Introduction
Gastric cancer is one of the most prevalent
malignant tumors worldwide (1). The
number of newly diagnosed cases of gastric cancer increased to
1.033 million worldwide in 2018, with 782,000 related deaths
(2). With aging and socioeconomic
development, cancer morbidity and mortality are rapidly increasing
globally (3). A variety of advanced
methods, including chemotherapy, surgical removal and radiotherapy,
have been used for gastric cancer treatment, however, mortality
remains high (4). An estimated
456,000 new cases of gastric cancer were diagnosed in China in
2018, with approximately 390,000 related deaths (5). Therefore, it is imperative to develop
effective therapeutic strategies for gastric cancer treatment and
gene therapy is a potentially beneficial option (6).
Kallikrein-related peptidase 6 (KLK6) is a member of
the KLK family that was initially identified based on its abnormal
expression in human ovarian and breast cancers (7). Accumulating evidence has demonstrated
the role of KLK6 in carcinogenesis and its potential as a cancer
biomarker (8). As a proteolytic
protein, KLK6 regulates the invasive phenotype of tumor cells by
degrading fibronectin, fibrinogen, collagen and laminin (9,10).
Previous research indicated that KLK6 promoted cancer cell
migration and invasion by modulating epithelial-mesenchymal
transition (EMT) (11). However,
the effect of KLK6 on EMT varies based on tumor classification.
KLK6 expression in non-KL6 expressing breast cancer cells
downregulated the expression of the EMT marker vimentin and
upregulated the expression of the epithelial markers cytokeratin 8
and 19, suggesting an inhibitory effect of KLK6 against EMT in
breast cancer cells (12).
Conversely, KLK6 overexpression or expression in KLK6-knockdown
colorectal cancer cells facilitated EMT (11).
Previous studies have demonstrated that KLK6 is
upregulated in gastric cancer (13-15).
However, the effect of KLK6 on the invasive phenotype and EMT
status of gastric cancer cells requires further study. In the
present study KLK6 was expressed at significantly higher levels in
metastatic gastric cancer cells (HGC-27) than in primary gastric
cancer cells (AGS and SNU-1). Subsequently, the effect of KLK6 on
the invasive phenotype and EMT status of gastric cancer cells lines
HGC-27 in vitro and in vivo in a mouse xenograft
model were investigated.
Materials and methods
Cell culture
Human gastric cancer cell line HGC-27 (derived from
a lymphatic metastasis) and primary gastric cancer cell lines AGS
and SNU-1 were purchased from The Cell Bank of Type Culture
Collection of The Chinese Academy of Sciences. The cells were
incubated in RPMI-1640 medium (Cytiva; cat. no. SH30809.01B)
containing 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.; cat. no. 10270-106) at 37˚C in an environment
containing 5% CO2. The protein and mRNA expression
levels of KLK6 in HGC-27, AGS and SNU-1 cells were measured using
western blotting and reverse transcription-quantitative polymerase
chain reaction (RT-qPCR), respectively.
Cell transfection
pSICOR interference vectors and pCDH-CMV-
MCS-EF1-CopGFP-T2A-Puro overexpression vectors were supplied by
Addgene, Inc. KLK6 mRNA was amplified via PCR (forward,
5'-GCTCTAGATGAAGAAGCTGATGGTG-3'; reverse,
5'-CGGGATCCTCACTTGGCCTGAATGGT-3') and inserted into the pCDH vector
to produce pCDH-KLK6 overexpression (OV) plasmids. The short
hairpin (sh) interference fragments (shKLK6-1,
5'-AGAATAAGTTGGTGCATGG-3'; shKLK6-2, 5'-CAGATGGTGATTTCCCTGAC-3';
shKLK6-3, 5'-GATCAAAGGAGAAGCCAGGA-3'; and shKLK6-4,
5'-CAGATACACGAACTGGATCC-3') were inserted into pSICOR vectors to
produce pSICOR-shKLK6 plasmids. HGC-27 cells were transfected for
48 h with 0.8 µg pCDH-KLK6 (OV-KLK6), pSICOR-shKLK6 (shRNA) or
their corresponding negative controls (OV-NC and sh-NC,
respectively) using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.; cat. no. 11668-027).
Non-transfected cells served as the control (CON).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
Cell viability was measured using the MTT assay.
HGC-27 cells were seeded in a 96-well plate at a density of
5x103 cells/well and maintained overnight at 37˚C with
5% CO2. A 20 µl volume of MTT reagent (Bioswamp Life
Science Lab; Wuhan Beinlai Biotechnology Co., Ltd.; cat. no. C1736)
was added to each well after 24, 48 and 72 h of transfection. The
cells were incubated at 37˚C for 4 h in an atmosphere containing 5%
CO2 and subsequently incubated with 150 µl of dimethyl
sulfoxide for 10 min. The absorbance of the wells was measured
using a microplate reader at 570 nm.
Flow cytometry
Flow cytometry was performed to evaluate apoptosis
and cell cycle progression in HGC-27 cells. To assess cell
apoptosis, 1x106 HGC-27 cells were resuspended in 1 ml
of phosphate-buffered saline (PBS) and centrifuged at 400 x g for 5
min at 4˚C. The cells were then resuspended in 200 µl of PBS,
stained with 10 µl of Annexin V-fluorescein isothiocyanate (BD
Biosciences), and 10 µl of propidium iodide (PI; BD Biosciences) in
the dark at 4˚C for 30 min. Thereafter, the cells were subjected to
flow cytometry. To assess cell cycle progression, 1x107
HGC-27 cells were fixed in a mixture of 300 µl of PBS and 700 µl of
absolute ethyl alcohol at -20˚C for 24 h. After two washes with
PBS, the cells were resuspended in 100 µl of RNase A (BD
Biosciences) and maintained at 37˚C for 30 min. The cells were then
stained with 400 µl of PI (50 µg/ml) in the dark at 4˚C for 5 min
and subjected to flow cytometry (NovoCyte, ACEA Biosciences, Inc.;
Agilent Technologies, Inc.). The data were analyzed using
NovoExpress software version 1.3.0 (ACEA Biosciences, Inc.; Agilent
Technologies, Inc.). For apoptosis analysis, both early- and
late-stage apoptosis (quadrants Q2-2 and Q2-4) were evaluated.
Wound healing
HGC-27 cells were cultured overnight in 6-well
plates at a density of 1x106 cells/well. After 4 h of
transfection, the cells were wounded by scratching the cell
monolayer with a sterile 200-µl plastic pipette tip. The scratches
were observed and imaged under an inverted fluorescence microscope
(magnification, x100; Leica Microsystems, GmbH) after incubation in
serum-free medium at 37˚C in an environment containing 5%
CO2 for 0 and 24 h.
Cell migration and invasion
assays
Cell migration and invasion were evaluated using
two-chamber Transwell inserts (Corning, Inc.). The cells were
starved in serum-free medium for 24 h and resuspended in RPMI-1640
medium (Cytiva; cat. no. SH30809.01B) containing 1% FBS. In the
upper chambers, 0.5 ml of treated cells were seeded at a density of
1x105 cells/ml and the lower chambers were filled with
0.75 ml of RPMI-1640 medium supplemented with 10% FBS. The inserts
for the cell invasion assay were pre-coated for 30 min at 37˚C with
Matrigel (BD Biosciences; cat. no. 354230) between the lower and
upper chambers. After 24 h of incubation at 37˚C, the cells were
fixed at 25˚C with 4% paraformaldehyde for 10 min and stained with
1 ml of 0.5% crystal violet (Life Science Lab; Wuhan Beinlai
Biotechnology Co., Ltd.; cat. no. C1701) for 30 min at room
temperature. Non-invading or non-migrating cells were wiped away
using cotton swabs, and the migrated or invaded cells were counted
under an inverted fluorescence microscope (magnification, x100;
Leica Microsystems GmbH).
RT-qPCR
RT-qPCR was performed to examine the mRNA expression
levels of KLK6 in HGC-27, AGS and SNU-1 cells, as well as
EMT-related genes in HGC-27 cells. Total RNA was extracted from
HGC-27, AGS and SNU-1 cells using TRIzol® reagent
(Thermo Fisher Scientific, Inc.) and reverse-transcribed into cDNA
using the M-MuLV kit (42˚C for 60 min; 70˚C for 15 min and held at
16˚C), according to the manufacturer's instructions (Takara Bio,
Inc). The collected cDNA was amplified using a SYBR Green PCR kit
(KAPA Biosystems, Inc.; Roche Diagnostics; cat. no. KM4101)
following the manufacturer's instructions in a CFX-Connect 96
apparatus (Bio-Rad Laboratories, Inc.). The thermocycling
conditions were as follows: 95˚C for 3 min; 39 cycles of
denaturation at 95˚C for 5 sec, annealing at 56˚C for 10 sec, and
extension at 72˚C for 25 sec; and final extension at 65˚C for 5 sec
and 95˚C for 50 sec. The primer sequences were as follows: KLK6
forward, 5'-GAACTCATCCAGCCCCTT-3' and reverse, 5'-CATCCCC
AGCACACAACA-3'; epithelial (E)-cadherin forward, 5'-GGC
AAGGTTTTCTACAGC-3' and reverse, 5'-ATGTGGCA ATGCGTTCT-3'; vimentin
forward, 5'-TTGAACGCAAAGT GGAATC-3'; and reverse,
5'-AGGTCAGGCTTGGAAACA-3'; GAPDH forward, 5'-CCACTCCTCCACCTTTG-3'
and reverse, 5'-CACCACCCTGTTGCTGT-3'. GAPDH served as an endogenous
control. Relative mRNA expression levels were calculated using the
2-∆∆Cq method (16).
Western blotting
Western blotting was performed to determine the
protein expression levels of KLK6 in HGC-27, AGS and SNU-1 cells
and EMT-related markers in HGC-27 cells. Total proteins were
collected using radioimmunoprecipitation assay lysis buffer
(Bioswamp Life Science Lab; Wuhan Beinlai Biotechnology Co., Ltd.;
cat. no. W1689) and quantified using a bicinchoninic acid assay kit
(Bioswamp Life Science Lab; Wuhan Beinlai Biotechnology Co., Ltd.;
cat. no. W1712). Proteins (20 µg) were separated by 12% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis and transferred
onto polyvinylidene fluoride membranes (EMD Millipore). The
membranes were blocked and incubated overnight at 4˚C with primary
antibodies against epithelial cell adhesion molecules (EP-CAM,
Bioswamp, cat. no. PAB30814, 1:1000), E-cadherin (Bioswamp Life
Science Lab; Wuhan Beinlai Biotechnology Co., Ltd.; cat. no.
PAB43792, 1:1000), vimentin (Bioswamp Life Science Lab; Wuhan
Beinlai Biotechnology Co., Ltd; cat. no. PAB40646; 1:1,000),
phosphorylated (p)-SMAD2 (Bioswamp Life Science Lab; Wuhan Beinlai
Biotechnology Co., Ltd; cat. no. PAB43294-P; 1:1,000), SMAD2
(Bioswamp Life Science Lab; Wuhan Beinlai Biotechnology Co., Ltd;
cat. no. PAB30712; 1:1,000), p-SMAD3 (Bioswamp Life Science Lab;
Wuhan Beinlai Biotechnology Co., Ltd; cat. no. PAB43521-P;
1:1,000), SMAD3 (Bioswamp Life Science Lab; Wuhan Beinlai
Biotechnology Co., Ltd; cat. no. PAB30705; 1:1,000), and GAPDH
(Bioswamp Life Science Lab; Wuhan Beinlai Biotechnology Co., Ltd;
cat. no. PAB36269; 1:1,000) and were subsequently incubated for 1 h
at room temperature with HRP-conjugated goat anti-rabbit IgG
secondary antibodies (Bioswamp Life Science Lab; Wuhan Beinlai
Biotechnology Co., Ltd; cat. no. SAB43714; 1:20,000). The membranes
were developed using ECL (EMD Millipore) and then observed using a
Tanon-5200 apparatus (Tanon Science & Technology Co., Ltd.) and
data were analyzed using Tanon GIS software version 4.2 (Tanon
Science & Technology Co., Ltd.). GAPDH served as an internal
reference.
Tumor xenografts in nude mice
Female BALB/c nude mice (age, 6 weeks; weight 18-20
g; n=15) were supplied by Changzhou Cavens Experimental Animal Co.
Ltd. (ref. no. 1107301911000025) and divided into five groups (n=3
per group): CON, sh-NC, sh-KLK6, OV-NC and OV-KLK6. All mice were
injected with 200 µl of HGC-27 cells at a density of
1x106 cells/m in the right axilla. When the tumor size
reached 80-100 mm3 (12 days after injection), the mice
were treated with 10 µg of PBS, sh-NC, sh-KLK6, OV-NC and OV-KLK6,
respectively, by intratumoral injections once every three days for
a total of four times. Animal health and behavior were monitored
every day. When the tumor could be clearly identified (4 days after
injection), tumor volume was measured every two days using the
following formula: volume (mm3) = length x
width2/2(17). After 10
days of treatment, the mice were euthanized with an intraperitoneal
injection of sodium pentobarbital at 100 mg/kg of body weight
(death was confirmed by the absence of heartbeat and breath), and
the tumors were removed for further analysis. The humane endpoint
was when the tumor maximum diameter reached >15 mm. No animals
died during the course of the experiment. All animal procedures
were approved by the ethics committee of Wuhan Myhalic
Biotechnology Co., Ltd. (approval no. HLK-20181102-01), who also
conducted the animal experiments.
Hematoxylin and eosin (H&E)
staining
Pathological changes in the tumors were evaluated by
H&E staining. The tissues were fixed in 10% formalin buffer at
25˚C for 48 h, embedded in paraffin and sectioned at a thickness of
4 µm. After dewaxing, the sections were stained with hematoxylin
(Bioswamp, cat. no. I1709) at 25˚C for 3 min and then stained with
eosin solution (Bioswamp Life Science Lab; Wuhan Beinlai
Biotechnology Co., Ltd; cat. no. I1703) for 3 min. Pathological
changes were assessed using a light microscope (magnification,
x100), Leica Microsystems GmbH).
Statistical analysis
Data are presented as the mean ± standard deviation
(SD). Differences between two groups were analyzed using unpaired
Student's t-test and those between more than two groups were
analyzed using one-way analysis of variance, followed by Tukey's
test. P<0.05 was considered statistically significant.
Results
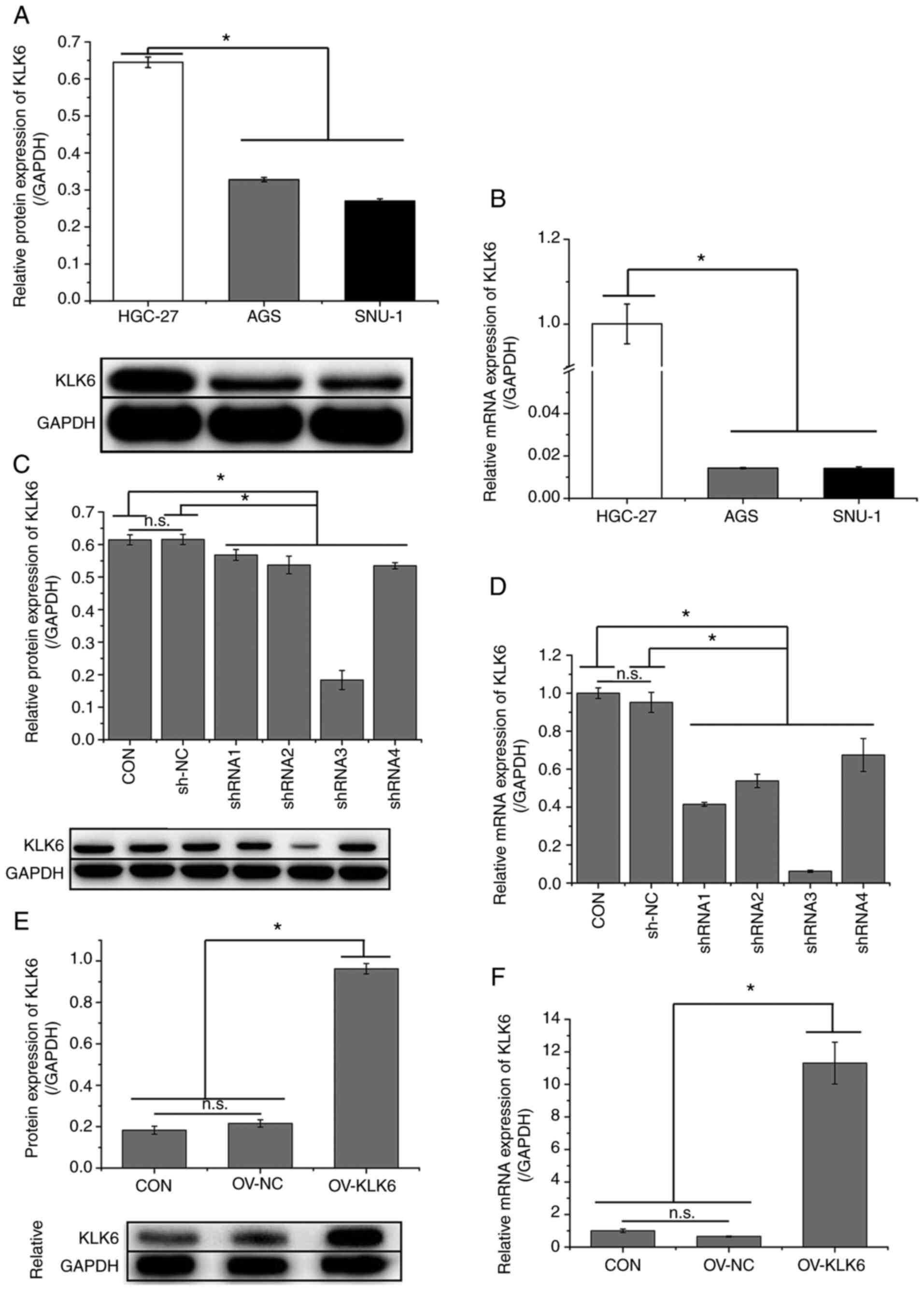
KLK6 is highly expressed in HGC-27
cells and increases HGC-27 cell viability
As indicated in Fig.
1A and B, the expression of
KLK6 was significantly higher in metastatic gastric cancer cells
(HGC-27) than in primary gastric cancer cells (AGS and SNU-1).
Thus, the effect of KLK6 on gastric cancer was investigated in
vitro and in vivo using HGC-27 cells. In HGC-27 cells,
KLK6 was inhibited by pSICOR-shKLK6 plasmids and overexpressed by
pCDH-KLK6 plasmids (Fig. 1C-F). As
shRNA3 interference resulted in the greatest reduction in the
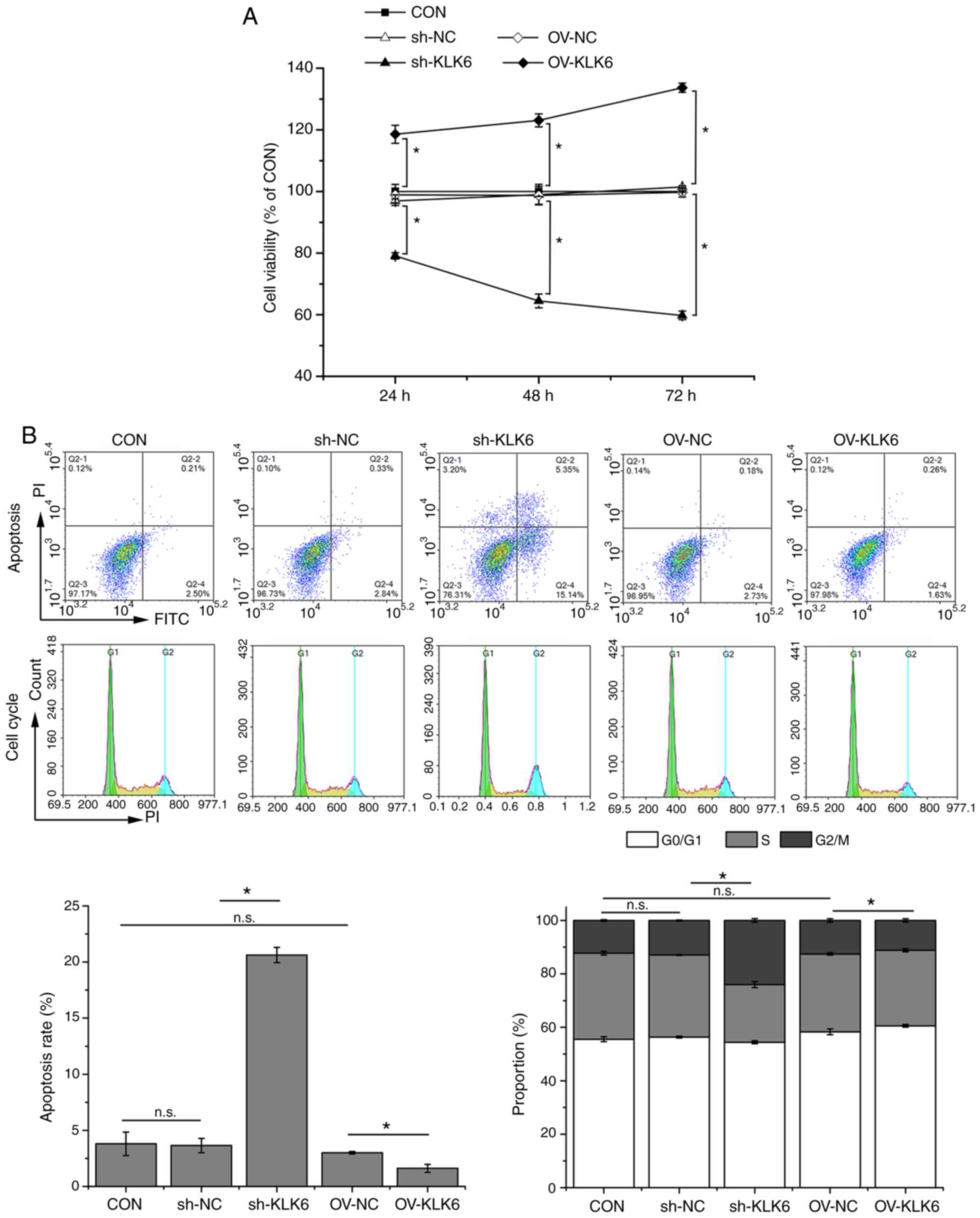
expression of KLK6, it was selected for subsequent experiments. MTT
assay demonstrated that KLK6 overexpression enhanced the viability
of HGC-27 cells, whereas KLK6 inhibition attenuated cell viability
in a time-dependent manner in comparison with controls (Fig. 2A). After 24 h of transfection, cell
viability was significantly different between the CON group and the
sh-KLK6 or OV-KLK6 groups. Therefore, subsequent experiments were
performed 24 h after transfection. Flow cytometry indicated that
KLK6 inhibition promoted apoptosis and G2/M phase arrest in HGC-27
cells, while KLK6 overexpression showed the opposite effect
(Fig. 2B).
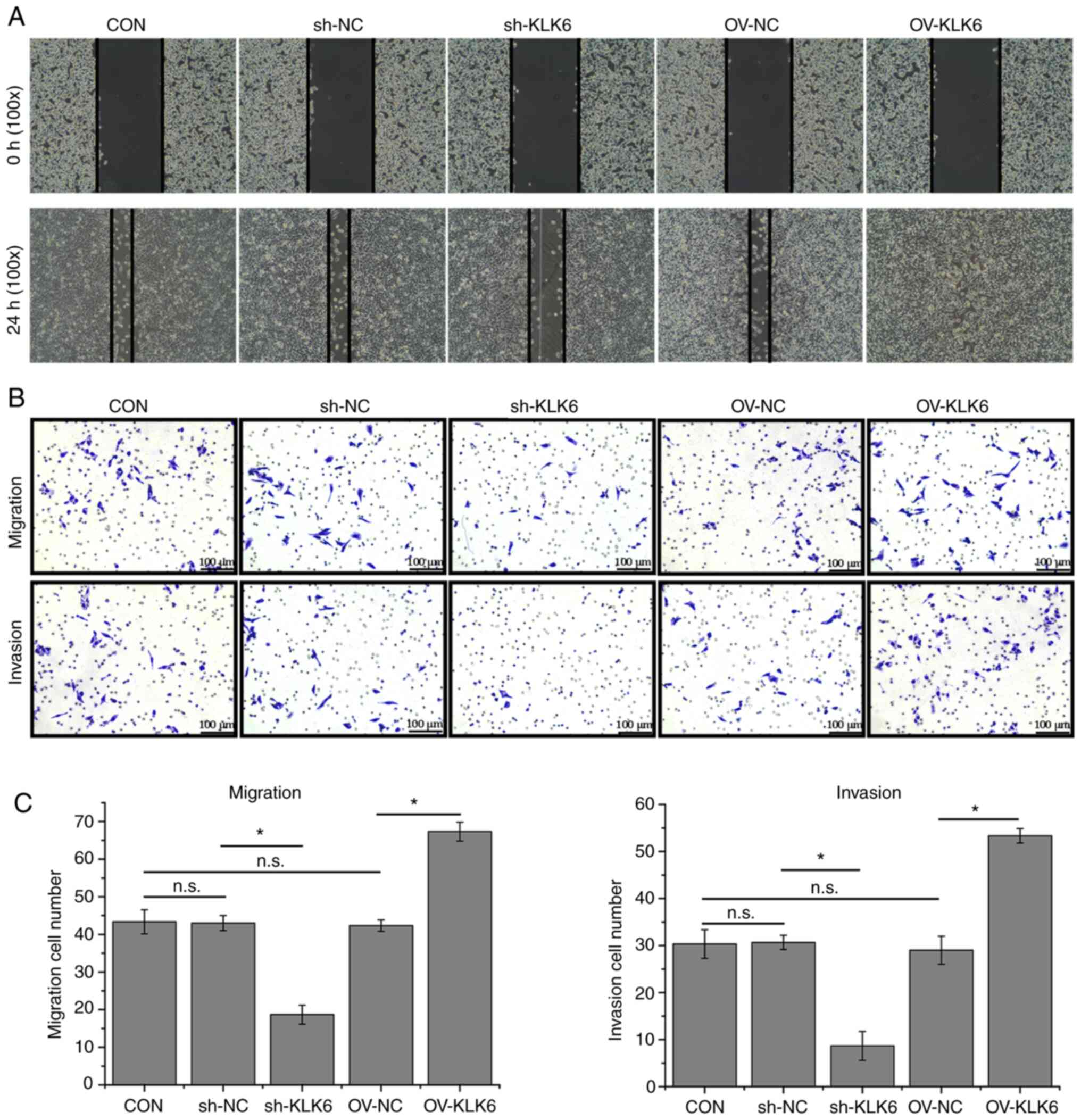
KLK6 inhibition attenuates HGC-27 cell
migration and invasion
The wound healing ability of sh-KLK6-transfected
HGC-27 cells was poorer than that of control cells, whereas it was
increased by KLK6 overexpression (Fig.
3A). In addition, the Transwell assay showed that KLK6
inhibition reduced the number of migratory and invading cells in
comparison with the control, whereas numbers were increased by KLK6
overexpression (Fig. 3B and
C).
KLK6 is involved in EMT regulation in
HGC-27 cells
Western blotting and RT-qPCR were performed to
evaluate the expression of EMT-related factors. As shown in
Fig. 4, compared to the CON group,
the protein and mRNA expression levels of E-cadherin were increased
by sh-KLK6 but decreased by OV-KLK6 in comparison with controls,
whereas vimentin showed the opposite trend as that of E-cadherin.
Additionally, KLK6 inhibition suppressed the protein expression of
EP-CAM and the phosphorylation of SMAD2 and SMAD3. These results
demonstrated the regulatory effect of KLK6 on EMT in HGC-27 cells
through modulation of EMT-related proteins.
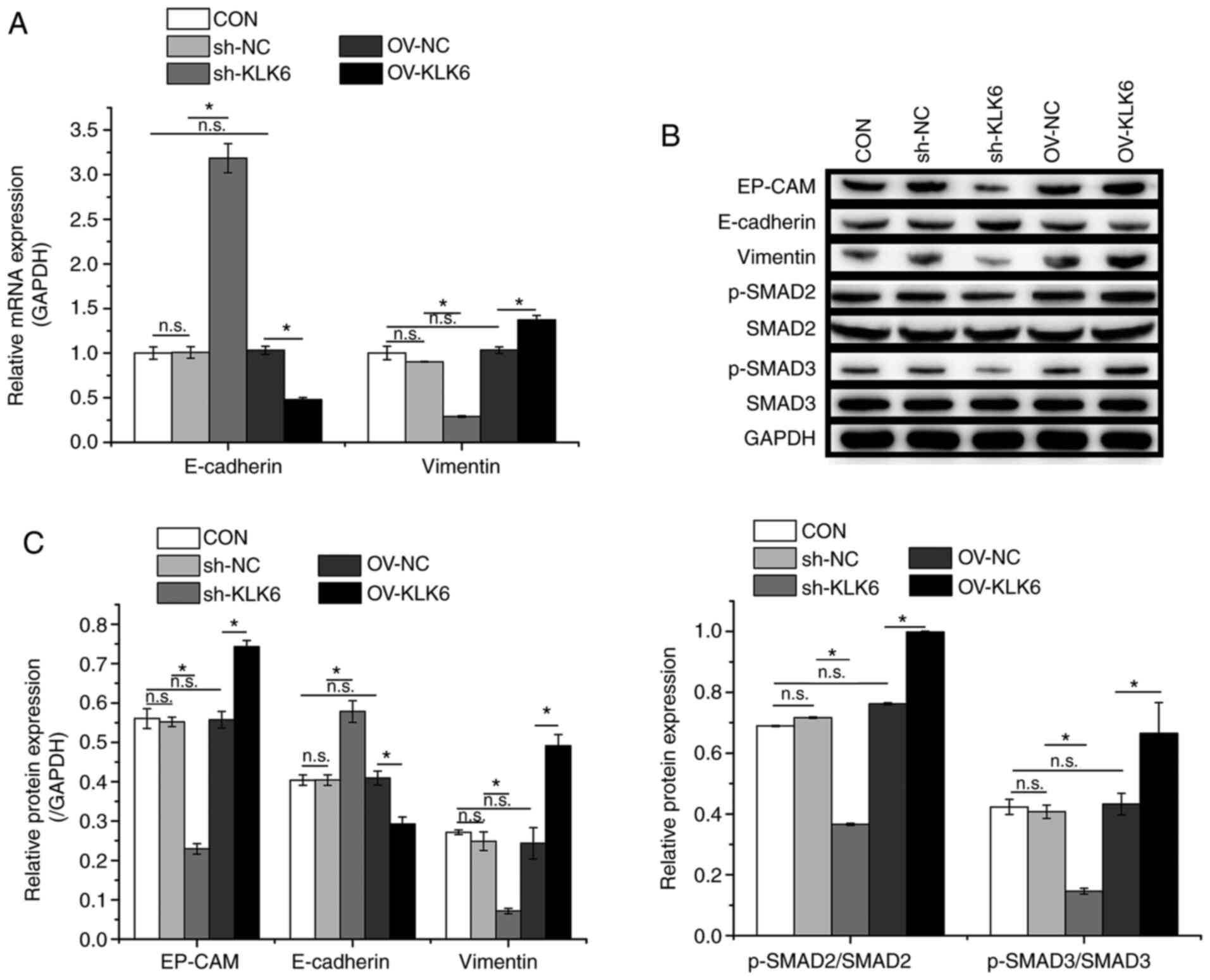 | Figure 4KLK6 inhibition suppresses EMT in
HGC-27 cells. (A) Relative mRNA expression levels of E-cadherin and
vimentin in HGC-27 cells. (B) Relative protein expression levels of
EP-CAM, E-cadherin and vimentin and phosphorylation levels of SMAD2
and SMAD3 in HGC-27 cells. (C) Western blotting densitometry
values. Data are presented as the mean ± SD (n=3). KLK6,
kallikrein-related peptidase 6; EMT, epithelial-to-mesenchymal
transition; E-cadherin, epithelial cadherin; EP-CAM, epithelial
cell adhesion molecule; p-, phosphorylated; Con, untreated control;
sh, short hairpin; NC, non-coding control; OV, overexpression
plasmid; n.s., not significant. *P<0.05. |
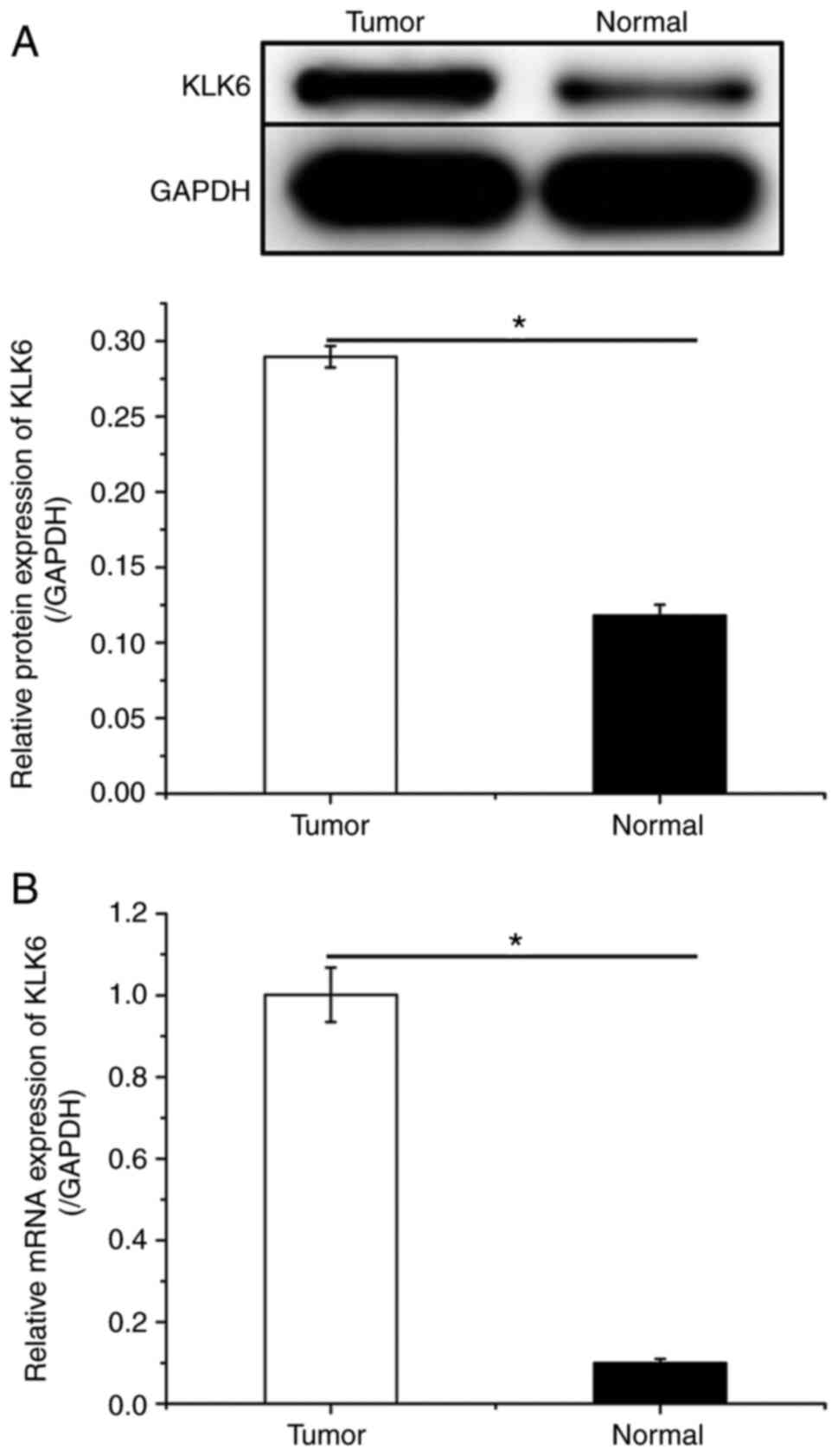
KLK6 inhibition suppresses gastric
cancer development in vivo
To investigate the effect of KLK6 on gastric cancer
in vivo, a xenograft mouse model was constructed by
injecting HGC-27 cells. Mice were treated with sh-KLK6, OV-KLK6 or
the corresponding negative controls. The results demonstrated that
both the mRNA and protein expression levels of KLK6 in tumor
tissues were increased compared to those in normal tissues
(Fig. 5). KLK6 overexpression
promoted the growth of gastric cancer, while KLK6 inhibition
blocked tumor growth (Fig. 6A-C).
H&E staining revealed that KLK6 increased tumor progression
(Fig. 6D). In addition, the
expression of EMT-related proteins in tumors was measured. Compared
to the CON and negative controls, the protein expression of EP-CAM
and vimentin and the phosphorylation of SMAD2, and SMAD3 was
downregulated by sh-KLK6, whereas that of E-cadherin was
upregulated, with OV-KLK6 showing the opposite trend (Fig. 6E).
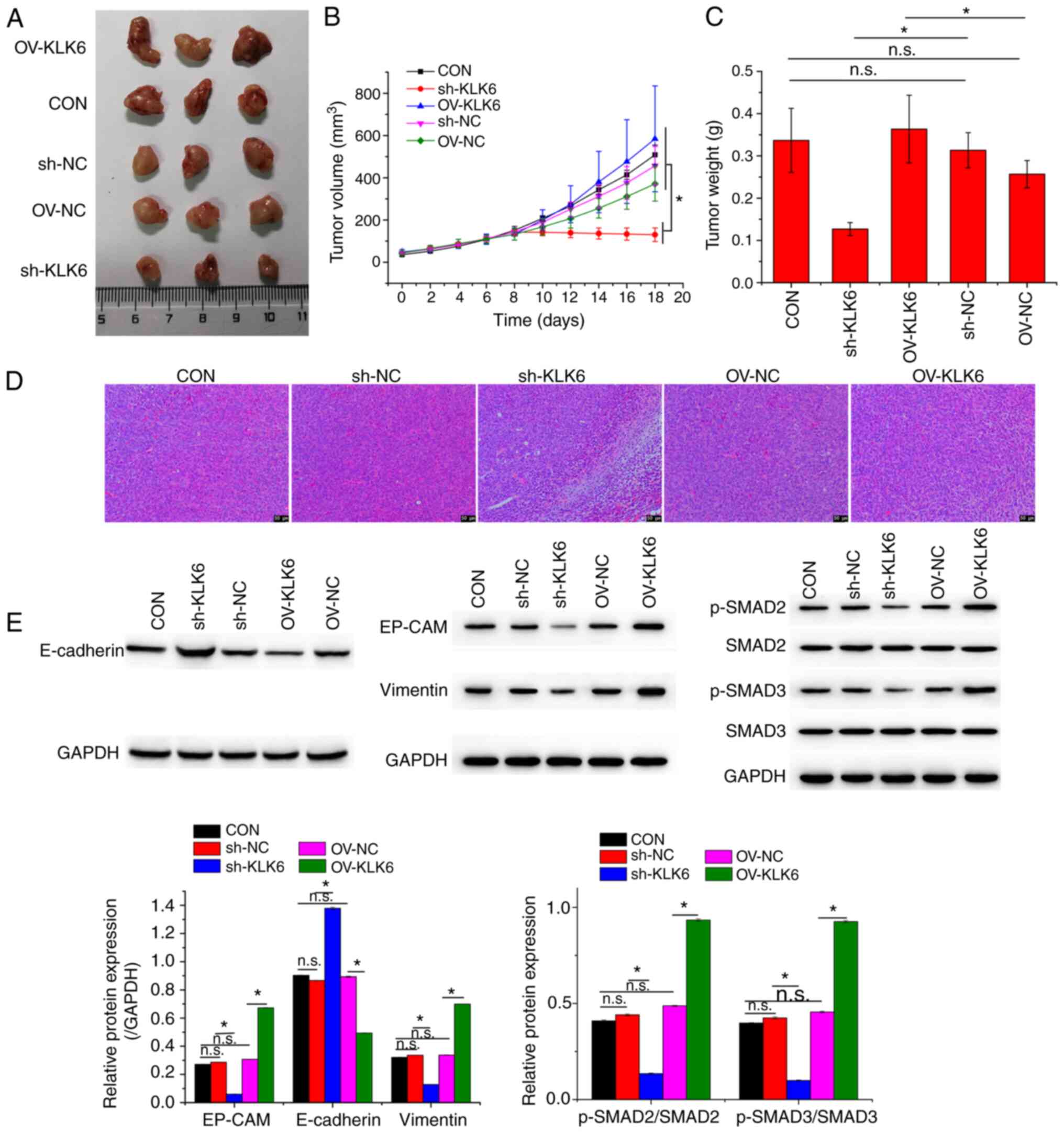 | Figure 6KLK6 inhibition suppresses tumor
growth. (A) Representative photographs, (B) volumes and (C) weights
of corresponding tumors. (D) Pathological morphology of xenografted
tumors. (E) Relative protein expression levels of EP-CAM,
E-cadherin and vimentin and levels of phosphorylation of SMAD2 and
SMAD3 in xenografted tumors. Data are presented as the mean ± SD
(n=3). KLK6, kallikrein-related peptidase 6; EMT,
epithelial-to-mesenchymal transition; E-cadherin, epithelial
cadherin; EP-CAM, epithelial cell adhesion molecule; p-,
phosphorylated; Con, untreated control; sh, short hairpin; NC,
non-coding control; OV, overexpression plasmid; n.s., not
significant. *P<0.05. |
Discussion
In the present study it was demonstrated that KLK6
was expressed at significantly higher levels in metastatic gastric
cancer cells (HGC-27) than in primary gastric cancer cells (AGS and
SNU-1). KLK6 interference inhibited the proliferation, migration
and invasion of gastric cancer cells in vitro and suppressed
the growth of gastric cancer cell line tumors in vivo. In
addition, KLK6 interference attenuated EMT in gastric cancer cells
by regulating the expression and phosphorylation of EMT-related
proteins (E-cadherin, vimentin, EP-CAM, SMAD2 and SMAD3). KLKs are
a family of serine proteases that contains 15 members, which are
associated with various physiological functions (18). Several KLKs, including KLK6, are
involved in neoplastic malignant progression and transformation,
through regulation of tumor chemoresistance, migration, invasion
and growth (18,19). Aberrant expression of KLK6 is common
in a number of malignancies, including head and neck squamous cell
carcinoma (20) and colorectal
(21), ovarian (22) and melanoma skin cancer (23). KLK6 is highly expressed in patients
with head and neck squamous cell carcinoma and modulates cancer
cell migration, invasion and chemoresistance by regulating EMT
(20). KLK6 is also associated with
drug resistance in gastric cancer. Kim et al (24) reported that KLK6 expression resulted
in chemoresistance by inhibiting auranofin-induced apoptosis via
autophagy activation in gastric cancer. In addition, KLK6 has been
reported to be highly expressed in gastric cancer (15) and acts as a prognostic indicator for
gastric cancer (25,26). Zhu et al (27) found that KLK6 facilitated gastric
cancer cell migration, invasion and growth, consistent with the
findings of the present study. Additionally, the present study
demonstrated that KLK6 enhanced EMT in gastric cancer cells, both
in vivo and in vitro, and promoted the progression of
gastric cancer cell tumors in vivo.
EMT is a biological process in which epithelial
cells transform to adopt a mesenchymal phenotype, resulting in the
loss of cell-cell adhesion and cell polarization and in the
acquisition of migration and invasion abilities (28). This process occurs during tissue
regeneration, organ fibrosis and wound healing (29). EMT has been demonstrated to be a
vital mechanism that causes epithelial cancer cells to acquire the
migratory and invasive properties associated with metastatic
characteristics (30). During EMT,
the expression of epithelial markers, including E-cadherin, a type
I classical cadherin that plays important roles in intercellular
interactions (31), is suppressed.
Meanwhile, mesenchymal markers, including vimentin, a cytoskeletal
protein involved in regulating cell motility (32), are upregulated (33). The present study revealed that KLK6
interference increased E-cadherin expression and suppressed that of
vimentin, suggesting an inhibitory effect of KLK6 on EMT in gastric
cancer.
In addition, the present study demonstrated that
KLK6 inhibition attenuated the phosphorylation of SMAD2 and SMAD3,
both of which are transcription factors that are activated by
transforming growth factor-β (29).
Several studies have reported that SMAD2/3 signaling is associated
with EMT. Tang et al (34)
revealed that SMAD2/3 activation promoted EMT in lung
adenocarcinoma. Kim et al (35) suggested that epidermal growth factor
enhanced the phosphorylation of SMAD2/3, thereby inducing EMT in
breast cancer cells. Wang et al (36) indicated that microfibril-associated
protein 2 promotes EMT in gastric cancer by activating the SMAD2/3
signaling pathway. In addition, it was previously demonstrated that
KLK6 activation altered the expression of EMT markers by promoting
SMAD2/3 phosphorylation (11).
These findings suggest that the effect of KLK6 on EMT in gastric
cancer may be associated with the regulation of SMAD2/3
signaling.
In conclusion, the present study demonstrated that
KLK6 is involved in the modulation of gastric cancer cell
proliferation, migration and invasion via EMT regulation. The
effect of KLK6 on EMT in gastric cancer is possibly mediated by
SMAD2/3 signaling. Whether there are other factors involved in the
mechanism of KLK6 in EMT regulation remains to be further
elucidated. A limitation of the present study is that only three
nude mice were used in each group for the in vivo studies,
which may have affected the statistical analysis. However, all
animal experiments were performed based on the findings of a
preliminary experiment. More animals (at least 6 in each group)
will be included in follow-up study designs, which will focus on
investigating the effect of KLK6 on EMT-associated metabolic
reprogramming in gastric cancer. The lack of co-immunoprecipitation
experiments for validating the signaling pathway and the use of
only one cell line are other limitations of this study, which will
be iresolved in the follow-up study. Overall, the present findings
suggest that KLK6 may be a future target for gastric cancer
therapy.
Acknowledgements
Not applicable
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
DZ was responsible for the study design, as well as
drafting and revision of the manuscript. YH, HL and WH performed
the experiments. YH analyzed the data. DZ and YH were responsible
for confirming the authenticity of raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal procedures were approved by the Ethics
Committee of Wuhan Myhalic Biotechnology Co., Ltd. (approval no.
HLK-20181102-01).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Seo GH, Kang HY and Choe EK: Osteoporosis
and fracture after gastrectomy for stomach cancer: A nationwide
claims study. Medicine (Baltimore). 97(e0532)2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Omran AR: The epidemiologic transition: A
theory of the epidemiology of population change 1971. Milbank Q.
83:731–757. 2005.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Tian Y, Li X, Li H, Lu Q, Sun G and Chen
H: Astragalus mongholicus regulate the Toll-like-receptor 4
meditated signal transduction of dendritic cells to restrain
stomach cancer cells. Afr J Tradit Complement Altern Med. 11:92–96.
2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Feng RM, Zong YN, Cao SM and Xu RH:
Current cancer situation in China: Good or bad news from the 2018
Global Cancer Statistics? Cancer Commun (Lond).
39(22)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gou WF, Yang XF, Shen DF, Zhao S, Liu YP,
Sun HZ, Takano Y, Su RJ, Luo JS and Zheng HC: The roles of BTG3
expression in gastric cancer: A potential marker for carcinogenesis
and a target molecule for gene therapy. Oncotarget. 6:19841–19867.
2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Anisowicz A, Sotiropoulou G, Stenman G,
Mok SC and Sager R: A novel protease homolog differentially
expressed in breast and ovarian cancer. Mol Med. 2:624–636.
1996.PubMed/NCBI
|
|
8
|
Yang F, Hu ZD, Chen Y and Hu CJ:
Diagnostic value of KLK6 as an ovarian cancer biomarker: A
meta-analysis. Biomed Rep. 4:681–686. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ghosh MC, Grass L, Soosaipillai A,
Sotiropoulou G and Diamandis EP: Human kallikrein 6 degrades
extracellular matrix proteins and may enhance the metastatic
potential of tumour cells. Tumour Biol. 25:193–199. 2004.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Klucky B, Mueller R, Vogt I, Teurich S,
Hartenstein B, Breuhahn K, Flechtenmacher C, Angel P and Hess J:
Kallikrein 6 induces E-cadherin shedding and promotes cell
proliferation, migration, and invasion. Cancer Res. 67:8198–8206.
2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Chen H, Sells E, Pandey R, Abril ER, Hsu
CH, Krouse RS, Nagle RB, Pampalakis G, Sotiropoulou G and Ignatenko
NA: Kallikrein 6 protease advances colon tumorigenesis viainduction
of the high mobility group A2 protein. Oncotarget. 10:6062–6078.
2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Pampalakis G, Prosnikli E, Agalioti T,
Vlahou A, Zoumpourlis V and Sotiropoulou G: A tumor-protective role
for human kallikrein-related peptidase 6 in breast cancer mediated
by inhibition of epithelial-to-mesenchymal transition. Cancer Res.
69:3779–3787. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Henkhaus RS, Gerner EW and Ignatenko NA:
Kallikrein 6 is a mediator of K-RAS-dependent migration of colon
carcinoma cells. Biol Chem. 389:757–764. 2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liu X, Xiong H, Li J, He Y and Yuan X:
Correlation of hK6 expression with tumor recurrence and prognosis
in advanced gastric cancer. Diagn Pathol. 8(62)2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kim JJ, Kim JT, Yoon HR, Kang MA, Kim JH,
Lee YH, Kim JW, Lee SJ, Song EY, Myung PK, et al: Upregulation and
secretion of kallikrein-related peptidase 6 (KLK6) in gastric
cancer. Tumour Biol. 33:731–738. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tang H, Xu L, Cen X, Yang L, Feng J, Li G,
Zhu H, Gao S, Yu Y, Zhao Y, et al: CDK5 inhibition in vitro
and in vivo induces cell death in myeloma and overcomes the
obstacle of bortezomib resistance. Int J Mol Med. 45:1661–1672.
2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ahmed N, Dorn J, Napieralski R, Drecoll E,
Kotzsch M, Goettig P, Zein E, Avril S, Kiechle M, Diamandis EP, et
al: Clinical relevance of kallikrein-related peptidase 6 (KLK6) and
8 (KLK8) mRNA expression in advanced serous ovarian cancer. Biol
Chem. 397:1265–1276. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Talieri M, Zoma M, Devetzi M, Scorilas A
and Ardavanis A: Kallikrein-related peptidase 6 (KLK6)gene
expression in intracranial tumors. Tumour Biol. 33:1375–1383.
2012.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Schrader CH, Kolb M, Zaoui K,
Flechtenmacher C, Grabe N, Weber KJ, Hielscher T, Plinkert PK and
Hess J: Kallikrein-related peptidase 6 regulates
epithelial-to-mesenchymal transition and serves as prognostic
biomarker for head and neck squamous cell carcinoma patients. Mol
Cancer. 14(107)2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ohlsson L, Lindmark G, Israelsson A,
Palmqvist R, Öberg Å, Hammarström ML and Hammarström S: Lymph node
tissue kallikrein-related peptidase 6 mRNA: A progression marker
for colorectal cancer. Br J Cancer. 107:150–157. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Wang P, Magdolen V, Seidl C, Dorn J,
Drecoll E, Kotzsch M, Yang F, Schmitt M, Schilling O, Rockstroh A,
et al: Kallikrein-related peptidases 4, 5, 6 and 7 regulate
tumour-associated factors in serous ovarian cancer. Br J Cancer.
119:1–9. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Krenzer S, Peterziel H, Mauch C, Blaber
SI, Blaber M, Angel P and Hess J: Expression and function of the
kallikrein-related peptidase 6 in the human melanoma
microenvironment. J Invest Dermatol. 131:2281–2288. 2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kim TW, Lee SJ, Kim JT, Kim SJ, Min JK,
Bae KH, Jung H, Kim BY, Lim JS, Yang Y, et al: Kallikrein-related
peptidase 6 induces chemotherapeutic resistance by attenuating
auranofin-induced cell death through activation of autophagy in
gastric cancer. Oncotarget. 7:85332–85348. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Nagahara H, Mimori K, Utsunomiya T,
Barnard GF, Ohira M, Hirakawa K and Mori M: Clinicopathologic and
biological significance of kallikrein 6 overexpression in human
gastric cancer. Clin Cancer Res. 11:6800–6806. 2005.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kolin DL, Sy K, Rotondo F, Bassily MN,
Kovacs K, Brezden-Masley C, Streutker CJ and Yousef GM: Prognostic
significance of human tissue kallikrein-related peptidases 6 and 10
in gastric cancer. Biol Chem. 395:1087–1093. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhu S, Shi J, Zhang S and Li Z: KLK6
Promotes Growth, Migration, and Invasion of Gastric Cancer Cells. J
Gastric Cancer. 18:356–367. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Gloushankova NA, Zhitnyak IY and Rubtsova
SN: Role of Epithelial-Mesenchymal Transition in Tumor Progression.
Biochemistry (Mosc). 83:1469–1476. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Liang X, He X, Li Y, Wang J, Wu D, Yuan X,
Wang X and Li G: Lyn regulates epithelial-mesenchymal transition in
CS-exposed model through Smad2/3 signaling. Respir Res.
20(201)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sun L and Fang J: Epigenetic regulation of
epithelial-mesenchymal transition. Cell Mol Life Sci. 73:4493–4515.
2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Bure IV, Nemtsova MV and Zaletaev DV:
Roles of E-cadherin and Noncoding RNAs in the
Epithelial-mesenchymal Transition and Progression in Gastric
Cancer. Int J Mol Sci. 20(20)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wang N, Liu D, Guo J, Sun Y, Guo T and Zhu
X: Molecular mechanism of Poria cocos combined with oxaliplatin on
the inhibition of epithelial-mesenchymal transition in gastric
cancer cells. Biomed Pharmacother. 102:865–873. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Cevenini A, Orrù S, Mancini A, Alfieri A,
Buono P and Imperlini E: Molecular Signatures of the Insulin-like
Growth Factor 1-mediated Epithelial-Mesenchymal Transition in
Breast, Lung and Gastric Cancers. Int J Mol Sci.
19(19)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tang Y, Xuan Y, Qiao G, Ou Z, He Z, Zhu Q,
Liao M and Yin G: MDM2 promotes epithelial-mesenchymal transition
through activation of Smad2/3 signaling pathway in lung
adenocarcinoma. OncoTargets Ther. 12:2247–2258. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kim J, Kong J, Chang H, Kim H and Kim A:
EGF induces epithelial-mesenchymal transition through
phospho-Smad2/3-Snail signaling pathway in breast cancer cells.
Oncotarget. 7:85021–85032. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wang JK, Wang WJ, Cai HY, Du BB, Mai P,
Zhang LJ, Ma W, Hu YG, Feng SF and Miao GY: MFAP2 promotes
epithelial-mesenchymal transition in gastric cancer cells by
activating TGF-β/SMAD2/3 signaling pathway. OncoTargets Ther.
11:4001–4017. 2018.PubMed/NCBI View Article : Google Scholar
|