Introduction
Chronic myeloid leukemia (CML) is a
myeloproliferative disorder caused by BCR-ABL-induced hematopoietic
stem cell transformation (1).
Although tyrosine kinase inhibitor (TKI) therapy has successfully
improved the long-term survival rates of patients with CML
(2), certain patients relapse due
to TKI resistance (3). BCR-ABL
tyrosine kinase constitutively activates multiple signaling
pathways, such as the PI3K/AKT/mTOR and JAK-STAT signaling
pathways, that dysregulate cell cycle progression, transforms
hematopoietic stem cells and induces drug resistance (4,5).
Therefore, targeting multiple pathways may provide novel
therapeutic effects for overcoming TKI resistance (6).
Dysregulation of lipid metabolism has been
demonstrated to be a major event in the progression of various
types of cancer, including hematopoietic malignancies (7-10).
Sphingosine 1-phosphate (S1P) is a bioactive sphingolipid
metabolite that mediates diverse cellular processes, including cell
proliferation, survival and migration (9,11).
Sphingosine kinase (Sphk) phosphorylates sphingosines to generate
S1P and is a critical signal regulator of sphingolipid metabolism
(12). There are two isoforms of
Sphk: Sphk1 and Sphk2(13). Sphk1
overexpression and aberrant activation have been detected in
various types of cancer, including hematopoietic malignancies such
as leukemia and multiple myeloma (14,15).
Extracellular stimuli and numerous genetic mutations, including
Fms-like tyrosine kinase 3 (FLT3), Kit receptor tyrosine kinase and
BCR-ABL fusion protein, aberrantly activate the Sphk1/S1P pathway
in leukemia cells (15-20).
In CML, the Sphk1/S1P pathway mediates BCR/ABL
activation-induced Mcl-1 upregulation (16). Furthermore, activation of Sphk1
contributes to imatinib resistance by modulating protein
phosphatase 2A (17). Previous
studies have revealed that targeting Sphk1 induces Mcl-1-dependent
cell death in acute myeloid leukemia (21) and inhibition of the Sphk1/S1P axis
is considered to be novel approach to overcome drug resistance
(22). These previous studies
support the hypothesis that Sphk1 is a novel therapeutic target for
the treatment of leukemia (15).
SIRT1 is a nicotine adenine dinucleotide-dependent
protein deacetylase which directly links transcriptional regulation
to intracellular energy and is involved in the coordination of
several distinct cellular functions, including cell survival,
apoptosis and metabolism (23).
SIRT1 is significantly increased in leukemia stem cells and
promotes leukemia development (24). Furthermore, SIRT1 activation
promotes the maintenance and drug resistance of human FLT3-internal
tandem duplication in acute myeloid leukemia stem cells (25). Inhibition of SIRT1 induces G1
arrest, apoptosis and inhibits the proliferation of acute myeloid
gene 1 and myeloid transforming gene 8)-positive cells (26). Furthermore, targeting SIRT1 induces
CML sensitivity to TKI treatment via the activation of p53(27). Therefore, SIRT1 inhibition may be a
promising novel approach for the ablation of leukemia stem cells in
leukemia therapy (25,28).
Previous research elucidated that SIRT1 mediated
Sphk1/S1P-induced neovascularization, including the proliferation
and migration of endothelial cells (13). Given the important regulatory roles
of Sphk1/S1P in leukemogenesis, it was hypothesized that Sphk1 and
SIRT1 may interact in leukemia cells and targeting them may offer a
novel and effective therapeutic approach. The present study
validated that the Sphk1/S1P/SIRT1 axis is activated in CML cell
lines and investigated the effects of Sphk1 and SIRT1 inhibition on
the growth, survival and drug resistance of leukemia cells, which
provided the basis of combined suppressive effects by targeting
dual molecules in leukemia cells.
Materials and methods
Leukemia cell lines and
inhibitors
The human leukemia cell lines K562 and KCL22 were
cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (HyClone; Cytiva). TF-1 cells were
obtained from America Type Culture Collection and transduced with
miGR1-BA-T315I retroviral vectors to establish the
imatinib-resistant cell line TF-1-T315I. These cells were cultured
in RPMI-1640 medium supplemented with 10% FBS and 5 ng/ml GM-CSF
(Sigma-Aldrich; Merck KGaA). The Sphk1-specific inhibitors DMS
(Sigma-Aldrich; Merck KGaA) and SKI-II (Sigma-Aldrich; Merck KGaA),
and the SIRT1-specific inhibitor EX527 (MedchemExpress) were
dissolved in DMSO.
Cell growth assay
The cell growth assay was performed using a Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.)
according to the manufacturer's instructions, which stains living
cells. K562, KCL22 or TF-1-T315I cells were seeded into 96-well
plates (5x103 cells/well) in 100 µl of culture medium
and treated with DMSO, SKI-II (5, 10 or 20 µM), EX527 (5, 10 or 20
µM) or SKI-II (20 µM) + EX527 (20 µM) for 24 or 48 h. Subsequently,
the cells were incubated with 10 µl of CCK-8 reagent in a 5%
CO2 atmosphere at 37˚C for 3 h and the optical density
at 450 nm was measured on Varioskan Flash (Thermo Fisher
Scientific, Inc.). All experiments were performed in
triplicate.
Cell colony forming assay
K562 or TF-1-T315I cells were seeded in triplicate
in 24-well plates (5x102 cells/well) and cultured in 1%
methylcellulose medium (Beijing Solarbio Science & Technology
Co., Ltd.) containing 10% FBS supplemented with or without 5 ng/ml
GM-CSF. The cells were then treated with DMSO, 20 µM EX527, 20 µM
SKI-II or 20 µM EX527 + 20 µM SKI-II. Following 7 days of culturing
at 37˚C, the number of colonies (>50 cells) were counted using
an inverted light microscope (Olympus Corporation) under normal
light at x40 magnification.
Cell cycle analysis
TF-1-T315I cells were seeded into 12-well plates
(1x105 cells/well) and cultured in RPMI-1640 medium
containing 10% FBS supplemented with 5 ng/ml GM-CSF. Then, DMSO, 20
µM EX527, 20 µM SKI-II or 20 µM EX527 + 20 µM SKI-II were added to
the medium. Following incubation in a 5% CO2 atmosphere
at 37˚C for 24 h, cells were collected, washed with PBS and fixed
in ice-cold 70% ethanol overnight at -20˚C. Cells were washed with
PBS and stained with 20 µg/ml propidium iodide (PI; BD Biosciences)
at 4˚C in the dark for 30 min. Data were acquired on a FACSCalibur
(BD Biosciences) and analyzed using FlowJo software version 7.6
(Becton-Dickinson & Company).
Cell apoptosis assay
K562, KCL22 or TF-1-T315I cells were plated in
12-well plates (1x105 cells/well) and cultured in
RMPI-1640 medium containing 10% FBS supplemented with or without 5
ng/ml GM-CSF. Subsequently, the cells were treated with DMSO, 20 µM
EX527, 20 µM SKI-II or 20 µM EX527 + 20 µM SKI-II. After culturing
in a 5% CO2 atmosphere at 37˚C for 24 h, the cells were
collected, washed with PBS and suspended in 100 µl of 1X binding
buffer containing 5 µl of Annexin V-APC (eBioscience; Thermo Fisher
Scientific, Inc.). Following incubation at room temperature for 20
min, samples were stained with PI at room temperature in the dark
for 15 min and detected by FACSCalibur (BD Biosciences) within 1 h.
The data were analyzed using FlowJo software version 7.6
(Becton-Dickinson & Company).
Lentivirus transduction
TF1 cells were plated in six-well plates
(2x105 cells/well) and transduced with lentiviral
vectors encoding BCR-ABL1 (T315I) or control vectors at a
multiplicity of infection of 20. The mutant BCR-ABL transduction
efficiency of lentiviral vectors, as indicated by GFP expression,
was detected by the intensity of green fluorescence at FITC channel
on FACSCalibur (BD Biosciences). The data were analyzed using
FlowJo software version 7.6 (Becton-Dickinson & Company).
Western blotting
K562 cells were treated with DMSO, S1P (0.5, 1, 1.5
or 2 µM for 8 h or 1 µM for 2, 4, 8, 12 or 24 h), DMS (5, 10 or 20
µM for 24 h), EX527 (5, 10, 20 or 40 µM for 24 h), SKI-II (20 µM
for 24 h) or SKI-II + EX527 (20 µM EX527 and 20 µM SKI-II for 24
h). TF-1-T315I cells were treated with DMSO, EX527 (5, 10, 20 or 40
µM for 24 h), SKI-II (20 µM for 24 h) or SKI-II + EX527 (20 µM
EX527 and 20 µM SKI-II for 24 h). Following this, the cells were
washed twice with 1X PBS and proteins were extracted from cells by
suspension in RIPA buffer (Sigma-Aldrich; Merck KGaA) containing
PMSF (Sigma-Aldrich; Merck KGaA) and 1% phosphatase inhibitors
(Sigma-Aldrich; Merck KGaA). Lysates were clarified by
centrifugation at 12,000 x g, 4˚C for 30 min and total protein was
determined using a BCA protein assay kit (Thermo Fisher Scientific,
Inc.). Then, 30 µg of sample protein was separated by 12% SDS-PAGE
and transferred to PVDF membranes. Following blocking with 5%
non-fat dry milk in TBS with 0.05% Tween-20 at room temperature for
2 h. The membranes were incubated with primary antibodies against
SIRT1 (cat. no. 2496; 1:1,000 dilution; Cell Signaling Technology,
Inc.), ERK (cat. no. 4695; 1:1,000 dilution; Cell Signaling
Technology, Inc.), phosphorylated (p-)ERK (cat. no. 9101; 1:1,000
dilution; Cell Signaling Technology, Inc.), STAT5 (cat. no. 25656;
1:1,000 dilution; Cell Signaling Technology, Inc.), p-STAT5 (cat.
no. 9359; 1:1,000 dilution; Cell Signaling Technology, Inc.),
β-actin (cat. no. 4970; 1:1,000 dilution; Cell Signaling
Technology, Inc.) and GAPDH (cat. no. 2118; 1:1,000 dilution; Cell
Signaling Technology, Inc.). Subsequently, the membranes were
incubated with an anti-rabbit secondary antibody conjugated with
peroxidase (cat. no. ZB-5301; 1:5,000 dilution; OriGene
Technologies, Inc.). Signals were detected using a chemiluminescent
detection system (Pierce; Thermo Fisher Scientific, Inc.).
Statistical analysis
Values were presented as the mean ± standard
deviation. All data were analyzed using GraphPad Prism software
(version no. 5; GraphPad Software, Inc.). Differences among ≥3
groups were compared using one-way ANOVA followed by the Bonferroni
post hoc test. P<0.05 was considered to indicate a statistically
significant difference. All experiments were performed three
times.
Results
Sphk1/S1P signaling upregulates SIRT1
expression in leukemia cells
To explore the interaction between Sphk1/S1P and
SIRT1 and signal integration in leukemia cells, SIRT1 expression
was determined in K562 cells treated with S1P. Treatment of K562
cells with 0.5, 1.0, 1.5 or 2.0 µM S1P for 8 h significantly
increased SIRT1 protein expression compared with the 0-µM group
(Fig. 1A). The time course of
S1P-induced SIRT1 expression is presented in Fig. 1B. SIP significantly upregulated the
protein levels of SIRT1 at 4 and 8 h. Furthermore, treatment of
K562 cells with 5, 10 or 20 µM DMS, a Sphk1 inhibitor,
significantly decreased SIRT1 expression compared with the 0-µM
group (Fig. 1C). Therefore, the
results demonstrated that the Sphk1/S1p/SIRT1 axis is functional in
leukemia cells and may serve an important role in regulating
leukemogenesis.
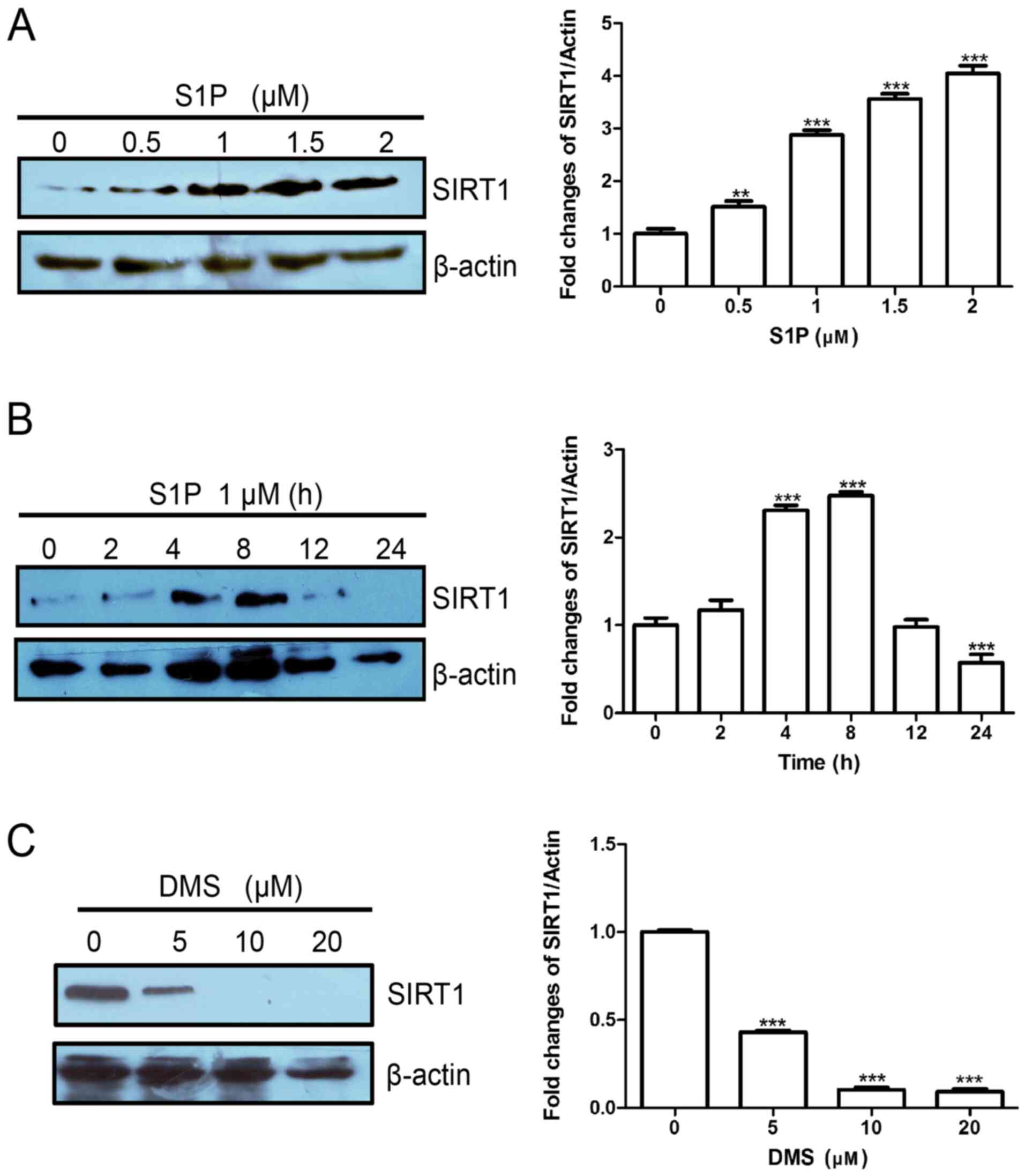 | Figure 1Sphingosine kinase 1/S1P upregulates
SIRT1 expression in leukemia cells. (A) K562 cells were treated
with S1P (0, 0.5, 1, 1.5 or 2 µM) for 8 h. (B) K562 cells were
treated with 1 µM S1P for various time-points (0, 2, 4, 8, 12 or 24
h). (C) K562 cells were treated with DMS (0, 5, 10 or 20 µM) for 24
h. SIRT1 expression was determined by western blotting. Data are
presented as the means ± standard deviation. **P<0.01
and ***P<0.001 compared with 0 µM or 0 h. S1P,
sphingosine 1-phosphate; SIRT1, sirtuin 1. |
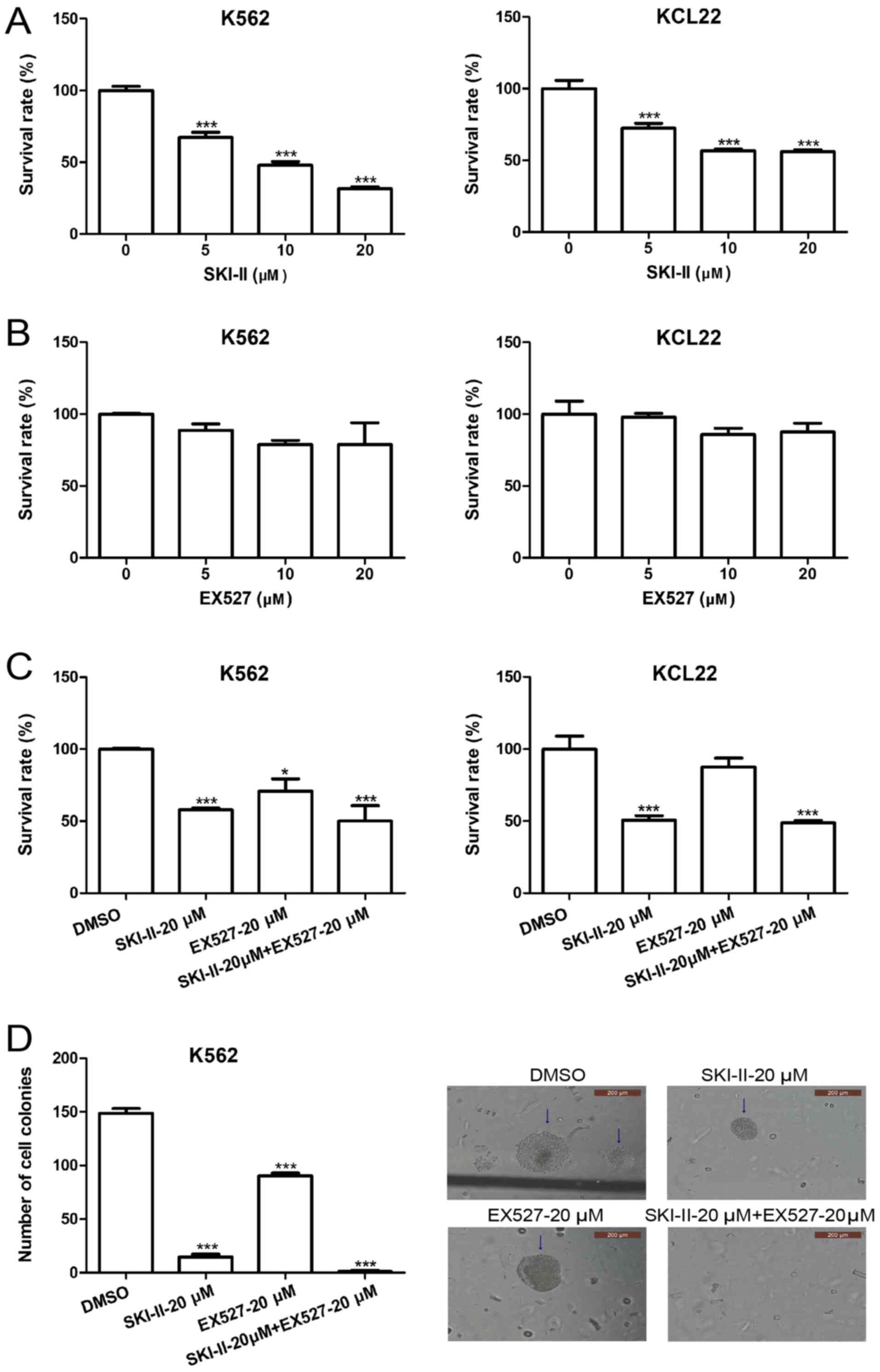
SKI-II and EX527 inhibit leukemia cell
growth
SKI-II is a novel specific inhibitor of Sphk
(29) and EX527 is a specific
inhibitor of SIRT1(30). To explore
the effect of Sphk1 and SIRT1 inhibition on the growth of leukemia
cells, K562 and KCL22 cells were treated with various
concentrations of SKI-II and EX527. Following incubation for 48 h,
cell proliferation was detected by CCK-8 assays. Treatment of K562
and KCL22 cells with 5, 10 or 20 µM SKI-II resulted in significant
growth inhibition compared with the 0-µM group (Fig. 2A). In contrast, EX527 treatment
slightly inhibited the growth of K562 and KCL22 cells; however,
this inhibition was not significant (Fig. 2B). SKI-II-induced growth inhibition
was enhanced by EX527 (Fig. 2C).
Additionally, the effect of combined treatment with SKI-II and
EX527 on the growth of leukemia cells was examined by evaluating
the in vitro colony-forming ability of K562 cells. The
results demonstrated that the number of cell colonies in the SKI-II
and EX527 combined treatment group was significantly lower compared
with the DMSO treatment group (Fig.
2D).
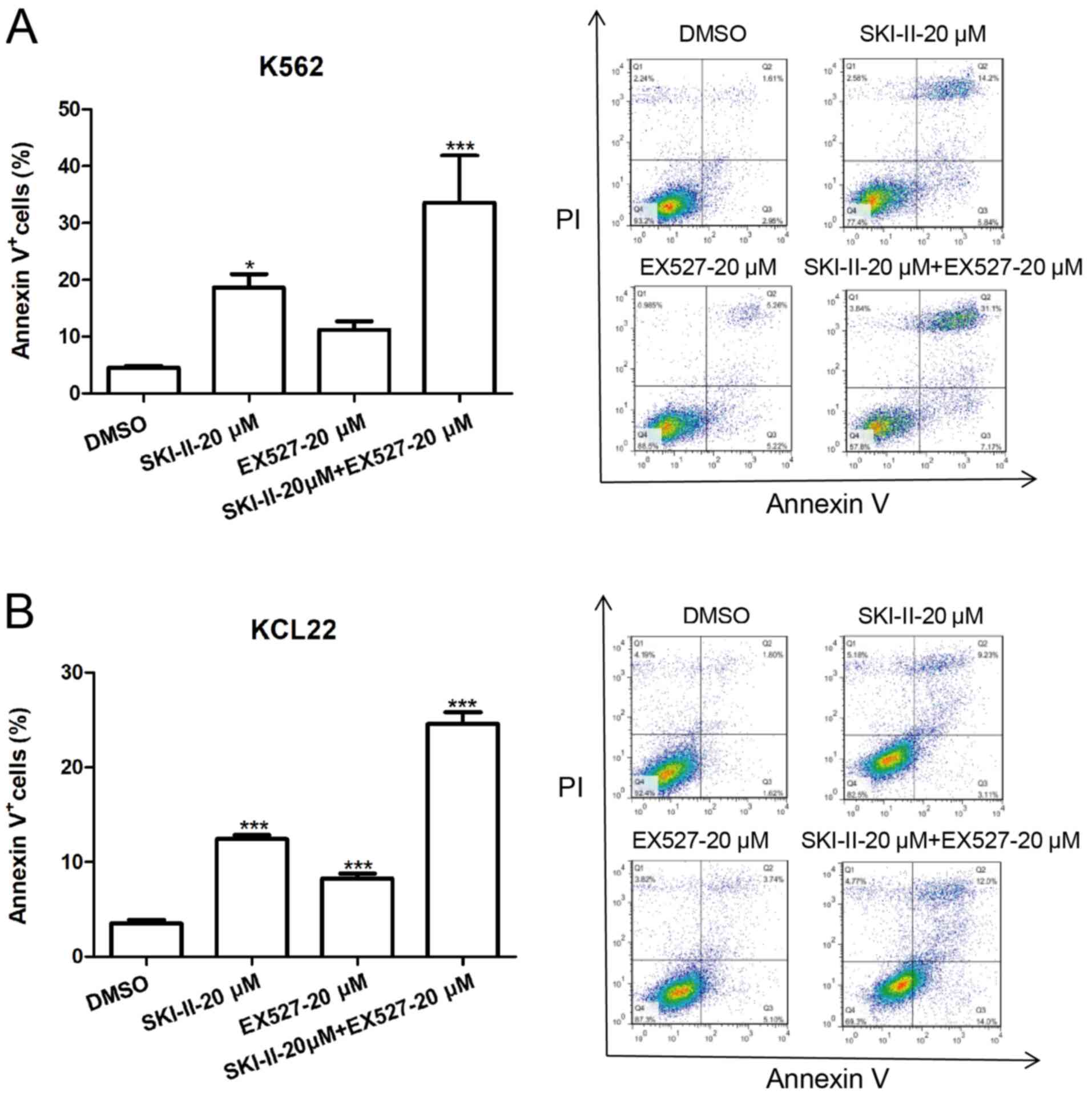
SKI-II and EX527 induce apoptosis in
leukemia cells
The apoptosis-promoting effects of SKI-II and EX527
were observed. K562 and KCL22 cells were incubated with DMSO, 20 µM
EX527, 20 µM SKI-II or 20 µM EX527 + 20 µM SKI-II for 24 h and
apoptosis was assessed. Treatment of K562 and KCL22 cells with 20
µM SKI-II resulted in significant cell apoptosis, whereas treatment
with EX527 moderately induced apoptosis of K562 and KCL22 cells,
compared with the DMSO group (Fig.
3A and B). Furthermore, the
combination of SKI-II and EX527 induced a significantly higher
apoptosis rate compared with DMSO treatment group.
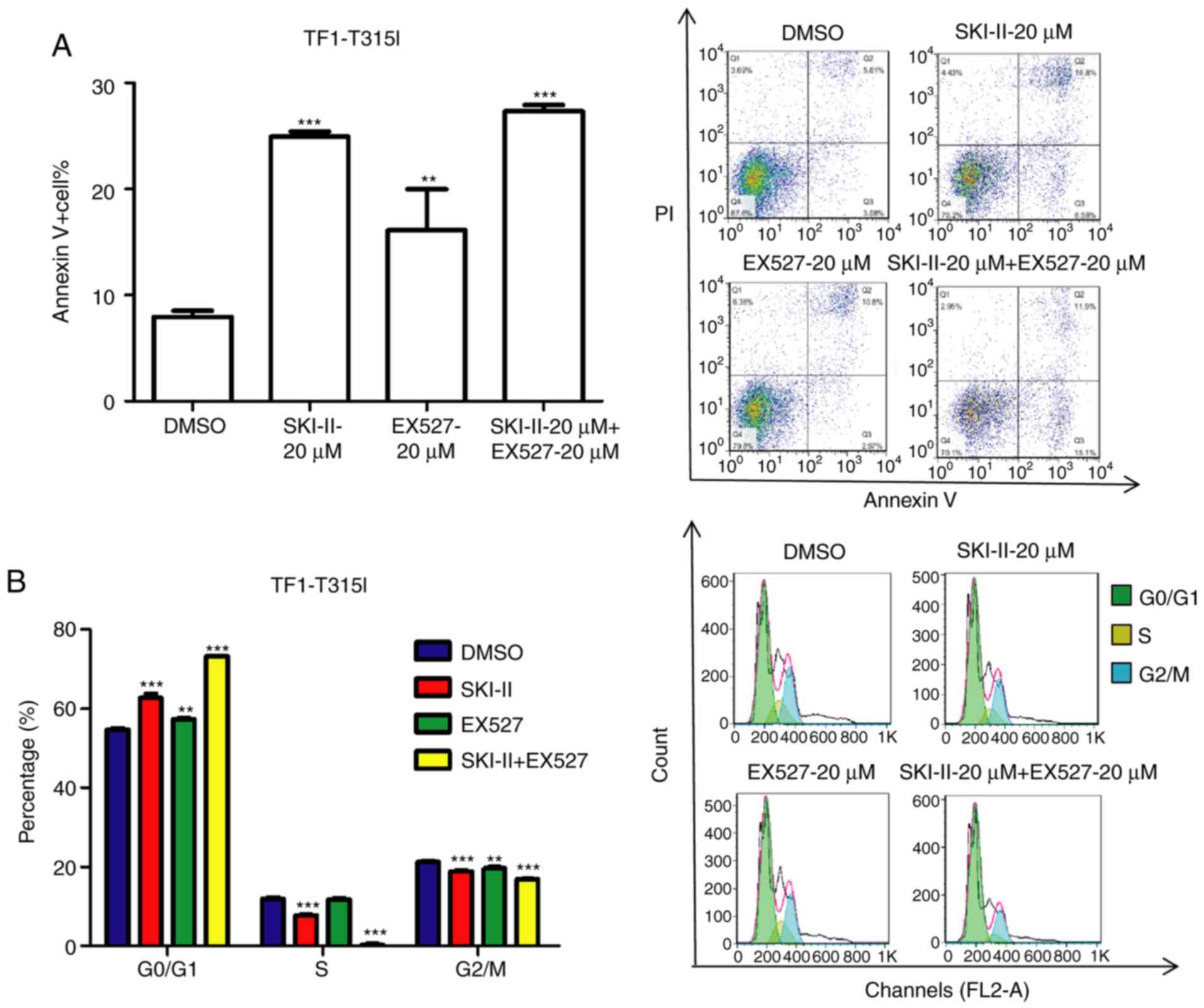
SKI-II and EX527 induce apoptosis and
growth arrest in imatinib-resistant leukemia cells
To evaluate the effect of SKI-II and EX527 treatment
on the growth of imatinib-resistant leukemia cells, TF-1-T315I
cells were established by introducing a lentiviral vector encoding
BCR-ABL1 (T315I). The mutant BCR-ABL transduction efficiency, as
indicated by GFP expression, was ~93.4% (Fig. 4A). TF-1-T315I cells were treated
with various concentrations of EX527 and SKI-II for 24 h.
Subsequently, cell growth was detected by CCK-8 assays and cell
apoptosis was measured by Annexin V/PI assays. Both SKI-II and
EX527 inhibited the proliferation of TF-1-T315I cells compared with
the 0-µM group (Fig. 4B and
C). Furthermore, the combination
treatment exhibited a marked synergistic growth-inhibiting effect
on TF-1-T315I cells compared with the DMSO group (Fig. 4D). Moreover, the combination
treatment with SKI-II and EX527 induced the lowest number of cell
colonies (Fig. 4E) and a higher
cell apoptosis rate (Fig. 5A) of
TF-1-T315I cells compared to the DMSO group. Additionally, the cell
cycle of TF-1-T315I cells was analyzed. Compared with the DMSO
treatment, the combination of SKI-II and EX527 significantly
increased the number of cells in the G0/G1 phase (Fig. 5B). This demonstrated that inhibition
of SIRT1 and Sphk1 induced apoptosis and overcame imatinib
resistance in leukemia cells.
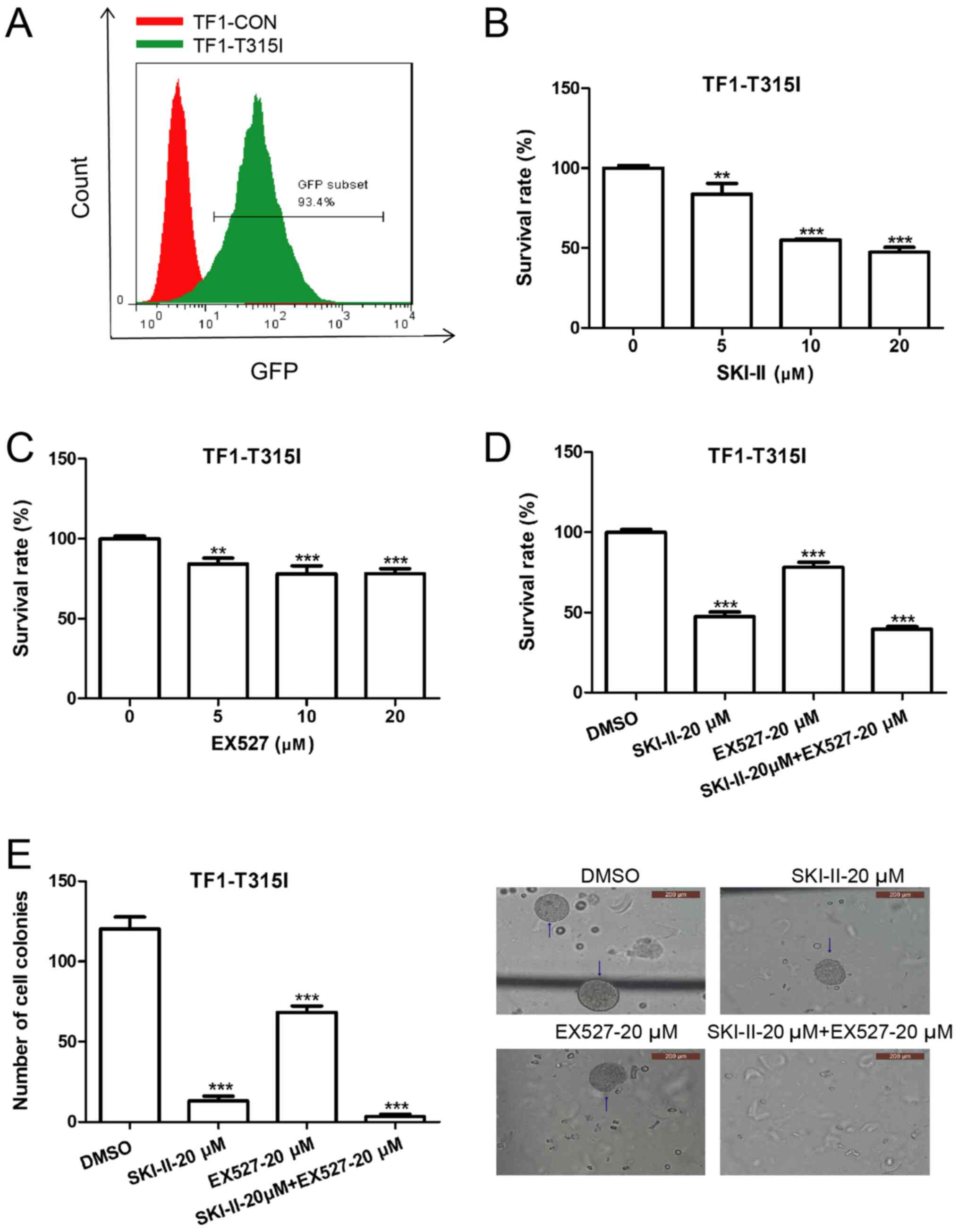 | Figure 4SKI-II and EX527 synergistically
inhibit TF-1-T315I growth. (A) TF-1 cells were transduced with
lentiviral vectors encoding BCR-ABL1 (T315I) and control vectors
for 48 h and the transduction efficiency was determined by flow
cytometry. Cell Counting Kit-8 assays were used to determine cell
proliferation in (B) TF-1-T315I cells treated with SKI-II (0, 5, 10
or 20 µM) for 24 h, (C) TF-1-T315I cells treated with EX527 (0, 5,
10 or 20 µM) for 24 h and (D) TF-1-T315I cells treated with DMSO,
20 µM SKI-II, 20 µM EX527 or 20 µM SKI-II + 20 µM EX527 for 24 h.
(E) TF-1-T315I cells were treated with DMSO, 20 µM SKI-II, 20 µM
EX527 or 20 µM SKI-II + 20 µM EX527 for 7 days and the number of
colonies (>50 cells) was counted under normal light at x40
magnification. Data are presented as the means ± standard
deviation. **P<0.01 and ***P<0.001
compared with 0 µM or DMSO. CON, control. |
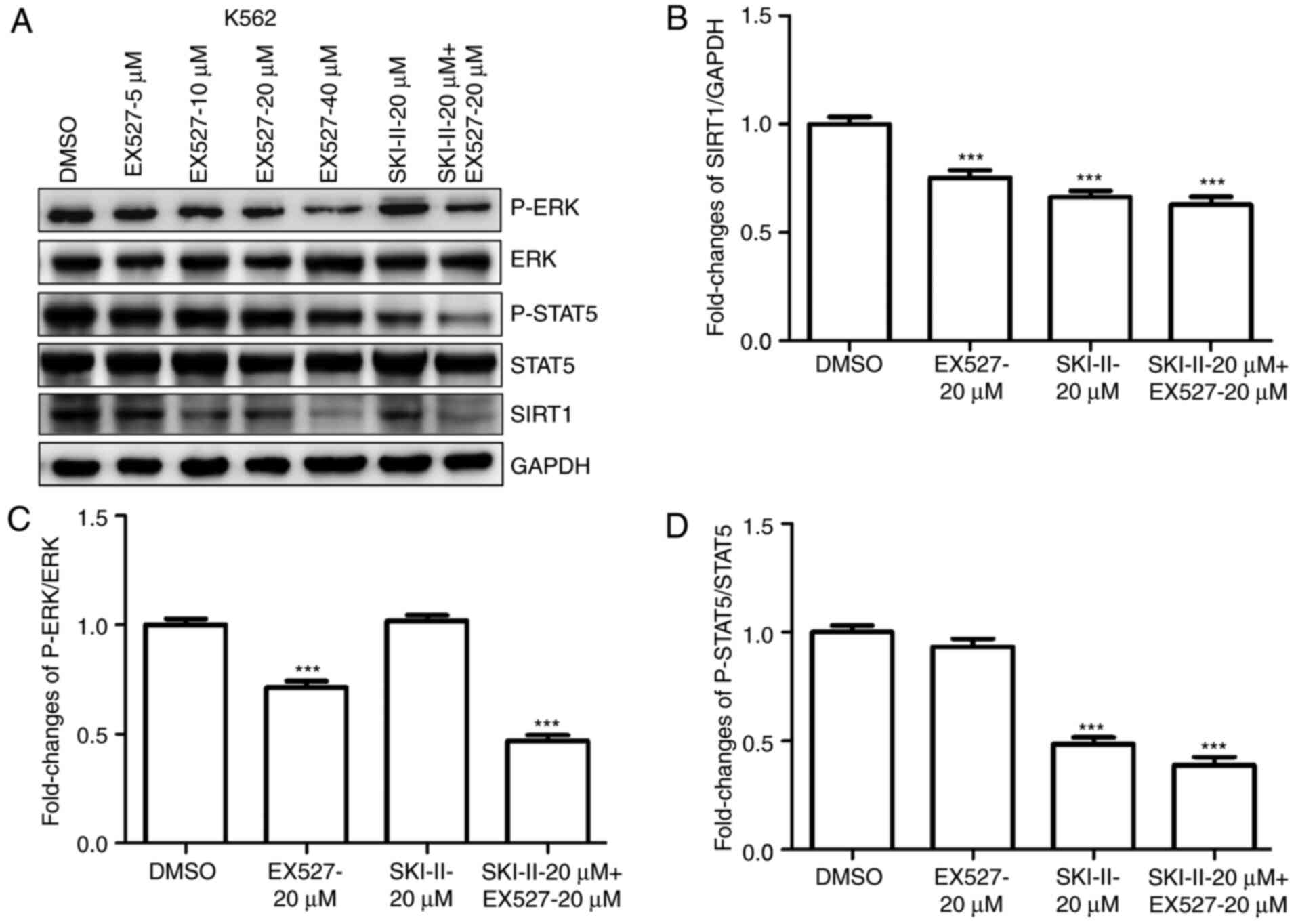
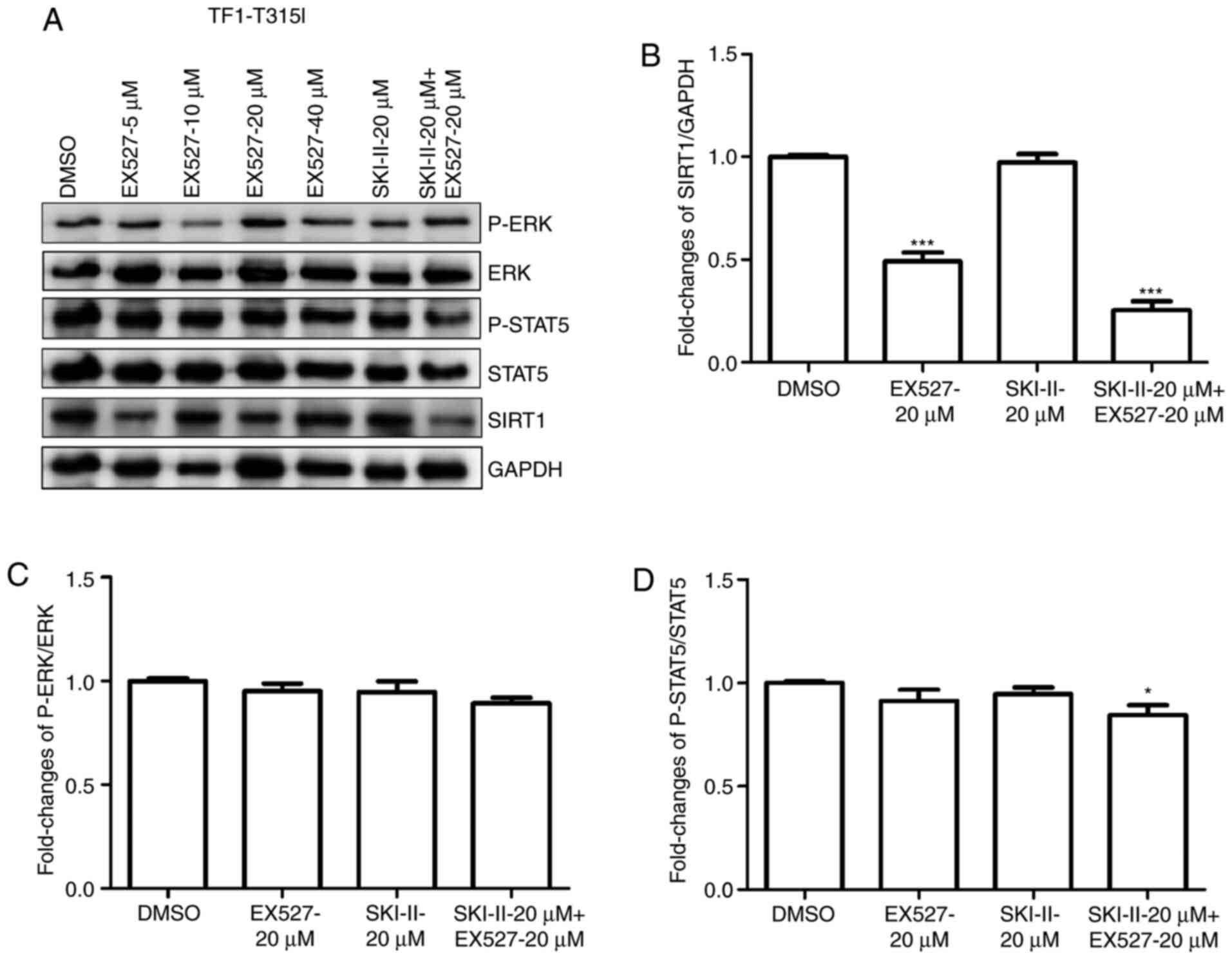
SKI-II and EX527 inhibit ERK and STAT5
signaling in leukemia cells
To clarify the mechanism underlying the combined
effects of SKI-II and EX527 on the growth and apoptosis of leukemia
cells, SIRT1, p-ERK and p-STAT5 protein levels were analyzed in
K562 (Fig. 6A-D) or TF-1-T315I
(Fig. 7A-D) cells treated with
DMSO, 20 µM SKI-II, 20 µM EX527 or 20 µM SKI-II + 20 µM EX527. The
results demonstrated that combination treatment with SKI-II and
EX527 had a synergistic inhibitory effect on the expression of
SIRT1, p-ERK and p-STAT5 protein in K562 cells compared to the DMSO
group. For TF-1-T315I cells, combination treatment with SKI-II and
EX527 inhibited the expression of SIRT1 and p-STAT5 compared with
the DMSO group. These results indicated that the STAT5 and ERK
pathways were involved in the growth inhibition and death of
leukemia cells treated with SKI-II and EX527.
Discussion
TKI therapy has markedly improved the long-term
survival rates of patients with leukemia (31,32).
However, certain patients exhibit resistance to TKIs (33). Furthermore, the molecular mechanisms
underlying TKI resistance in leukemia are not fully understood.
Dysregulated lipid metabolism pathways have been considered to be
novel targets for overcoming drug resistance in cancer therapy
(34,35). Sphk1, S1P and S1P receptors are key
players in lipid metabolism signaling networks and their
dysregulation contributes to tumorigenesis (36,37).
SIRT1 is an important regulator of genomic integrity and cell
processes, including the cell cycle, DNA damage response,
metabolism, apoptosis and autophagy (38). Considering CML cells constitutively
express high levels of Sphk1 and SIRT1, the present study detected
their interaction and integration in CML cells. The bioactive lipid
S1P induced SIRT1 expression, while the inhibition of Sphk1 induced
SIRT1 downregulation in leukemia cells. These data confirmed that
the Sphk1/S1P/SIRT1 axis was active in leukemia cells. Since SIRT1
enhances the progression of leukemia and promotes drug resistance
by increasing genetic instability (28), inhibition of SIRT1 was involved in
the synergistic anti-leukemic effects of divalproex sodium and
imatinib in CML cells (39). It has
been hypothesized that targeting the Sphk1 and SIRT1 pathways may
have synergistic inhibitory effects on leukemia cell growth and
drug resistance.
Considering stable transfection of Sphk shRNA
induced cell death (13), the
present study used chemical inhibitors to explore the effects of
Sphk1 and SIRT1 inhibition on the growth and survival of leukemia
cells. Treatment of K562 and KCL22 cells with SKI-II, a highly
selective, non-ATP-competitive Sphk1 inhibitor (29), reduced cell viability, inhibited
cell proliferation and induced apoptosis. EX527 is a potential
novel and specific small-molecule inhibitor of SIRT1 activity
(30). EX527 had a modest
suppressive effect on the growth of leukemia cells; however, in
combination with SKI-II, EX527 exhibited a synergistic inhibitory
effect on cell growth and a synergistic positive effect on
apoptosis in leukemia cells. Furthermore, the cell cycle changes of
K562 and KCL22 cells were examined. Unfortunately, the cycle data
of each cell line did not provide consistent results across
experimental repeats, as such the cell cycle data for these two
cell lines was omitted from the present study. However, the
combination of SKI-II and EX527 significantly increased the
percentage of the G0/G1 phase in TF-1-T315I cells. SKI-II inhibits
the growth of acute myelogenous leukemia cells in vitro and
in vivo (40) and the
combination of EX527 with SKI-II exhibited synergistic
anti-leukemic activity, which could possibly be therapeutically
exploited.
TKI therapies are rendered ineffective due to the
survival of leukemia stem cells and drug resistance (41). Primitive quiescent CML stem cells
are particularly resistant to imatinib-induced apoptosis (42). Since Sphk1/S1P signaling is
dysregulated by stimuli originating from tumor microenvironments,
the inhibition of this pathway is crucial in overcoming TKI
resistance in leukemia cells (11,19).
T315I is the most frequent mutation causing imatinib resistance in
patients with advanced CML or Ph+ acute lymphocytic
leukemia (43). It has been
demonstrated that targeting Sphk1 and SIRT1 individually enhanced
the sensitivity to TKI in various cancer cells, including acute
myeloid leukemia, CML, renal cancer and lung adenocarcinoma
(25,27,44,45),
indicated its potential as a novel agent for the treatment of
TKI-resistant leukemia. The present study lacked data on the
treatment for leukemia cells with TKI in combination with SKI-II
and EX527.
Although SKI-II and EX527 exhibited significant
suppressive effects on the growth of leukemia cells, their
mechanisms remain unknown. SKI-II treatment reduced SIRT1 protein
levels and presented cytotoxic effects in leukemia cells. Since
constitutively active STAT5 and MAPK/ERK signaling have been
demonstrated in CML cell lines and primary CML CD34+
cells (46,47), STAT5 and MAPK/ERK appears to be a
critical determinant of the TKI sensitivity of CML progenitor cells
(48,49). Combined treatment with SKI-II and
EX527 decreased SIRT1, p-ERK and p-STAT5 levels in K562 cells.
Furthermore, the combination treatment inhibited SIRT1 and p-STAT5
expression in TF-1-T31rI. Considering EX527 exhibited a marked
inhibitory effect on SIRT1 expression, the combined effect with
SKI-II was not apparent over that of EX527 treatment alone.
Occasionally, p-ERK expression demonstrated an inhibitor-induced
feedback regulation in K562 cells; however, this effect was not
evident in TF-1-T315I cells. Therefore, the suppression of the ERK
and STAT5 pathways may contribute to SKI-II and EX527-induced
apoptosis and growth arrest in leukemia cells.
In conclusion, the present study confirmed a novel
axis in leukemias cells in which Sphk1/S1P signaling mediated SIRT1
upregulation. Inhibition of Sphk1 and SIRT1 exhibited synergistic
suppressive effects on growth and overcame leukemias TKI
resistance. Therefore, targeting the Sphk1/S1P/SIRT1 axis may be a
novel therapeutic strategy for the treatment of leukemia.
Acknowledgements
Not applicable.
Funding
The present study was partially supported by the Key
R&D Project of Shandong Province (grant no. 2019GSF108144).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LW conceived and designed the present study. YL, YG,
BL and WN performed the experiments. YL, YG, BL, LZ and LW were
involved in the acquisition, analysis and/or interpretation of
data. YL and LW wrote the manuscript. YL, YG, BL and LW reviewed
and edited the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kumari A, Brendel C, Hochhaus A, Neubauer
A and Burchert A: Low BCR-ABL expression levels in hematopoietic
precursor cells enable persistence of chronic myeloid leukemia
under imatinib. Blood. 119:530–539. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
O'Hare T, Deininger MW, Eide CA, Clackson
T and Druker BJ: Targeting the BCR-ABL signaling pathway in
therapy-resistant Philadelphia chromosome-positive leukemia. Clin
Cancer Res. 17:212–221. 2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Corbin AS, Agarwal A, Loriaux M, Cortes J,
Deininger MW and Druker BJ: Human chronic myeloid leukemia stem
cells are insensitive to imatinib despite inhibition of BCR-ABL
activity. J Clin Invest. 121:396–409. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Cilloni D and Saglio G: Molecular
pathways: BCR-ABL. Clin Cancer Res. 18:930–937. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sattler M and Griffin JD: Molecular
mechanisms of transformation by the BCR-ABL oncogene. Semin
Hematol. 40 (Suppl 2):S4–S10. 2003.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sinclair A, Latif AL and Holyoake TL:
Targeting survival pathways in chronic myeloid leukemia stem cells.
Br J Pharmacol. 169:1693–1707. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Liu Q, Luo Q, Halim A and Song G:
Targeting lipid metabolism of cancer cells: A promising therapeutic
strategy for cancer. Cancer Lett. 401:39–45. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tang X, Benesch MG and Brindley DN: Lipid
phosphate phosphatases and their roles in mammalian physiology and
pathology. J Lipid Res. 56:2048–2060. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Patmanathan SN, Wang W, Yap LF, Herr DR
and Paterson IC: Mechanisms of sphingosine 1-phosphate receptor
signalling in cancer. Cell Signal. 34:66–75. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Tabasinezhad M, Samadi N, Ghanbari P,
Mohseni M, Saei AA, Sharifi S, Saeedi N and Pourhassan A:
Sphingosin 1-phosphate contributes in tumor progression. J Cancer
Res Ther. 9:556–563. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Nagahashi M, Takabe K, Terracina KP, Soma
D, Hirose Y, Kobayashi T, Matsuda Y and Wakai T:
Sphingosine-1-phosphate transporters as targets for cancer therapy.
Biomed Res Int. 2014(651727)2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Mendelson K, Evans T and Hla T:
Sphingosine 1-phosphate signalling. Development. 141:5–9.
2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gao Z, Wang H, Xiao FJ, Shi XF, Zhang YK,
Xu QQ, Zhang XY, Ha XQ and Wang LS: SIRT1 mediates
Sphk1/S1P-induced proliferation and migration of endothelial cells.
Int J Biochem Cell Biol. 74:152–160. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li QF, Wu CT, Guo Q, Wang H and Wang LS:
Sphingosine 1-phosphate induces Mcl-1 upregulation and protects
multiple myeloma cells against apoptosis. Biochem Biophys Res
Commun. 371:159–162. 2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Evangelisti C, Evangelisti C, Buontempo F,
Lonetti A, Orsini E, Chiarini F, Barata JT, Pyne S, Pyne NJ and
Martelli AM: Therapeutic potential of targeting sphingosine kinases
and sphingosine 1-phosphate in hematological malignancies.
Leukemia. 30:2142–2151. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li QF, Huang WR, Duan HF, Wang H, Wu CT
and Wang LS: Sphingosine kinase-1 mediates BCR/ABL-induced
upregulation of Mcl-1 in chronic myeloid leukemia cells. Oncogene.
26:7904–7908. 2007.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Salas A, Ponnusamy S, Senkal CE,
Meyers-Needham M, Selvam SP, Saddoughi SA, Apohan E, Sentelle RD,
Smith C, Gault CR, et al: Sphingosine kinase-1 and sphingosine
1-phosphate receptor 2 mediate Bcr-Abl1 stability and drug
resistance by modulation of protein phosphatase 2A. Blood.
117:5941–5952. 2011.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Baran Y, Salas A, Senkal CE, Gunduz U,
Bielawski J, Obeid LM and Ogretmen B: Alterations of
ceramide/sphingosine 1-phosphate rheostat involved in the
regulation of resistance to imatinib-induced apoptosis in K562
human chronic myeloid leukemia cells. J Biol Chem. 282:10922–10934.
2007.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ricci C, Onida F, Servida F, Radaelli F,
Saporiti G, Todoerti K, Deliliers GL and Ghidoni R: In vitro
anti-leukaemia activity of sphingosine kinase inhibitor. Br J
Haematol. 144:350–357. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Marfe G, Di Stefano C, Gambacurta A,
Ottone T, Martini V, Abruzzese E, Mologni L, Sinibaldi-Salimei P,
de Fabritis P, Gambacorti-Passerini C, et al: Sphingosine kinase 1
overexpression is regulated by signaling through PI3K, AKT2, and
mTOR in imatinib-resistant chronic myeloid leukemia cells. Exp
Hematol. 39:653–665. 2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Powell JA, Lewis AC, Zhu W, Toubia J,
Pitman MR, Wallington-Beddoe CT, Moretti PA, Iarossi D, Samaraweera
SE, Cummings N, et al: Targeting sphingosine kinase 1 induces
MCL1-dependent cell death in acute myeloid leukemia. Blood.
129:771–782. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sobue S, Nemoto S, Murakami M, Ito H,
Kimura A, Gao S, Furuhata A, Takagi A, Kojima T, Nakamura M, et al:
Implications of sphingosine kinase 1 expression level for the
cellular sphingolipid rheostat: Relevance as a marker for
daunorubicin sensitivity of leukemia cells. Int J Hematol.
87:266–275. 2008.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Vachharajani VT, Liu T, Wang X, Hoth JJ,
Yoza BK and McCall CE: Sirtuins link inflammation and metabolism. J
Immunol Res. 2016(8167273)2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Roth M, Wang Z and Chen WY: Sirtuins in
hematological aging and malignancy. Crit Rev Oncog. 18:531–547.
2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li L, Osdal T, Ho Y, Chun S, McDonald T,
Agarwal P, Lin A, Chu S, Qi J, Li L, et al: SIRT1 activation by a
c-MYC oncogenic network promotes the maintenance and drug
resistance of human FLT3-ITD acute myeloid leukemia stem cells.
Cell Stem Cell. 15:431–446. 2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhou L, Wang Q, Chen X, Fu L, Zhang X,
Wang L, Deng A, Li D, Liu J, Lv N, et al: AML1-ETO promotes SIRT1
expression to enhance leukemogenesis of t(8;21) acute myeloid
leukemia. Exp Hematol. 46:62–69. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Li L, Wang L, Li L, Wang Z, Ho Y, McDonald
T, Holyoake TL, Chen W and Bhatia R: Activation of p53 by SIRT1
inhibition enhances elimination of CML leukemia stem cells in
combination with imatinib. Cancer Cell. 21:266–281. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Li L and Bhatia R: Role of SIRT1 in the
growth and regulation of normal hematopoietic and leukemia stem
cells. Curr Opin Hematol. 22:324–329. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Giusto K, Patki M, Koya J, Ashby CR Jr,
Munnangi S, Patel K and Reznik SE: A vaginal nanoformulation of a
SphK inhibitor attenuates lipopolysaccharide-induced preterm birth
in mice. Nanomedicine (Lond). 14:2835–2851. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang T, Li X and Sun SL: EX527, a Sirt-1
inhibitor, induces apoptosis in glioma via activating the p53
signaling pathway. Anticancer Drugs. 31:19–26. 2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Peters GJ: From ‘targeted therapy’ to
targeted therapy. Anticancer Res. 39:3341–3345. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Bhalla S, Tremblay D and Mascarenhas J:
Discontinuing tyrosine kinase inhibitor therapy in chronic
myelogenous leukemia: Current understanding and future directions.
Clin Lymphoma Myeloma Leuk. 16:488–494. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Holyoake TL and Vetrie D: The chronic
myeloid leukemia stem cell: Stemming the tide of persistence.
Blood. 129:1595–1606. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Gao Y, Gao F, Chen K, Tian ML and Zhao DL:
Sphingosine kinase 1 as an anticancer therapeutic target. Drug Des
Devel Ther. 9:3239–3245. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Wallington-Beddoe CT, Bradstock KF and
Bendall LJ: Oncogenic properties of sphingosine kinases in
haematological malignancies. Br J Haematol. 161:623–638.
2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Shida D, Takabe K, Kapitonov D, Milstien S
and Spiegel S: Targeting SphK1 as a new strategy against cancer.
Curr Drug Targets. 9:662–673. 2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Xie Z, Liu H and Geng M: Targeting
sphingosine-1-phosphate signaling for cancer therapy. Sci China
Life Sci. 60:585–600. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Qiu G, Li X, Che X, Wei C, He S, Lu J, Jia
Z, Pang K and Fan L: SIRT1 is a regulator of autophagy:
Implications in gastric cancer progression and treatment. FEBS
Lett. 589:2034–2042. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wang W, Zhang J, Li Y, Yang X, He Y, Li T,
Ren F, Zhang J and Lin R: Divalproex sodium enhances the
anti-leukemic effects of imatinib in chronic myeloid leukemia cells
partly through SIRT1. Cancer Lett. 356:791–799. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yang L, Weng W, Sun ZX, Fu XJ, Ma J and
Zhuang WF: SphK1 inhibitor II (SKI-II) inhibits acute myelogenous
leukemia cell growth in vitro and in vivo. Biochem Biophys Res
Commun. 460:903–908. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Braun TP, Eide CA and Druker BJ: Response
and resistance to BCR-ABL1-targeted therapies. Cancer Cell.
37:530–542. 2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Holtz MS, Forman SJ and Bhatia R:
Nonproliferating CML CD34+ progenitors are resistant to apoptosis
induced by a wide range of proapoptotic stimuli. Leukemia.
19:1034–1041. 2005.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Roche-Lestienne C and Preudhomme C:
Mutations in the ABL kinase domain pre-exist the onset of imatinib
treatment. Semin Hematol. 40 (Suppl 2):S80–S82. 2003.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhang L, Wang X, Bullock AJ, Callea M,
Shah H, Song J, Moreno K, Visentin B, Deutschman D, Alsop DC, et
al: Anti-S1P antibody as a novel therapeutic strategy for VEGFR
TKI-resistant renal cancer. Clin Cancer Res. 21:1925–1934.
2015.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Sun J, Li G, Liu Y, Ma M, Song K, Li H,
Zhu D, Tang X, Kong J and Yuan X: Targeting histone deacetylase
SIRT1 selectively eradicates EGFR TKI-resistant cancer stem cells
via regulation of mitochondrial oxidative phosphorylation in lung
adenocarcinoma. Neoplasia. 22:33–46. 2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Kaymaz BT, Selvi N, Gündüz C, Aktan C,
Dalmızrak A, Saydam G and Kosova B: Repression of STAT3, STAT5A,
and STAT5B expressions in chronic myelogenous leukemia cell line
K-562 with unmodified or chemically modified siRNAs and induction
of apoptosis. Ann Hematol. 92:151–162. 2013.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Chorzalska A, Ahsan N, Rao RSP, Roder K,
Yu X, Morgan J, Tepper A, Hines S, Zhang P, Treaba DO, et al:
Overexpression of Tpl2 is linked to imatinib resistance and
activation of MEK-ERK and NF-κB pathways in a model of chronic
myeloid leukemia. Mol Oncol. 12:630–647. 2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Schafranek L, Nievergall E, Powell JA,
Hiwase DK, Leclercq T, Hughes TP and White DL: Sustained inhibition
of STAT5, but not JAK2, is essential for TKI-induced cell death in
chronic myeloid leukemia. Leukemia. 29:76–85. 2015.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Nambu T, Araki N, Nakagawa A, Kuniyasu A,
Kawaguchi T, Hamada A and Saito H: Contribution of
BCR-ABL-independent activation of ERK1/2 to acquired imatinib
resistance in K562 chronic myeloid leukemia cells. Cancer Sci.
101:137–142. 2010.PubMed/NCBI View Article : Google Scholar
|