Introduction
Chronic obstructive pulmonary disease (COPD)
involves persistent airway inflammation and emphysema; it is
primarily caused by cigarette smoke (CS) and genetic predisposition
(1). It is characterized by tissue
remodeling, emphysematous alveolar destruction, enlarged air spaces
and chronic airway inflammation. The two major pathological changes
of COPD include emphysema and airway inflammation. Previous
research has focused on the underlying molecular mechanisms that
lead to COPD progression, and some of the pathological processes
are mediated by the abnormal activation of certain signaling
pathways (2); however, the
mechanisms that initiate the disease are not fully understood.
Hedgehog (Hh) signaling is one of the pathways
involved in early embryogenesis, serving an important role in cell
growth, differentiation, tissue patterning and vascularization
(3). Sonic hedgehog (SHH), desert
hedgehog (DHH) and Indian hedgehog (IHH) are three ligands that
initiate the canonical Hh signaling pathway. SHH is a predominant
ligand in the adult lung, which is expressed by the proximal airway
and distal alveolar epithelium. The ligand binding of Hh to its
repressive receptor, patched (Ptc; a twelve-transmembrane protein),
causes the suppression of signal transduction, relieving smoothened
(Smo; a seven-transmembrane protein) from Ptc and subsequently
activating the signaling cascade (3). This leads to the translocation of
certain transcription factors, including glioma-associated oncogene
homolog (Gli) 1, Gli2 and Gli3, to the nucleus, therefore involving
many downstream target genes that initiate transcription (4). In particular, Gli1 acts as the most
important molecule in the human body. Cyclopamine is a steroid
alkaloid that inhibits Hh signaling by directly binding to Smo
(4).
Previous studies (3-5)
have demonstrated that Hh signaling maintains mesenchymal
quiescence in the adult lung; however, repeated exposure to CS
causes the aberrant activation of Hh signaling, leading to its
overexpression in bronchial epithelial cells. Additionally, Hh
signaling is capable of promoting the airway inflammatory process,
inducing abnormal alveolar and bronchial epithelial cell apoptosis,
which may facilitate COPD initiation and progression (5). The interruption of Hh signaling at
earlier time points (embryon) results in enlarged alveolar
airspaces. Signaling is also dysregulated in diseases such as COPD
(6). Hedgehog-interacting protein
(HIP) is a feedback inhibitor of Hh. It competitively binds to
three Hh ligands and has an affinity equal to that of Ptc, thus
leading to the repression of the Hh signaling pathway (7). HIP has a protective role in COPD
pathogenesis, and according to studies by Lao et al
(7,8), 32% of patient COPD lung tissues
exhibited a reduced expression of the HIP gene compared with
smokers without airflow limitation.
Our previous study indicated that Hh served a role
in cigarette-induced airway inflammation through the regulation of
certain inflammatory cytokines in an A549 cell model (9); however, the role of Hh in emphysema
was not elucidated. The current study therefore assessed the effect
of the Hh signaling pathway on COPD progression, which is a
possible molecular mechanism of cigarette-induced emphysema and
airway inflammation. This was evaluated by measuring the expression
of SHH, Gli1, HIP and various airway inflammatory mediators using
an animal model.
Materials and methods
Animals
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Shanghai Rui Jin
Hospital, Shanghai Jiao Tong University (Shanghai, China; approval
no. 105). A total of 30 12-week-old C57BL/6J male mice (31.7±4.2 g)
were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. These
mice were housed in a pathogen-free, temperature-controlled
environment with 24˚C and 40% humidity under a 12:12 h light-dark
cycle, with wood chip bedding, food and water were available ad
libitum throughout experiments. After 2 weeks of adaptation,
mice were randomly divided into three groups (n=10 mice/group):
Control, CS and CS + cyclopamine (CSC). In the control group,
C57BL/6J mice were exposed to normal room air. The mice in CS group
were exposed to the tobacco smoke produced from five cigarettes,
four times per day for 30 min, for a total of 5 days per week for
24 weeks. Mice were subsequently anesthetized with an
intraperitoneal injection of ketamine (60 mg/kg) and xylazine (8
mg/kg) (10), after which
lipopolysaccharide (LPS; 0.9 mg/kg body weight; Sigma-Aldrich;
Merck KGaA) was administrated intratracheally each day in the last
week, as previously described (11). One week following LPS injections,
mice in CSC group were administered intraperitoneal injections of
50 mg/kg cyclopamine (12).
Histopathological examination
Mice were euthanized by an intraperitoneal injection
of sodium pentobarbital (100 mg/kg), and lung tissues were
carefully removed. Hematoxylin and eosin (H&E) staining was
performed to observe the pathological changes of the lung. Lung
tissues were obtained from the mice of each group and fixed using
4% formaldehyde in PBS in 4˚C overnight, after which tissues were
dehydrated using a Leica TP 1020 Rapid Tissue Processor (Leica,
Inc.). Samples were then embedded in paraffin and serial sections (4
µm thick) were stained H&E staining (Servicebio, Inc.) in room
temperature. All experiments were performed according to the
manufacturers' instructions. To observe the morphology of lung
tissues, slides were evaluated under a Leica photograph light
microscope (Leica Microsystems GmbH; magnification, x200).
Reverse transcription-quantitative PCR
(RT-qPCR) analysis of SHH, Gli1, HIP and other inflammatory
mediators
A total of ~3 g tissues of each samples were used
for PCR analysis. Tissues were ground into fragments in PBS. Total
RNA was extracted using TRIzol® reagent (Ambion; Thermo
Fisher Scientific, Inc.) from the dissolved lung tissues. cDNA was
synthesized using Moloney Murine Leukemia Virus Reverse (M-MLV)
reverse transcriptase (Takara Biotechnology Co., Ltd.). PCR was
performed using ChamQ SYBR Color qPCR Master Mix (Vazyme Biotech
Co., Ltd.). Briefly, 2.5 µl of 20 µM oligo (dT) primer stock was
added to the RNA sample, after which RNase-free H2O was
also added to a final volume of 11.5 µl. The mixture was incubated
at 70˚C for 3 min. Next, 4 µl 5X Reverse Transcription Buffer, 2 µl
dNTP Mix, 2 µl 100 mM DTT and 0.5 µl M-MLV Reverse Transcriptase
was added. Samples were then incubated at 42˚C for 60 min and the
reaction was terminated by incubation at 70˚C for 15 min. An ABI
7900 Real-Time PCR System (Thermo Fisher Scientific, Inc.) was used
to monitor amplification reactions in real-time. The primers used
for PCR were as follows: SHH forward, 5'-AAGCGCGCTCTTTGCCA-'3 and
reverse, 5'-TTGATGAGAATGGTGCCGTG-3'; Gli1 forward,
5'-TTGCAGCCAGGAGTTCGATT-3' and reverse, 5'-TGTGCACCACCAGCATGTAT-3';
HIP forward, 5'-GAAGAACGCAGAGGTGACCA-3' and reverse,
5'-CCTGTTGCTCTTGAGTCCGT-3'; intracellular adhesion molecule
(ICAM-1) forward, 5'-CTGGGCTTGGAGACTCAGTG-3' and reverse,
5'-CCACACTCTCCGGAAACGAA-3'; IL-6 forward,
5'-CACTTCACAAGTCGGAGGCT-3' and reverse,
5'-CTGCAAGTGCATCATCGTTGT-3'; IL-8 forward,
5'-CTAGGCATCTTCGTCCGTCC-3' and reverse, 5'-TTCACCCATGGAGCATCAGG-3';
TNF-α forward, 5'-AGGCACTCCCCCAAAAGATG-3' and reverse,
5'-TGAGGGTCTGGGCCATAGAA-3'; GAPDH forward,
5'-CTCCTCCTGGCCTCGCTGT-3' and reverse, 5'-GCTGTCACCTTCACCGTTCC-3'.
Data were evaluated using the 2-∆∆Cq method (13).
Western blotting of SHH, Gli1, HIP and
inflammatory mediators
RIPA buffer (Sigma-Aldrich; Merck KgaA) containing
protease inhibitors was used to lyse and extract protein from lung
tissue (3 g tissues of each samples). A Bradford assay was
subsequently performed to detect the total protein concentration of
lung tissue. Equal amounts of protein were separated by TGX
Stain-Free FastCast Acrylamide Kit 12% gel (Bio-Rad, Inc.), and
subsequently transferred onto a 0.45 µm PVDF membrane (GE
Healthcare, Inc.). Membranes were then incubated with primary
antibodies (Table SI) overnight at
4˚C, after which membranes were probed with secondary antibodies
for 1 h at room temperature as described previously (Table SI) (14). After further washing,
Chemiluminescent HRP Substrate; EMD Millipore) was used to
visualize the protein bands. Finally, target protein levels
relative to the internal control were calculated using Image J
1.52v software (National Institutes of Health).
Statistical analysis
Data were analyzed using SPSS (version 20.0; IBM
Corp.) and were presented as the mean ± SD. One-way ANOVA was
performed to evaluate the differences between groups, followed by
Scheffe's post hoc test for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference. The
experiments were performed in triplicate.
Results
Lung histology and measurement of
emphysema
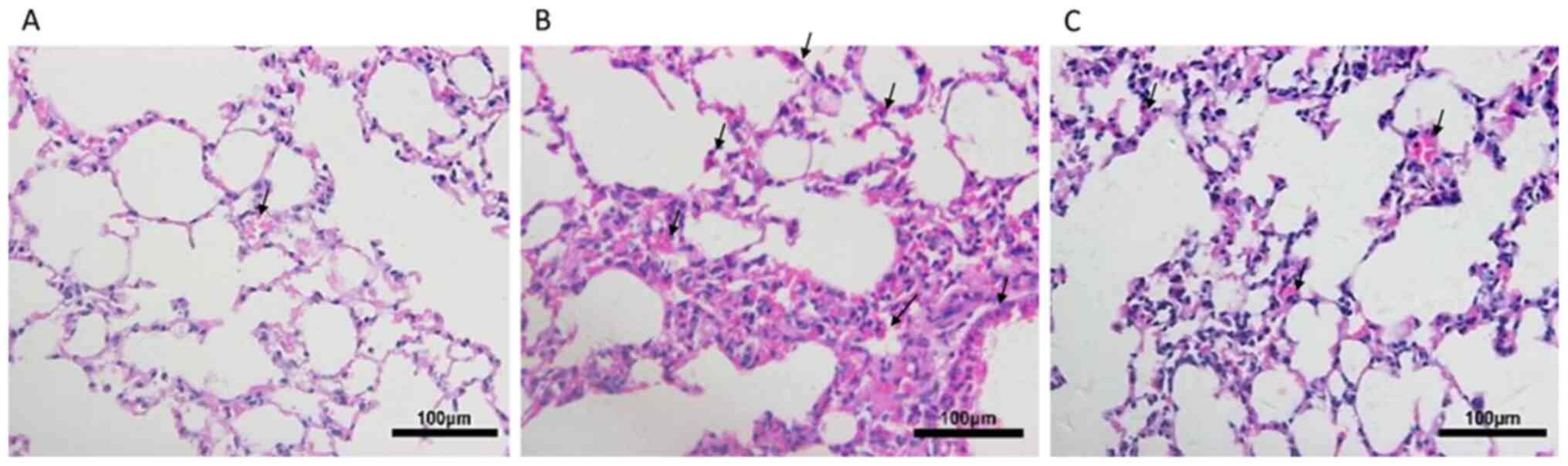
Following exposure to CS for 24 weeks, the lungs of
mice were histologically analyzed (Fig.
1). Compared with the control group, mice in the CS group
exhibited focal enlargement of the alveoli and inflammatory cell
infiltration, exhibiting neutrophil, lymphocyte and macrophage
accumulation in lung parenchyma and interstitial spaces, with
thickening of certain septa. By contrast, emphysema was partially
ameliorated, as demonstrated by decreased inflammatory cell
infiltration, in the lung tissues of CSC mice.
mRNA expression levels of SHH, Gli1,
HIP and inflammatory mediators in the three groups
The mRNA expression levels of SHH, Gli1 and HIP in
the lung tissue of mice were compared among the three groups. The
results revealed that SHH and Gli1 levels were significantly
increased in the CS group compared with the control group (both
P<0.05), whereas their expression levels in the CSC group was
lower than the CS group, significantly for SHH. HIP levels in the
CS group were significantly lower compared with the control, but
their levels in the CSC group were significantly higher than that
in the in the CS (P<0.05) (Fig.
2A).
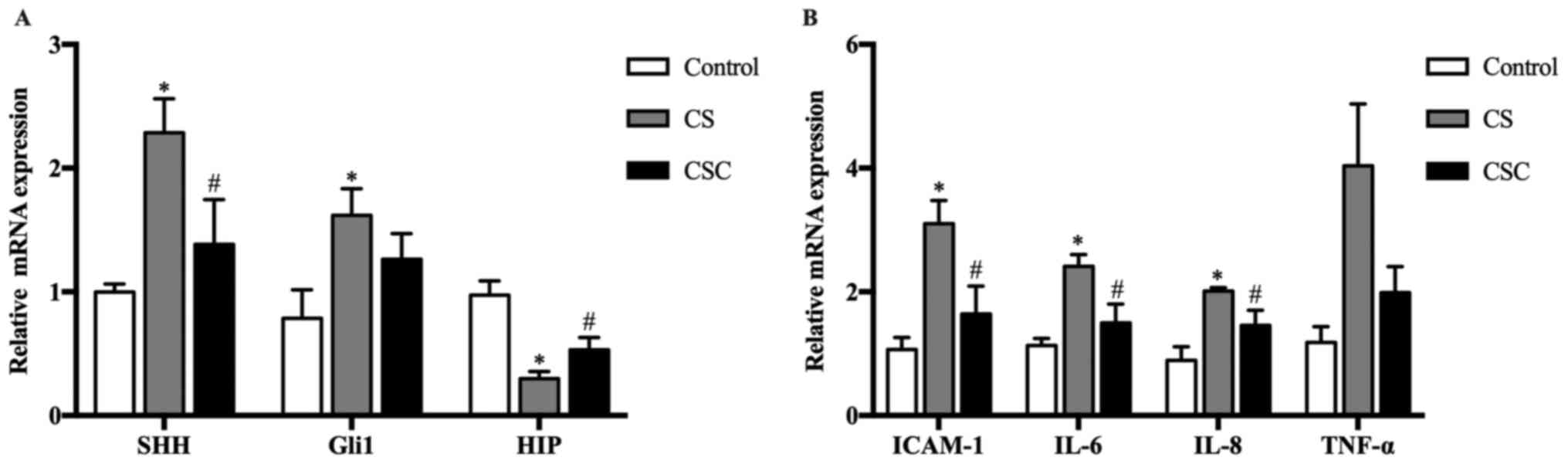 | Figure 2SHH, Gli1, HIP and inflammatory
mediator mRNA expression in the three groups. (A) SHH, Gli1 and HIP
mRNA expression levels. (B) ICAM-1, IL-6, IL-8 and TNF-α mRNA
expression levels. *P<0.05 vs. control;
#P<0.05 vs. CS. CS, cigarette smoke; CSC, CS +
cyclopamine; Gli1, glioma-associated oncogene homolog 1; HIP,
hedgehog-interacting protein; ICAM-1, intracellular adhesion
molecule-1; SHH, sonic hedgehog. |
The mRNA expression levels of inflammatory mediators
ICAM-1, IL-6 and IL-8 were significantly increased in the CS group
compared with the control group (P<0.05; Fig. 2B), and these levels were
significantly decreased following cyclopamine treatment, but not
for TNF-α.
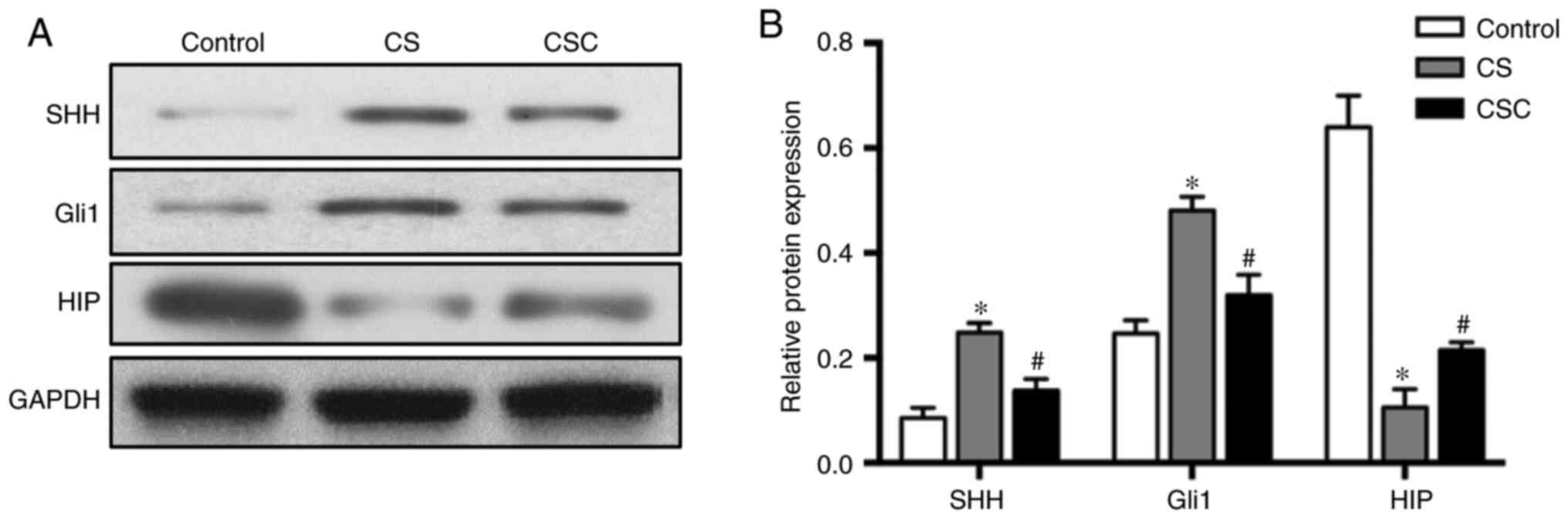
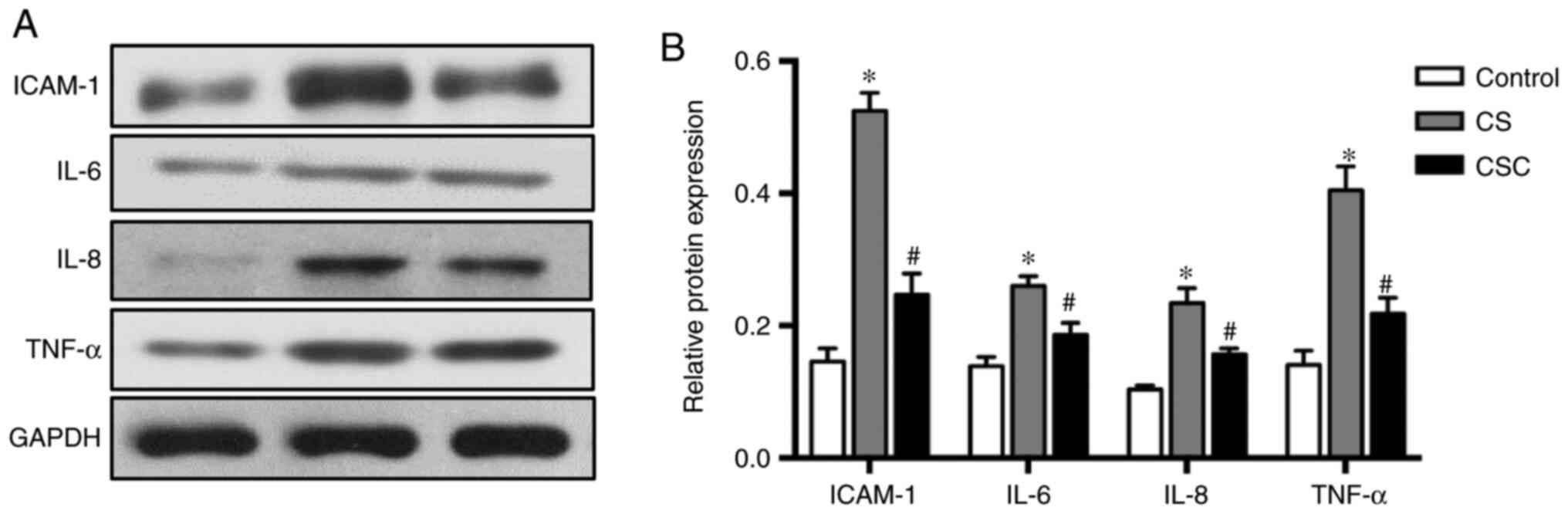
Protein levels of SHH, Gli1, HIP and
inflammatory mediators in the three groups
The protein expression levels of SHH, Gli1 and HIP
in lung tissue were compared among the three groups. SHH and Gli1
levels in the CS group were significantly increased compared with
the control group (both P<0.05; Fig.
3). Furthermore, these expression levels were significantly
decreased in the CSC group compared with the CS group (both
P<0.05). HIP levels were significantly decreased in the CS group
compared with the control group, but significantly increased in the
CSC group compared with the CS group (both P<0.05; Fig. 3).
The results of inflammatory mediators ICAM-1, IL-6,
IL-8 and TNF-α followed similar trends. Protein expression levels
were higher in the CS group compared with the control, but
significantly lower following cyclopamine treatment (all P<0.05;
Fig. 4).
Discussion
The Hh signaling pathway serves a critical role in
development, cell fate decisions and tissue growth (3). In adult tissues, the activation of the
Hh pathway maintains homeostasis (15), which is typically regarded as a
mitogenic cue during tissue development and injury or repair
(16). A previous study
demonstrated that Hh interacts with certain factors, such as MMPs,
demonstrating an association with chronic lung inflammation
(17). In the current study, the
underlying mechanisms of Hh signaling were investigated in
CS-induced emphysema and airway inflammation using an experimental
animal model.
The mRNA and protein expression levels of SHH and
Gli1 were significantly increased in the CS group when compared
with the control, suggesting that CS stimulation aberrantly
activated the Hh pathway, which is in line with a previous study
(5). As a result of lung
histopathological examination, the mice of the CS group presented
with an emphysematous phenotype when compared with the controls, an
observation that was partially ameliorated in the CSC group. This
indicated that Hh signaling may have an important effect on
cigarette-induced chronic airway disease. Therefore, the Hh
signaling pathway must be strictly controlled to prevent
inappropriate re-activation, which could be involved in the
initiation and progression of inflammation and emphysema. Wang
et al (14) revealed that
the expansion of Hh activation in the alveolar mesenchyme may
result in typical emphysema owing to alveolar damage and airspace
enlargement (14). This suggested
that the restriction of Hh activation in the alveolar mesenchyme is
important for maintaining alveolar regeneration, and that emphysema
can be caused by hyperactivation of the Hh pathway.
HIP, which is a negative regulator of Hh,
competitively binds to all three Hh ligands: SHH, IHH and DHH
(3). In the current study, HIP
levels were decreased when mice were exposed to CS, suggesting that
HIP may exert a protective role in COPD pathogenesis. These results
were congruent to that of a previous study (7). Mice with HIP haplo-insufficiency
(HIP+/-) are susceptible to severe airspace enlargement
and emphysema with increasing lymphoid aggregates, enhancing the
CS-induced lymphocyte activation pathways in the lungs (8). During early lung development, loss of
HIP increases Hh activation, leading to defective branching
morphogenesis (18). In adults, HIP
expression persists in the lung tissue for the maintenance of lung
homeostasis (7). HIP may regulate
the function of the alveolar mesenchyme by restricting Hh
signaling. Genome-wide association studies have identified certain
loci, including the HIP gene, that is associated with COPD
susceptibility (19,20). Our previous study also identified
various single nucleotide polymorphisms on the HIP gene in the
Chinese Han population, which were associated with the
susceptibility of COPD (21).
The effect of the Hh pathway in cigarette-induced
airway inflammatory disease is not well established. Certain
pro-inflammatory cytokines, such as IL-6, IL-8 and TNF-α, which act
as biomarkers of airway inflammation, have been involved in
systemic oxidative inflammation, promoting COPD development
(2). Hence, the mRNA and protein
expression of various airway inflammatory mediators in the lung
tissues of mice in the CS or CSC groups, including ICAM-1, IL-6,
IL-8 and TNF-α, were was measured in the current study. The results
revealed that inflammation mediator expression levels were
significantly increased in the CS group compared with the control
group, but decreased following cyclopamine treatment. Therefore,
aberrant activation of the Hh signaling pathway may partly enhance
airway inflammation, whereas airway inflammatory reactions might be
downregulated through decreased cytokine levels when Hh signaling
is blocked. These pro-inflammatory molecules have also been
reported to be regulated by Hh in other inflammatory diseases
(22). Additional studies have
demonstrated that the Hh pathway participates in inflammatory
processes, not only in lung diseases, but also in other organ
diseases. For example, the cyclopamin-induced inhibition of Hh
signaling in rats with arthritis led to decreased TNF-α, IL-1β and
IL-6 levels (22). In gastritis,
Gli1 upregulated IL-6, IL-1β and TNF-α, and was regarded as the
main promoter of inflammatory response (23). As such, previous studies have
demonstrated that Hh signaling is capable of promoting the
inflammatory process in the microenvironment (23). The results of the present study
revealed that the protein level of TNF-α in the CS group was
significantly higher compared with the control; however, while an
increase was also observed in mRNA levels, this effect was not
significant. Further studies should be performed to confirm the
repeatability of the results obtained for this inflammatory
mediator.
Hh signaling may regulate hepatocyte growth factor
(HGF), which is a target gene of Hh in COPD. HGF is secreted by
macrophage and mesenchyme cells in the airway, and it has been
demonstrated to serve a crucial role in emphysema. Deficient HGF
expression leads to emphysematous alveolar destruction, enlarged
air spaces and chronic airway inflammation (24). However, further studies are required
to clarify the potential pathways by which Hh signaling regulates
inflammatory cytokines in COPD.
Notably, our previous study (9) indicated that Hh served a role in
cigarette-induced airway inflammation through the regulation of
certain inflammatory cytokines. Using the A549 cell model, we
demonstrated the role of Hh in regulating some inflammatory
mediators, but not emphysema (histological analysis of lung
tissues). However, in the current study, another experiment was
perforned using an animal model, with a focus on lung histology and
measurement of emphysema, in addition to some inflammatory
mediators, with the aim of clarifying the effect of Hh signaling on
emphysema.
The current study had certain limitations.
Additional evidence including cell or animal experiments is
required to clarify the underlying mechanism with regard to the
impact of the Hh pathway on COPD development. Furthermore, the
current study focused on smoking-related airway inflammation, but
in certain patients with COPD and no history of smoking, emphysema
and airway inflammation would not be associated with cigarette
smoke. Thus, further research is warranted to determine whether Hh
signaling also participates in COPD development in these
patients.
The present study revealed that Hh signaling might
serve an important role in cigarette-induced emphysema and airway
inflammation through the regulation of inflammatory cytokines.
Therefore, diminishing the activation of Hh signaling may serve as
a novel therapeutic strategy for patients with smoking-related
COPD.
Supplementary Material
Antibodies used during western
blotting.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FYL and RC performed experiments, analyzed the data
and wrote the manuscript. YG and MZ proposed the hypothesis,
revised the manuscript and approved the version to be submitted. YG
and MZ confirmed the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Shanghai Rui Jin
Hospital, Shanghai Jiao Tong University (Shanghai, China; approval
no. 105).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McCloskey SC, Patel BD, Hinchliffe SJ,
Reid ED, Wareham NJ and Lomas DA: Siblings of patients with severe
chronic obstructive pulmonary disease have significant risk of
airflow obstruction. Am J Respir Crit Care Med. 164:1419–1424.
2001.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bames PJ, Burney PG, Silverman EK, Celli
BR, Vestbo J, Wedzicha JA and Wouters EFM: Chronic obstructive
pulmonary disease. Nat Rev Dis Primers. 1(15076)2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Briscoe J and Therond PP: The mechanisms
of Hedgehog signalling and its roles in development and disease.
Nat Rev Mol Cell Biol. 14:416–429. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Kasper M, Regl G, Frischauf AM and Aberger
F: GLI1 transcription factors: Mediators of oncogenic Hedgehog
signaling. Eur J Cancer. 42:437–445. 2006.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lemjabbar-Alaoui H, Dasari V, Sidhu SS,
Mengistab A, Finkbeiner W, Gallup M and Basbaum C: Wnt and Hedgehog
are critical mediators of cigarette smoke-induced lung cancer. PLoS
One. 1(e93)2006.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kugler MC, Loomis CA, Zhao Z, Cushman JC,
Liu L and Munger JS: Sonic Hedgehog signaling regulates
myofibroblast function during alveolar septum formation in murine
postnatal lung. Am J Respir Cell Mol Biol. 57:280–293.
2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lao T, Jiang Z, Yun J, Qiu W, Guo F, Huang
C, Mancini JD, Gupta K, Laucho-Contreras ME, Naing ZZC, et al: Hhip
haploin sufficiency sensitizes mice to age-related emphysema. Proc
Natl Acad Sci USA. 113:E4681–E4687. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lao T, Glass K, Qiu W, Polverino F, Gupta
K, Morrow J, Dominic Mancini J, Vuong L, Perrella MA, Hersh CP, et
al: Haploinsufficiency of Hedgehog interacting protein causes
increased emphysema induced by cigarette smoke through network
rewiring. Genome Med. 7(12)2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Guo Y, Shi GC, Wan HY and Zhou M: Hedgehog
signaling regulates the expression levels of inflammatory mediators
in cigarette-induced airway inflammation. Mol Med Rep.
17:8557–8563. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wu F, He ZQ, Ding R, Huang ZG, Jiang QX,
Cui HM, Lin Y, Huang SB, Dai XL, Zhang JY, et al: Danhong promotes
angiogenesis in diabetic mice after critical limb ischemia by
activation of CSE-H2S-VEGF axis. Evid Based Complement Alternat
Med. 2015(276263)2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
D'hulst AI, Vermaelen KY, Brusselle GG,
Joos GF and Pauwels RA: Time course of cigarette smoke-induced
pulmonary inflammation in mice. Eur Respir J. 26:204–213.
2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chen X, Jin YT, Hou XM, Liu FQ and Wang
YL: Sonic Hedgehog signaling: Evidence for its protective role
protective role in endotoxin induced acute lung injury in mouse
model. PLoS One. 10(e0140886)2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang C, de Mochel NSR, Christenson SA,
Cassandras M, Moon R, Brumwell AN, Byrnes L, Li A, Yokosaki Y, Shan
P, et al: Expansion of Hedgehog disrupts mesenchymal identity and
induces emphysema phenotype. J Clin Invest. 128:4343–4358.
2018.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Solanas G and Benitah SA: Regenerating the
skin: A task for the heterogeneous stem cell pool and surrounding
niche. Nat Rev Mol Cell Biol. 14:737–748. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Varjosalo M and Taipale J: Hedgehog:
Functions and mechanisms. Genes Dev. 22:2454–2472. 2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Onishi H, Morisaki T, Nakao F, Odate S,
Morisaki T and Katano M: Protein-bound polysaccharide decreases
invasiveness and proliferation in pancreatic cancer by inhibition
of Hedgehog signaling and HIF-1α pathways under hypoxia. Cancer
Lett. 335:289–298. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chuang PT, Kawcak T and McMahon AP:
Feedback control of mammalian Hedgehog signaling by the
Hedgehog-binding protein, Hip1, modulates Fgf signaling during
branching morphogenesis of the lung. Genes Dev. 17:342–347.
2003.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hancock DB, Eijgelsheim M, Wilk JB, Gharib
SA, Loehr LR, Marciante KD, Franceschini N, van Durme YMTA, Chen
TH, Barr RG, et al: Meta-analyses of genome-wide association
studies identify multiple loci associated with pulmonary function.
Nat Genet. 42:45–52. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Pillai SG, Ge DL, Zhu GH, Kong XY, Shianna
KV, Need AC, Feng S, Hersh CP, Bakke P, Gulsvik A, et al: A
genome-wide association study in chronic obstructive pulmonary
disease (COPD): Identification of two major susceptibility loci.
PLoS Genet. 5(e1000421)2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Guo Y, Gong Y, Pan C, Qian Y, Shi G, Cheng
Q, Li QY, Ren L, Weng QL, Chen Y, et al: Association of genetic
polymorphisms with chronic obstructive pulmonary disease in the
chinese han population: A case-control study. BMC Med Genomics.
5(64)2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Li R, Cai L, Ding J, Hu CM, Wu TN and Hu
XY: Inhibition of Hedgehog signal pathway by cyclopamine attenuates
inflammation and articular cartilage damage in rats with
adjuvant-induced arthritis. J Pharm Pharmacol. 67:963–971.
2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
El-Zaatari M, Kao JY, Tessier A, Bai L,
Hayes MM, Fontaine C, Eaton KA and Merchant JL: Gli1 deletion
prevents helicobacter-induced gastric metaplasia and expansion of
myeloid cell subsets. PLoS One. 8(e58935)2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang CQ, Cassandras M and Peng T: The role
of Hedgehog signaling in adult lung regeneration and maintenance. J
Dev Biol. 7(14)2019.PubMed/NCBI View Article : Google Scholar
|