is required for the activation of NF-κB
(43), and that NF-κB negatively
regulates autophagy in specific cell types in vitro,
including RAW 264.7 cells and bone marrow-derived macrophages upon
brief coculture with E. coli (44). A previous study has suggested that
rapamycin may suppress the generation of IL-1β and IL-18 in
LPS-treated RAW264.7 cells by decreasing NF-κB signaling and
increasing autophagy (45). In the
present study, the NF-κB/IκBα signaling pathway was activated by
LPS. The effects of 3-MA and rapamycin on the expression levels of
IκBα and p-NF-κB were reversed by BAY11-7082, which is an NF-κB
pathway inhibitor. These results demonstrated that autophagy may
inhibit the LPS-induced pyrolysis death of dental pulp cells by
regulating the NF-κB signaling pathway.
Rapamycin affects cell cycle, proliferation,
autophagy and protein synthesis by suppressing mammalian target of
rapamycin (mTOR) activity (46,47).
Previous studies have demonstrated that mTOR signaling serves a key
role in mediating chronic inflammation and is involved in
regulating inflammatory factors, including IL-1β and TNF-α
(48,49). Rapamycin-induced inhibition of mTOR
has been reported to significantly reduce the inflammation induced
by various substances (50,51). Previous studies have demonstrated
that rapamycin exhibits anti-inflammatory actions by affecting
NF-κB activity (52,53). In the current study, rapamycin
induced autophagy by regulating autophagy-related genes, such as
LC3-II and p62. Rapamycin may inhibit the LPS-induced pyroptotic
cell death by inhibiting the expression of IL-1β and regulating the
NF-κB/IκBα signaling pathway.
However, the current study had certain limitations.
For example, the effect of rapamycin-induced autophagy on
LPS-mediated dental pulp cell pyroptosis was not verified in
vivo. In addition, the present study was conducted on dental
pulp cells from healthy teeth without comorbidities common in teeth
susceptible to caries infection. Although the aim of the current
study was to explore whether rapamycin-induced autophagy may
produce protective effects against LPS-mediated dental pulp cell
pyroptosis, the safety and efficacy of rapamycin in clinical
treatment still requires further investigation. Based on the
current experimental design, it is difficult to exclude the direct
protective effect of rapamycin-induced autophagy independent of
pyroptosis. Autophagy may be one of several relevant secondary
effects of mTOR inhibition, and the role of rapamycin in this
system needs to be further studied. In addition, the current study
only compared the effects of LPS on cells with the baseline, but
not with controls incubated with medium over equivalent time.
Finally, the photomicrographs of the LPS treated cells were not
presented.
In conclusion, the present study demonstrated that
LPS treatment may induce a low level of autophagy, which may be a
self-help strategy in dental pulp cells. These results highlighted
for the first time the potential of rapamycin in pulpitis
therapy.
Acknowledgements
Not applicable.
Funding
This work was supported by Jiangsu Provincial Key
Research Development Program (Social Development) (grant no.
BE2016796).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YG and XY made substantial contributions to
conception and design; YL, FG, JY, CY and YZ were responsible for
data acquisition, analysis and interpretation; and JY and CY
drafted the article or critically revising it for important
intellectual content. All authors agreed to be accountable for all
aspects of the work in ensuring that questions related to the
accuracy or integrity of the work are appropriately investigated
and resolved and all authors gave final approval of the version to
be published.
Ethics approval and consent to
participate
The study protocol was approved by the Ethical
Committee Department of Nanjing Medical University (cat. no:
NM20180510). Written informed consents were obtained from the
children's parents or legal representatives. All procedures
involving human participants were in accordance with the ethical
standards of the institutional and/or national research committee
and with the 1964 Helsinki declaration and its later amendments or
comparable ethical standards.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hahn CL and Liewehr FR: Relationships
between caries bacteria, host responses, and clinical signs and
symptoms of pulpitis. J Endod. 33:213–219. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jontell M, Okiji T, Dahlgren U and
Bergenholtz G: Immune defense mechanisms of the dental pulp. Crit
Rev Oral Biol Med. 9:179–200. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang A, Wang P, Ma X, Yin X, Li J, Wang
H, Jiang W, Jia Q and Ni L: Mechanisms that lead to the regulation
of NLRP3 inflammasome expression and activation in human dental
pulp fibroblasts. Mol Immunol. 66:253–262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sood AK, Burbank AJ, Duran CG, Enders K,
Zhou H, Peden DB and Hernandez ML: Gamma tocopherol effect on
LPS-induced sputum neutrophilia is not modified by BMI or GSTM1
genotype. J Allergy Clin Immunol. 143:1937–1939. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tang Y, Wang C, Wang Y, Zhang J, Wang F,
Li L, Meng X, Li G, Li Y and Wang L: Isoliquiritigenin attenuates
LPS-induced AKI by suppression of inflammation involving NF-κB
pathway. Am J Transl Res. 10:4141–4151. 2018.PubMed/NCBI
|
|
6
|
Li S, Ning LG, Lou XH and Xu GQ:
Necroptosis in inflammatory bowel disease and other intestinal
diseases. World J Clin Cases. 6:745–752. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chimenti MS, Sunzini F, Fiorucci L, Botti
E, Fonti GL, Conigliaro P, Triggianese P, Costa L, Caso F, Giunta
A, et al: Potential role of cytochrome c and tryptase in psoriasis
and psoriatic arthritis pathogenesis: Focus on resistance to
apoptosis and oxidative stress. Front Immunol. 9:23632018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brough D and Rothwell NJ:
Caspase-1-dependent processing of pro-interleukin-1beta is
cytosolic and precedes cell death. J Cell Sci. 120:772–781. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He Y, Hara H and Núñez G: Mechanism and
regulation of NLRP3 inflammasome activation. Trends Biochem Sci.
41:1012–1021. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang W, Lv H, Wang H, Wang D, Sun S, Jia
Q, Wang P, Song B and Ni L: Activation of the NLRP3/caspase-1
inflammasome in human dental pulp tissue and human dental pulp
fibroblasts. Cell Tissue Res. 361:541–555. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang H and Sun SC: NF-κB in inflammation
and renal diseases. Cell Biosci. 5:632015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bhatt D and Ghosh S: Regulation of the
NF-κB-mediated transcription of inflammatory genes. Front Immunol.
5:712014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Denton D, Xu T and Kumar S: Autophagy as a
pro-death pathway. Immunol Cell Biol. 93:35–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guan JL, Simon AK, Prescott M, Menendez
JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A and Zhang
J: Autophagy in stem cells. Autophagy. 9:830–849. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nikoletopoulou V, Papandreou ME and
Tavernarakis N: Autophagy in the physiology and pathology of the
central nervous system. Cell Death Differ. 22:398–407. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levine B, Mizushima N and Virgin HW:
Autophagy in immunity and inflammation. Nature. 469:323–335. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Levine B and Klionsky DJ: Development by
self-digestion: Molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Deretic V, Jiang S and Dupont N: Autophagy
intersections with conventional and unconventional secretion in
tissue development, remodeling and inflammation. Trends Cell Biol.
22:397–406. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Couve E, Osorio R and Schmachtenberg O:
The amazing odontoblast: Activity, autophagy, and aging. J Dent
Res. 92:765–772. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang J, Wan C, Nie S, Jian S, Sun Z, Zhang
L and Chen Z: Localization of Beclin1 in mouse developing tooth
germs: Possible implication of the interrelation between autophagy
and apoptosis. J Mol Histol. 44:619–627. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mankowski RT, Ahmed S, Beaver T, Dirain M,
Han C, Hess P, Martin T, Smith BK, Someya S, Leeuwenburgh C and
Martin AD: Intraoperative hemidiaphragm electrical stimulation
reduces oxidative stress and upregulates autophagy in surgery
patients undergoing mechanical ventilation: Exploratory study. J
Transl Med. 14:3052016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Caramés B, Taniguchi N, Otsuki S, Blanco
FJ and Lotz M: Autophagy is a protective mechanism in normal
cartilage, and its aging-related loss is linked with cell death and
osteoarthritis. Arthritis Rheum. 62:791–801. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pei F, Lin H, Liu H, Li L, Zhang L and
Chen Z: Dual role of autophagy in lipopolysaccharide-induced
preodontoblastic cells. J Dent Res. 94:175–182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meng N, Wu L, Gao J, Zhao J, Su L, Su H,
Zhang S and Miao J: Lipopolysaccharide induces autophagy through
BIRC2 in human umbilical vein endothelial cells. J Cell Physiol.
225:174–179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hickson-Bick DL, Jones C and Buja LM:
Stimulation of mitochondrial biogenesis and autophagy by
lipopolysaccharide in the neonatal rat cardiomyocyte protects
against programmed cell death. J Mol Cell Cardiol. 44:411–418.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu Y, Kim SO, Li Y and Han J: Autophagy
contributes to caspase-independent macrophage cell death. J Biol
Chem. 281:19179–19187. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jung JY, Woo SM, Kim WJ, Lee BN, Nör JE,
Min KS, Choi CH, Koh JT, Lee KJ and Hwang YC: Simvastatin inhibits
the expression of inflammatory cytokines and cell adhesion
molecules induced by LPS in human dental pulp cells. Int Endod J.
50:377–386. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li H, Gao A, Feng D, Wang Y, Zhang L, Cui
Y, Li B, Wang Z and Chen G: Evaluation of the protective potential
of brain microvascular endothelial cell autophagy on blood-brain
barrier integrity during experimental cerebral ischemia-reperfusion
injury. Transl Stroke Res. 5:618–626. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim MJ, Kim EH, Pun NT, Chang JH, Kim JA,
Jeong JH, Choi DY, Kim SH and Park PH: Globular adiponectin
inhibits lipopolysaccharide-primed inflammasomes activation in
macrophages via autophagy induction: The critical role of AMPK
signaling. Int J Mol Sci. 18:E12752017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhan FL, Liu XY and Wang XB: The role of
MicroRNA-143-5p in the differentiation of dental pulp stem cells
into odontoblasts by targeting Runx2 via the OPG/RANKL signaling
pathway. J Cell Biochem. 119:536–546. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dupont N, Jiang S, Pilli M, Ornatowski W,
Bhattacharya D and Deretic V: Autophagy-based unconventional
secretory pathway for extracellular delivery of IL-1β. EMBO J.
30:4701–4711. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Murai H, Okazaki S, Hayashi H, Kawakita A,
Hosoki K, Yasutomi M, Sur S and Ohshima Y: Alternaria extract
activates autophagy that induces IL-18 release from airway
epithelial cells. Biochem Biophys Res Commun. 464:969–974. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin YC, Huang DY, Wang JS, Lin YL, Hsieh
SL, Huang KC and Lin WW: Syk is involved in NLRP3
inflammasome-mediated caspase-1 activation through adaptor ASC
phosphorylation and enhanced oligomerization. J Leukoc Biol.
97:825–835. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu J, Fernandes-Alnemri T and Alnemri ES:
Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in
caspase-1 activation by Listeria monocytogenes. J Clin Immunol.
30:693–702. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Miao EA, Rajan JV and Aderem A:
Caspase-1-induced pyroptotic cell death. Immunol Rev. 243:206–214.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Couve E and Schmachtenberg O: Autophagic
activity and aging in human odontoblasts. J Dent Res. 90:523–528.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Couve E, Osorio R and Schmachtenberg O:
Mitochondrial autophagy and lipofuscin accumulation in aging
odontoblasts. J Dent Res. 91:696–701. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li L, Zhu YQ, Jiang L and Peng W:
Increased autophagic activity in senescent human dental pulp cells.
Int Endod J. 45:1074–1079. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee YH, Lee HY, Kim TG, Lee NH, Yu MK and
Yi HK: PPARγ maintains homeostasis through autophagy regulation in
dental pulp. J Dent Res. 94:729–737. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Park SY, Sun EG, Lee Y, Kim MS, Kim JH,
Kim WJ and Jung JY: Autophagy induction plays a protective role
against hypoxic stress in human dental pulp cells. J Cell Biochem.
119:1992–2002. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pantovic A, Krstic A, Janjetovic K, Kocic
J, Harhaji-Trajkovic L, Bugarski D and Trajkovic V: Coordinated
time-dependent modulation of AMPK/Akt/mTOR signaling and autophagy
controls osteogenic differentiation of human mesenchymal stem
cells. Bone. 52:524–531. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chang X, He H, Zhu L, Gao J, Wei T, Ma Z
and Yan T: Protective effect of apigenin on Freund's complete
adjuvant-induced arthritis in rats via inhibiting P2X7/NF-κB
pathway. Chem Biol Interact. 236:41–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Criollo A, Chereau F, Malik SA,
Niso-Santano M, Mariño G, Galluzzi L, Maiuri MC, Baud V and Kroemer
G: Autophagy is required for the activation of NFkappaB. Cell
Cycle. 11:194–199. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schlottmann S, Buback F, Stahl B,
Meierhenrich R, Walter P, Georgieff M and Senftleben U: Prolonged
classical NF-kappaB activation prevents autophagy upon E. coli
stimulation in vitro: A potential resolving mechanism of
inflammation. Mediators Inflamm. 2008:7258542008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jia X, Cao B, An Y, Zhang X and Wang C:
Rapamycin ameliorates lipopolysaccharide-induced acute lung injury
by inhibiting IL-1β and IL-18 production. Int Immunopharmacol.
67:211–219. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Meijer AJ and Codogno P: Regulation and
role of autophagy in mammalian cells. Int J Biochem Cell Biol.
36:2445–2462. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu YC, Gao XX, Chen L and You XQ:
Rapamycin suppresses Aβ25-35- or LPS-induced neuronal
inflammation via modulation of NF-κB signaling. Neuroscience.
355:188–199. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huang HY, Chang HF, Tsai MJ, Chen JS and
Wang MJ: 6-Mercaptopurine attenuates tumor necrosis factor-α
production in microglia through Nur77-mediated transrepression and
PI3K/Akt/mTOR signaling-mediated translational regulation. J
Neuroinflammation. 13:782016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Vangan N, Cao Y, Jia X, Bao W, Wang Y, He
Q, Binderiya U, Feng X, Li T, Hao H and Wang Z: mTORC1 mediates
peptidoglycan induced inflammatory cytokines expression and NF-κB
activation in macrophages. Microb Pathog. 99:111–118. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fielhaber JA, Carroll SF, Dydensborg AB,
Shourian M, Triantafillopoulos A, Harel S, Hussain SN, Bouchard M,
Qureshi ST and Kristof AS: Inhibition of mammalian target of
rapamycin augments lipopolysaccharide-induced lung injury and
apoptosis. J Immunol. 188:4535–4542. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yue Y, Wang Y, Li D, Song Z, Jiao H and
Lin H: A central role for the mammalian target of rapamycin in
LPS-induced anorexia in mice. J Endocrinol. 224:37–47. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mengke NS, Hu B, Han QP, Deng YY, Fang M,
Xie D, Li A and Zeng HK: Rapamycin inhibits
lipopolysaccharide-induced neuroinflammation in vitro and
in vivo. Mol Med Rep. 14:4957–4966. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Okamoto T, Ozawa Y, Kamoshita M, Osada H,
Toda E, Kurihara T, Nagai N, Umezawa K and Tsubota K: The
neuroprotective effect of rapamycin as a modulator of the
mTOR-NF-κB axis during retinal inflammation. PLoS One.
11:e01465172016. View Article : Google Scholar : PubMed/NCBI
|