1. Introduction
Arrhythmias, particularly ventricular arrhythmias
(VAs), have a relatively high morbidity and mortality among the
population, with ~250,000 deaths reported annually in the USA alone
(1). Similarly to ventricular
fibrillation (VF), VA has been reported to occur in >10% of all
patients with acute myocardial infarction (AMI) prior to
hospitalization, and survival in these patients remains poor
(2). A total of 17 million deaths
occur per year, worldwide, as a result of cardiovascular disease,
50% of which are attributable to sudden cardiac death (SCD)
(2). The major cause of SCD is VA,
particularly ventricular tachycardia (VT) and VF, which account for
~85% of all SCD events (3,4).
VA is an arrhythmia that originates in the
ventricles that does not require any myocardial tissue above the
His bundle to maintain (5). VA is
particularly common in clinical practice and includes premature
ventricular contraction, VT and VF (6,7).
Reentry and triggered activity are the two main mechanisms of
tachyarrhythmia. Reentry occurs when a beat encounters ventricular
myocardium modified by fibrosis, scarring or conduction
abnormalities (6). Triggered
activity is caused by early afterdepolarizations (EADs), which are
induced by reducing the repolarization reserve, either due to
increas-ing inward currents, reducing outward currents or both,
occurring in the second and third stages of the action potential
(AP) (6,8). Delayed afterdepolarizations (DADs) are
mediated by Ca2+ dysregulation after the fourth stage of
the AP. Abnormal depolarizations reach the membrane potential
threshold and further give rise to a spontaneous AP between two
regular APs (6,8,9).
According to mechanistic studies (10,11),
the occurrence and development of VA events during the acute phase
of AMI can be attributed to diastolic Ca2+ leak and
disturbed Ca2+ homeostasis. This can be induced by
enhanced sympathetic tone and is accompanied by the formation of
reentry circuits, further increasing vulnerability to VT (12).
Calcium/calmodulin-dependent protein kinase II
(CaMKII) is a versatile serine/threonine kinase that is found
widely in muscle, nerve and immune tissues (13). CaMKII serves multiple regulatory
effects, including excitation-contraction coupling,
excitation-transcription coupling, Ca2+ handling and
mitochondrial function in cardiomyocytes (14,15).
Chronic activation of CaMKII causes significant cardiomyocyte
remodelling and alterations in Ca2+ handling, ion
channels, cell-to-cell coupling and metabolism, leading to
increased susceptibility to VA (15-21).
The present review aimed to assess the participation of CaMKII in
the occurrence of EADs and DADs by targeting L-type Ca2+
channels (LTCCs), phospholamban (PLB), ryanodine receptors (RyRs),
voltage-gated Na+ (Nav) channels and multiple
voltage-gated K+ channels, which further result in VA
(18,19).
2. Molecular structure, function, subtypes
and distribution of CaMKII
Molecular structure and function of
CaMKII
CaMKII is a serine/threonine kinase that is composed
of two stacked hexamers assembled from 12 monomers (22,23).
Each monomer is composed of an N-terminal catalytic region, an
intermediate regulatory do-main and a C-terminal associated region
(15,23). The catalytic region contains an ATP
and target substrate binding site, which is responsible for the
regulation of kinase activity (23). Under basic conditions, the function
of the catalytic region is inhibited by interacting with the
intermediate regulatory region (23). The intermediate regulatory region
interacts with Ca2+/calmodulin (CaM) at a KD
of 10-50 nM, which not only activates CaMKII by preventing the
inhibitory effect of the catalytic region, but also increases the
activity of CaMKII by phosphorylating threonine 287 (Thr287)
(18,23). The C-terminal associated domain is
responsible for the oligomerization of individual CaMKII molecules
to form a mature dodecameric-holoenzyme (Fig. 1) (18).
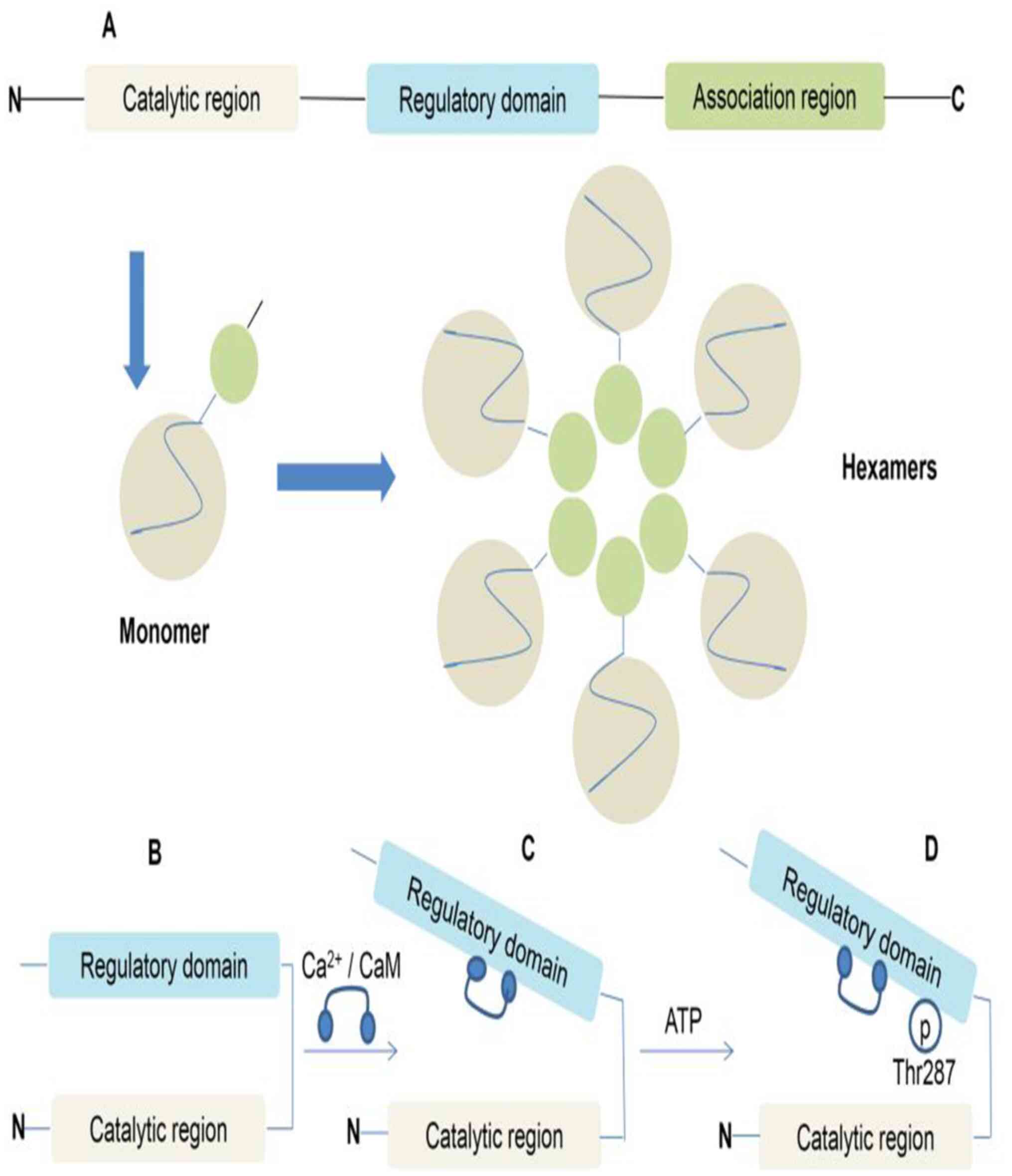 | Figure 1CaMKII structural domains and
regulation. (A) CaMKII monomers are com-posed of an N-terminal
catalytic region, an intermediate regulatory domain and a
C-terminal associated region. Two stacked hexamers assembled from
12 monomers form CaMKII. (B) Under basal conditions, the catalytic
domain of CaMKII is inhibited through direct interaction with the
regulatory domain. (C) CaMKII is activated by the binding of
Ca2+/CaM. (D) Ca2+/CaM binding also exposes
sites in the regulatory domain, resulting in alternative activation
modes. For example, the autophosphorylation of Thr287 by a
neighbouring active subunit (autophosphorylation) induces a high
activity mode subunit. Similar autonomy is observed with oxidation
at the exposed Met281/282 site, O-linked glycosylation at Ser280 or
NO-dependent nitrosation at Cys290. CaMKII,
calcium/calmodulin-dependent protein kinase II;
Ca2+/CaM, calcium/calmodulin; Thr287, threonine 287; p,
phosphorylation; N, N-terminus; C, C-terminus. |
CaMKII subtypes and distribution
CaMKII has four subtypes (α, β, γ and δ), and each
subtype has a different basic affinity for Ca2+/CaM (in
order of highest to lowest, γ, β, δ and α) (15,18).
The CaMKIIδ and CaMKIIγ subtypes are mainly present in myocardial
tissue (18). CaMKIIδ has four
splice variants (δA, δB, δC, and δ9), among which CaMKIIδB and
CaMKIIδC are observed primarily expressed in the heart (18,23).
CaMKIIδB contains an 11-amino acid nuclear localization sequence,
which is preferentially localized in the nucleus, thereby exerting
an important influence on the transcriptional activity of genes
involved in cardiac hypertrophy (18,23).
CaMKIIδC is the main cytoplasmic form, which is involved in
membrane excitability and regulation of intracellular
Ca2+ homeostasis (15,23).
The ratio of δB to δC in the multimer can regulate the localization
of holoenzymes, and stable hetero-oligomers are formed by these
CaMKII subtypes (18,23,24).
3. CaMKII activation mechanism
Ca2+/CaM dependent CaMKII
activation pathway
In the presence of ATP, the pseudo-substrate section
of the intermediate regulatory region of CaMKII can inhibit the
func-tion of the N-terminal catalytic region, resulting in the
inactivation of CaMKII (23). When
Ca2+ content increases, Ca2+ combines with
CaM (a ubiquitous intracellular Ca2+ binding protein) to
form Ca2+/CaM (24). The
intermediate regulatory region binds to Ca2+/CaM, which
causes conformational changes in the pseudosubstrate region and
releases the catalytic domain, exposing the substrate and ATP
binding sites, further resulting in CaMKII activation (Fig. 1) (23,24).
Ca2+/CaM independent CaMKII
activation pathway
In the presence of ATP, continuously increasing
Ca2+/CaM sustainably combines with the intermediate
regulatory region of CaMKII, which results in the
autophosphorylation of Thr287. Thr287 autophosphorylation
significantly increases the affinity of Ca2+/CaM to the
intermediate regulatory region, slowing the release of
Ca2+/CaM and retaining residual activity even after the
dissociation of Ca2+/CaM, further resulting in CaMKII
activation (3,16,24). A
previous study by Erickson et al (25) showed that the methionine 281/282
(Met281/282) site is oxidized in the presence of reactive oxygen
species (ROS). Oxidation of Met281/282 can not only lead to the
autonomous activation of CaMKII by preventing the recombination of
the catalytic domain and the intermediate regulatory region, but
also promote CaMKII activation at low intracellular Ca2+
concentrations by increasing the capability of CaMKII to be
activated by Ca2+/CaM (3,18). In
addition, O-linked glycosylation at serine 280 (Ser280) and nitric
oxide (NO)-dependent nitrosation at cysteine 290 (Cys290) can
activate CaMKII. Ser280 O-linked-glycosylation of CaMKII has been
demonstrated to promote Thr287 autophosphorylation (Fig. 1) (18,26).
4. CaMKII regulates cardiac Nav
channels to induce VA
Nav channels and sodium ion
current
Under normal conditions, Nav channels
rapidly activate and inactivate, resulting in a transient
Na+ current (INa,T), which allows for AP
depolarization (phase 0 of the AP). However, even under
physiological conditions, a minor population of Nav
channels may fail to inactivate, giving rise to a late
Na+ current (INa,L) that persists throughout
the AP. Importantly, amplification of INa,L in disease
set-tings has been demonstrated to increase arrhythmia
susceptibility (27).
CaMKII regulates Nav
channels
CaMKII has a VA-inducing effect by regulating
Nav channels. Previous studies have demonstrated that
acute CaMKII overexpression may shift Nav channel
resting potential to more negative membrane potentials, enhancing
in-termediate inactivation and slowing recovery from inactivation,
thereby reducing the fraction of available Na+ channels.
However, this also slows INa,T inactivation, enhances
INa,L and increases intracellular Na+
concentrations. These effects increase susceptibility to arrhythmia
(27,28) Additionally, serine 571 of
Nav1.5 is in the Nav pore-forming subunit and
is a key site of CaMKII phosphorylation. Nav channels
can be continuously opened or reopened to produce long-lasting
INa,L via phosphorylation at this subunit (16,29).
Increased INa,L can significantly prolong the AP
duration (APD) and increase the Na+ load in
cardiomyocytes, which can enhance the
Na+-Ca2+ exchanger (NCX) activity in the
reverse mode (3 Na+ extruded from the cell in exchange
for 1 Ca2+), further increasing the Ca2+ load
in cardiomyocytes (Fig. 2)
(30-33).
A prolonged APD in combination with an increased Ca2+
load can induce EADs and DADs, eventually lead-ing to VA. In
addition, INa,L can enhance the Ca2+
regulation capacity through the feed-back regulation of CaMKII,
thereby participating in the occurrence of VA (16).
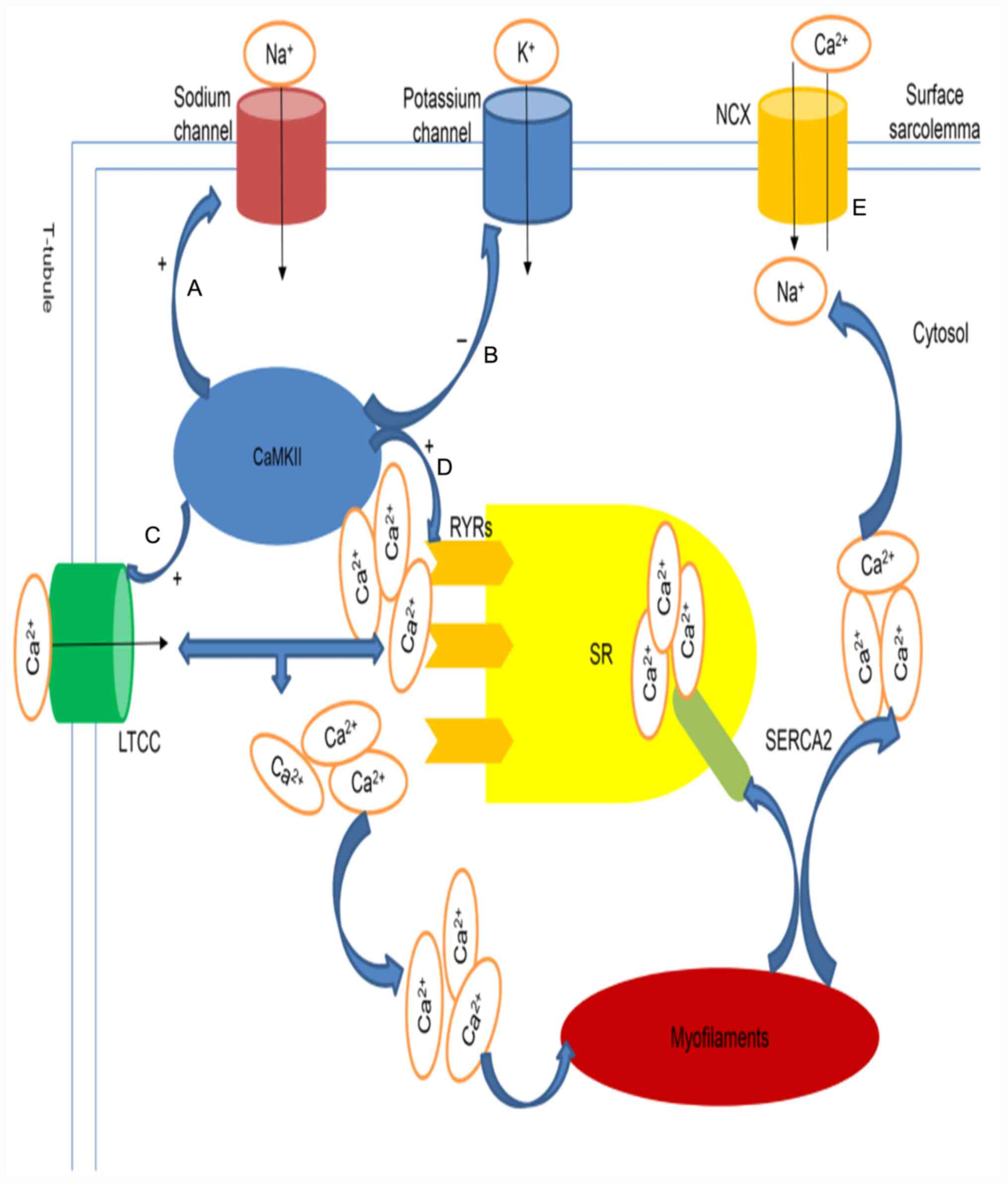 | Figure 2Proposed model of CaMKII-induced
ventricular arrhythmia. (A and E) CaMKII increases late
Na+ currents by phosphorylation at the Serine 571 site,
further prolonging the APD and decreasing NCX function, which
results in increased Ca2+ load (B). CaMKII reduces the
outward K+ current, inward rectifier K+
current and delayed rectifier K+ current intensity,
further prolonging the APD (C-E). CaMKII increases Ca2+
overload in the cytosol by phosphorylating LTCCs and RyRs. LTCCs
coupled with Ca2+ induces further Ca2+
release from RyRs. Ca2+ is returned to the SR by SERCA2
and extruded via the NCX after participating in myofilament
contraction. CaMKII, calcium/calmodulin dependent protein kinase
II; APD, action potential duration; NCX,
Na+-Ca2+ exchanger; LTCCs, L-type
Ca2+ channels; RyRs, ryanodine receptors; SR,
sarcoplasmic reticulum; SERCA2, sarco(endo)plasmic reticulum
calcium ATPase 2. |
5. CaMKII regulates K+ channels
to induce VA
K+ channels and
K+ current
The K+ current formed by the
K+ channels of the heart is a key determinant of heart
excitability. There are three types of K+ currents in
the heart: Transient outward K+ current
(Ito), inward rectifier K+ current
(IK1) and delayed rectifier K+ current
(IK). Ito is mainly generated by the
activation of voltage-gated K+ channels with subunits
that mainly consist of KV4.2, KV4.3 and KV1.4. Ito
produced by KV4.3 is primarily involved in the formation of the
first phase of the AP (the early stage of rapid repolarization)
(34). IK1 is primarily
produced by activation of the inward rectifier K+
channel, which is important for maintaining the resting cell
membrane potential and the third phase of the AP (end of rapid
repolarization). The inward rectifier K+ channel
pore-forming subunit is composed of Kir2.1 and Kir6.2.
IK1 is generally considered to be antiarrhythmic as it
stabilizes the resting membrane potential (35). IK is mainly produced by
the activation of delayed rectifier K+ channel groups
with pore-forming subu-nits consisting of Kv1.5, human
ether-a-go-go-related gene and Kv7.1, participating in
the formation of the second and third phases of the AP (36).
CaMKII regulates K+
channels
CaMKII induces VA by participating in the regulation
of Ito, IK1 and IK. Chronic CaMKII
activation reduces Ito intensity by reducing the mRNA
and protein expression levels of the KV4.2 and KV4.3 subunits. In
addition, de-creased expression of the KV4.3 subunit can cause
feedback that activates CaMKII. The KV4.3 subunit can also bind to
the Ca2+/CaM binding site of CaMKII (34). Activation of a large amount of
CaMKII can increase its ability to regulate K+ channels.
Chronic CaMKII activation also reduces the intensity of
IK1 by reducing the mRNA and protein expression levels
of the Kir2.1 and Kir6.2 subunits (36,37). A
slow change in IK1 intensity causes the resting membrane
potential to be unstable, such that the depolarization current can
be transformed into larger DADs, leading to the occurrence of VA
(37,38). Chronic activation of CaMKII can
phosphorylate the serine 484 site of the KV7.1 subunit, leading to
a decrease in IK intensity (39). It has been suggested that reduction
in Ito, IK1 and IK intensity can
lead to prolongation of the APD, which promotes the occurrence of
VA (Fig. 2) (36).
6. CaMKII regulates Ca2+ channels
to induce VA
Ca2+ cycle
The excitation-contraction coupling of
cardiomyocytes is a highly coordinated process that links
electrical signals with mechanical contractions. LTCCs can produce
an L-type Ca2+ current (ICa,L) that
participates in the formation of the second phase of the AP. LTCCs
coupled with Ca2+ induces Ca2+ release from
RyR channels. Increased Ca2+ binds to troponin and
triggers myofilament contraction. When ventricular myocytes enter
the diastolic phase, Ca2+ in the cytoplasm is returned
to the sarcoplasmic reticulum (SR) through sarco(endo)plasmic
reticulum calcium ATPase 2 (SERCA2) (40,41).
CaMKII regulates Ca2+
homeostasis
CaMKII serves an important role in the regulation of
Ca2+ homeostasis and has a VA-causing effect. CaMKII
activation can increase LTCC phosphorylation, which generates a
greater ICa,L (32). The
serine 2814 site of RyR2 is phosphorylated upon CaMKII
activation, which occurs when the release of Ca2+ stored
in the diastolic SR abnormally increases (31,42-44).
Abnormally released Ca2+ propagates along adjacent
RyRS on the SR and activates them to trigger further
Ca2+ release (8,12). Increased intracellular
Ca2+ concentrations can participate in the regulation of
Nav channel function through CaMKII activation, thereby
adjusting the flow of Na+ (45). Excess Ca2+ in the
cytoplasm is extruded via the NCX, which produces an inward current
(Iti; Fig. 2). When
Iti is sufficient to depolarize the myocardial cell
membrane, Nav channels can be activated, which triggers
additional APs and further results in DADs (8,29,31,40,43).
When ICa,L or Iti is greater than the outward
current (mainly K+ current) during the later period of
the AP, the APD can be prolonged, which leads to the occurrence of
EADs (8). The occurrence of DADs
and EADs will eventu-ally lead to VA. However, the threonine 17
(Thr17) site of PLB, which is mainly expressed in the SR to
regulate SERCA2 activity, is a specific target of CaMKII
phosphorylation. PLB phosphorylation at Thr17 helps to limit
cytosolic Ca2+ overload by increasing SERCA2 activity
and accelerating SR Ca2+ reuptake, which is beneficial
for improving Ca2+ cycle dysfunction and reducing the
risk of VA (46-48).
7. Summary and outlook
In summary, VA is a highly fatal arrhythmia,
involving the regulation of multiple ion channels. CaMKII serves an
important regulatory role in the mechanism of VA. Overexpression of
CaMKII can promote the occurrence of DADs and EADs by increasing
the extent of INa,L, decreasing the intensity of
Ito, IK1 and IK, and increasing
Ca2+ in the cytoplasm, thereby inducing VA.
Additionally, CaMKII activation is closely related to connexin 43
dysregulation; however, CaMKII activation also indirectly decreases
the expression and subcellular localization of connexin 43 in
intercalated discs. Both effects potentially increase
arrhythmogenic susceptibility (49-53).
CaMKII inhibition also has a potential proarrhythmic
effect. Early ischemia may increase CaMKII activation due to a
progressive increase in Ca2+ concentration and excessive
formation of ROS (54,55). CaMKII activity is detrimental in
this process; however, it is beneficial during the first minutes of
ischemia, as it has a regulative effect on conduction and can avoid
ischemia-mediated conduction block (55). Previous studies have demonstrated
that CaMKII upregulation is of great significance to maintaining
conduction during ischemia. Therefore, intervening through CaMKII
activity can cause the heterogeneous depression of conduction
during ischemia, exacerbating the arrhythmia substrate and
resulting in a proarrhythmic condition (20,55).
It is necessary to develop novel drugs based on
mechanistic research. Currently, effective clinical treatments for
VA include non-pharmacological treatments, such as defibrillation,
radiofrequency catheter ablation and pharmacological interventions
that include blockers of Na+ channels (class I),
β-receptors (class II), K+ channels (class III) and
Ca2+ channels (class IV), as well as miscellaneous
agents such as digoxin and adenosine. However, each treatment has
specific limitations. For example, the pharmacological treatment of
VA results in substantial toxicities and the potential for
proarrhythmic side effects (35).
Therefore, it is necessary to develop novel antiarrhythmic drugs
based on a comprehensive understanding of the proarrhythmic
mechanisms of CaMKII. At present, pharmacological inhibitors of
CaMKII (such as KN93 and GS-680), peptide inhibitors (such as
CN19o) and CaMKII-targeted interference drugs (such as RNAi) have
been developed, though these inhibitors are associated with
bioavailability limitations and poorly understood in vivo
effects (23). Therefore, the
molecular mechanism underlying the role of CaMKII in VA requires
further examination. For example, whether there are other sites of
CaMKII phosphorylation in Na+, K+,
Ca2+ and other ion channels still requires further
study. Related VA-specific drugs, such as targeted inhibi-tors of
CaMKII phosphorylation sites on ion channels, also require further
development.
Acknowledgements
Not applicable.
Funding
Funding: The present review was supported by Hebei
Administration of Traditional Chinese Medicine, China (grant no.
5000 RMB) and The First Hospital of Hebei Medical University.
(grant no. 203777117D)
Availability of data and materials
Not applicable.
Authors' contributions
KM searched literature and further analysed the
data, and wrote, revised and finalized the manuscript. GM analyzed
the data from literature, drafted the article and produced the
final manuscript. ZG conceived the current review and revised the
manuscript. WL and GL conceived and designed the study, revised the
manuscript and produced the final version. All authors agree to be
responsible for all aspects of the article. All authors have read
and approved the final manuscript. LW and LG confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bernardi J, Aromolaran KA, Zhu H and
Aromolaran AS: Circadian Mechanisms: Cardiac Ion Channel Remodeling
and Arrhythmias. Front Physiol. 11(611860)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Sattler SM, Skibsbye L, Linz D, Lubberding
AF, Tfelt-Hansen J and Jespersen T: Ventricular Arrhythmias in
First Acute Myocardial Infarction: Epidemiology, Mecha-nisms, and
Interventions in Large Animal Models. Front Cardiovasc Med.
6(158)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Donahue JK: Current state of the art for
cardiac arrhythmia gene therapy. Pharmacol Ther. 176:60–65.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jazayeri MA and Emert MP: Sudden Cardiac
Death: Who Is at Risk? Med Clin North Am. 103:913–930.
2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Haqqani HM, Chan KH, Kumar S, Denniss AR
and Gregory AT: The Contemporary Era of Sudden Cardiac Death and
Ventricular Arrhythmias: Basic Concepts, Recent Developments and
Future Directions. Heart Lung Circ. 28:1–5. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
AlMahameed ST and Ziv O: Ventricular
Arrhythmias. Med Clin North Am. 103:881–895. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Markman TM and Nazarian S: Treatment of
ventricular arrhythmias: What's New? Trends Cardiovasc Med.
29:249–261. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Skogestad J and Aronsen JM:
Hypokalemia-Induced Arrhythmias and Heart Failure: New Insights and
Implications for Therapy. Front Physiol. 9(1500)2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Weiss JN, Garfinkel A, Karagueuzian HS,
Chen PS and Qu Z: Early afterdepolariza-tions and cardiac
arrhythmias. Heart Rhythm. 7:1891–1899. 2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Di Diego JM and Antzelevitch C: Ischemic
ventricular arrhythmias: Experimental models and their clinical
relevance. Heart Rhythm. 8:1963–1968. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Landstrom AP, Dobrev D and Wehrens XHT:
Calcium Signaling and Cardiac Arrhythmias. Circ Res. 120:1969–1993.
2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Mollenhauer M, Mehrkens D, Klinke A, Lange
M, Remane L, Friedrichs K, Brau-mann S, Geißen S, Simsekyilmaz S,
Nettersheim FS, et al: Nitro-fatty acids suppress ischemic
ventricular arrhythmias by preserving calcium homeostasis. Sci Rep.
10(15319)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Joviano-Santos JV, Santos-Miranda A,
Botelho AFM, de Jesus ICG, Andrade JN, de Oliveira Barreto T,
Magalhães-Gomes MPS, Valadão PAC, Cruz JDS, Melo MM, et al:
Increased oxidative stress and CaMKII activity contribute to
electro-mechanical defects in cardiomyocytes from a murine model of
Huntington's disease. FEBS J. 286:110–123. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bers DM: Calcium cycling and signaling in
cardiac myocytes. Annu Rev Physiol. 70:23–49. 2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hegyi B, Bers DM and Bossuyt J: CaMKII
signaling in heart diseases: Emerging role in diabetic
cardiomyopathy. J Mol Cell Cardiol. 127:246–259. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Greer-Short A, Musa H, Alsina KM, Ni L,
Word TA, Reynolds JO, Gratz D, Lane C, El-Refaey M, Unudurthi S, et
al: Calmodulin kinase II regulates atrial myocyte late sodium
current, calcium handling, and atrial arrhythmia. Heart Rhythm.
17:503–511. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Pyun JH, Kim HJ, Jeong MH, Ahn BY, Vuong
TA, Lee DI, Choi S, Koo SH, Cho H and Kang JS: Cardiac specific
PRMT1 ablation causes heart failure through CaMKII dysregulation.
Nat Commun. 9(5107)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wood BM, Simon M, Galice S, Alim CC,
Ferrero M, Pinna NN, Bers DM and Bossuyt J: Cardiac CaMKII
activation promotes rapid translocation to its extra-dyadic
targets. J Mol Cell Cardiol. 125:18–28. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yoo S, Aistrup G, Shiferaw Y, Ng J, Mohler
PJ, Hund TJ, Waugh T, Browne S, Gussak G, Gilani M, et al:
Oxidative stress creates a unique, CaMKII-mediated sub-strate for
atrial fibrillation in heart failure. JCI Insight.
3(3)2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Howard T, Greer-Short A, Satroplus T,
Patel N, Nassal D, Mohler PJ and Hund TJ: CaMKII-dependent late
Na+ current increases electrical dispersion and
arrhythmia in ischemia-reperfusion. Am J Physiol Heart Circ
Physiol. 315:H794–H801. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Motloch LJ, Cacheux M, Ishikawa K, Xie C,
Hu J, Aguero J, Fish KM, Hajjar RJ and Akar FG: Primary Effect of
SERCA 2a Gene Transfer on Conduction Reserve in Chronic Myocardial
Infarction. J Am Heart Assoc. 7(e009598)2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Johnson CN, Pattanayek R, Potet F, Rebbeck
RT, Blackwell DJ, Nikolaienko R, Sequeira V, Le Meur R, Radwański
PB, Davis JP, et al: The CaMKII inhibitor KN93-calmodulin
interaction and implications for calmodulin tuning of NaV1.5 and
RyR2 function. Cell Calcium. 82(102063)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Nassal D, Gratz D and Hund TJ: Challenges
and Opportunities for Therapeutic Targeting of Calmodulin Kinase II
in Heart. Front Pharmacol. 11(35)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wong MH, Samal AB, Lee M, Vlach J, Novikov
N, Niedziela-Majka A, Feng JY, Koltun DO, Brendza KM, Kwon HJ, et
al: The KN-93 Molecule Inhibits Calcium/Calmodulin-Dependent
Protein Kinase II (CaMKII) Activity by Binding to
Ca2+/CaM. J Mol Biol. 431:1440–1459. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Erickson JR, Joiner ML, Guan X, Kutschke
W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE,
Aykin-Burns N, et al: A dynamic pathway for calcium-independent
activation of CaMKII by methionine oxidation. Cell. 133:462–474.
2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Erickson JR, Pereira L, Wang L, Han G,
Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, et
al: Diabetic hyperglycaemia activates CaMKII and arrhythmias by
O-linked glycosylation. Nature. 502:372–376. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hegyi B, Bányász T, Izu LT, Belardinelli
L, Bers DM and Chen-Izu Y: β-adrenergic regulation of late
Na+ current during cardiac action potential is mediated
by both PKA and CaMKII. J Mol Cell Cardiol. 123:168–179.
2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wagner S, Dybkova N, Rasenack EC,
Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss
G, Brown JH, et al: Ca2+/calmodulin-dependent protein
ki-nase II regulates cardiac Na+ channels. J Clin
Invest. 116:3127–3138. 2006.PubMed/NCBI View Article : Google Scholar
|
|
29
|
El Refaey M, Musa H, Murphy NP, Lubbers
ER, Skaf M, Han M, Cavus O, Koenig SN, Wallace MJ, Gratz D, et al:
Protein Phosphatase 2A Regulates Cardiac Na+ Channels.
Circ Res. 124:737–746. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hegyi B, Morotti S, Liu C, Ginsburg KS,
Bossuyt J, Belardinelli L, Izu LT, Chen-Izu Y, Bányász T, Grandi E,
et al: Enhanced Depolarization Drive in Failing Rabbit Ventricular
Myocytes: Calcium-Dependent and β-Adrenergic Effects on Late
Sodium, L-Type Calcium, and Sodium-Calcium Exchange Currents. Circ
Arrhythm Electrophysiol. 12(e007061)2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Valverde CA, Mazzocchi G, Di Carlo MN,
Ciocci Pardo A, Salas N, Ragone MI, Felice JI, Cely-Ortiz A,
Consolini AE, Portiansky E, et al: Ablation of phospholamban
rescues reperfusion arrhythmias but exacerbates myocardium
infarction in hearts with Ca2+/calmodulin kinase II
constitutive phosphorylation of ryanodine receptors. Cardio-vasc
Res. 115:556–569. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Coppini R, Ferrantini C, Mugelli A,
Poggesi C and Cerbai E: Altered Ca2+ and Na+
Homeostasis in Human Hypertrophic Cardiomyopathy: Implications for
Arrhythmogenesis. Front Physiol. 9(1391)2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Nie J, Duan Q, He M, Li X, Wang B, Zhou C,
Wu L, Wen Z, Chen C, Wang DW, et al: Ranolazine prevents pressure
overload-induced cardiac hypertrophy and heart failure by restoring
aberrant Na+ and Ca2+ handling. J Cell
Physiol. 234:11587–11601. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Alday A, Ahyayauch H, Fernández-López V,
Echeazarra L, Urrutia J, Casis O and Gallego M: CaMKII Modulates
the Cardiac Transient Outward K+ current through its
Association with Kv4 Channels in Non-Caveolar Membrane Rafts. Cell
Physiol Bio-chem. 54:27–39. 2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhai X, Qiao X, Zhang L, Wang D, Zhang L,
Feng Q, Wu B, Cao J and Liu Q: IK1 channel agonist zacopride
suppresses ventricular arrhythmias in conscious rats with healing
myocardial infarction. Life Sci. 239(117075)2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Hegyi B, Bossuyt J, Ginsburg KS, Mendoza
LM, Talken L, Ferrier WT, Pogwizd SM, Izu LT, Chen-Izu Y and Bers
DM: Altered Repolarization Reserve in Failing Rabbit Ventricular
Myocytes: Calcium and β-Adrenergic Effects on Delayed- and
Inward-Rectifier Potassium Currents. Circ Arrhythm Electrophysiol.
11(e005852)2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Liu QH, Qiao X, Zhang LJ, Wang J, Zhang L,
Zhai XW, Ren XZ, Li Y, Cao XN, Feng QL, et al: IK1 Channel Agonist
Zacopride Alleviates Cardiac Hypertrophy and Failure via
Alterations in Calcium Dyshomeostasis and Electrical Remodeling in
Rats. Front Pharmacol. 10(929)2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Elnakish MT, Canan BD, Kilic A, Mohler PJ
and Janssen PM: Effects of zacopride, a moderate IK1 channel
agonist, on triggered arrhythmia and contractility in human
ventricular myocardium. Pharmacol Res. 115:309–318. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Shugg T, Johnson DE, Shao M, Lai X,
Witzmann F, Cummins TR, Rubart-Von-der Lohe M, Hudmon A and
Overholser BR: Calcium/calmodulin-dependent protein kinase II
regulation of IKs during sustained β-adrenergic receptor
stimulation. Heart Rhythm. 15:895–904. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Park SJ, Zhang D, Qi Y, Li Y, Lee KY,
Bezzerides VJ, Yang P, Xia S, Kim SL, Liu X, et al: Insights Into
the Pathogenesis of Catecholaminergic Polymorphic Ventricular
Tachycardia From Engineered Human Heart Tissue. Circulation.
140:390–404. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Lee TI, Chen YC, Lin YK, Chung CC, Lu YY,
Kao YH and Chen YJ: Empagliflozin Attenuates Myocardial Sodium and
Calcium Dysregulation and Reverses Cardiac Remodeling in
Streptozotocin-Induced Diabetic Rats. Int J Mol Sci.
20(20)2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Kamada R, Yokoshiki H, Mitsuyama H,
Watanabe M, Mizukami K, Tenma T, Takahashi M, Takada S and Anzai T:
Arrhythmogenic β-adrenergic signaling in cardiac hypertrophy: The
role of small-conductance calcium-activated potassium channels via
activation of CaMKII. Eur J Pharmacol. 844:110–117. 2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Popescu I, Yin G, Velmurugan S, Erickson
JR, Despa F and Despa S: Lower sarcoplasmic reticulum
Ca2+ threshold for triggering afterdepolarizations in
diabetic rat hearts. Heart Rhythm. 16:765–772. 2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Soliman H, Nyamandi V, Garcia-Patino M,
Zhang PC, Lin E, Jia ZP, Tibbits GF, Hove-Madsen L and MacLeod KM:
ROCK2 promotes ryanodine receptor phosphorylation and arrhythmic
calcium release in diabetic cardiomyocytes. Int J Cardiol.
281:90–98. 2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Johnson CN: Calcium modulation of cardiac
sodium channels. J Physiol. 598:2835–2846. 2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhong P, Quan D, Huang Y and Huang H:
CaMKII Activation Promotes Cardiac Electrical Remodeling and
Increases the Susceptibility to Arrhythmia Induction in High-fat
Diet-Fed Mice With Hyperlipidemia Conditions. J Cardiovasc
Pharmacol. 70:245–254. 2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Sun L, Chen Y, Luo H, Xu M, Meng G and
Zhang W: Ca2+/calmodulin-dependent protein kinase II
regulation by inhibitor 1 of protein phosphatase 1 alleviates
necropto-sis in high glucose-induced cardiomyocytes injury. Biochem
Pharmacol. 163:194–205. 2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Tzimas C, Terrovitis J, Lehnart SE,
Kranias EG and Sanoudou D: Calcium/calmodulin-dependent protein
kinase II (CaMKII) inhibition ameliorates arrhythmias elicited by
junctin ablation under stress conditions. Heart Rhythm.
12:1599–1610. 2015.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Cao L, Chen Y, Lu L, Liu Y, Wang Y, Fan J
and Yin Y: Angiotensin II upregu-lates fibroblast-myofibroblast
transition through Cx43-dependent CaMKII and TGF-β1 signaling in
neonatal rat cardiac fibroblasts. Acta Biochim Biophys Sin
(Shanghai). 50:843–852. 2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Himelman E, Lillo MA, Nouet J, Gonzalez
JP, Zhao Q, Xie LH, Li H, Liu T, Wehrens XH, Lampe PD, et al:
Prevention of connexin-43 remodeling protects against Duchenne
muscular dystrophy cardiomyopathy. J Clin Invest. 130:1713–1727.
2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Huang RY, Laing JG, Kanter EM, Berthoud
VM, Bao M, Rohrs HW, Townsend RR and Yamada KA: Identification of
CaMKII phosphorylation sites in Connexin43 by high-resolution mass
spectrometry. J Proteome Res. 10:1098–1109. 2011.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Li W, Gao H, Gao J and Wang Z:
Upregulation of MMP-9 and CaMKII prompts cardiac
electrophysiological changes that predispose denervated
transplanted hearts to arrhythmogenesis after prolonged cold
ischemic storage. Biomed Pharmacother. 112(108641)2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Takanari H, Bourgonje VJ, Fontes MS,
Raaijmakers AJ, Driessen H, Jansen JA, van der Nagel R, Kok B, van
Stuijvenberg L, Boulaksil M, et al: Calmodulin/CaMKII inhibition
improves intercellular communication and impulse propagation in the
heart and is antiarrhythmic under conditions when fibrosis is
absent. Cardiovasc Res. 111:410–421. 2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Pasdois P, Beauvoit B, Tariosse L, Vinassa
B, Bonoron-Adèle S and Dos Santos P: Effect of diazoxide on
flavoprotein oxidation and reactive oxygen species generation
during ischemia-reperfusion: A study on Langendorff-perfused rat
hearts using optic fibers. Am J Physiol Heart Circ Physiol.
294:H2088–H2097. 2008.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Warren M, Sciuto KJ, Taylor TG, Garg V,
Torres NS, Shibayama J, Spitzer KW and Zaitsev AV: Blockade of
CaMKII depresses conduction preferentially in the right ventricular
outflow tract and promotes ischemic ventricular fibrillation in the
rabbit heart. Am J Physiol Heart Circ Physiol. 312:H752–H767.
2017.PubMed/NCBI View Article : Google Scholar
|














