Introduction
Lymphatic metastasis is one of the crucial routes of
metastasis in cervical cancer (1)
and treatment failure in cervical cancer is often associated with
lymph node metastasis (2).
Lymphatic vessels are usually considered to serve a passive role in
metastasis, acting merely as a channel for tissue-invading tumor
cells (3). Previous studies have
focused on the immunoregulatory function of lymphatic endothelial
cells (LECs), which are active players in lymphatic metastasis
(3,4). However, it remains unknown whether and
how LECs actively regulate lymphatic metastasis in cancer.
Endothelial-mesenchymal transition (EndMT) is a
process via which endothelial cells (ECs) display considerable
plasticity in the transition to mesenchymal cells, which is crucial
for ECs to obtain migratory abilities similar to those of tumor
cells (5,6). During this process, ECs lose their
endothelial markers, such as E-cadherin and CDs, and acquire a
migratory phenotype and mesenchymal markers, such as vimentin and
fibroblast-specific protein 1 (5,6). LECs
also express E-cadherin and can be induced to enter
endothelial-mesenchymal transition (EndMT) via WNT5B and Kaposi's
sarcoma-associated herpesvirus (7,8).
E-cadherin is a major component of epithelial cell junctions, as
well as a hallmark of EndMT (6,9).
However, the current understanding of the regulatory mechanism
underlying E-cadherin expression in EndMT remains limited. In EMT,
which is analogous to EndMT, epidermal growth factor is suggested
to induce the downregulation of E-cadherin via the ERK pathway
(10), which may activate a similar
signaling pathway in EndMT.
Semaphorins (Semas) are a large family of
extracellular signaling proteins that regulate the motility of
cells during the development of nevi (Sema3A, 3F, 6C and 6D), the
neuroendocrine system (Sema7A and 4D), the immune system (Sema4D),
the reproductive system (Sema3) and cancer progression (Sema4D, 3A
and 6D) (11-13).
Interestingly, Semas antagonize the effect of VEGF (13). In addition, several studies have
reported that Sema3F may affect lymphangiogenesis (14,15).
In a previous study, tumor-associated LECs (TLECs) were isolated
and normal LECs were obtained from tissues using in situ
laser capture microdissection, and it was determined that synthetic
membrane-bound Sema4C functioned as an autocrine factor that
promoted lymphangiogenesis (16).
However, the underlying mechanism of Sema4C in regulating TLEC
biological characteristics is largely unknown.
It has been revealed that tumors or metastasis may
rely, at least in part, on the gene regulatory events in cells that
constitute the primary tumor, as well as the tumor microenvironment
(TME) (17). LECs, which are one of
the most important components in the TME, also exhibit a unique
gene expression pattern compared with non-tumor LECs, such as the
expression of VEGFR3, which is activated under pathological
conditions, including cancer and inflammation (18). LECs bought from
ScienCell™ (cat. no. 2500) are separated from human
lymph nodes, according to the instructions of the cell line. The
differential expression level of genes between normal LECs and
TLECs prompted the present study to assess TLECs from tumors. Flow
cytometry has been indicated to be the most effective method for
isolating cells, as well as analyzing the TME (19,20).
To the best of our knowledge, the present study for the first time
used primary TLECs separated from mouse cervical tumors to study
LEC biological characterization.
The present study evaluated the role of Sema4C in
the transdifferentiation of primary TLECs to determine the tumor
cell-like invasive characteristics. The aim of the present study
was to investigate the effect of Sema4C on the migration of primary
TLEC isolated from tumor tissues by flow cytometry and its
mechanism using lentivirus infection to modulate the expression of
Sema4C. These findings indicated that regulation of the
Sema4C/ERK/E-cadherin pathway may be a novel target for cancer
therapy, which may potentially inhibit endothelial
transdifferentiation into tumor-like cells, thereby preventing the
lymphatic metastasis of cancer. The current preclinical study
provided novel insights into the role of Sema4C in TLECs and
demonstrated the active participation of TLECs in lymphatic
metastasis.
Materials and methods
Antibodies and cell line
All reagents used in the present study were of
analytical grade and are commercially available. The primary
antibodies used were as follows: Sema4C (cat. no. sc-136445; Santa
Cruz Biotechnology, Inc.); lymphatic vessel endothelial hyaluronan
receptor 1 (LYVE1) antibody-Alexa Fluor® 488 for flow
cytometry (cat. no. 53-0433-82; eBioscience; Thermo Fisher
Scientific, Inc.); LYVE1 antibody for immunohistochemistry (cat.
no. ab14917; Abcam); VEGFR3 (cat. no. ab51874; Abcam); E-cadherin
(cat. no. ab181296; Abcam); ERK1/2 (cat. no. ab17942; Abcam),
phosphorylated (p)-ERK1/2 (cat. no. ab214362; Abcam) and β-actin
(cat. no. ab8227; Abcam). Biotinylated anti-rabbit immunoglobulin
for DAB staining (cat. no. K406511-2) was purchased from Agilent
Technologies, Inc. The ERK inhibitor PD98059 (cat. no. HY-12028)
was purchased from MedChemExpress and cells were treated with 30 mM
for 24 h at room temperature.
The mouse cervical cancer cell line U14 (cat. no.
YB-M060) was purchased from Shanghai Yubo Biotechnology Co., Ltd.
The cells were cultured in RPMI-1640 medium (cat. no. 21870076;
Gibco; Thermo Fisher Scientific, Inc.) 37˚C with 5%
CO2.with 10% (v/v) FBS (cat. no. 16140071; Gibco; Thermo
Fisher Scientific, Inc.) NIH3T3 cells (mouse embryonic fibroblast
cell line) was purchased from ATCC (cat. no. CRL-1658), cultured in
Dulbecco's Modified Eagle's Medium (cat. no. 11885084; Gibco;
Thermo Fisher Scientific, Inc.) with 10% (v/v) FBS (cat. no.
16140071; Gibco; Thermo Fisher Scientific, Inc.) at 37˚C with 5%
CO2. No antibiotics were used in the culture medium. The
use of primary LECs was approved by the Scientific Research Ethics
Committee of Shandong University.
Lentivirus for delivery of full-length
Sema4C and small interfering (si)RNA against Sema4C
All the lentiviruses used in the present study were
purchased from Wester Biological Technology Co., Ltd. (serial no.
WST201710015). Each lentiviral packaging system included a
four-plasmid system (Fig. S1). A
full-length mouse Sema4C open reading frame was obtained via PCR
(PrimeSTAR® HS DNA Polymerase; cat. no. R010A; Takara
BioInc.) (thermocycling conditions were as follows: 95˚C for 3 min
1 cycle, followed by 30 cycles of 94˚C for 30 sec, -55˚C for 40 sec
and -72˚C for 30 sec and 72˚C for 5 min for 1 cycle), using cDNA
(from TLECs) by agarose electrophoresis and gel-cutting recovery as
template (cat. no. DP219-03; Tiangen Biotech Co. Ltd.) and the
primer sequences were as follows: Forward,
5'-AGGGTTCCAAGCTTAAGCGGCCGCGCCACCATGGCCCCACACTGGGCTGTCTGG-3' and
reverse, 5'-GATCCATCCCTAGGTAGATGCATTCATACTGAAGACTCCTCTGGGTTG-3'.
The lentivirus used for delivery of full-length Sema4C was LV5. The
final construction was termed LV5-Sema4C. The control construction
was LV5 negative control (empty vector, LV5NC).
The lentivirus used for delivery of siRNA against
Sema4C was LV3. The Sema4C siRNA sense sequence was
5'-CCUAUGCCUUCCAGCCCAADTDT-3' and antisense sequence was
5'-UUGGGCUGGAAGGCAUAGGDTDT-3', and the final construction was
termed LV3-siRNA. The sequence for the control siRNA was a
non-targeting sequence, sense, 5'-UUCUCCGAACGUGUCACGU-3' and
antisense, 5'-ACGUGACACGUUCGGAGAA-3', and the lentiviral construct
was termed LV3NC. The Sema4C siRNA has been verified in a previous
study (10).
Animals
C57BL/6 female mice (age, 4-6 weeks; weight, 18-22
g) were purchased from Tengxin Biomedical Technology. The mice were
maintained in the accredited animal facility of Shandong
University. The animal facility had a controlled temperature
(23-25˚C), a 12/12 h light/dark cycle with lights on from 8:00 to
20:00, and a relative humidity (50-60%). The mice had free access
to standard food and water. Animals were cared for by qualified
personnel every day, including weekends and holidays, to monitor
their health and behavior. The hygienic status of the mice was
specific pathogen-free, according to the Association for Assessment
and Accreditation of Laboratory Animal Care International
(Laboratory Animal Center of Tsinghua University) recommendations.
Each group contained 3 mice, and there were three time points (days
5, 7 and 14 after the injection of U14 cells) and 9 mice in total.
No mouse died until the end of the experiments. All the experiments
were carried out in accordance with the National Institute of
Health Guide of the Care and Use of Laboratory Animals. All the
experimental protocols were approved by the Animal Care and Use
Committee of Shandong University for animal ethics [approval no.
KYLL-2015(KS)-079].
Mouse xenograft tumor model
The mice were anesthetized using
isoflurane/O2 flow (4% induction and 2% maintenance
dose) in an induction chamber. Forceps were used to touch the leg
of the mouse, which was used to confirm that the mouse was fully
unconscious. The mice received 100 µl (~1x107) U14
cells, resuspended in RPMI-1640 medium, subcutaneously into the
left shoulder. A total of 2 weeks after the injection, the animals
was euthanized and the tumor volume was calculated according to the
following formula: Length x width2/2(21). The cage was placed in a chamber with
CO2, and CO2 was turned on for ≥20 min at 2
l/min. The percentage of the chamber volume/min displaced by the
flow rate of CO2 was 20% (April 2019). CO2
was maintained for ≥1 min after the mice stopped breathing. The
sacrificed mice were placed on a polystyrene board, and the tumor
was removed and measured.
Separation of TLECs via flow
cytometry
Tumor tissue cells were dissociated using 1%
collagenase (cat. no. C2139; Sigma-Aldrich; Merck KGaA) for 30 min
at 37˚C and 0.25% trypsin/EDTA solution, (cat. no. 25200056; Gibco;
Thermo Fisher Scientific, Inc.) for 5-8 min at room temperature.
Cell suspensions were filtered through cell sieves (200-µm mesh).
After incubation for 48 h at 37˚C with 5% CO2, the
suspended cells were removed and incubated with a monoclonal
antibody against mouse LYVE1-Alexa Fluor 488 or IgG1 kappa Isotype
Control-Alexa Fluor 488 (1:25; cat. no. 53-4714-80; EBioscience;
Thermo Fisher Scientific, Inc.) at room temperature for 30 min,
after adjusting the cell density to 5-8x106/ml. The
samples were analyzed on a FACSCalibur apparatus (BD Biosciences)
and cells that expressed LYVE1 were isolated. LECs were cultured in
EBM-2 medium (Lonza Group Ltd.) with 2% (v/v) FBS (Gibco; Thermo
Fisher Scientific, Inc.) at 37˚C with 5% CO2.
Immunohistochemistry for the
quantification of lymphatic microvessel density (LMVD)
Immunohistochemical analysis of LYVE1 was performed
using the avidin-biotin-peroxidase complex method. Fresh tissue was
fixed by 4% paraformaldehyde for 24 h and embedded using paraffin
at room temperature. Dewaxed and rehydrated 5-µm mouse tumor tissue
sections were incubated with 3% H2O2 for 10
min at room temperature and then rinsed with PBS for 3 times.
Sections were blocked with 5% BSA (w/v) in PBS (cat. no. A1993;
MilliporeSigma) for 20 min at room temperature and with a rabbit
polyclonal anti-mouse LYVE1 antibody (1:100 dilution) or with PBS
as the negative control overnight at 4˚C. The detection system was
Dako LSAB KIT (cat. no. K406511-2; Agilent Technologies, Inc).
Sections were then washed with PBS and biotinylated anti-rabbit
immunoglobulin (1:200) was added to the sections for 30 min at room
temperature. Peroxidase-conjugated avidin (1:400) was then applied
after the sections were washed with PBS. The peroxidase activity
was detected by exposing the sections to a solution of 0.05%
3,3'-diaminobenzidine and 0.01% H2O2 in
Tris-HCl buffer (3,3'-diaminobenzidine solution) for 10 min at room
temperature. The sections were counterstained with hematoxylin for
30 sec at room temperature. The stained sections were then analyzed
using standard light microscopy (Eclipse 200; Nikon Corporation).
Under a low magnification, the most vascularized intratumoral areas
were selected (hot spots). The number of immunostained lymphatic
vessels observed in three hot spot areas at x400 magnification was
counted. LMVD was expressed as the mean value (total number of
vessels in three hot spot microscopic fields/3).
Confocal microscopy imaging
Frozen tissue sections (5-µm, solidified in liquid
nitrogen) were blocked with 5% BSA (w/v) in PBS for 20 min at room
temperature. The LYVE1 (1:50) or Sema4C (1:20) primary antibodies
were applied to the slides at 4˚C overnight. After incubation with
the primary antibodies, the slides were washed three times with
cold PBS and incubated with FITC-conjugated goat anti-mouse (cat.
no. A5608; Beyotime Institute of Biotechnology) or Cy3-conjugated
goat anti-rabbit IgG (cat, no. A0516; Beyotime Institute of
Biotechnology) at a 1:50 dilution in PBS for 30 min at room
temperature. The nuclei were stained for 5 min at room temperature
with DAPI. The slides were rinsed with PBS and observed using a
confocal microscope and 3 fields for analyzed per sample (Olympus
Corporation) (magnification, x600).
Immunohistochemistry for
identification of TLECs
Immunohistochemical identification analysis of TLECs
was performed using the avidin-biotin-peroxidase complex method.
TLECs were incubated on coverslips (2x104/ml) in a
12-well plate and were fixed with 4% paraformaldehyde for 20 min at
room temperature. The cells were blocked with 5% BSA (cat. no.
A1993; MilliporeSigma w/v) in PBS for 60 min at room temperature,
and then incubated overnight at 4˚C with monoclonal anti-mouse
VEGFR3 antibody (1:30) and then washed with PBS. Goat-anti-rat
HRP-immunoglobulin (cat. no. ab97057; Abcam) was then added to the
sections for 30 min at 37˚C. After the sections were washed with
PBS, peroxidase-conjugated avidin (Dako; Agilent Technologies,
Inc.) was applied. The peroxidase activity was detected by exposing
the sections to a 3,3'-diaminobenzidine solution for 10 min at room
temperature. The sections were counterstained with hematoxylin at 5
min at room temperature and analyzed using an inverted light
microscope (Olympus Corporation) (magnification, x100).
Cell transduction
The primary TLECs were cultured in 24-well tissue
culture plates or flasks at 37˚C with 5% CO2 in a
humidified incubator (Heraeus Group). For the lentiviral infection,
the cells (105/ml/well; 200 µl) were incubated with
lentivirus (MOI=40) when the cells were 70% confluent, according to
the preliminary experiment. The cells were infected for 72 h for
the subsequent experiments. The package system was a four-plasmid
system, including LV5-GFP/LV3-GFP, PG-p1-VSVG, PG-P2-REV and
PG-P3-RR, purchased from Wester Biological Technology Co., Ltd. The
results of transduction were detected using a confocal microscope
(Olympus Corporation; magnification, x100).
Reverse transcription-quantitative
(RT-q)PCR
For RT-qPCR, total RNA was isolated from transduced
TLECs using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). First-strand cDNA synthesis was performed via RT
using a Transcriptor First Strand cDNA synthesis kit (Amersham;
Cytiva) according to the manufacturer's instructions. qPCR was
performed on an ABI PRISM 7700 Sequence Detection system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) using a QuantiTect SYBR
Green kit (Qiagen, Inc.). The primers were designed using Primer
Express 3.0 software (Applied Biosystems; Thermo Fisher Scientific
Inc.), as identified using Basic Local Alignment Search Tool
(http://www.ncbi.nlm.nih.gov/BLAST),
and were purchased from Invitrogen (Thermo Fisher Scientific,
Inc.). Each sample was run in triplicate. The conditions for the
qPCR reaction were as follows: One cycle at 50˚C for 15 min; one
cycle at 94˚C for 4 min; and 35 cycles at 94˚C for 20 sec, 60˚C for
30 sec and 70˚C for 35 sec. At the end of the PCR reaction, the
samples were subjected to a melting curve analysis to confirm the
specificity of the amplification. The Sema4C primer sequences were:
Forward, 5'-CCTCCCATCTGTATGTCTGCG-3' and reverse,
5'-GCTGGGTCATATGGGCATTTAC-3'. E-cadherin, forward
5'-TCCTTGGGTGGTCTTGAT-3' and reverse 5'-CATATCGCTGTTCTTCGTT-3'; ERK
1/2 forward, 5'-TCACACAGGGTTCTTGACAG-3' and reverse
5'-GGAGATCCAAGAATACCCA-3'. For the internal standard, the following
β-actin primer sequences were used: Forward,
5'-GAGACCTTCAACACCCCAGC-3' and reverse, 5'-ATGTCACGCACGATTTCCC-3'.
The method of quantification used was 2-ΔΔCq (22).
Immunoblotting
For western blot analyses, the TLEC cells treated
with lentivirus or/and PD98059 were lysed in RIPA buffer (cat. no.
P0013C; Beyotime Institute of Biotechnology) [50 mM Tris/HCl, pH
7.2, 150 mM NaCl, 1% NP-40, 0.1% SDS and 0.5% (w/v) sodium
deoxycholate]. Equivalent amounts of the cell extracts (BCA method,
20 µg) were separated via 10% SDS-PAGE and transferred onto a PVDF
membrane. The membranes were blocked in 25 mM Tris (pH 8.0)
containing 125 mM NaCl, 0.1% Tween-20 and 5% skimmed milk for 1 h
at room temperature, and then incubated with the diluted primary
antibodies (Sema4C, 1:500; E-cadherin, ERK1/2 and p-ERK1/2, all
1:2,000; β-actin, 1:1,000) at 4˚C overnight. After incubation with
the primary antibodies, the alkaline phosphatase-conjugated
anti-rabbit (cat. no. A9919; Sigma-Aldrich; Merck KGaA) and
anti-mouse (cat. no. A4312; Sigma-Aldrich; Merck KGaA) were added
at a 1:1,000 dilution. The immunoreactive bands were visualized
using the ECL western blotting technique. The software used for
densitometry was Image J v.1.8.0 (National Institutes of
Health).
Migration assay
For the migration assay, 5x103 TLEC
cells, treated with lentivirus or/and PD98059 were seeded in 100 µl
EBM-2 media with 1% FBS on the top of polyethylene terephthalate
membranes within Transwell cell culture inserts (24-well inserts;
pore size, 8 µm; Corning, Inc.). The bottom chamber was filled with
600 µl EBM-2 media containing 2% FBS and 100 µl supernatant from
NIH3T3 cells (mouse embryonic fibroblast cell line) to act as a
chemotactic factor. The cells were incubated for 48 h at 37˚C with
5% CO2. Subsequently, the cells were fixed in 2.5% (v/v)
glutaraldehyde for 20 min at room temperature and stained for 15
min with crystal violet at room temperature. Cells on the bottom
were visualized under an inverted light microscope (Leica
MicroSystems GmbH) and were quantified by counting the number of
cells in three randomly selected fields at a x100 magnification
(Olympus Corporation).
Statistical analysis
Data are presented as the mean ± SEM, and
statistical analysis was performed using SPSS version 13.0 software
(SPSS, Inc.). The experiments were repeated three times.
Statistical comparisons were performed using one-way ANOVA followed
by Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Separation of TLECs from a mouse
xenograft cervical tumor model
Our previous study detected the co-localization of
Sema4C and LYVE1 in human breast carcinoma (16). Firstly, the present study determined
the expression level of Sema4C in TLECs in mouse cervical tumor
tissue. Using a confocal microscope, LYVE1 was used to detect
lymphatic vessels, and co-localization (merge) was identified
between Sema4C (green) and LYVE1 (red; Fig. 1A). Primary TLECs were used to mimic
the entire TME of cervical tumors, including the immune and
proliferative conditions, as well as the interactions among cells,
using C57BL/6 mice with U14 tumor xenografts. U14 cells were
injected subcutaneously into the left shoulder, and LMVD was
examined using LYVE1 immunohistochemistry (Fig. 1B). As determined via LMVD analysis,
when the tumor grew to 1.0-1.5 cm3 [long diameter
(length), 1.4-1.8 cm; short diameters (width), 1.2-1.8 cm], ~1 week
after injection, the lymphatic vessel density notably increased,
compared with the 5 days group (Fig.
1C). After 14 days, massive necrosis occurred, and the density
of lymphatic vessels was significantly decreased, compared with the
7 days group. Therefore, the appropriate time for the isolation of
TLECs was considered to be day 7 after tumor cell injection.
Moreover, LYVE1-positive cells were separated via flow cytometry
(Fig. 1D). The cells were cultured
successfully and appeared as a typical monolayer (Fig. 1E). VEGFR3 was used to identify the
separated cells (Fig. 1F), and it
was observed that the majority of the cells were positive for
VEGFR3. Thus, the separated cells were positive for both LYVE1 and
VEGFR3.
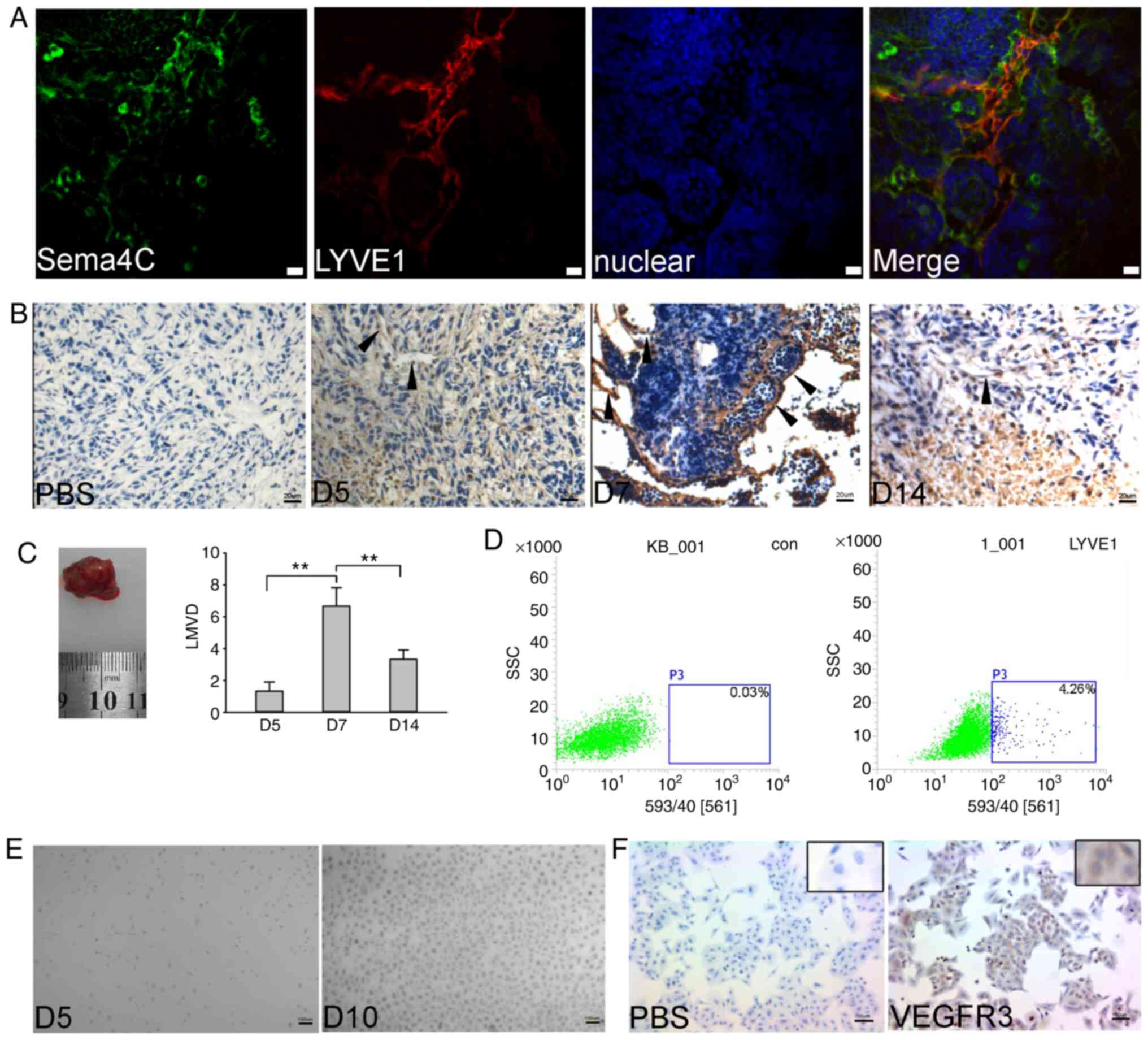 | Figure 1TLECs isolated from a mouse xenograft
tumor. (A) Localization of Sema4C was detected in mouse tumor
tissue. In lymphatic vessels, Sema4C (green) was co-localized with
LYVE1 (red). Nuclei were identified via DAPI staining (blue). Scale
bars, 20 µm. (B) LYVE1 immunohistochemical analysis indicated that
day 7 after U14 cell injection was the most suitable time for
isolation of TLECs. Arrowheads demonstrate TLEC positive staining.
Scale bars, 20 µm. (C) Tumor image indicated the tumor size at day
7 after injection, and quantification of the numbers of lymphatic
vessels on day 5, 7 and 14 was performed. (D) Tumor tissues were
dissociated into a cell suspension and then stained for
LYVE1-phycoerythrin. LYVE1-positive cells were separated via flow
cytometry. The control group was used non-specific IgG. (E) TLECs
were cultured in EBM-2 medium for 5 and 10 days. Scale bars, 10 µm.
(F) VEGFR3-positive TLECs were identified via immunohistochemistry.
Scale bars, 100 µm. The experiments were repeated three times.
**P<0.01. TLECs, tumor-associated lymphatic
endothelial cells; LYVE1, lymphatic vessel endothelial hyaluronan
receptor 1; Sema4C, semaphorin 4C; LMVD, lymphatic microvessel
density; SSC, side-scattered light; con, control. |
Sema4C regulates the migratory ability
of TLECs
The expression levels of the target gene Sema4C were
altered via lentiviral infection, and non-infected cells were used
as blank controls (Fig. 2A).
LV3-siRNA and full-length LV5-Sema4C, as well as the control
lentiviruses LV3NC and LV5NC, all expressed green fluorescent
protein (GFP) at a MOI of 40 at 72 h after transduction. The
efficiency of the silencing lentivirus and the overexpression
lentivirus infection was ~90%, according to GFP expression assessed
via fluorescence microscopy. The qPCR results demonstrated a ~70%
reduction and a 3.5-fold increase in the Sema4C mRNA expression
levels in the LV3-siRNA and LV5-Sema4C groups, respectively,
compared with the control (Fig.
2B), both of which were statistically significant.
Subsequently, the effects of Sema4C on the invasiveness of the
TLECs were examined using a classical Transwell model. The number
of cells at the bottom of the membrane, which reflects the
migration of cells, was 56±9, 52±8, 51±6, 40±8 and 73±13 for the
control, LV3NC, LV5NC, LV3-siRNA and LV5-Sema4C groups,
respectively. Cells at the bottom of the membrane were
significantly reduced in the LV3-siRNA compared with the LV3NC
group, and significantly increased in the LV5-Sema4C compared with
the LV5NC group (Fig. 2C). Thus,
silencing of Sema4C inhibited, and overexpression of Sema4C
induced, the migratory ability of TLECs.
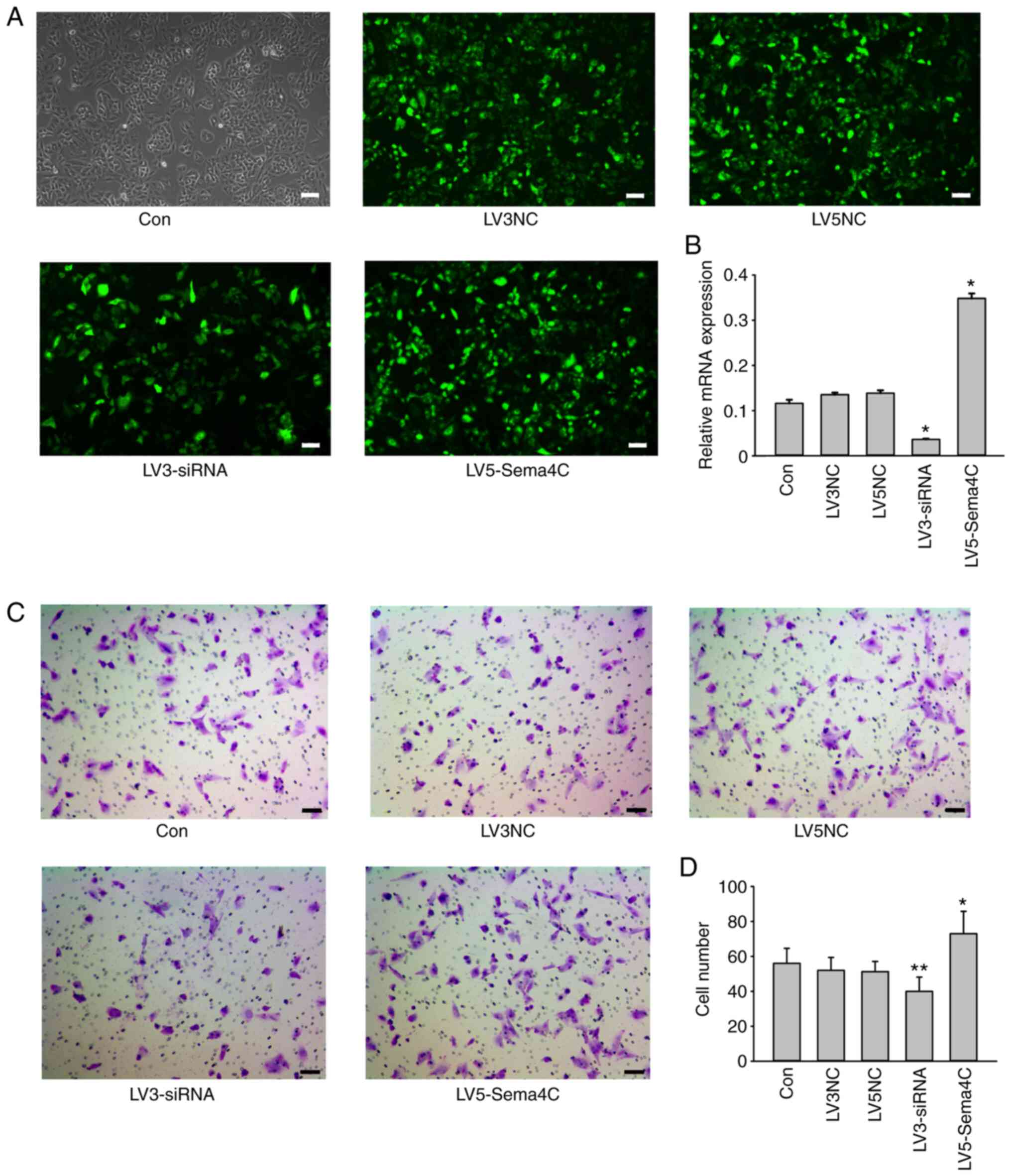 | Figure 2Role of Sema4C expression in the
migratory ability of TLECs. (A) TLECs treated with lentiviral
medium only, lentiviral control vector for Sema4C siRNA, lentiviral
control vector for full-length Sema4C, Sema4C siRNA and full-length
Sema4C. Scale bars, 10 µm. (B) TLECs with LV3-siRNA or LV5-Sema4C
were generated, and Sema4C mRNA expression was measured via reverse
transcription-quantitative PCR. (C) Migratory ability of cells was
assessed using a Transwell assay. (D) Quantification of the number
of cells at the bottom of the Transwell chamber. The experiments
were repeated three times. *P<0.05,
**P<0.01 LV3-siRNA group vs. LV3NC group; LV5-Sema4C
group vs. LV5NC group. Sema4C, semaphorin 4C; siRNA, small
interfering RNA; NC, negative control; TLECs, tumor-associated
lymphatic endothelial cells. |
Sema4C regulates E-cadherin and ERK1/2
in TLECs
Based on the previously reported literature
(10), a possible signaling pathway
(Sema4C-E-caherin) underlying cell migration was investigated. It
was revealed that Sema4C overexpression significantly inhibited the
expression level of E-cadherin in the LV5-Sema4C group compared
with LV5NC, whereas silencing of Sema4C significantly promoted its
expression in the LV3-siRNA group compared with the LV3NC group.
Moreover, the expression level of p-ERK1/2, a protein
phosphorylation of ERK1/2 which constitutes the active form of
ERK1/2, was significantly altered after lentiviral infection of the
Sema4C full-length gene or Sema4C siRNA, compared with the LV5NC or
LV3NC group, respectively. p-ERK could not be tested at the RNA
level, so total ERK was tested at the RNA level to reveal the
regulation of Sema4C (Fig. 3A). At
the protein level, there were no significantly changes in total ERK
(Fig. 3B and C). Therefore, it was indicated that Sema4C
stimulated the phosphorylation of ERK. A similar pattern was
observed for mRNA and protein expression levels of Sema4C and
E-cadherin using RT-qPCR (Fig. 3A)
and western blotting (Fig. 3B and
C), respectively. Fig. 3D presents the p-ERK/ERK ratio in the
five groups, and it was indicated that the activation of ERK was
significantly reduced in the LV3-siRNA group compared with LV3NC,
while it was significantly enhanced in the LV5-Sema4C group
compared with LV5NC.
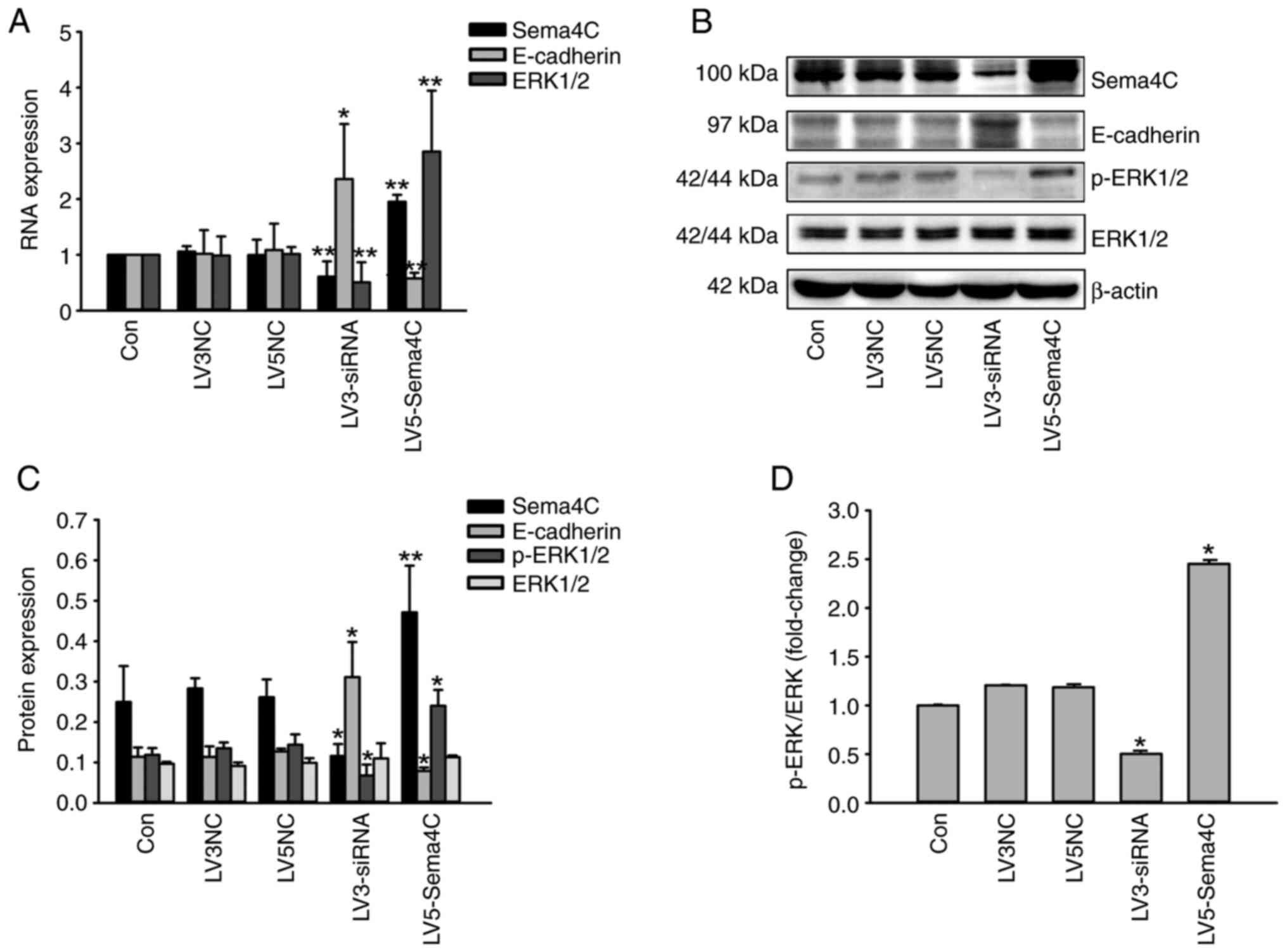 | Figure 3Role of Sema4C expression on
E-cadherin and ERK1/2 expression of tumor-associated lymphatic
endothelial cells. (A) Assessment of the mRNA expression levels of
Sema4C, E-cadherin and ERK1/2 using reverse
transcription-quantitative PCR. (B) Protein expression levels of
Sema4C, E-cadherin, p-ERK1/2 and total ERK1/2 were detected using
western blot analysis. (C) Semiquantitative analysis of protein
expression levels of Sema4C, E-cadherin, p-ERK1/2 and total ERK1/2.
(D) Expression of p-ERK1/2 was normalized to total ERK1/2, and
represented as fold change over the control group. The experiments
were repeated three times. *P<0.05 LV5-Sema4C group
vs. LV5 NC group, **P<0.01 LV3-siRNA group vs. LV3NC
group. Sema4C, semaphorin 4C; p, phosphorylated; siRNA, small
interfering RNA; NC, negative control. |
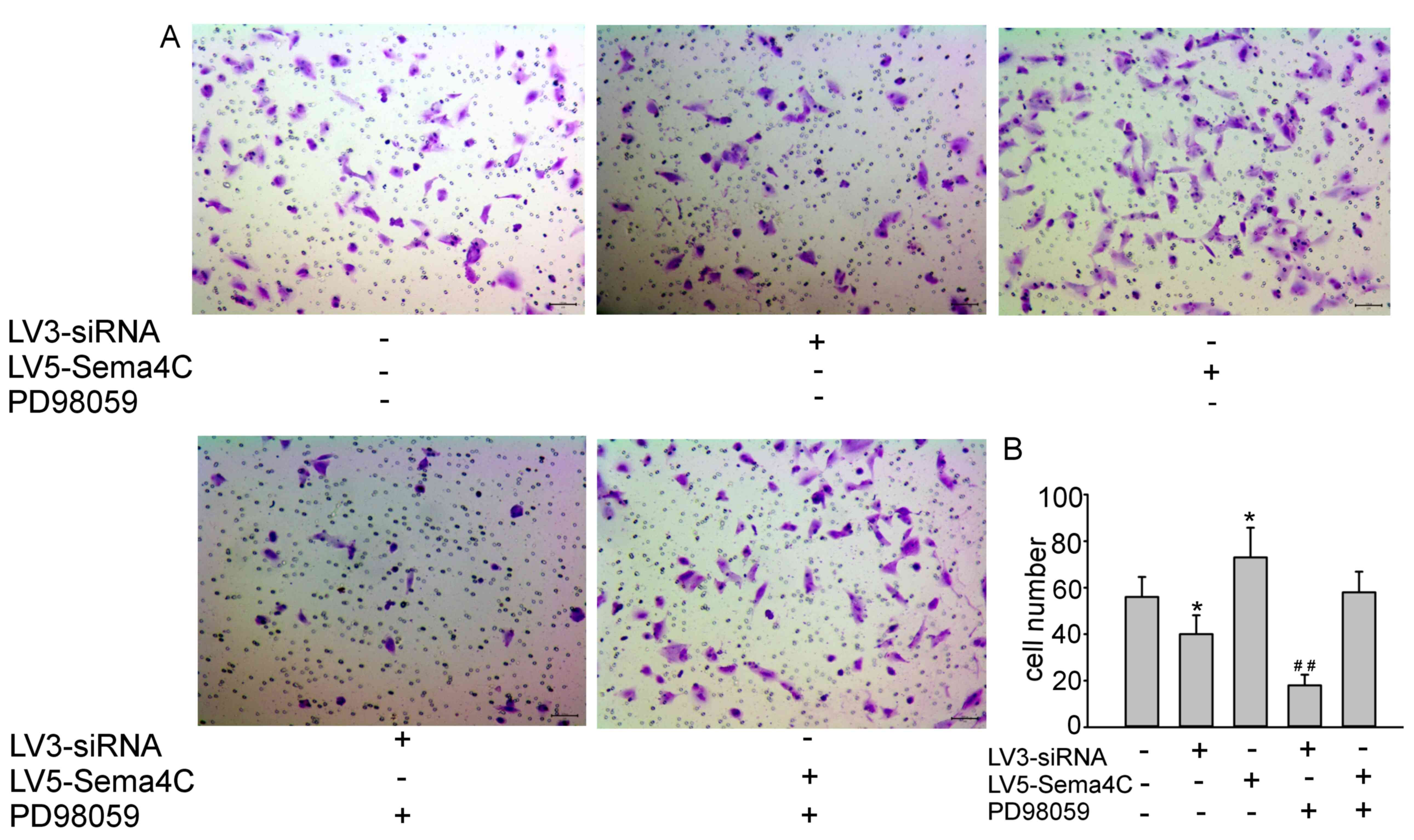
Sema4C regulates migration in TLECs
via ERK activation
It was hypothesized that Sema4C regulated EndMT
partially via the regulation of E-cadherin, which is one of the
most important characteristics of EndMT. The ERK inhibitor PD98059
(30 mM for 24 h at room temperature) was employed to determine the
alterations in the cell migratory ability following Sema4C
silencing or overexpression via lentiviral infection. The results
demonstrated that PD98059 enhanced the inhibition of cell migration
induced by Sema4C siRNA compared with the control group (Fig. 4A). Furthermore, the addition of
PD98059 reversed the promotion of TLEC migration induced following
overexpression of Sema4C (Fig. 4A).
The cell numbers at the bottom of the membrane, as presented in the
bar graph, were 56±9, 40±8, 73±13, 18±5 and 58±9 for control,
LV3-siRN-, LV5-Sema4C, LV3-siRNA + PD98059 and LV5-Sema4C + PD98059
groups, respectively, reflecting the migration of the cells
(Fig. 4B).
ERK inhibitor stimulates E-cadherin
expression by blocking Sema4C
The ERK inhibitor was used to determine whether the
Sema4C-mediated regulation of E-cadherin was dependent on the
phosphorylation level of ERK1/2. It was revealed that Sema4C
silencing significantly inhibited the phosphorylation level of ERK
compared with control group (no treatment), and the introduction of
PD98059 further enhanced the upregulation of E-cadherin expression
induced by Sema4C siRNA infection, compared with the LV3-siRNA
group. However, inhibition of the phosphorylation of ERK1/2
partially reversed the downregulation of E-cadherin that was
mediated by Sema4C overexpression, compared with the LV5-Sema4C
group. The observed mRNA expression levels of Sema4C and E-cadherin
were in accordance with the protein expression levels (Fig. 5A, B
and D-F). As the phosphorylation of
ERK1/2 is indicative of the regulation of ERK1/2 at the protein
level, the mRNA expression levels of ERK1/2 were also measured
(Fig. 5C). Recovery of p-ERK1/2
expression was observed in the LV5-Sema4C + PD98059 group compared
with the LV5-Sema4C group, indicating that PD98059 can effectively
block the function of Sema4C (Fig.
5D and G). Moreover, ERK1/2
mRNA expression was significantly increased following Sema4C
overexpression, but ERK1/2 protein expression did not change
significantly, compared with the control group (Fig. 5C and H), the alteration of p-ERK/ERK ratio
showed the same alteration (Fig.
5I). Of note, the protein expression level of p-ERK1/2
exhibited the same tendency as the ERK1/2 mRNA expression. Thus,
the phosphorylation level of ERK1/2 was an effective modification
in the Sema4C signaling pathway.
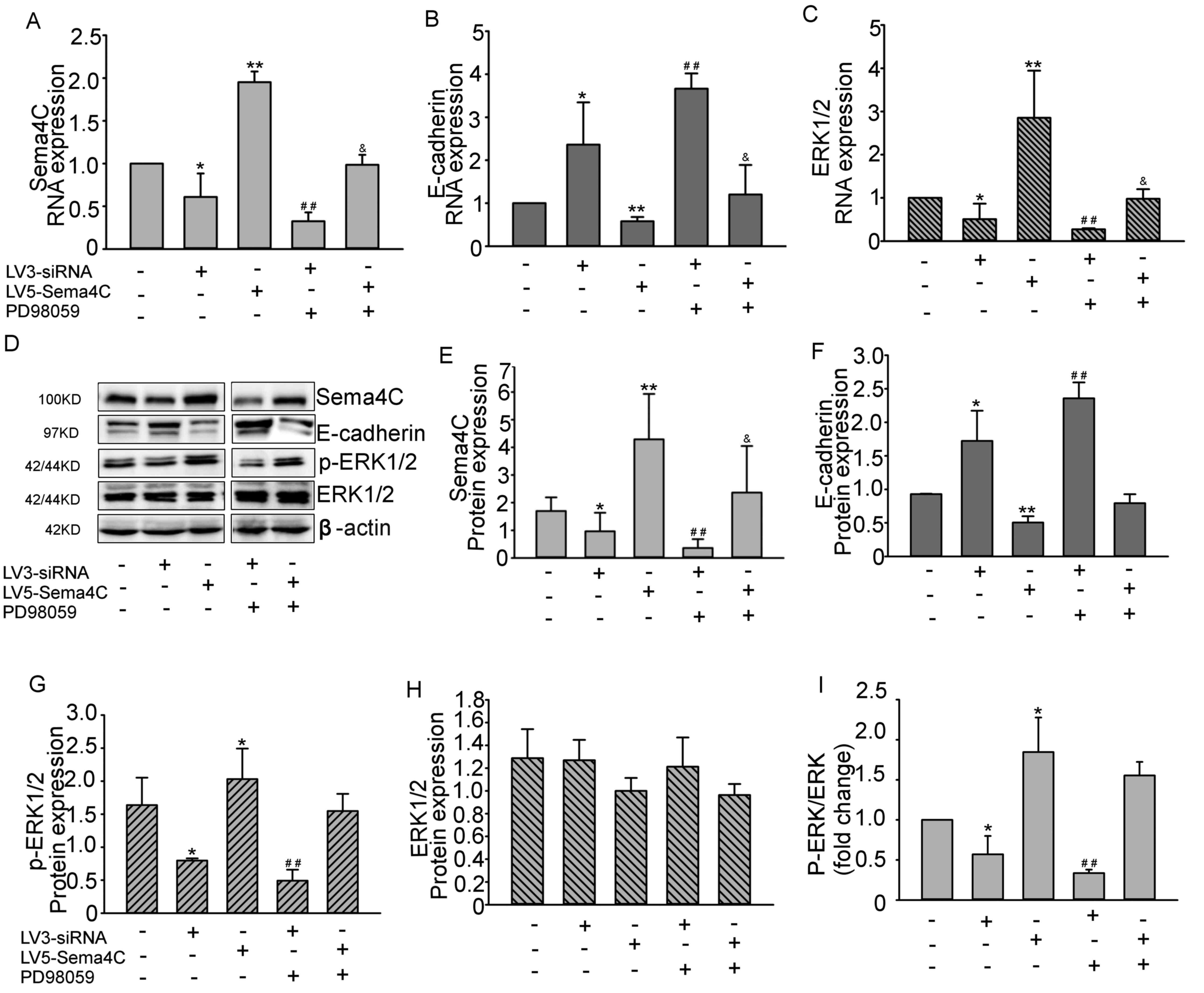 | Figure 5Sema4C regulates E-cadherin
expression via the ERK pathway. mRNA expression levels of (A)
Sema4C, (B) E-cadherin and (C) total ERK1/2 in TLECs treated with
lentiviral medium only (control group), LV3-siRNA, LV5-Sema4C,
LV3-siRNA + PD98059 and LV5-Sema4C + PD98059. (D) Protein
expression levels of Sema4C, E-cadherin, p-ERK1/2 and total ERK1/2
in TLECs treated with lentiviral medium only, LV3-siRNA,
LV5-Sema4C, LV3-siRNA + PD98059 and LV5-Sema4C + PD98059 detected
using western blot analysis. Semiquantitative analysis of protein
expression levels of (E) Sema4C, (F) E-cadherin, (G) p-ERK1/2 and
(H) total ERK1/2. (I) Expression of p-ERK1/2 was normalized to
total ERK1/2, and represented as fold change over the control
group. The experiments were repeated three times.
*P<0.05, **P<0.01 vs. control group;
##P<0.01 vs. LV3-siRNA group;
&P<0.05 LV5-Sema4C group. Sema4C, semaphorin 4C;
p, phosphorylated; siRNA, small interfering RNA; TLECs,
tumor-associated lymphatic endothelial cells. |
Discussion
Tumor-associated lymphangiogenesis is a key
modulator of tumor metastasis, although the underlying mechanism
remains unknown. It was previously reported that the lymphatic
system actively participates in tumor metastasis (23), and that LECs serve important roles
in inducing immune tolerance (24).
The ability of LECs to promote immunosuppression may induce tumor
cell metastasis (24). A previous
study using in situ laser capture microdissection revealed
that the gene expression profile of human tumor LECs differed from
that of normal LECs, and demonstrated that Sema4C was
differentially expressed between LECs in tumor and normal tissues
(16). The present study focused on
the effect of LECs in attaining the cell invasive ability, which
was attributed, at least in part, to the higher expression of
Sema4C in tumor LECs.
There seem to be shared mechanisms between EMT and
EndMT, and E-cadherin appeared in both of them (5,25). It
has been reported that EndMT occurs in cancer and tissue fibrosis
(26), but the regulation of this
process requires further investigation. In addition, whether and
how LECs participate in EndMT to induce tumorous characteristics
during cancer development is yet to be elucidated. The biological
characteristics of LECs markedly change in tumors in vivo
compared with normal tissues (17).
Thus, in the present study, TLECs were isolated from mouse cervical
tumor tissues via flow cytometry. LYVE1 was used as the marker for
LEC separation (19), and VEGFR3 as
an identification marker (27).
It has been reported that certain factors exhibit
differences in their molecular mechanism between their membrane and
soluble forms. For example, full-length Sema3C is a tumor
angiogenesis inhibitor, whereas cleaved Sema3C is a tumor
progression promoter (28). A
previous study has revealed that biological properties differed
between the soluble and membrane forms of Sema4C (sSema4C and
mSema4C, respectively). For instance, sSema4C promoted
lymphangiogenesis, whereas mSema4C directed cell-cell contacts,
thereby providing the possibility of a new mechanism of mSema4C
functioning in LECs (16).
Therefore, the current study used lentiviral transfection for
consistent overexpression or silencing of Sema4C in TLECs.
In a previous study, Ras homolog family member A
(RhoA) was identified to be critical for Sema4C-mediated signaling
(17). One of the distinct cell
migration models, which is named amoeboid-type migration, is
characterized by a spherical or elongated cell shape and is
strongly dependent on Rho kinase activity (29). Thus, it was suggested that LECs
migrate and undergo EndMT via amoeboid-type migration, which could
be promoted by Sema4C, as indicated by the Transwell assay. A
spherical cell shape with a small number of short protrusions was
revealed in the cells after passing through the polyethylene
terephthalate membranes (29). Wu
et al (10) reported a p38
MAPK-dependent effect of Sema4C in terminal myogenic
differentiation, including ERK regulation, indicating a potential
signaling pathway of Sema4C. The present study revealed that the
promotion of the migratory ability of Sema4C-overexpressing LECs
was in part attributed to ERK activation-induced repression of
E-cadherin expression.
E-cadherin is a cell adhesive molecule that serves a
key role in cellular adhesion and migration, constitutes one of the
most important players in EndMT and can also be regulated by RhoA
(30). Previous studies reported a
novel role for E-cadherin in regulating LEC progeny in newly
synthesized lymphatic vessels (28,31).
In particular, forced disruption of E-cadherin-mediated
intercellular adhesion has been indicated to open the intercellular
junctions in LEC monolayers, as determined by a previous study
using Transwell assays (32). The
present study demonstrated that repression of E-cadherin was an
important molecular event for the promotion of the migratory
ability in Sema4C-overexpressing LECs. This finding was in line
with that of a previous study, which revealed that the
overexpression of Sema4C suppressed E-cadherin, induced vimentin
and promoted fibronectin secretion in human kidney cells (33). While the current results indicated
that E-cadherin was an important molecular target of Sema4C in
EndMT function, the downstream signaling pathways that were
impacted by the loss of E-cadherin expression and promoted the cell
migratory ability, along with the overexpression of Sema4C, are yet
to be determined.
The expression of E-cadherin is regulated by an
ERK-dependent mechanism (34,35).
Therefore, it will be interesting to determine whether the
regulation of the Sema4C-mediated E-cadherin expression and
migratory ability in LECs are activated by ERK. We focused on
protein phosphorylation, a post-translational modification (PTM)
with a prevalent role in the control of protein activity and signal
transduction) (36). p-ERK could
not be tested at the RNA level, so total ERK was tested at the RNA
level to reveal the regulation of Sema4C in the present study. The
application of an ERK inhibitor demonstrated that Sema4C regulated
E-cadherin, and this process was dependent on ERK activation. It
was revealed that the application of an ERK inhibitor could also
affect the expression levels of Sema4C, and therefore, further
studies are required to clarify whether other molecules are
involved in this pathway and the feedback loop. Wei et al
(16) revealed that Sema4C could be
expressed in TLECs and promote the proliferation and migration of
tumor cells by activating plexin-B2/MET signaling; however, the
study by Wei et al (16)
lacked molecular investigation in primary LECs. A previous study on
primary lymphatic endothelial cells has demonstrated that there are
differences between cultured cells and cells (37). Therefore, it is important to
understand the molecular characteristic of primary TLECs.
Subsequent studies should focus on additional
molecular markers of EndMT in LECs altered by Sema4C, as well as
the cell morphology alterations, such as cytoskeletal changes, to
demonstrate the role of Sema4C in regulating EndMT. Also, the
therapeutic effect of lentivirus-mediated inhibition should be
examined.
In conclusion, the present study demonstrated that
Sema4C was expressed in TLECs via co-localization with LVYE1, and
identified that upregulation of Sema4C was an important molecular
mechanism that contributed to the induction of tumor-like
characteristics by suppressing E-cadherin expression.
Mechanistically, the process involved the activation of the ERK
pathway, which functioned upstream of E-cadherin and downstream of
Sema4C. Studies on TLECs are limited, and therefore, it will be
important to determine whether enhanced ERK activation during the
upregulation of Sema4C, as observed in TLECs, can induce tumor
lymphatic metastasis, which in turn could be targeted using
pharmacological approaches.
Supplementary Material
Lentiviral packaging systems. (A)
Four-plasmid system for LV5 lentivirus. (B) Four-plasmid system for
LV3 lentivirus.
Acknowledgements
The authors would like to thank Dr Qingliang Wang
(Department of Medical Affairs, Qilu Hospital, Cheeloo College of
Medicine, Shandong University, Jinan, China) for his assistance
with the statistical analysis of the present study.
Funding
Funding: The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81502238
and 81602286), the Department of Medical and Health Science
Technology of Shandong Province (grant no. 2016w0345), the
Department of Science Technology of Jinan City (grant no.
201705051), the China Postdoctoral Science Foundation (grant no.
2019T120594) and Natural Science Foundation General Project of
Shandong Province (grant no. ZR2020MH230).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HW designed the experiments and wrote the paper. JP
and XL conceived the experiments, performed the molecular
experiments and analyzed the data. CL established the mouse model,
performed the histological examination of tumor and separated the
cells. MG performed the statistical analysis. HW and JP confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
All the experiments were conducted in accordance
with the National Institute of Health Guide of the Care and Use of
Laboratory Animals. All the animal experimental protocols were
approved by the Animal Care and Use Committee of Shandong
University for Animal Ethics [approval no. KYLL-2015(KS)-079]. The
use of primary LECs was approved by the Scientific Research Ethics
Committee of Shandong University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Frumovitz M: Sentinel lymph node biopsy
for cervical cancer patients-what's it gonna take? Gynecol Oncol.
144:3–4. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Li H, Wu X and Cheng X: Advances in
diagnosis and treatment of metastatic cervical cancer. J Gynecol
Oncol. 27(e43)2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hoshino A and Lyden D: Metastasis:
Lymphatic detours for cancer. Nature. 546:609–610. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Lucas ED and Tamburini BAJ: Lymph node
lymphatic endothelial cell expansion and contraction and the
programming of the immune response. Front Immunol.
10(36)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Saito A: EMT and EndMT: Regulated in
similar ways? J Biochem. 153:493–495. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kourtidis A, Lu R, Pence LJ and
Anastasiadis PZ: A central role for cadherin signaling in cancer.
Exp Cell Res. 358:78–85. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang SH, Chang JS, Hsiao JR, Yen YC, Jiang
SS, Liu SH, Chen YL, Shen YY, Chang JY and Chen YW: Tumour
cell-derived WNT5B modulates in vitro lymphangiogenesis via
induction of partial endothelial-mesenchymal transition of
lymphatic endothelial cells. Oncogene. 36:1503–1515.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lee HR, Li F, Choi UY, Yu HR, Aldrovandi
GM, Feng P, Gao SJ, Hong YK and Jung JU: Deregulation of HDAC5 by
viral interferon regulatory factor 3 plays an essential role in
Kaposi's sarcoma-associated herpesvirus-induced lymphangiogenesis.
mBio. 9:e02217–17. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Jiang L, Hao C, Li Z, Zhang P, Wang S,
Yang S, Wei F and Zhang J: miR-449a induces EndMT, promotes the
development of atherosclerosis by targeting the interaction between
AdipoR2 and E-cadherin in Lipid Rafts. Biomed Pharmacother.
109:2293–2304. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wu H, Wang X, Liu S, Wu Y, Zhao T, Chen X,
Zhu L, Wu Y, Ding X, Peng X, et al: Sema4C participates in myogenic
differentiation in vivo and in vitro through the p38 MAPK pathway.
Eur J Cell Biol. 86:331–344. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Elder AM, Tamburini BAJ, Crump LS, Black
SA, Wessells VM, Schedin PJ, Borges VF and Lyons TR: Semaphorin 7A
promotes macrophage-mediated lymphatic remodeling during postpartum
mammary gland involution and in breast cancer. Cancer Res.
78:6473–6485. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Acker DWM, Wong I, Kang M and Paradis S:
Semaphorin 4D promotes inhibitory synapse formation and suppresses
seizures in vivo. Epilepsia. 59:1257–1268. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sakurai A, Doci CL and Gutkind JS:
Semaphorin signaling in angiogenesis, lymphangiogenesis and cancer.
Cell Res. 22:23–32. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Doçi CL, Mikelis CM, Lionakis MS, Molinolo
AA and Gutkind JS: Genetic identification of SEMA3F as an
antilymphangiogenic metastasis suppressor gene in head and neck
squamous carcinoma. Cancer Res. 75:2937–2948. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Nasarre P, Gemmill RM and Drabkin HA: The
emerging role of class-3 semaphorins and their neuropilin receptors
in oncology. Onco Targets Ther. 7:1663–1687. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wei JC, Yang J, Liu D, Wu MF, Qiao L, Wang
JN, Ma QF, Zeng Z, Ye SM, Guo ES, et al: Tumor-associated lymphatic
endothelial cells promote lymphatic metastasis by highly expressing
and secreting SEMA4C. Clin Cancer Res. 23:214–224. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Albini A, Mirisola V and Pfeffer U:
Metastasis signatures: Genes regulating tumor-microenvironment
interactions predict metastatic behavior. Cancer Metastasis Rev.
27:75–83. 2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Olmeda D, Cerezo-Wallis D,
Riveiro-Falkenbach E, Pennacchi PC, Contreras-Alcalde M, Ibarz N,
Cifdaloz M, Catena X, Calvo TG, Cañón E, et al: Whole-body imaging
of lymphovascular niches identifies pre-metastatic roles of
midkine. Nature. 546:676–680. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Young YK, Bolt AM, Ahn R and Mann KK:
Analyzing the tumor microenvironment by flow cytometry. Methods Mol
Biol. 1458:95–110. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Lukacs-Kornek V: The role of lymphatic
endothelial cells in liver injury and tumor development. Front
Immunol. 7(548)2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Faustino-Rocha A, Oliveira PA,
Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, da Costa RG,
Colaço B, Pires MJ, Colaço J, Ferreira R and Ginja M: Estimation of
rat mammary tumor volume using caliper and ultrasonography
measurements. Lab Anim (NY). 42:217–224. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Humbert M, Hugues S and Dubrot J: Shaping
of peripheral T cell responses by lymphatic endothelial cells.
Front Immunol. 7(684)2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ji RC: Lymph nodes and cancer metastasis:
New perspectives on the role of intranodal lymphatic sinuses. Int J
Mol Sci. 18(51)2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Potenta S, Zeisberg E and Kalluri R: The
role of endothelial-to-mesenchymal transition in cancer
progression. Br J Cancer. 99:1375–1379. 2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Cho JG, Lee A, Chang W, Lee MS and Kim J:
Endothelial to mesenchymal transition represents a key link in the
interaction between inflammation and endothelial dysfunction. Front
Immunol. 9(294)2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Harris AR, Perez MJ and Munson JM:
Docetaxel facilitates lymphatic-tumor crosstalk to promote
lymphangiogenesis and cancer progression. BMC Cancer.
18(718)2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mumblat Y, Kessler O, Ilan N and Neufeld
G: Full-length semaphorin-3C Is an inhibitor of tumor
lymphangiogenesis and metastasis. Cancer Res. 75:2177–2186.
2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lawson CD and Ridley AJ: Rho GTPase
signaling complexes in cell migration and invasion. J Cell Biol.
217:447–457. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lee G, Kim HJ and Kim HM: RhoA-JNK
regulates the E-cadherin junctions of human gingival epithelial
cells. J Dent Res. 95:284–291. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Connor AL, Kelley PM and Tempero RM:
Lymphatic endothelial lineage assemblage during corneal
lymphangiogenesis. Lab Invest. 96:270–282. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hou WH, Liu IH, Tsai CC, Johnson FE, Huang
SS and Huang JS: CRSBP-1/LYVE-1 ligands disrupt lymphatic
intercellular adhesion by inducing tyrosine phosphorylation and
internalization of VE-cadherin. J Cell Sci. 124:1231–1244.
2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhou QD, Ning Y, Zeng R, Chen L, Kou P, Xu
CO, Pei GC, Han M and Xu G: Erbin interacts with Sema4C and
inhibits Sema4C-induced epithelial-mesenchymal transition in HK2
cells. J Huazhong Univ Sci Technolog Med Sci. 33:672–679.
2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Marchese V, Juarez J, Patel P and
Hutter-Lobo D: Density-dependent ERK MAPK expression regulates
MMP-9 and influences growth. Mol Cell Biochem. 456:115–122.
2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Tang FY, Chiang EP, Chung JG, Lee HZ and
Hsu CY: S-allylcysteine modulates the expression of E-cadherin and
inhibits the malignant progression of human oral cancer. J Nutr
Biochem. 20:1013–1020. 2009.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kawata K, Yugi K, Hatano A, Kokaji T,
Tomizawa Y, Fujii M, Uda S, Kubota H, Matsumoto M, Nakayama KI and
Kuroda S: Reconstruction of global regulatory network from
signaling to cellular functions using phosphoproteomic data. Genes
Cells. 24:82–93. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wick N, Saharinen P, Saharinen J,
Gurnhofer E, Steiner CW, Raab I, Stokic D, Giovanoli P, Buchsbaum
S, Burchard A, et al: Transcriptomal comparison of human dermal
lymphatic endothelial cells ex vivo and in vitro. Physiol Genomics.
28:179–192. 2007.PubMed/NCBI View Article : Google Scholar
|