Introduction
Age-related cataract (ARC) causes ~50% of blindness
worldwide (1). Posterior capsule
opacification (PCO) is a common complication of cataract, which can
result in secondary loss of vision (2). Increasing evidence has suggested that
residual lens epithelial cells (LECs) can be transformed into
myofibroblasts via epithelial-mesenchymal transition (EMT) and
accumulation of extracellular matrix (ECM) components (3-5).
Previous studies have also reported that transforming growth
factor-β2 (TGF-β2), a TGF-β homology isomer, promoted EMT and ECM
synthesis in LECs (6,7).
Long non-coding RNAs (lncRNAs) are a class of RNAs
>200 nucleotides in length that lack translational capacity
(8) and have been reported to
participate in numerous different diseases, including PCO. For
example, lncRNA HOX transcript antisense RNA upregulation mediated
TGF-β2-induced EMT in SRA01/04 cell lines (9). lncRNA KCNQ1OT1, taurine upregulated 1
and FEX family zinc finger 1-antisense RNA have also been reported
to facilitated LEC progression (10-12).
Moreover, certain lncRNAs have been reported to be associated with
oxidative stress during cataract (13-15).
LncRNA myocardial infarction associated transcript (MIAT) aberrant
expression has been identified in a number of diseases, including
coronary artery disease (16,17),
ischemic stroke (18) and ARC
(19). However, the biological
mechanism underlying MIAT during ARC is not completely
understood.
MicroRNAs (miRNAs), a class of short non-coding RNAs
~22 nucleotides in length, affect gene expression by inhibiting
mRNA translation or mediating mRNA degradation (20). miRNA dysregulation has been
identified in a number of diseases, including ARC. For example,
miR-221 accelerated LEC apoptosis by regulating sirtuin 1 and E2F
transcription factor 3 expression (21). Furthermore, miR-181a has been
reported to be involved in ARC development (22). Connective tissue growth factor
(CTGF), a downstream effector of TGF-β2, has been reported to be
associated with several cellular functions, Including
proliferation, migration and adhesion in LECs (7,23,24).
However, the regulatory mechanisms underlying miR-181a and CTGF
during ARC have not been investigated.
The aim of the present study was to investigate the
role and mechanism underlying MIAT during ARC development. The
results of the present study may provide a theoretical basis for
further investigation of ARC.
Materials and methods
Tissue samples
A total of 20 ARC posterior capsular tissue samples
(cataract) were collected from 12 female patients and 8 male
patients (age, 58-75 years; mean age, 65 years) recruited from the
Department of Ophthalmology, Renmin Hospital between January 2017
and July 2018. A further 20 normal posterior capsule tissue samples
were obtained from 10 female patients and 10 male patients from the
Department of Ophthalmology; Renmin Hospital (age, 49-71 years;
mean age, 57.7 years) who had been in an accident but did not
exhibit eye damage between March 2017 and November 2018. All
tissues were frozen at -80˚C until further analysis. All
participants or their guardians provided written informed consent.
The present study was approved by the Ethics Committee of the
Department of Ophthalmology, Renmin Hospital, Hubei University of
Medicine.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from ARC tissue samples and
SRA01/04 cells using TRIzol® reagent (Thermo Fisher
Scientific, Inc.). Subsequently, reverse transcription was
performed using the miScript RT kit (Takara Biotechnology Co.,
Ltd.) at 37˚C for 60 min, according to the manufacturer's protocol.
qPCR was performed using the ABI Prism 7700 Sequence Detection
system (Thermo Fisher Scientific, Inc.) and TaqMan miRNA assay (for
miRNA; Applied Biosystems; Thermo Fisher Scientific, Inc.) and SYBR
Premix Ex Taq II (for lncRNA and mRNA; Takara) were used, according
to their manufacturer's protocols. The thermocycling conditions for
qPCR were initial denaturation at 95˚C for 10 min followed by 40
cycles of 95˚C for 15 sec and 60˚C for 1 min. The following primer
pairs were synthesized by Sangon Biotech Co., Ltd. and used for
qPCR: MIAT forward, 5'-GGACGTTCACAACCACACTG-3' and reverse,
5'-TCCCACTTTGGCATTCTAGG-3'; miR-181a forward,
5'-GCGGTAACATTCAACGCTGTCG-3' and reverse, 5'-GTGCAGGGTCCGAGGT-3';
CTGF forward, 5'-GGAAATGCTGTGAGGAGTGGGTGT-3' and reverse,
5'-TGTCTTCCAGTCGGTAGGCAGCTA-3'; GAPDH forward,
5'-TGTTCGTCATGGGTGTGAAC-3' and reverse, 5'-ATGGCATGGACTGTGGTCAT-3';
and U6 forward, 5'-CTCGCTTCGGCAGCACA-3' and reverse,
5'-AACGCTTCACGAATTTGCGT-3'. mRNA and miRNA levels were quantified
using the 2-ΔΔCq method (25). mRNA levels of MIAT and CTGF were
normalized to the internal reference gene GAPDH. miR-181a levels
were normalized to the internal reference gene U6.
Western blot assay
Total protein was extracted from ARC tissues or
TGF-β2-treated SRA01/04 cells by RIPA buffer (Thermo Fisher
Scientific, Inc.). Protein concentration was detected using BCA
Protein Assay Kit (Beyotime Institute of Biotechnology).
Subsequently, total protein was boiled with loading buffer
(Beyotime Institute of Biotechnology) for 10 min at 95˚C. Proteins
(30 µg/lane) were separated by 10% SDS-PAGE and transferred onto
PVDF membranes (EMD Millipore). Subsequently, the membranes were
blocked with skim milk for 4 h at 37˚C and incubated overnight at
4˚C with primary antibodies targeted against: CTGF (1:1,000;
ab6992), α-smooth muscle actin (α-SMA; 1:5,000; ab32575),
fibronectin (FN; 1:5,000; ab2413), collagen I (COL-1; 1:1,000;
ab34710), extracellular signal-regulated kinase (ERK; 1:1,000;
ab17942), phosphorylated-ERK (p-ERK; 1:500; ab214362),
mitogen-activated protein kinase kinase (MEK; 1:10,000; ab32091),
p-MEK (1:1,000; ab96379) and GAPDH (1:5,000; ab9485). Following
primary incubation, the membranes were incubated with a horseradish
peroxidase-conjugated anti-rabbit lgG secondary antibody (1:10,000;
ab205718) for 3 h at 37˚C. All antibodies were purchased from
Abcam. Protein bands were visualized using the RapidStep ECL
reagent (EMD Millipore) and analyzed by Imagequant software
(version 5.1; Amersham-Pharmacia's Biotech). GAPDH was used as the
loading control.
Cell culture and transfection
The human LEC line SRA01/04 was purchased from the
Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences. Cells were cultured in DMEM (Invitrogen; Thermo Fisher
Scientific, Inc.) containing 10% FBS (Thermo Fisher Scientific,
Inc.) and 1% penicillin/streptomycin (Invitrogen; Thermo Fisher
Scientific, Inc.). TGF-β2 (Sigma-Aldrich; Merck KGaA) was dissolved
in PBS to make a 5 ng/ml stock solution for subsequent
experiments.
Small interfering (si)RNAs targeting MIAT (si-MIAT,
5'-CCUUACCAUUCCUCCACUUTT-3') or CTGF (si-CTGF,
5'-GCUGACCUGGAAGAGAACATT-3'), a negative control (NC) siRNA (si-NC,
5'-UUCUCCGAACGUGUCA-3'), MIAT overexpression vector (MIAT), CTGF
overexpression vector (CTGF), an NC vector (pcDNA), miR-181a mimic
(miR-181a, 5'-AACAUUCAACGCUGUCGGUGAGU-3'), an NC mimic (miR-NC,
5'-UUCUCCGAACGUGUCACGUTT-3'), miR-181a inhibitor (anti-miR-181a,
5'-ACUCACCGACAGCGUUGAAUGUU-3') and a NC inhibitor (anti-miR-NC,
5'-CAGUACUUUUGUGUAGUACAA-3') were obtained from Shanghai GenePharma
Co., Ltd. SRA01/04 cells were seeded into 6-well plates, si-RNAs
(50 nM), miRNA mimics (50 nM), miRNA inhibitors (100 nM) and
plasmids (100 nM) were transfected into SRA01/04 cells when the
cell density reached 70% using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. At 24 h post-transfection, cells were used
for subsequent experiments.
Cell viability assay
An MTT assay (Sigma-Aldrich; Merck KGaA) was used to
detect SRA01/04 cell viability, according to the manufacturer's
protocol. SRA01/04 cells (6x103 cells/well) were seeded
into 96-well plates. Following transfection for 24 h, cells were
incubated with 5 ng/ml TGF-β2 for another 48 h. Subsequently, MTT
was added to each well and incubated for 4 h at 37˚C. DMSO was used
to dissolve the purple formazan crystals for 15 min in the dark at
37˚C. Cell viability was measured at a wavelength of 570 nm using
an Evolution™ 350 UV-Vis spectrophotometer (Thermo
Fisher Scientific, Inc.).
Transwell assay
The Transwell assay was conducted using Transwell
plates (Corning Life Sciences). Following a 24 h culture and
treatment with TGF-β2 for another 48 h at 37˚C, SRA01/04cells
(5x105 cells/ml) were seeded into the upper chambers of
the Transwell plates with serum-free DMEM. DMEM containing 10% FBS
was plated in the lower chambers of the Transwell plates. Following
incubation for 24 h, cells on the lower surface of the Transwell
membrane were fixed with 4% methanol at room temperature for 30 min
and stained with 0.1% crystal violet at room temperature for 20
min. Migratory cells were counted in 10 random fields of view using
a light microscope a magnification of x100 (Olympus
Corporation).
Dual-luciferase reporter assay
The interaction between miR-181a and MIAT or CTGF
was predicted using the StarBase online database (version 2;
starbase.sysu.edu.cn). The wild-type (WT) and mutant (MUT)
sequences of MIAT and 3'-untranslated regions (3'-UTRs) of CTGF
were amplified and inserted into the pGL3 vector (Promega
Corporation), WT-MIAT, MUT-MIAT, CTGF 3'UTR-WT and CTGF 3'UTR-MUT,
respectively. The pGL3 vector and miR-181a mimic or miR-NC were
co-transfected into SRA01/04 cells using Lipofectamine®
2000 transfection reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. Luciferase
activity was detected, according to the manufacturer's protocol,
using the Dual-Luciferase Reporter assay kit (Promega Corporation)
48 h post-transfection. Firefly luciferase activity was normalized
to Renilla luciferase activity.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism software (version 7; GraphPad Software, Inc.). Data from
three repeated independent experiments are presented as the mean ±
standard deviation. Comparisons between two groups or among
multiple groups were assessed using the Student's t-test or one-way
ANOVA with Tukey's post hoc test, respectively. The correlation
between CTGF or miR-181a and MIAT was analyzed by Pearson's
correlation analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
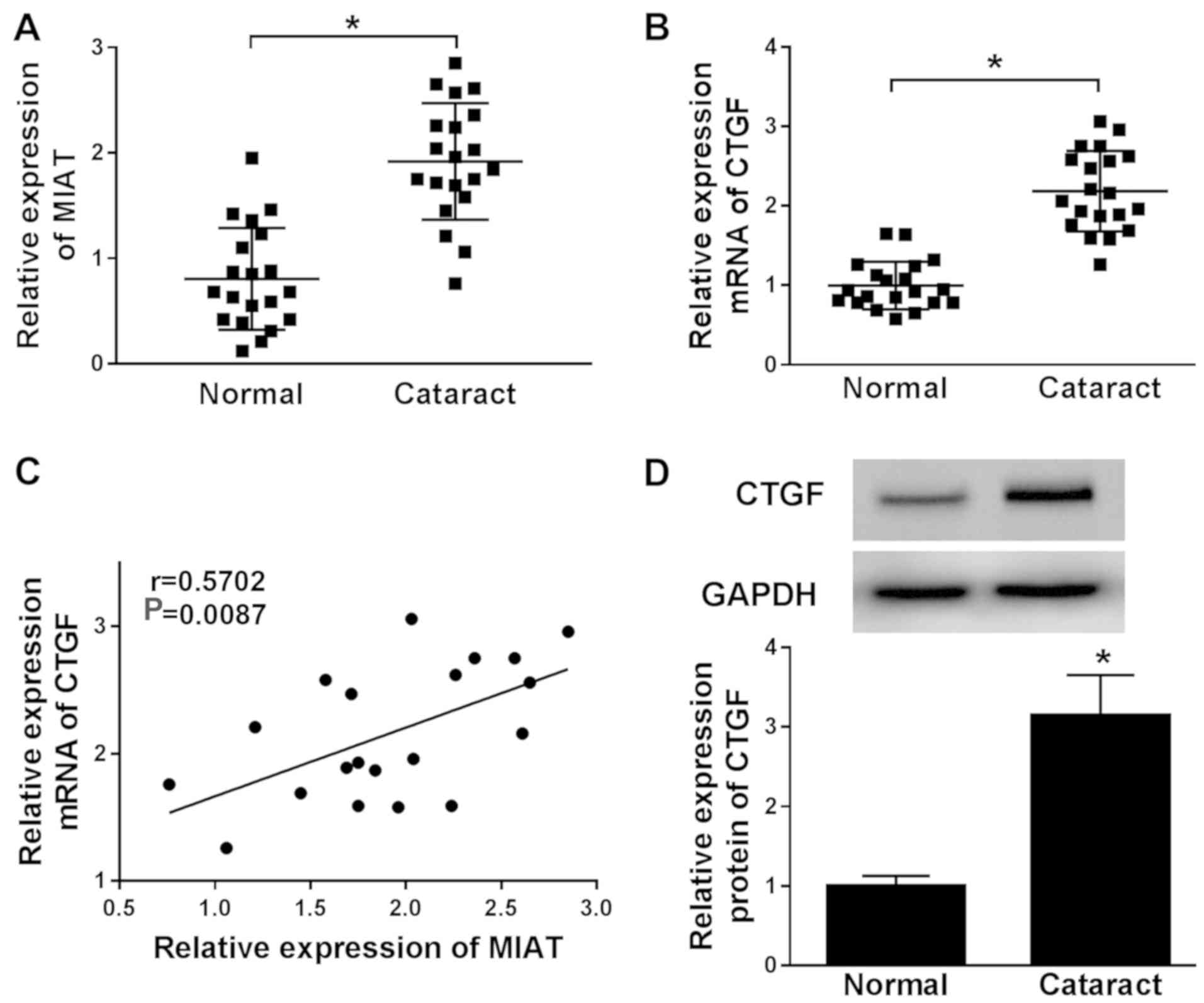
MIAT and CTGF are upregulated in ARC
tissues
To investigate the roles of MIAT and CTGF during
ARC, the levels of MIAT and CTGF in ARC tissues were detected. The
levels of MIAT and CTGF were upregulated in ARC tissue samples
compared with the normal posterior capsule tissue samples (Fig. 1A and B). The correlation analysis indicated that
the level of CTGF mRNA expression was positively correlated with
the level of MIAT mRNA expression in ARC tissues (Fig. 1C). In addition, the western blotting
results also indicated that the protein level of CTGF was
upregulated in ARC tissues compared with the normal tissues
(Fig. 1D). Therefore, the data
suggested that MIAT and CTGF might play an important role during
ARC, and a relationship between the two factors may be present.
MIAT knockdown reverses TGF-β2-induced
effects on cell viability, migration, EMT and ECM
productioninSRA01/04 cells
To further investigate the effects of MIAT in ARC,
MIAT expression was knocked down in SRA01/04 cells using si-MIAT
and the efficiency of MIAT knockdown was verified using RT-qPCR
(Fig. S1A). The RT-qPCR results
indicated that TGF-β2 elevated the expression levels of MIAT in
SRA01/04 cells compared with the control group and this effect was
reversed by MIAT knockdown (Fig.
2A). Furthermore, TGF-β2 increasedSRA01/04 cell viability and
migration compared with the control group, whereas si-MIAT reversed
the TGF-β2-induced effects (Fig. 2B
and C). α-SMA is a marker of EMT
(26), and FN and COL-1 are ECM
markers (27). Therefore, the effect
of MIAT knockdown on the protein expression levels of α-SMA, FN and
COL-1 was investigated. TGF-β2 significantly upregulated the
protein expression levels of α-SMA, FN and COL-1 in SRA01/04 cells
compared with the control group, while si-MIAT decreased
TGF-β2-induced protein expression (Fig.
2D). Additionally, MIAT overexpression increased SRA01/04 cell
viability, migration, EMT and ECM production compared with the
control group (Fig. S1B and
S2A-C). The results suggested that
MIAT expression was critical for the development of ARC.
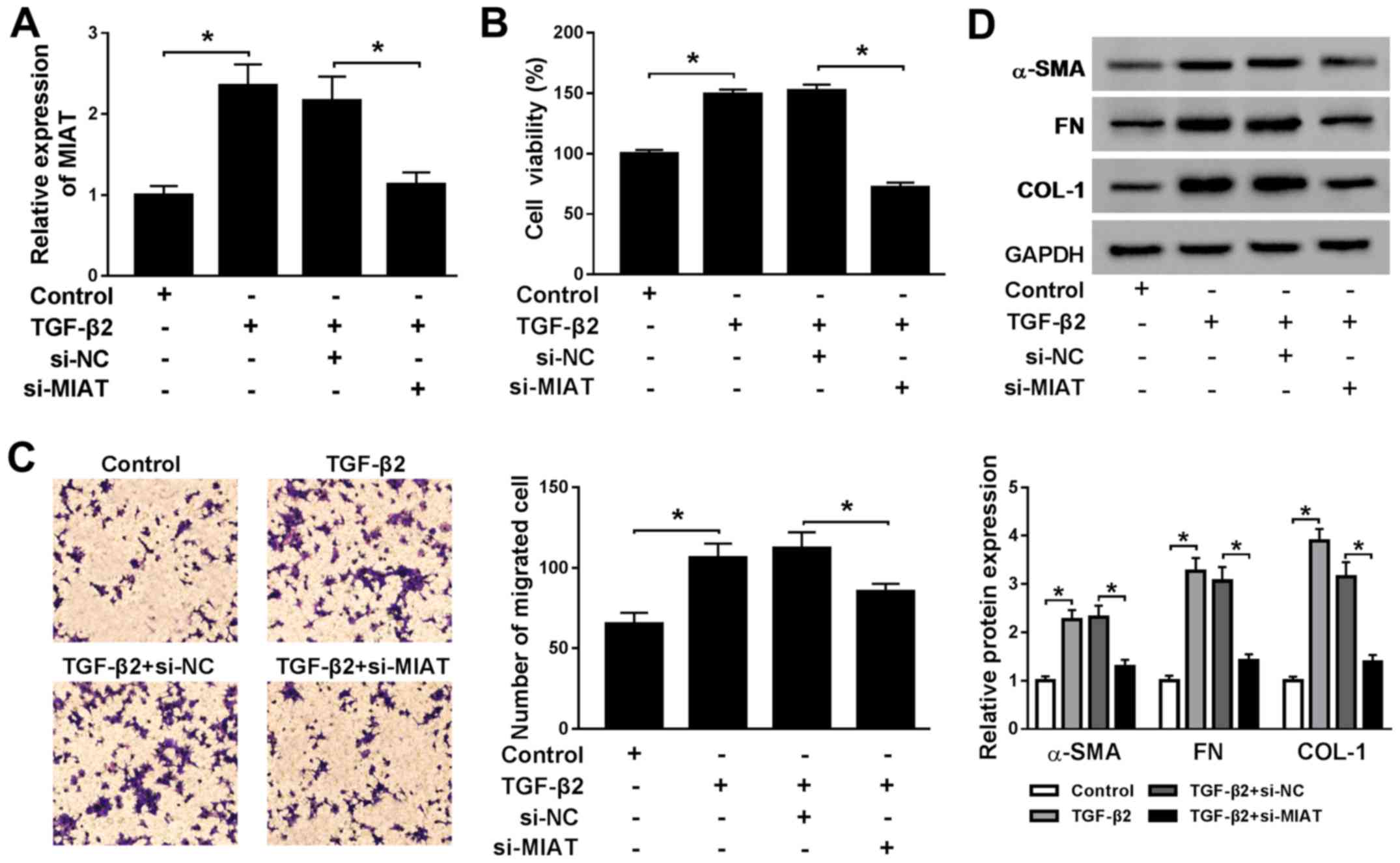 | Figure 2MIAT knockdown reverses
TGF-β2-induced upregulation of SRA01/04 cell viability, migration,
EMT and ECM production. SRA01/04 cells were treated with TGF-β2 and
subsequently transfected with si-NC or si-MIAT. (A) The expression
levels of MIAT in SRA01/04 cells were measured by reverse
transcription-quantitative PCR. (B) SRA01/04 cell viability was
assessed by the MTT assay. (C) The number of migratory SRA01/04
cells was examined using the Transwell assay. (D) The protein
expression levels of α-SMA, FN and COL-1 in SRA01/04 cells were
detected by western blotting. *P<0.05, as indicated.
MIAT, myocardial infarction associated transcript; TGF-β2,
transforming growth factor-β2; EMT, epithelial-mesenchymal
transition; ECM, extracellular matrix; si, small interfering RNA;
NC, negative control; α-SMA, α-smooth muscle actin; FN,
fibronectin; COL-1, collagen 1. |
MIAT promotes cell viability,
migration, EMT and ECM production in TGF-β2-treated SRA01/04 cells
by sponging miR-181a
To further explore the biological mechanism
underlying MIAT during ARC, the putative target of MIAT was
predicted using the StarBase online database. miR-181a displayed a
complementary base pairing with MIAT (Fig. 3A). By investigating miR-181a
expression, it was indicated that miR-181a was downregulated in ARC
tissue samples compared with normal tissue samples (Fig. 3B). Furthermore, correlation analysis
indicated that the level of miR-181a expression was negatively
correlated with MIAT expression in ARC tissues (Fig. 3C). The dual-luciferase reporter assay
results indicated that the luciferase activity of the WT-MIAT
reporter was suppressed by miR-181a overexpression and enhanced by
miR-181a inhibition compared with the control group; however, the
luciferase activity of the MUT-MIAT reporter was not significantly
altered by either transfection (Fig.
3D). The efficiency of miR-181a overexpression and miR-181a
inhibition experiments are presented in Fig. S1C and D. Moreover, miR-181a expression was
upregulated by MIAT knockdown and downregulated by MIAT
overexpression, compared with the control group (Fig. 3E). To explore the role of miR-181a
during ARC, si-MIAT1 and anti-miR-181a were co-transfected into
TGF-β2-treated SRA01/04 cells. The results of the MTT, Transwell
and western blot assays suggested that miR-181a inhibitor reversed
the inhibitory effects of MIAT1 knockdown on TGF-β2-treated
SRA01/04 cell viability, migration, and the protein expression
levels of α-SMA, FN and COL-1 (Fig.
3F-H). Therefore, the results suggested that miR-181a was
sponged by MIAT and participated in the regulation of MIAT during
ARC progression.
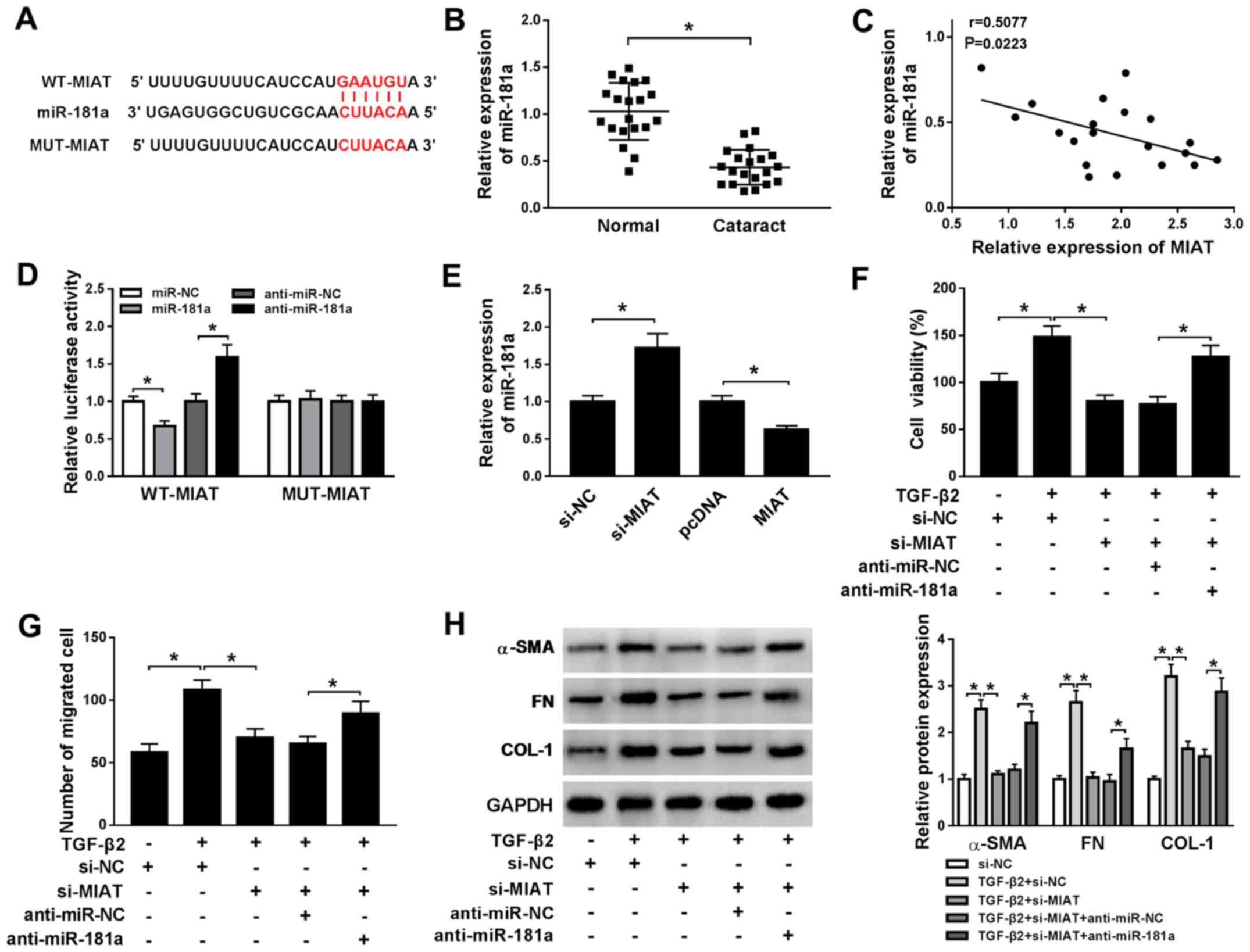 | Figure 3MIAT promotes cell viability,
migration, EMT and ECM production in TGF-β2-treated SRA01/04 cells
by sponging miR-181a. (A) The putative complementary binding
sequences or mutant sequences between MIAT and miR-181a. (B) The
level of miR-181a expression in ARC tissue samples (cataract) and
normal posterior capsule tissue samples (normal) was measured by
RT-qPCR. (C) The correlation between miR-181a and MIAT expression
was assessed by Pearson's correlation coefficient. (D) The
luciferase activities of the WT-MIAT and MUT-MIAT reporters were
detected using the dual-luciferase reporter assay. (E) The level of
miR-181a expression in TGF-β2-treated SRA01/04 cells transfected
with si-MIAT, MIAT overexpression plasmid or the corresponding
matched controls was measured by RT-qPCR. SRA01/04 cells were
transfected with si-NC, si-MIAT, si-MIAT + anti-miR-NC or si-MIAT +
anti-miR-181a and incubated with TGF-β2 for 48 h. (F) SRA01/04 cell
viability was assessed by the MTT assay. (G) The number of
migratory SRA01/04 cells was analyzed using the Transwell assay.
(H) The expression levels of α-SMA, FN and COL-1 in SRA01/04 cells
were detected by western blotting. *P<0.05, as
indicated. MIAT, myocardial infarction associated transcript; EMT,
epithelial-mesenchymal transition; ECM, extracellular matrix; miR,
microRNA; ARC, age-related cataract; RT-qPCR, reverse
transcription-quantitative PCR; WT, wild-type; MUT, mutant; TGF-β2,
transforming growth factor-β2; si, small interfering RNA; NC,
negative control; α-SMA, α-smooth muscle actin; FN, fibronectin;
COL-1, collagen 1. |
CTGF knockdown diminishes
TGF-β2-induced cell viability, migration, EMT and ECM production in
SRA01/04 cells
The expression of CTGF in TGF-β2-treated SRA01/04
cells was also investigated. CTGF expression was significantly
increased in TGF-β2-treated SRA01/04 cells compared with the
control group, which further indicated that the cell model of ARC
was successfully established (Fig.
4A and B). The RT-qPCR and
western blotting results demonstrated that si-CTGF successfully
knocked down CTGF expression in SRA01/04 cells compared with the
control group (Fig. 4C and D). To assess the effects of CTGF on ARC
development, si-CTGF was transfected into TGF-β2-treated SRA01/04
cells. CTGF knockdown reversed TGF-β2-induced promotion of SRA01/04
cell viability, migration, EMT and ECM production (Fig. 4E-G). The results indicated that CTGF
may have an active role during ARC development.
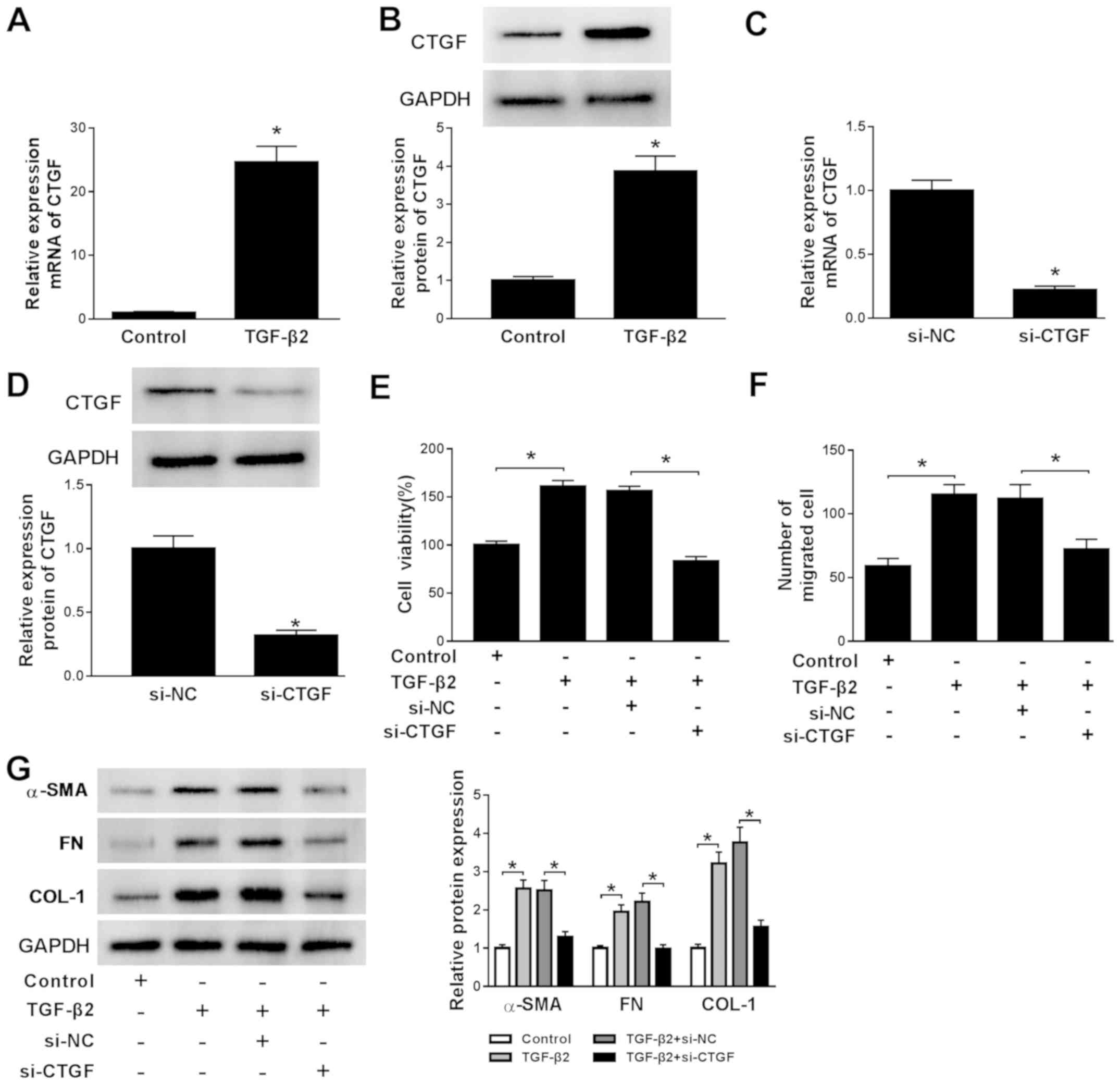 | Figure 4CTGF knockdown reverses
TGF-β2-induced cell viability, migration, epithelial-mesenchymal
transition and extracellular matrix in SRA01/04 cells. mRNA and
protein expression levels of CTGF in TGF-β2-treated SRA01/04 cells
were measured by (A) RT-qPCR and (B) western blotting,
respectively. mRNA and protein expression levels of CTGF in
SRA01/04 cells transfected with si-CTGF or si-NC were measured by
(C) RT-qPCR and (D) western blotting, respectively. SRA01/04 cells
were incubated with TGF-β2 and transfected with si-NC or si-CTGF.
(E) SRA01/04 cell viability was examined using the MTT assay. (F)
The number of migratory SRA01/04 cells was assessed by the
Transwell assay. (G) The protein expression levels of α-SMA, FN and
COL-1 in SRA01/04 cells were detected by western blotting.
*P<0.05 vs. the control group. CTGF, connective
tissue growth factor; TGF-β2, transforming growth factor-β2;
RT-qPCR, reverse transcription-quantitative PCR; si, small
interfering RNA; NC, negative control; α-SMA, α-smooth muscle
actin; FN, fibronectin; COL-1, collagen 1. |
CTGF is targeted by miR-181a
The StarBase online database indicated that the
3'-UTR of CTGF had complementary binding sites with miR-181a
(Fig. 5A). Subsequently, the
dual-luciferase reporter assay demonstrated that the luciferase
activity of the CTGF 3'-UTR-WT reporter was significantly decreased
by miR-181a mimic compared with the control group, while the
luciferase activity of the CTGF 3'UTR-MUT reporter was not altered
by miR-181a overexpression (Fig.
5B). Furthermore, anti-miR-181a resulted in the upregulation of
luciferase activity of the CTGF 3'UTR-WT reporter compared with the
control group, but the luciferase activity of the CTGF 3'UTR-MUT
reporter also displayed no alteration following transfection with
anti-miR-181a (Fig. 5C). To further
explore the effect of miR-181a expression on CTGF expression,
miR-181a mimic or miR-181a inhibitor were transfected into
TGF-β2-treated SRA01/04 cells. CTGF expression was decreased by
miR-181a overexpression and increased by miR-181a inhibition,
compared with the control group (Fig.
5D and E). The results indicated
that miR-181a may target CTGF in ARC.
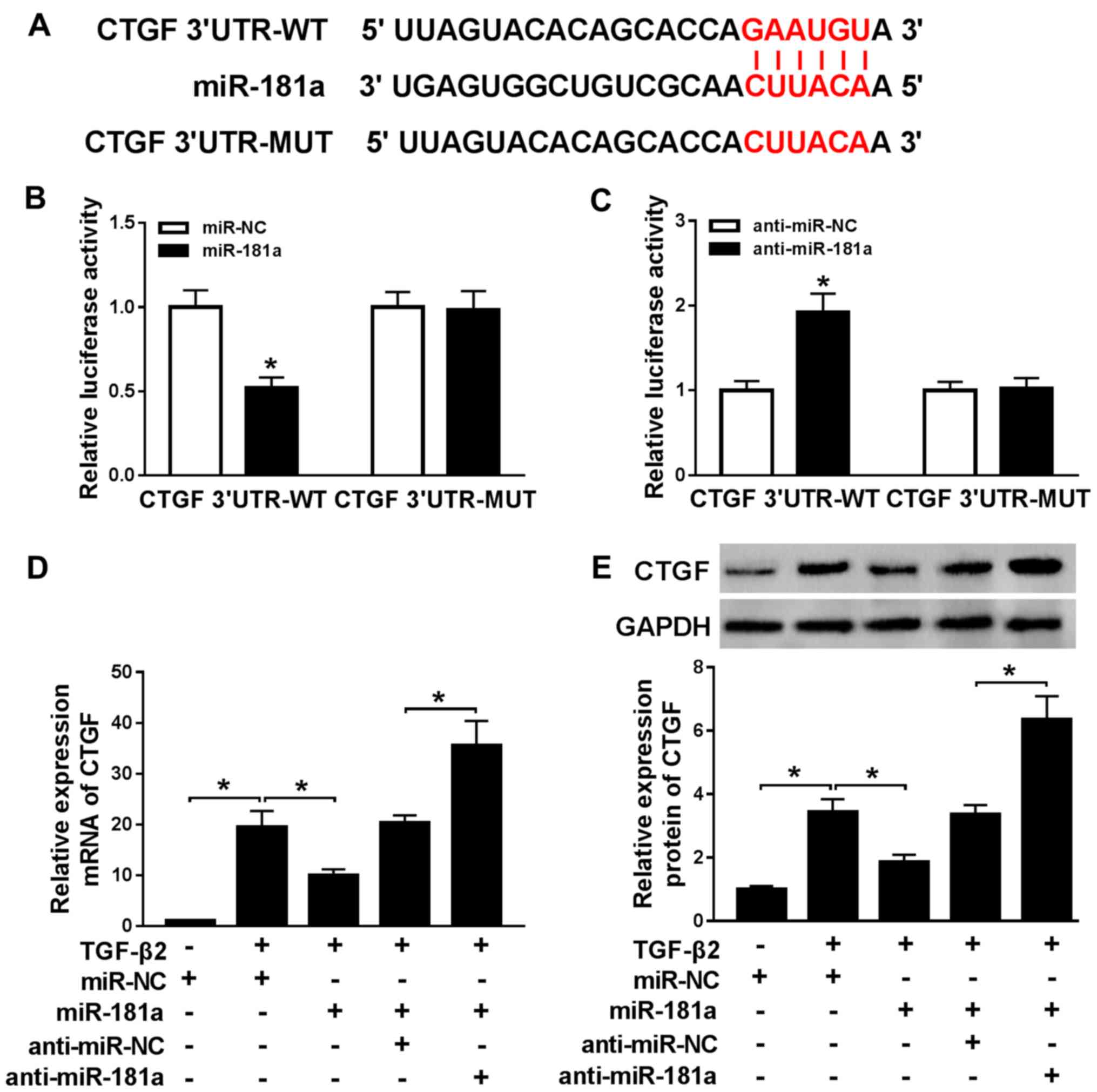 | Figure 5CTGF is targeted by miR-181a. (A) The
putative complementary or mutant sequences between miR-181a and
CTGF were identified. The luciferase activities of the CTGF
3'UTR-WT and CTGF 3'UTR-MUT reporters following transfection with
(B) miR-NC or miR-181a and (C) anti-miR-NC or anti-miR-181a were
estimated using the dual-luciferase reporter assay. mRNA and
protein expression levels of CTGF in TGF-β2-treated SRA01/04 cells
transfected with miR-181a, anti-miR-181a, miR-NC or anti-miR-NC
were measured by (D) reverse transcription-quantitative PCR and (E)
western blotting, respectively. *P<0.05, vs. the
control group. CTGF, connective tissue growth factor; miR,
microRNA; UTR, untranslated region; WT, wild-type; MUT, mutant; NC,
negative control. |
MIAT increases CTGF expression to
promote cell viability, migration, EMT and ECM production in
TGF-β2-treated SRA01/04 cells via miR-181a
To explore the biological mechanism linking MIAT,
miR-181a and CTGF, TGF-β2-treated SRA01/04 cells were
co-transfected with miR-181a mimic and MIAT overexpression plasmid
to evaluate the effect of MIAT expression on CTGF expression. The
mRNA and protein expression levels of CTGF were increased by MIAT
overexpression in TGF-β2-treated SRA01/04 cells compared with the
control group. miR-181a overexpression reversed the enhancing
effects of MIAT overexpression, indicating that CTGF expression was
regulated by MIAT and miR-181a. To further verify the hypothesis,
MIAT overexpression plasmid and si-CTGF were co-transfected into
TGF-β2-treated SRA01/04 cells to detect cell viability, migration,
EMT and ECM production. CTGF knockdown reversed the effects of MIAT
overexpression on TGF-β2-treated SRA01/04 cell viability, migration
and protein expression levels of α-SMA, FN and COL-1 (Fig. 6C-E). The results indicated that MIAT
modulated CTGF to regulate the progression of ARC by sponging
miR-181a.
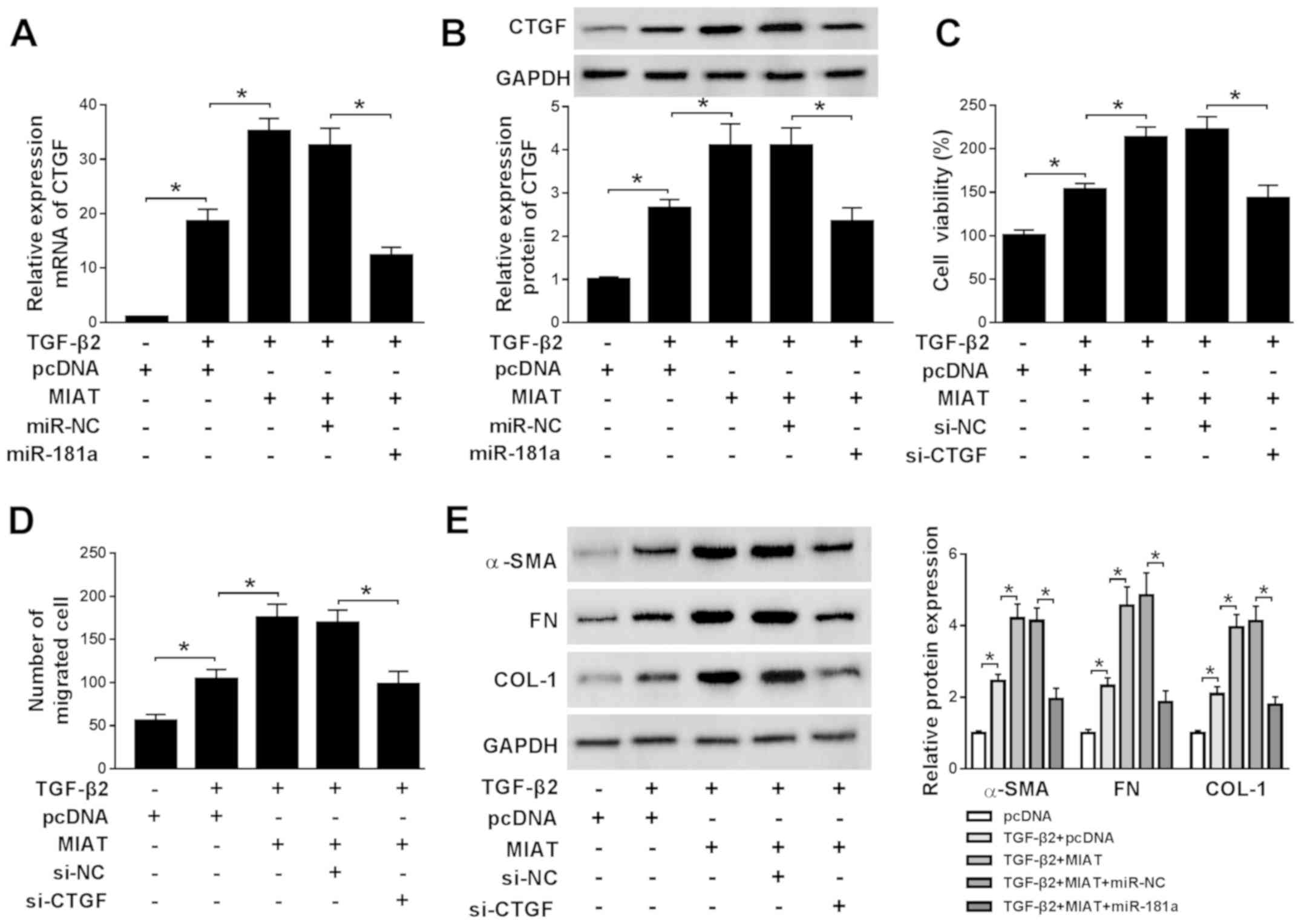 | Figure 6MIAT increases CTGF expression to
promote cell viability, migration, epithelial-mesenchymal
transition and extracellular matrix in TGF-β2-treated SRA01/04
cells via miR-181a. SRA01/04 cells were treated with TGF-β2 and
subsequently transfected with pcDNA, MIAT, MIAT + miR-NCor MIAT +
miR-181a. mRNA and protein expression levels of CTGF in SRA01/04
cells were measured by (A) reverse transcription-quantitative PCR
and (B) western blotting, respectively. SRA01/04 cells were treated
with TGF-β2 and subsequently transfected with pcDNA, MIAT, MIAT +
si-NC or MIAT + si-CTGF. (C) SRA01/04 cell viability was assessed
using the MTT assay. (D) The number of migratory SRA01/04 cells was
examined by the Transwell assay. (E) The protein expression levels
of α-SMA, FN and COL-1 in SRA01/04 cells were assessed by western
blotting. *P<0.05, as indicated. MIAT, myocardial
infarction associated transcript; CTGF, connective tissue growth
factor; TGF-β2, transforming growth factor-β2; miR, microRNA; si,
small interfering RNA; NC, negative control; α-SMA, α-smooth muscle
actin; FN, fibronectin; COL-1, collagen 1. |
MIAT and CTGF regulate the activity of
the ERK signaling pathway
ERK signaling is related to the development of a
number of diseases (24); therefore,
si-MIAT and CTGF overexpression plasmid were co-transfected into
TGF-β2-treated SRA01/04 cells to detect the expression levels of
ERK signaling pathway-related proteins. The efficiency of CTGF
overexpression transfection is presented in Fig. S1C and D. si-MIAT successfully decreased the
expression of CTGF and CTGF overexpression plasmid successfully
reversed si-MIAT-mediated downregulation of CTGF expression
compared with the control group (Fig.
7A and B). TGF-β2 increased the
protein expression levels of p-MEK and p-ERK compared with the
control group, while MIAT knockdown reversed TGF-β2-induced effects
on protein expression. Additionally, CTGF overexpression reversed
the effects of MIAT knockdown on the protein expression levels of
p-MEK and p-ERK (Fig. 7A, C and D). The
results indicated that the ERK signaling pathway was involved in
MIAT-mediated regulation of ARC.
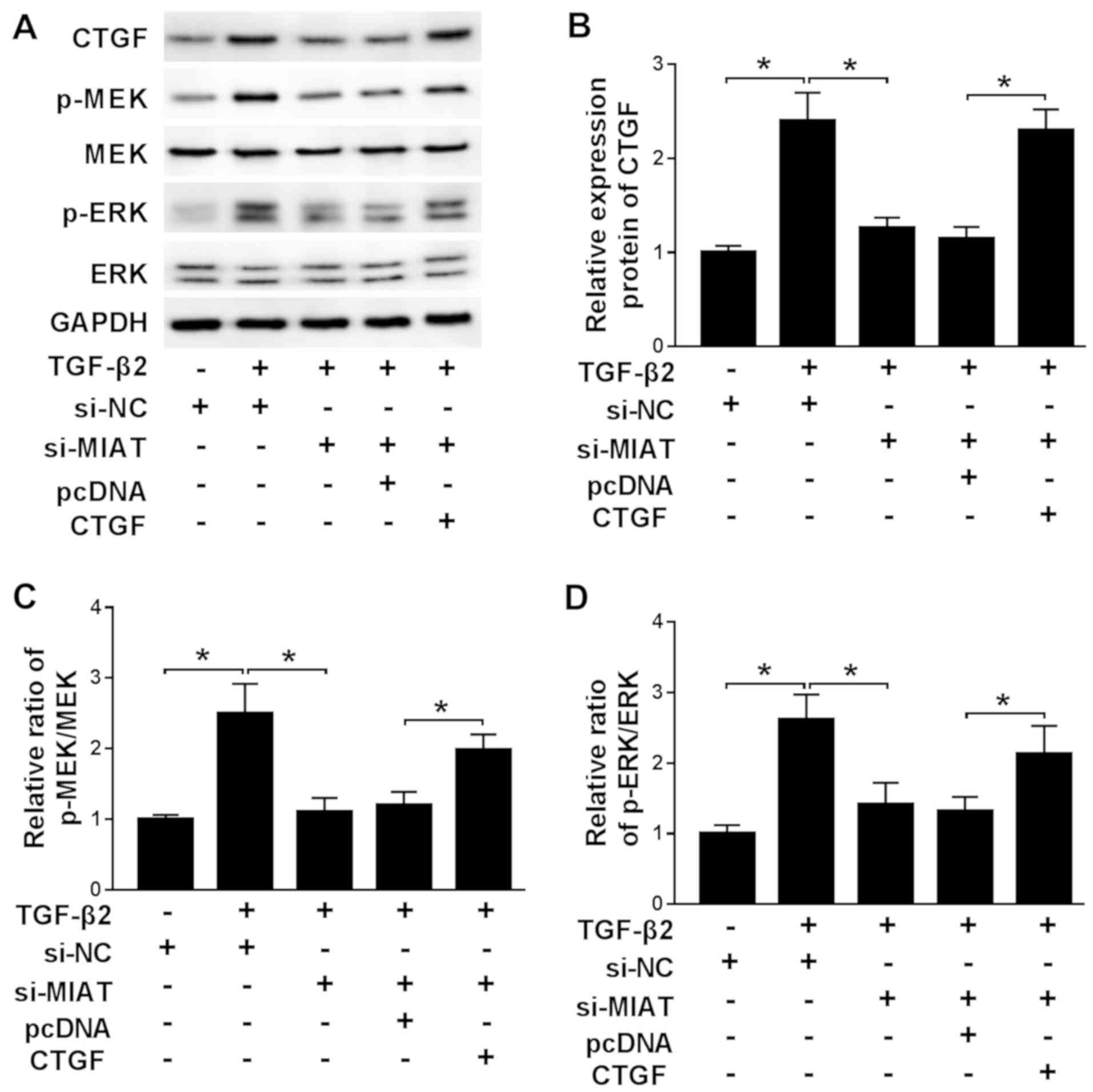 | Figure 7MIAT and CTGF regulate the activity
of the ERK signaling pathway. TGF-β2-treated SRA01/04 cells were
transfected with si-NC, si-MIAT, si-MIAT + pcDNA or si-MIAT + CTGF.
Protein expression levels were (A) determined by western blotting
and quantified for (B) CTGF, (C) p-MEK/MEK and (D)
p-ERK(p-ERK1/2)/ERK (ERK1/2). Fig. 7A presents two bands for p-ERK
as anti-erk1/2 and anti-p-ERK1/2 were used. *P<0.05,
as indicated. MIAT, myocardial infarction associated transcript;
CTGF, connective tissue growth factor; TGF-β2, transforming growth
factor-β2; si, small interfering RNA; NC, negative control; p,
phosphorylated; MEK, mitogen-activated protein kinase. |
Discussion
ARC can lead to blindness in elderly individuals
(28). Increasing evidence suggests
that lncRNAs affect a number of biological processes in numerous
different diseases, including ARC (9-12).
The present study aimed to explore the biological mechanism
underlying MIAT during ARC development. Collectively, the results
indicated that MIAT regulated CTGF expression to facilitate cell
viability, migration, EMT and ECM production during ARC via the ERK
signaling pathway by sponging miR-181a.
MIAT dysregulation has been identified in a number
of different diseases. For example, previous studies investigating
coronary artery disease indicated that MIAT expression was
significantly elevated in the serum of patients with coronary
artery disease (16,17). Furthermore, MIAT upregulation has
been reported in ischemic stroke (18). In the present study, MIAT was
upregulated in ARC tissues and TGF-β2-treated SRA01/04 cells
compared with the corresponding control groups. MIAT knockdown
reversed TGF-β2-induced cell viability, migration, EMT and ECM
production in SRA01/04 cells. The aforementioned results were
consistent with a previous report (19), indicating that MIAT may play an
active role during the development of ARC.
Accumulating evidence has suggested that miRNAs are
associated with the development of a number of diseases (20,29). For
example, MIAT knockdown inhibited the viability, migration and
invasion of pancreatic carcinoma cells by targeting
miR-133(30). Zhang et al
(31) reported that MIAT promoted
cell viability and migration, and inhibited cell apoptosis in
osteosarcoma via sponging miR-128-3p in vitro. The present
study further indicated that miR-181a was downregulated in ARC
tissues compared with normal tissues. Similar results were also
reported in a previous study (22).
Emerging evidence indicated that MIAT acted as a molecular sponge
to regulate miRNA expression, resulting in altered target gene mRNA
expression (32). In the present
study, the results indicated that MIAT sponged miR-181a.
Furthermore, miR-181a inhibitor reversed the inhibitory effects of
MIAT knockdown on cell viability, migration, EMT and ECM in
TGF-β2-treated SRA01/04 cells. Overall, the results suggested that
MIAT facilitated ARC progression by sponging miR-181a.
Previous studies revealed that CTGF is involved in a
number of biological processes, including ECM production, cell
proliferation, adhesion, migration, fibrosis and differentiation
(33-35).
Wang et al (36) reported
that CTGF was increased in vivo and in vitro during
hypertension, and its overexpression promoted vascular smooth
muscle cell proliferation. In the present study, CTGF expression
was enhanced in ARC samples and TGF-β2-treated SRA01/04 cells
compared with the control groups. CTGF knockdown reduced cell
viability, migration, EMT and ECM production in TGF-β2-treated
SRA01/04 cells. Furthermore, CTGF was a direct candidate target of
miR-181a. MIAT promoted cell viability, migration, EMT and ECM
production in TGF-β2-treated SRA01/04 cells by modulating CTGF
expression. The results suggested that CTGF expression was crucial
for MIAT-mediated regulation of ARC progression.
Accumulating evidence has suggested that the ERK
signaling pathway is associated with the development of multiple
diseases (26). For example, one
study reported that ERK signaling was activated during the
development of Kashin-Beck disease (37). Another study reported that Notch1
expression relieved cigarette smoke extract-induced apoptosis by
inhibiting the ERK signaling pathway during chronic obstructive
pulmonary disease (38). In the
present study, MIAT knockdown inhibited the activity of the ERK
signaling pathway and CTGF overexpression reversed the inhibitory
effect of MIAT. The results suggested that MIAT was involved in the
development of ARC by activating the ERK signaling pathway.
The present study had a number of limitations. A
previous study has reported that MIAT affects the progression of
PCO by altering the proliferation, metastasis and EMT of LECs
(19); however, the role of MIAT in
cataract also remains largely unclear. Therefore, the effect of
MIAT on the progression of other types of cataract requires further
investigation. In addition, the inhibitory effect of miR-181a
inhibitor on MIAT-induced effects was partial; therefore, other
miRNAs may be involved in MIAT-mediated regulation of ARC
progression, which requires further investigation.
To conclude, MIAT and CTGF expression were
increased, while miR-181a was decreased in ARC tissues and
TGF-β2-treated SRA01/04 cells. Mechanical and functional
experiments indicated that MIAT increased cell viability,
migration, EMT and ECM production during ARC by activating the ERK
signaling pathway via the miR-181a/CTGF axis. Therefore, the novel
regulatory network identified in the present study may serve as a
therapeutic target for ARC and provide theoretical basis for the
mechanism underlying ARC development.
Supplementary Material
Cell transfection efficiency. Relative
expression of MIAT was assessed by RT-qPCR in SRA01/04 cells
transfected with (A) si-NC, si-MIAT, (B) pcDNA and MIAT. Relative
expression of miR-181a was assessed by RT-qPCR in SRA01/04 cells
transfected with (C) miR-NC, miR.181a, (D) anti-miR-NC and
anti-miR-181a. Relative expression of CTGF was assessed by (E)
RT-qPCR and (F) western blotting in SRA01/04 cells transfected with
pcDNA or CTGF. *P<0.05 vs. the control group. MIAT,
myocardial infarction associated transcript; RT-qPCR, reverse
transcription-quantitative PCR; si, small interfering RNA; NC,
negative control; miR, microRNA; CTGF, connective tissue growth
factor.
MIAT overexpression facilitates
SRA01/04 cell proliferation, migration, epithelial-mesenchymal
transition and extracellular matrix production. SRA01/04 cells were
transfected with pcDNA or MIAT. (A) Cell viability was assessed
using the MTT assay. (B) SRA01/04 cell migration was analysed using
the Transwell assay. (C) Protein expression levels of α-SMA, FN and
COL-1 were detected by western blotting. *P<0.05 vs.
the control group. MIAT, myocardial infarction associated
transcript; α-SMA, α-smooth muscle actin; FN, fibronectin; COL-1,
collagen I.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL and KT conceptualized the current study. JL, KT,
LLu and FY acquired data. All authors contributed to writing and
reviewing the manuscript. JL and LLian supervised the current
study. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All participants or their guardians provided written
informed consent. The present study was approved by the Ethics
Committee of the Department of Ophthalmology, Renmin Hospital,
Hubei University of Medicine.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lim JC, Umapathy A and Donaldson PJ: Tools
to fight the cataract epidemic: A review of experimental animal
models that mimic age related nuclear cataract. Exp Eye Res.
145:432–443. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Raj SM, Vasavada AR, Johar SR and Vasavada
VA and Vasavada VA: Post-operative capsular opacification: A
review. Int J Biomed Sci. 3:237–250. 2007.PubMed/NCBI
|
|
3
|
Peng Q, Hennig A, Vasavada AR and Apple
DJ: Posterior capsular plaque: A common feature of cataract surgery
in the developing world. Am J Ophthalmol. 125:621–626.
1998.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lovicu FJ, Ang S, Chorazyczewska M and
McAvoy JW: Deregulation of lens epithelial cell proliferation and
differentiation during the development of TGFbeta-induced anterior
subcapsular cataract. Dev Neurosci. 26:446–455. 2004.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Marcantonio JM, Syam PP, Liu CS and Duncan
G: Epithelial transdifferentiation and cataract in the human lens.
Exp Eye Res. 77:339–346. 2003.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Guo R, Meng Q, Guo H, Xiao L, Yang X, Cui
Y and Huang Y: TGF-β2 induces epithelial-mesenchymal transition in
cultured human lens epithelial cells through activation of the
PI3K/Akt/mTOR signaling pathway. Mol Med Rep. 13:1105–1110.
2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ma B, Kang Q, Qin L, Cui L and Pei C:
TGF-β2 induces transdifferentiation and fibrosis in human lens
epithelial cells via regulating gremlin and CTGF. Biochem Biophys
Res Commun. 447:689–695. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Mathieu EL, Belhocine M, Dao LT, Puthier D
and Spicuglia S: Functions of lncRNA in development and diseases.
Med Sci (Paris). 30:790–796. 2014.PubMed/NCBI View Article : Google Scholar : (In French).
|
|
9
|
Zhang Z, Zhu H, Liu Y, Quan F, Zhang X and
Yu L: LncRNA HOTAIR mediates TGF-β2-induced cell growth and
epithelial-mesenchymal transition in human lens epithelial cells.
Acta Biochim Biophys Sin (Shanghai). 50:1028–1037. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chen B, Ma J, Li C and Wang Y: Long
noncoding RNA KCNQ1OT1 promotes proliferation and
epithelial-mesenchymal transition by regulation of SMAD4 expression
in lens epithelial cells. Mol Med Rep. 18:16–24. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li G, Song H, Chen L, Yang W, Nan K and Lu
P: TUG1 promotes lens epithelial cell apoptosis by regulating
miR-421/caspase-3 axis in age-related cataract. Exp Cell Res.
356:20–27. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang Y, Chen L, Gu Y, Wang Y, Yuan Y, Zhu
Q, Bi M and Gu S: LncRNA FEZF1-AS1 promotes TGF-β2-mediated
proliferation and migration in human lens epithelial cells
SRA01/04. J Ophthalmol. 2019(4736203)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gong W, Zhu G, Li J and Yang X: LncRNA
MALAT1 promotes the apoptosis and oxidative stress of human lens
epithelial cells via p38MAPK pathway in diabetic cataract. Diabetes
Res Clin Pract. 144:314–321. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Qi D, Wang M, Zhang D and Li H: Tanshinone
IIA protects lens epithelial cells from
H2O2-induced injury by upregulation of lncRNA
ANRIL. J Cell Physiol (Epub ahead of print).
|
|
15
|
Cheng T, Xu M, Qin B, Wu J, Tu Y, Kang L,
Wang Y and Guan H: lncRNA H19 contributes to oxidative damage
repair in the early age-related cataract by regulating miR-29a/TDG
axis. J Cell Mol Med. 23:6131–6139. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Toraih EA, El-Wazir A, Alghamdi SA,
Alhazmi AS, El-Wazir M, Abdel-Daim MM and Fawzy MS: Association of
long non-coding RNA MIAT and MALAT1 expression profiles in
peripheral blood of coronary artery disease patients with previous
cardiac events. Genet Mol Biol. 42:509–518. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tan J, Liu S, Jiang Q, Yu T and Huang K:
LncRNA-MIAT increased in patients with coronary atherosclerotic
heart disease. Cardiol Res Pract. 2019(6280194)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhu M, Li N, Luo P, Jing W, Wen X, Liang C
and Tu J: Peripheral blood leukocyte expression of lncRNA MIAT and
its diagnostic and prognostic value in ischemic stroke. J Stroke
Cerebrovasc Dis. 27:326–337. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Shen Y, Dong LF, Zhou RM, Yao J, Song YC,
Yang H, Jiang Q and Yan B: Role of long non-coding RNA MIAT in
proliferation, apoptosis and migration of lens epithelial cells: A
clinical and in vitro study. J Cell Mol Med. 20:537–548.
2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Korhan P, Erdal E and Atabey N:
MiR-181a-5p is downregulated in hepatocellular carcinoma and
suppresses motility, invasion and branching-morphogenesis by
directly targeting c-Met. Biochem Biophys Res Commun.
450:1304–1312. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Gong W, Li J, Wang Y, Meng J and Zheng G:
miR-221 promotes lens epithelial cells apoptosis through
interacting with SIRT1 and E2F3. Chem Biol Interact. 306:39–46.
2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Dong N, Tang X and Xu B: miRNA-181a
inhibits the proliferation, migration, and epithelial-mesenchymal
transition of lens epithelial cells. Invest Ophthalmol Vis Sci.
56:993–1001. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ramazani Y, Knops N, Elmonem MA, Nguyen
TQ, Arcolino FO, van den Heuvel L, Levtchenko E, Kuypers D and
Goldschmeding R: Connective tissue growth factor (CTGF) from basics
to clinics. Matrix Biol. 68-69:44–66. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Pei C, Ma B, Kang QY, Qin L and Cui LJ:
Effects of transforming growth factor β2 and connective tissue
growth factor on induction of epithelial mesenchymal transition and
extracellular matrix synthesis in human lens epithelial cells. Int
J Ophthalmol. 6:752–757. 2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lv J, Sun B, Mai Z, Jiang M and Du J:
CLDN-1 promoted the epithelial to migration and mesenchymal
transition (EMT) in human bronchial epithelial cells via Notch
pathway. Mol Cell Biochem. 432:91–98. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ma B, Yang L, Jing R, Liu J, Quan Y, Hui
Q, Li J, Qin L and Pei C: Effects of Interleukin-6 on posterior
capsular opacification. Exp Eye Res. 172:94–103. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Asbell PA, Dualan I, Mindel J, Brocks D,
Ahmad M and Epstein S: Age-related cataract. Lancet. 365:599–609.
2005.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Li Z, Qu L, Luo W, Tian Y, Zhai H, Xu K
and Zhong H: Mig-6 is down-regulated in HCC and inhibits the
proliferation of HCC cells via the P-ERK/Cyclin D1 pathway. Exp Mol
Pathol. 102:492–499. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Li TF, Liu J and Fu SJ: The interaction of
long non-coding RNA MIAT and miR-133 play a role in the
proliferation and metastasis of pancreatic carcinoma. Biomed
Pharmacother. 104:145–150. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhang C, Xie L, Liang H and Cui Y: LncRNA
MIAT facilitates osteosarcoma progression by regulating
miR-128-3p/VEGFC Axis. IUBMB Life. 71:845–853. 2019.PubMed/NCBI View
Article : Google Scholar
|
|
32
|
Esteller M: Non-coding RNAs in human
disease. Nat Rev Genet. 12:861–874. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
33
|
Jun JI and Lau LF: Taking aim at the
extracellular matrix: CCN proteins as emerging therapeutic targets.
Nat Rev Drug Discov. 10:945–963. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Hong L, Lai HL, Fang Y, Tao Y and Qiu Y:
Silencing CTGF/CCN2 inactivates the MAPK signaling pathway to
alleviate myocardial fibrosis and left ventricular hypertrophy in
rats with dilated cardiomyopathy. J Cell Biochem. 119:9519–9531.
2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Liu F, Chen WW, Li Y, Zhang JQ and Zheng
QB: MiR-6836-3p promotes proliferation of hypertrophic scar
fibroblasts by targeting CTGF. Eur Rev Med Pharmacol Sci.
22:4069–4074. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wang WB, Li HP, Yan J, Zhuang F, Bao M,
Liu JT, Qi YX and Han Y: CTGF regulates cyclic stretch-induced
vascular smooth muscle cell proliferation via microRNA-19b-3p. Exp
Cell Res. 376:77–85. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Dai X, Song R and Xiong Y: The expression
of ERK and JNK in patients with an endemic osteochondropathy,
Kashin-Beck disease. Exp Cell Res. 359:337–341. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zong D, Li J, Cai S, He S, Liu Q, Jiang J,
Chen S, Long Y, Chen Y, Chen P and Ouyang R: Notch1 regulates
endothelial apoptosis via the ERK pathway in chronic obstructive
pulmonary disease. Am J Physiol Cell Physiol. 315:C330–C340.
2018.PubMed/NCBI View Article : Google Scholar
|