1. Introduction
Sodium/potassium-dependent ATPase
(Na+/K+-ATPase) is a ubiquitous protein
embedded between the phospholipid layers of the cell membrane
(1). When the levels of
intracellular Na+ or extracellular K+
increase, Na+/K+-ATPase becomes activated,
and its main function is to regulate the transmembrane transport of
Na+ and K+. In each cycle, the energy
released by hydrolyzing ATP can pump three Na+ out of
and two K+ into the cell. This transport across
membranes is essential for maintaining cellular resting potential
and adjusting the excitability of neurons (2,3).
In recent years, there has been ongoing research on
the functions of the Na+/K+-ATPase (4-6),
and the regulation of this enzyme appears to be influenced by
multiple factors through several pathways, among which hormones
play an important role in both short-term and long-term regulation
(Fig. 1). Although there have been
several previous studies on the regulatory effect of hormones on
Na+/K+-ATPase (7-10),
the mechanism underlying the regulatory effect of insulin on
Na+/K+-ATPase in various organs and the
related metabolic pathways have been attracting increasing
attention. The aim of the present study was to review the
structure, biochemical characteristics, function and metabolism of
Na+/K+-ATPase and, subsequently, to discuss
in depth all the possible mechanisms through which insulin may
regulate Na+/K+-ATPase and investigate the
possibility of new therapies for related diseases.
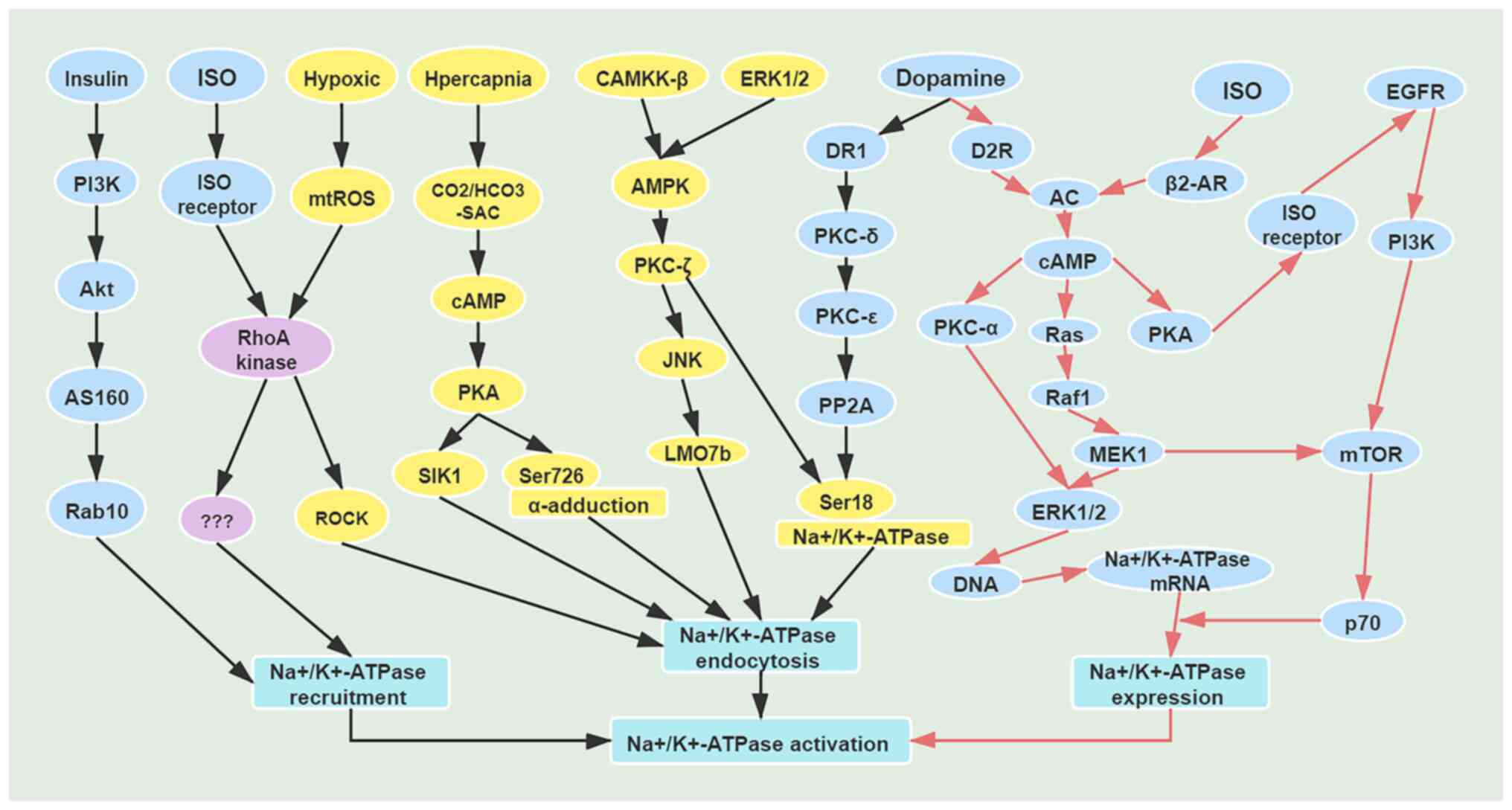 | Figure 1Mechanisms of short-term and
long-term regulation of Na+/K+-ATPase. Black
arrows indicate mechanisms of short-term regulation of
Na+/K+-ATPase; red arrows indicate mechanisms
of long-term regulation of Na+/K+-ATPase.
AS160, AKT substrate protein of 160 kDa; Rab10, Ras-related
GTP-binding protein; ISO, isoprenaline; RhoA, Ras homolog family
member A; mtROS, mitochondrial reactive oxygen species; ROCK,
Rho-associated kinase; PKA, protein kinase A; SIK1, salt-inducible
kinase 1; CAMKK-β, Ca2+/calmodulin-dependent protein
kinase β; AMPK, AMP-activated protein kinase; JNK, c-Jun N-terminal
kinase; LMO7b, LIM domain only protein 7-like; DR1, dopamine D1
receptor; D2R, dopamine D2 receptor; PP2A, protein phosphatase 2A;
AC, adenylate cyclase; Raf1, Raf-1 proto-oncogene; MEK1,
mitogen-activated protein kinase kinase 1. |
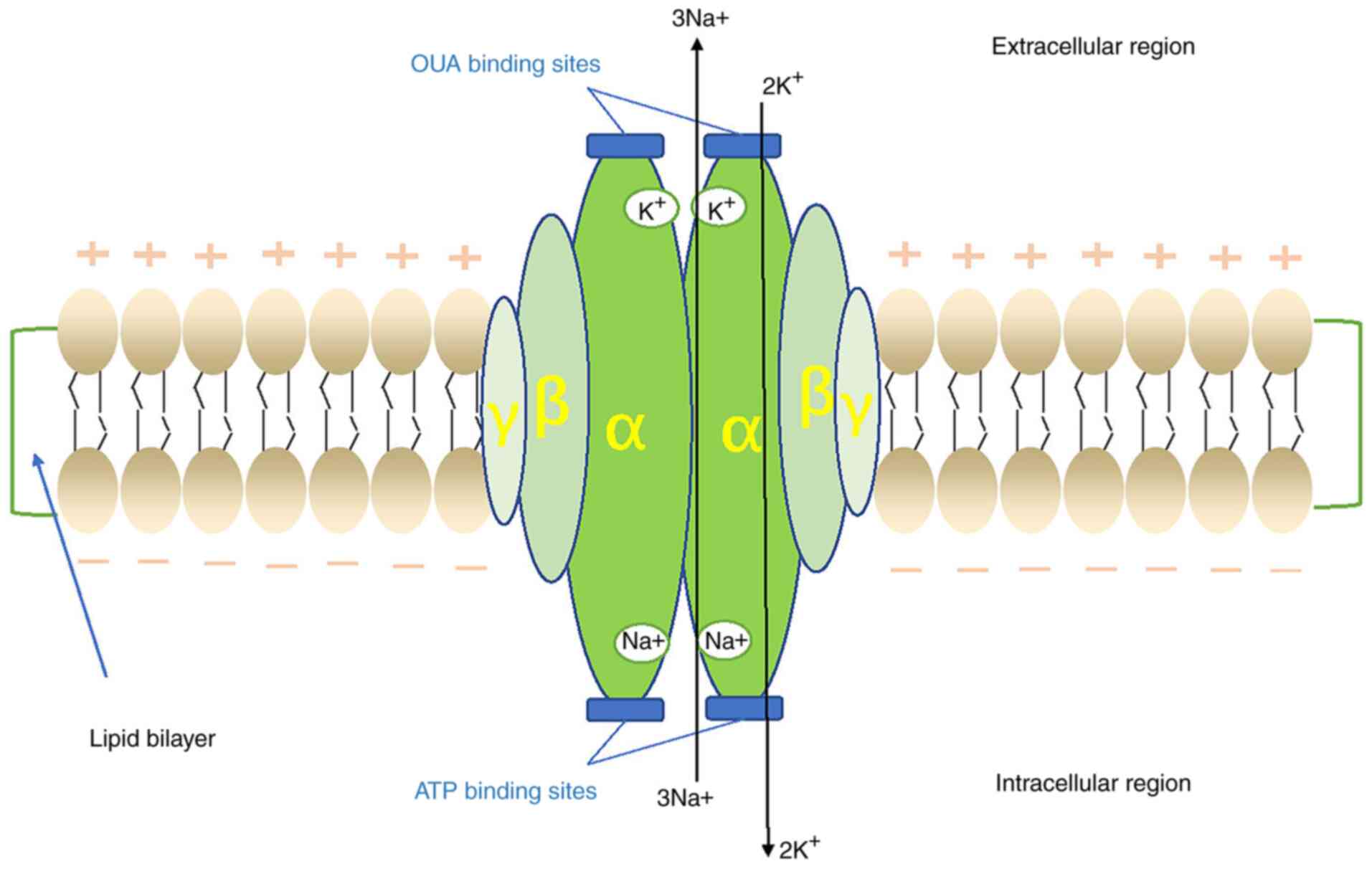
2. Structure and biochemistry of
Na+/K+-ATPase
Na+/K+-ATPase is a ubiquitous
enzyme consisting of three subunits, namely α-, β- and γ-subunits
(Fig. 2). The α-subunit is a
catalytic subunit composed of 10 transmembrane helices (m1-10) with
a total molecular mass of 110 kD, which has binding sites for
Na+ and K+ (11). Its main function is to transfer
Na+ out of the cells and K+ into the cells,
and it functions as an ATPase by hydrolyzing ATP (12). The α-subunit has four isoforms
(α1-4), which are differentially expressed in different tissues
throughout the development of the organism: α1 is widely expressed
in various tissues; α2 is mainly expressed in the brain, heart and
muscle; α3 is mainly distributed in the brain, retina and heart;
and α4 is mainly expressed in the testes (11,13,14).
The β-subunit consists of a transmembrane fragment
and a highly glycosylated extracellular domain with a molecular
mass of 55 kD, the role of which is regulatory, enabling the
α-subunit to accurately fold and translocate from the endoplasmic
reticulum (ER) to the plasma membrane (4), stabilizing the protein configuration
and regulating its activity on the plasma membrane (4,7,15).
The β-subunit has three isoforms (β1-3); among these, β1 is
distributed throughout all tissues, β2 is concentrated in nervous
tissue, heart, cartilage and erythrocytes, whereas β3 is found
predominantly in nervous tissue, as well as in skeletal muscle and
the lung (7,15).
The γ subunit has only one transmembrane domain with
a molecular mass of 15 kD, and it is a member of the FXYD protein
family, which has seven isoforms (FXYD1-7) (5,13).
Its function appears to be associated with the modulation of the
enzyme affinity for different ligands, with a direct and positive
effect on the maximum rate of ATP hydrolysis; thus, the γ-subunit
is also considered to be regulatory in addition to the β-subunit
(8,13,15-17).
Furthermore, relevant clinical trials have demonstrated that the
expression of FXYD1, 3 and 5 is upregulated in lung epithelial
cells of patients with acute respiratory distress syndrome (ARDS),
and FXYD5 is the key mediator (9),
whereas FXYD1, 6 and 7 are mainly expressed in brain tissue, where
they regulate the affinity between
Na+/K+-ATPase and substrate and the maximum
response rate (13).
3. Function of
Na+/K+-ATPase
Through pumping Na+ into and
K+ out of the cells, Na+/K+-ATPase
plays an important role in maintaining cells in a resting state by
preserving the balance of electrolytes and fluids, regulating the
active transport of carbohydrates, amino acids, bile acids,
neurotransmitters and ions, and regulating membrane potential, cell
volume, energy metabolism and signal transmission (3). Therefore, the abnormal regulation and
dysfunction of Na+/K+-ATPase may lead to
serious pathophysiological changes, and maintaining its stability
is crucial.
The mechanism through which cardiac glycosides
increase myocardial contractility is increasing intracellular
Na+ levels by inhibiting
Na+/K+-ATPase, then increasing intracellular
Ca+ levels through Na+-Ca+
exchange, ultimately enhancing myocardial contractility (10,11).
Consequently, Na+/K+-ATPase activity appears
to be closely associated with myocardial contractility.
In order to achieve pulmonary edema clearance in
ARDS, Na+ in the alveolar space enters the cells through
the epithelial Na+ channel in the apical membrane of
alveolar type I and alveolar type II (ATII) epithelial cells, and
then enters the pulmonary interstitium through the
Na+/K+-ATPase on the basal side of the cells,
forming a local osmotic pressure gradient, thus driving fluid
clearance from the alveolar space (18). Over a decade ago,
Na+/K+-ATPase downregulation was reported in
several acute lung injury models (19-22).
Current research is mainly focusing on the role of
Na+/K+-ATPase in promoting pulmonary edema
clearance. Disruption in the function of
Na+/K+-ATPase is likely to aggravate the
formation of pulmonary edema, which may be caused by limited
Na+ transport as well as disrupted alveolar barrier
function (19). Therefore,
regulating the function of Na+/K+-ATPase in
the alveolar epithelium is crucial for relieving pulmonary
edema.
4. Metabolic pathway of
Na+/K+-ATPase
Ubiquitin proteasome pathway (UPP) of
Na+/K+-ATPase
The UPP is a highly efficient protein decomposition
pathway, which has a wide range of biological functions and is
closely associated with several diseases. Ubiquitin must bind to
the relevant substrate proteins in the form of polyubiquitin chains
to label the substrate proteins for further degradation (23). The UPP system comprises ubiquitin,
E1 ubiquitin-activating enzyme, E2 ubiquitin-binding enzyme, E3
ubiquitin ligase, deubiquitinases (DUBs) and proteasome (24). During ubiquitin modification,
ubiquitin is first activated by E1 ubiquitin activator, and then
the activated ubiquitin binds to the E2 ubiquitin-binding enzyme.
Then, under the action of E3 ubiquitin ligase, ubiquitin is
transferred to the target protein to complete the process of
ubiquitination (25). E3 ubiquitin
ligase plays an important role in this process, which determines
the timing and specificity of ubiquitination.
In 1997, Coppi and Guidotti (26) first proposed the ubiquitination of
Na+/K+-ATPase as a possible regulatory
mechanism. It was reported that the αl and α2 isoforms of the
Na+/K+-ATPase α subunit are modified by the
covalent attachment of ubiquitin polymers in COS-7 cells (an
African green monkey kidney fibroblast-like cell line), and
polyubiquitination of the Na+/K+-ATPase α
subunit may play a role in regulating its degradation, namely by
promoting the ER-associated degradation of unassembled and
misfolded α subunits, and by participating in the internalization
and subsequent degradation of cell surface
Na+/K+-ATPase molecules (26). Moreover, the physiological effect
of ubiquitination on Na+/K+-ATPase was first
demonstrated in alveolar epithelial cells during hypoxia (27). Severe short-term hypoxia resulted
in the endocytosis and degradation of
Na+/K+-ATPase in alveolar epithelial cells,
whereas the phosphorylation of the protein kinase C
(PKC)ζ-dependent-Na+/K+-ATPase catalytic
subunit Ser-18 triggered the ubiquitination and endocytosis of
plasma membrane Na+/K+-ATPase (27). Furthermore, long-term exposure of
alveolar epithelial cells to hypoxia may lead to a decrease in
total Na+/K+-ATPase levels. Further studies
demonstrated that, if the four lysine residues on Ser-18
(KK18SKK) side are immediately mutated to arginine,
hypoxia-induced ubiquitination and endocytosis may be prevented
(22,27). Hypoxia-induced
Na+/K+-ATPase degradation may be prevented by
inhibiting its ubiquitination on the plasma membrane and treatment
with lysosome inhibitors, which indicates that
Na+/K+-ATPase is ubiquitinated on the plasma
membrane, but its degradation occurs in the lysosome (28). Accordingly,
Na+/K+-ATPase may serve as a vector for
ubiquitin-dependent intracellular transport.
The separation of the basolateral membrane revealed
the presence of Na+/K+-ATPase/ubiquitin
conjugates, suggesting that ubiquitination occurs on the plasma
membrane. However, when Ser-18 was mutated to alanine,
ubiquitination was inhibited, suggesting that phosphorylation may
be a prerequisite for ubiquitination. Therefore,
Na+/K+-ATPase appears to be regulated by a
mechanism involving phosphorylation, ubiquitination, recognition,
endocytosis and degradation (29).
Phosphorylation, as a signal that triggers ubiquitination, provides
a ‘pass check’ for endocytosis and degradation. Hoxhaj et al
(30) confirmed that the E3
ubiquitin ligases ZNRF1 and ZNRF2 are new participants in the
regulation of Na+/K+-ATPase. In human 293
cells, insulin was shown to increase the phosphorylation of ZNRF1
and ZNRF2, thereby increasing its binding to 14-3-3 and reducing
polyubiquitination modification of the
Na+/K+-ATPase α1 protein and subsequent
degradation by proteases (30).
Phosphorylation may alter the subcellular localization of proteins,
thus regulating the availability of interaction between the target
protein and E3 ligase (23). In
conclusion, the ubiquitination of
Na+/K+-ATPase is one of the metabolic
pathways of this enzyme (i.e., one of the scavenging pathways).
However, ubiquitinase or DUBs do not appear to regulate the
degradation of Na+/K+-ATPase itself or its
related pathway proteins. Therefore, the exact function and
underlying mechanism of DUBs in this field have yet to be fully
elucidated.
Autophagy-lysosomal pathway of
Na+/K+-ATPase
The autophagy-lysosomal pathway is a process through
which macromolecules, damaged organelles and residual metabolites
are transported to the lysosomes for degradation. During this
process, materials may be reused for biosynthesis or energy
production (31). Abnormalities in
the autophagic process may adversely affect the development of
various organs in the body. Few previous studies have mentioned
whether Na+/K+-ATPase actively participates
in autophagy or endosome cycling by altering the Na+ and
K+ contents. However, Na+ and K+
are all located in the α-subunit, which is considered as the
catalytic subunit (32,33). Therefore, the α subunit may be used
to investigate whether Na+/K+-ATPase is
actively involved in autophagy or endosome cycling by altering the
cell Na+ and K+ content. It has been
demonstrated that the association between
Na+/K+-ATPase and the autophagy-lysosomal
pathway requires the α1 subunit (34). Recently, it was also reported that
Na+/K+-ATPase α1 and AMP-dependent protein
kinase may represent the ‘on’ and ‘off’ states of the autophagic
pathway, respectively (34).
Na+/K+-ATPase α1 may serve as a new mechanism
of signal transduction and autophagy in the process of
ischemia/reperfusion, which may provide a new approach to
therapeutic intervention in ischemic stroke (32,33).
Importantly, Na+/K+-ATPase can
be degraded by the UPP and the autophagy-lysosomal pathway.
Sequestosome 1 (SQSTM1)/p62, is an autophagic protein (35), and defects in autophagy may lead to
the accumulation of SQSTM1 and may induce cell stress and disease
states. SQSTM1 regulates multiple signaling pathways by binding to
different proteins to form an important cellular signaling hub
(36). Thus, SQSTM1 is involved in
the UPP and autophagy-lysosomal degradation processes, and appears
to be an important regulatory molecule connecting ubiquitinated
proteins to the autophagy mechanism (37). Hancock et al (38) demonstrated that insulin
significantly reduced SQSTM1 mRNA expression. Proteomic analysis
demonstrated that Na+/K+-ATPase α1 was able
to bind with SQSTM1, which was verified by endogenous protein
interaction analysis. Therefore, the decreased expression of SQSTM1
mRNA is likely to reduce the transport of the polyubiquitinated
Na+/K+-ATPase α1 protein to the
autophagy-lysosomal system for degradation.
In conclusion, few studies on the degradation of
Na+/K+-ATPase through these two pathways have
been conducted to date, and the relationship between the two may
require further investigation in the future to fully elucidate the
role of Na+/K+-ATPase α1 abundance and
enzymatic activity.
5. Insulin-mediated regulation of ATPase
pumps
Insulin is a key hormonal factor that has been
extensively investigated in the context of various metabolic
disorders, such as obesity and non-insulin-dependent diabetes
mellitus, as well as cardiovascular disorders, such as essential
hypertension and atherosclerosis (39). The signaling pathways that are
activated by insulin are summarized in Fig. 3. Understanding the biochemical and
cellular properties of insulin receptor signalling constitutes a
priority in biomedical research (40). Borge et al (41) reported that the existence of a
novel positive-feedback pathway in which insulin may regulate
insulin secretion in the β-cells of the pancreas by interaction
between the insulin receptor substrate 1 (IRS-1) protein and
sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA). IRS-1
is present in the ER and can directly bind to the β-cell isoforms
of SERCA, specifically SERCA3b. Insulin stimulation results in
increased binding of IRS-1 to SERCA3b, which inhibits the
Ca2+-ATPase, increases cytosolic Ca2+, and
augments fractional insulin secretion. The rat pancreatic β-cell
expresses 6 splice variants of the plasma membrane
Ca2+-ATPase and two splice variants of the
Na+/Ca2+ exchanger 1. In the β-cell, the
Na+/Ca2+ exchanger displays a high capacity,
contributing to both Ca2+ outflow and influx, and
participating in the control of insulin release (42,43).
In addition, vacuolar-type ATPase (V-ATPase) is present in unique
organelles, such as insulin secretory granules, neural synaptic
vesicles and acrosomes of spermatozoa. Futai et al (44) reported that the V-ATPase α3 isoform
was found to be highly expressed in pancreatic Langerhans islets,
and ~80% of the isoform was shown to be localized to
insulin-containing secretory granules of pancreatic islet β-cells.
This evidence indicates that insulin may serve an important role in
ATPases.
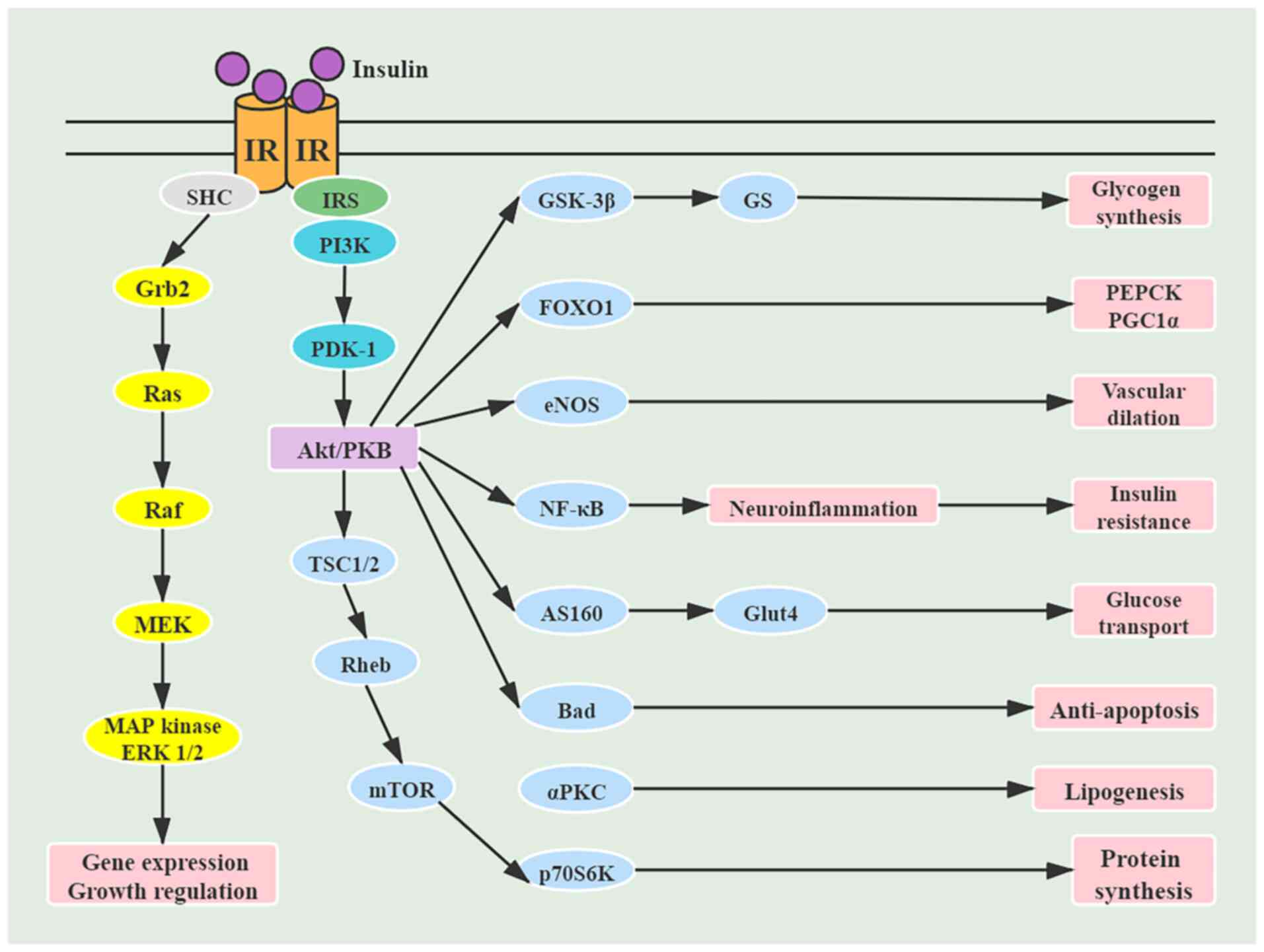 | Figure 3Signaling pathways activated by
insulin. IR, insulin receptor; IRS, insulin receptor substrate;
SHC, Src homology collagen protein; Grb2, growth factor
receptor-bound protein 2; PDK-1, protein
3-phosphoinositide-dependent protein kinase-1; TSC, tuberous
sclerosis complex; Rheb, Ras homolog enriched in brain; GSK,
glycogen synthase kinase; FOXO1, Forkhead box protein O1; eNOS,
endothelial nitric oxide synthase; AS160, AKT substrate protein of
160 kDa; PKC, protein kinase C; GS, glycogen synthase; Glut4,
glucose transporter 4; PEPCK, phosphoenolpyruvate carboxykinase;
PGC1, PPARγ-coactivator 1. |
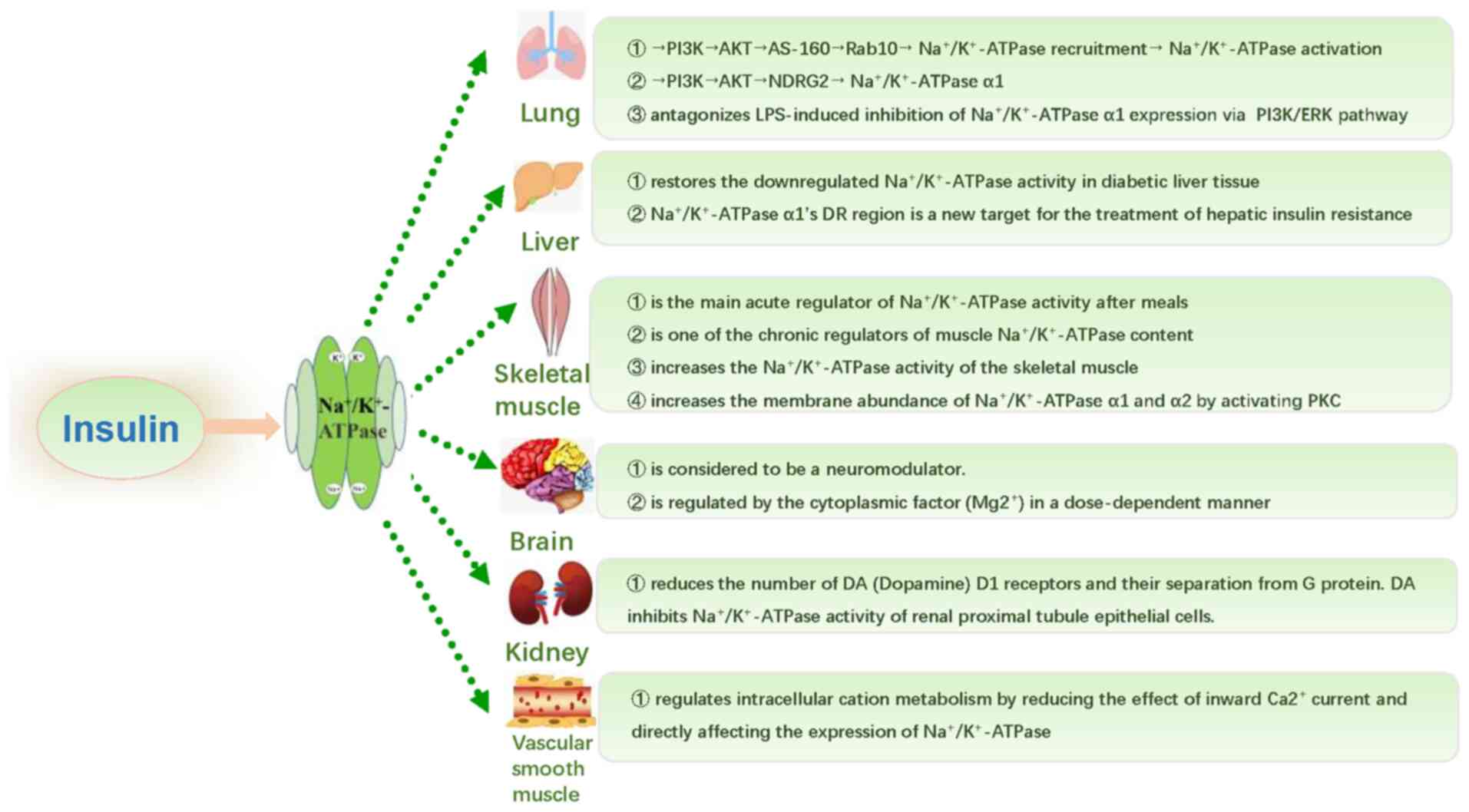
6. Regulatory effect of insulin on
Na+/K+-ATPase
It is well known that Na+ regulates
Na+/K+-ATPase in most mammalian cells
(45). Under insulin stimulation,
increasing Na+ influx may stimulate
Na+/K+-ATPase. There are tissue-specific
differences in this mechanism, which may be a receptor-mediated
process (46). As regards the
mechanism through which insulin regulates
Na+/K+-ATPase, it was found to promote a
relative abundance of Na+/K+-ATPase and
restore the transport efficiency in multiple in vitro and
vivo experiments (47-51).
However, in most studies, the effect of insulin on
Na+/K+-ATPase mainly manifests as short-term
regulation, i.e., only Na+/K+-ATPase α1 is
translocated to the plasma membrane and internalized into the
cytoplasm, and insulin changes its molecular conformation and
regulates the transport efficiency (47). Moreover,
Na+/K+-ATPase activity may also be subject to
long-term regulation, i.e., insulin may promote its transcription
and translation, or reduce degradation of the α1 subunit. The
mechanism underlying the regulation of
Na+/K+-ATPase by insulin in the human body is
summarized in Fig. 4.
Lung
Insulin can prevent or reduce lipopolysaccharide
(LPS)-induced acute lung injury in rats (48) and can also reduce the in-hospital
mortality of patients with ARDS (49). Insulin is considered to be of
potential therapeutic value in ARDS. However, the specific
mechanism through which it regulates the activity and relative
abundance of Na+/K+-ATPase on ATII cells
remains unclear (49). A549 cells,
which were isolated from human lung adenocarcinoma, have been
widely used as a model of ATII cells due to the presence of
lamellar bodies and surfactant proteins. A study on ATII and A549
cells reported that insulin increased
Na+/K+-ATPase activity and pulmonary edema
clearance by recruiting Na+/K+-ATPase to the
cell membrane of ATII epithelial cells within 5 min (50). This short-term regulation was
induced by phosphorylation of AKT substrate protein of 160 kDa
(AS160). It should be noted that the promoting effect of insulin
was found to be fully mediated via the PI3K/AKT pathway upstream of
AS160(50). AKT mediates
insulin-induced glucose transporter type 4 exocytosis by
phosphorylating AS160, which contains a Rab GTPase-activating
protein (GAP) domain (51). In
addition, Rab proteins switch between a GTP-bound active state and
GDP-bound inactive state; the phosphorylation of AS160 by AKT
inhibits GAP activity and stabilizes the Rab proteins in their
GTP-bound form, which has been described to promote vesicle
trafficking (52). Specifically,
they described that i) insulin increases
Na+/K+-ATPase abundance at the plasma
membrane; ii) the activation of AKT is both necessary and
sufficient to recruit Na+/K+-ATPase molecules
to the cell surface; and iii) the effects of AKT on
Na+/K+-ATPase are mediated in part by the
phosphorylation of AS160 and the activity of Rab10(52). Thus, AS160 may be a new substrate
of AKT.
Furthermore, insulin regulates the expression of
Na+/K+-ATPase α1 through the PI3K/AKT/N-Myc
downstream-regulated gene 2 protein and PI3K/ERK pathways. It has
been reported that insulin upregulates
Na+/K+-ATPase activity and α1 subunit protein
abundance on cell membranes through the PI3K pathway, which can
prevent the occurrence and development of ARDS and improve patient
prognosis (53,54). In recent years, it was also
reported that insulin increases Na+/K+-ATPase
α1 mRNA level and enzymatic activity (55). In our previous research, insulin
was found to antagonize LPS-induced inhibition of
Na+/K+-ATPase α1 expression via the PI3K/ERK
pathway. LPS induced a decrease in
Na+/K+-ATPase α1 protein levels in ATII
cells, while insulin significantly increased the protein levels of
Na+/K+-ATPase α1 inhibited by LPS for at
least 24 h (56). Therefore,
insulin appears to promote pulmonary edema clearance through
long-term regulation of Na+/K+-ATPase.
However, the specific mechanism remains to be further
investigated.
Liver
Obesity is associated with hyperglycemia and
hyperinsulinemia, which may inhibit or inactivate
Na+/K+-ATPase (57). In diabetic rats, the activity of
Na+/K+-ATPase in the liver was also found to
be decreased, and antidiabetic compounds were able to restore the
downregulated Na+/K+-ATPase activity in
diabetic liver tissue (58). It
was also recently demonstrated that
Na+/K+-ATPase α1 is a physiological regulator
of glucose homeostasis, and its DR region may represent a new
target for the treatment of hepatic insulin resistance (59). Therefore,
Na+/K+-ATPase α1 may be a key gene involved
in the homeostasis of glucose and lipid metabolism. However, the
role and exact underlying mechanism of
Na+/K+-ATPase α1 in obesity and insulin
resistance remain unclear.
Skeletal muscle
In skeletal muscle tissue, insulin is the main
acute regulator of postprandial Na+/K+-ATPase
activity (60). In addition, it is
one of the chronic regulators of muscle
Na+/K+-ATPase content. Insulin can
significantly increase the Na+/K+-ATPase
activity of skeletal muscle, thus reducing extracellular and
increasing intracellular K+ concentration. The
mechanisms for increasing Na+/K+-ATPase
transport to the plasma membrane include insulin regulation and
skeletal muscle contraction (60).
In cultured human skeletal muscle cells, insulin treatment was
found to increase the membrane abundance of the α1 and α2 subunits,
which required activation of PKC (61).
Brain
Insulin is considered to function as a
neuromodulator in the brain, and its inhibitory effect is regulated
by the cytoplasmic factor Mg2+ in a dose-dependent
manner (62).
Kidney
In kidney tissue, Banday et al (63) reported that long-term exposure to
insulin can reduce the number of dopamine (DA) D1 receptors and
their separation from G protein, which may be the mechanism through
which DA inhibits Na+/K+-ATPase activity in
renal proximal tubule epithelial cells in hyperinsulinemic
hypertensive rats.
Vascular smooth muscle
Vascular smooth muscle is a type of
insulin-insensitive tissue. Insulin regulates intracellular cation
metabolism by reducing the effect of inward Ca2+ current
and directly affecting the expression of
Na+/K+-ATPase (64).
Carcinogenesis
Na+/K+-ATPase has been
proposed as a signal transducer involved in various pathobiological
processes, including carcinogenesis (65). Lu et al (66) found upregulation of the mRNA
expression of the Na+/K+-ATPase α1, β1 and β3
subunits in hepatocellular carcinoma (HCC) using The Cancer Genome
Atlas (https://cancergenome.nih.gov/),
International Cancer Genome Consortium (https://icgc.org/daco) and Gene Expression Omnibus
(https://www.ncbi.nlm.nih.gov/gds)
databases, and indicated that Na+/K+-ATPase
β3 may serve as an oncogene; it was also demonstrated to be an
independent prognostic factor and was associated with immune cell
infiltration in HCC. In addition to its prominent metabolic action,
insulin has a well-known mitogenic effect, promoting proliferation
of both non-cancerous and malignant cells (67,68).
It is a known fact that several types of cancer cells require
insulin for optimal growth in vitro. Recent data (67,68)
have demonstrated that: i) Insulin stimulates growth mainly through
its own receptor rather than insulin-like growth factor-1 receptor
(IGF-1R). By employing blocking monoclonal antibodies specific to
both the insulin receptor (IR) and IGF-1R, it was demonstrated that
the growth response of breast cancer cell lines to insulin could be
specifically blocked by an anti-IR, but not by the anti-IGF-IR
blocking antibody (67); and ii)
several types of cancer cells overexpress the IR as shown using
immunostaining; it was observed that overexpression of IR was not
unique to breast cancer, but a common phenomenon across several
types of human cancers. Increased IR levels were observed in colon,
lung, ovarian and thyroid cancer (69,70).
Furthermore, the a-genotype is more efficient than the b-genotype
in promoting mitosis. When exposed to insulin, these
characteristics confer a selective growth advantage to malignant
cells. However, the evidence on the combined effect of insulin and
Na+/K+-ATPase on carcinogenesis is
insufficient, and extensive further research is required to explore
and confirm their relationship in the future.
7. Conclusions and perspectives
Na+/K+-ATPase has been
implicated in the development of various diseases. In the present
study, the structure, biochemical characteristics, function and
metabolism of Na+/K+-ATPase were reviewed to
further elucidate the mechanism underlying the regulatory effect of
insulin on Na+/K+-ATPase in the whole body.
The mRNA transcription and protein expression levels of
Na+/K+-ATPase can be regulated by insulin
(55). The
Na+/K+-ATPase α1 subunit is regulated by the
ERK1/mTORC1, PI3K/AKT/ZNRF and other pathways (30,54).
Furthermore, it is known that Na+/K+-ATPase
may be degraded by UPP and autophagy-lysosomal pathway (26,27,33,34).
However, to the best of our knowledge, no in-depth research of
these mechanisms has been conducted and there have been no
breakthroughs to date.
Based on the present review, it appears that
insulin may affect the abundance of
Na+/K+-ATPase α1 through the UPP and
autophagy-lysosomal system, and it may also affect the expression
of Na+/K+-ATPase α1 through a variety of
pathways. Therefore, it is crucial to study the dynamic process
underlying the regulatory action of insulin on
Na+/K+-ATPase. Although there remain several
unresolved issues, more factors that may be implicated in the
development of several diseases through regulating
Na+/K+-ATPase must be discovered and
explored, as Na+/K+-ATPase may represent a
promising potential target for the clinical treatment of ARDS and
other diseases.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Science and
Technology Department of Hunan Province, China (grant no.
2020JJ4851).
Availability of data and materials
Not applicable.
Authors' contributions
XPW was responsible for integrating all the
information and wrote and edited this review. Supervisor QQW made
substantial contributions to conception and design and revised the
manuscript. All authors have read and approved the final version of
the article. Data sharing is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liu J, Lilly MN and Shapiro JI: Targeting
Na/K-ATPase signaling: A new approach to control oxidative stress.
Curr Pharm Des. 24:359–364. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kaplan JH: Biochemistry of Na,K-ATPase.
Annu Rev Biochem. 71:511–535. 2002.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Suhail M: Na, K-ATPase: Ubiquitous
multifunctional transmembrane protein and its relevance to various
pathophysiological conditions. J Clin Med Res. 2:1–17.
2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Shrivastava AN, Triller A and Melki R:
Cell biology and dynamics of Neuronal
Na+/K+-ATPase in health and diseases.
Neuropharmacology. 169(107461)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Yang P, Cartwright C, Efuet E, Hamilton
SR, Wistuba II, Menter D, Addington C, Shureiqi I and Newman RA:
Cellular location and expression of Na+, K+
-ATPase α subunits affect the anti-proliferative activity of
oleandrin. Mol Carcinog. 53:253–263. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Clausen MV, Hilbers F and Poulsen H: The
structure and function of the Na,K-ATPase isoforms in health and
disease. Front Physiol. 8(371)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Tidow H, Aperia A and Nissen P: How are
ion pumps and agrin signaling integrated? Trends Biochem Sci.
35:653–659. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Li Z and Langhans SA: Transcriptional
regulators of Na,K-ATPase subunits. Front Cell Dev Biol.
3(66)2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Brazee PL, Soni PN, Tokhtaeva E, Magnani
N, Yemelyanov A, Perlman HR, Ridge KM, Sznajder JI, Vagin O and
Dada LA: FXYD5 Is an essential mediator of the inflammatory
response during lung injury. Front Immunol. 8(623)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Marck PV and Pierre SV: Na/K-ATPase
signaling and cardiac pre/postconditioning with cardiotonic
steroids. Int J Mol Sci. 19(2336)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lingrel JB: The physiological significance
of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase.
Annu Rev Physiol. 72:395–412. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Johnson MD, Widdicombe JH, Allen L, Barbry
P and Dobbs LG: Alveolar epithelial type I cells contain transport
proteins and transport sodium, supporting an active role for type I
cells in regulation of lung liquid homeostasis. Proc Natl Acad Sci
USA. 99:1966–1971. 2002.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Blanco G: Na,K-ATPase subunit
heterogeneity as a mechanism for tissue-specific ion regulation.
Semin Nephrol. 25:292–303. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang HY and O'doherty GA: Modulators of
Na/K-ATPase: A patent review. Expert Opin Ther Pat. 22:587–605.
2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Geering K: Functional roles of Na,K-ATPase
subunits. Curr Opin Nephrol Hypertens. 17:526–532. 2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Toyoshima C, Kanai R and Cornelius F:
First crystal structures of Na+,K+-ATPase:
New light on the oldest ion pump. Structure. 19:1732–1738.
2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Mijatovic T, Dufrasne F and Kiss R:
Na+/K+-ATPase and cancer. Pharm Pat Anal.
1:91–106. 2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Herold S, Gabrielli NM and Vadász I: Novel
concepts of acute lung injury and alveolar-capillary barrier
dysfunction. Am J Physiol Lung Cell Mol Physiol. 305:L665–L681.
2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Laffey JG and Matthay MA: Fifty years of
research in ARDS. Cell-based therapy for acute respiratory distress
syndrome. Biology and potential therapeutic value. Am J Respir Crit
Care Med. 196:266–273. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Vadász I, Raviv S and Sznajder JI:
Alveolar epithelium and Na,K-ATPase in acute lung injury. Intensive
Care Med. 33:1243–1251. 2007.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Mutlu GM and Sznajder JI: Mechanisms of
pulmonary edema clearance. Am J Physiol Lung Cell Mol Physiol.
289:L685–L695. 2005.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lecuona E, Trejo HE and Sznajder JI:
Regulation of Na,K-ATPase during acute lung injury. J Bioenerg
Biomembr. 39:391–395. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hunter T: The age of crosstalk:
Phosphorylation, ubiquitination, and beyond. Mol Cell. 28:730–738.
2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Calistri A, Munegato D, Carli I, Parolin C
and Palù G: The ubiquitin-conjugating system: Multiple roles in
viral replication and infection. Cells. 3:386–417. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Heaton SM, Borg NA and Dixit VM: Ubiquitin
in the activation and attenuation of innate antiviral immunity. J
Exp Med. 213:1–13. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Coppi MV and Guidotti G: Ubiquitination of
Na,K-ATPase alpha1 and alpha2 subunits. FEBS Lett. 405:281–284.
1997.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dada LA, Welch LC, Zhou G, Ben-Saadon R,
Ciechanover A and Sznajder JI: Phosphorylation and ubiquitination
are necessary for Na,K-ATPase endocytosis during hypoxia. Cell
Signal. 19:1893–1898. 2007.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Comellas AP, Dada LA, Lecuona E, Pesce LM,
Chandel NS, Quesada N, Budinger GR, Strous GJ, Ciechanover A and
Sznajder JI: Hypoxia-mediated degradation of Na,K-ATPase via
mitochondrial reactive oxygen species and the ubiquitin-conjugating
system. Circ Res. 98:1314–1322. 2006.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Helenius IT, Dada LA and Sznajder JI: Role
of ubiquitination in Na,K-ATPase regulation during lung injury.
Proc Am Thorac Soc. 7:65–70. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hoxhaj G, Najafov A, Toth R, Campbell DG,
Prescott AR and Mackintosh C: ZNRF2 is released from membranes by
growth factors and, together with ZNRF1, regulates the
Na+/K+ATPase. J Cell Sci. 125:4662–4675.
2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ryter SW, Bhatia D and Choi ME: Autophagy:
A lysosome-dependent process with implications in cellular redox
homeostasis and human disease. Antioxid Redox Signal. 30:138–159.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhu M, Cao L, Xiong S, Sun H, Wu Z and
Bian JS: Na+/K+-ATPase-dependent autophagy
protects brain against ischemic injury. Signal Transduct Target
Ther. 5(55)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Felippe Gonçalves-de-Albuquerque C,
Ribeiro Silva A, Ignácio da Silva C, Caire Castro-Faria-Neto H and
Burth P: Na/K pump and beyond: Na/K-ATPase as a modulator of
apoptosis and autophagy. Molecules. 22(578)2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Liu Y, Shoji-Kawata S, Sumpter RM Jr, Wei
Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, et al:
Autosis is a Na+,K+-ATPase-regulated form of
cell death triggered by autophagy-inducing peptides, starvation,
and hypoxia-ischemia. Proc Natl Acad Sci USA. 110:20364–20371.
2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu
HL, Yang C and Liu HF: p62 links the autophagy pathway and the
ubiqutin-proteasome system upon ubiquitinated protein degradation.
Cell Mol Biol Lett. 21(29)2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lin X, Li S, Zhao Y, Ma X, Zhang K, He X
and Wang Z: Interaction domains of p62: A bridge between p62 and
selective autophagy. DNA Cell Biol. 32:220–227. 2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wurzer B, Zaffagnini G, Fracchiolla D,
Turco E, Abert C, Romanov J and Martens S: Oligomerization of p62
allows for selection of ubiquitinated cargo and isolation membrane
during selective autophagy. Elife. 4(e08941)2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hancock ML, Meyer RC, Mistry M, Khetani
RS, Wagschal A, Shin T, Ho Sui SJ, Naar AM and Flanagan JG: Insulin
receptor associates with promoters genome-wide and regulates gene
expression. Cell. 177:722–736.e22. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Tokarz VL, MacDonald PE and Klip A: The
cell biology of systemic insulin function. J Cell Biol.
217:2273–2289. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Haeusler RA, McGraw TE and Accili D:
Biochemical and cellular properties of insulin receptor signalling.
Nat Rev Mol Cell Biol. 19:31–44. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Borge PD, Moibi J, Greene SR, Trucco M,
Young RA, Gao Z and Wolf BA: Insulin receptor signaling and
sarco/endoplasmic reticulum calcium ATPase in beta-cells. Diabetes.
51 (Suppl 3):S427–S433. 2002.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Herchuelz A and Pachera N: The
Na+/Ca2+ exchanger and the Plasma Membrane
Ca2+-ATPase in β-cell function and diabetes. Neurosci
Lett. 663:72–78. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Herchuelz A, Nguidjoe E, Jiang L and
Pachera N: Na(+)/Ca (2+) exchange and the plasma membrane
Ca(2+)-ATPase in β-cell function and diabetes. Adv Exp Med Biol.
961:385–394. 2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Futai M, Sun-Wada GH, Wada Y, Matsumoto N
and Nakanishi-Matsui M: Vacuolar-type ATPase: A proton pump to
lysosomal trafficking. Proc Jpn Acad Ser B Phys Biol Sci.
95:261–277. 2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Lichtstein D, Ilani A, Rosen H, Horesh N,
Singh SV, Buzaglo N and Hodes A: Na+,
K+-ATPase signaling and bipolar disorder. Int J Mol Sci.
19(2314)2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Therien AG and Blostein R: Mechanisms of
sodium pump regulation. Am J Physiol Cell Physiol. 279:C541–C566.
2000.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Shahidullah M, Mandal A and Delamere NA:
Src family kinase links insulin signaling to short term regulation
of Na,K-ATPase in nonpigmented ciliary epithelium. J Cell Physiol.
232:1489–1500. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Chen HI, Yeh DY, Liou HL and Kao SJ:
Insulin attenuates endotoxin-induced acute lung injury in conscious
rats. Crit Care Med. 34:758–764. 2006.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Brunkhorst FM, Engel C, Bloos F,
Meier-Hellmann A, Ragaller M, Weiler N, Moerer O, Gruendling M,
Oppert M, Grond S, et al: Intensive insulin therapy and pentastarch
resuscitation in severe sepsis. N Engl J Med. 358:125–139.
2008.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Comellas AP, Kelly AM, Trejo HE, Briva A,
Lee J, Sznajder JI and Dada LA: Insulin regulates alveolar
epithelial function by inducing Na+/K+-ATPase
translocation to the plasma membrane in a process mediated by the
action of Akt. J Cell Sci. 123:1343–1351. 2010.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Bruss MD, Arias EB, Lienhard GE and Cartee
GD: Increased phosphorylation of Akt substrate of 160 kDa (AS160)
in rat skeletal muscle in response to insulin or contractile
activity. Diabetes. 54:41–50. 2005.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Ishikura S, Bilan PJ and Klip A: Rabs 8A
and 14 are targets of the insulin-regulated Rab-GAP AS160
regulating GLUT4 traffic in muscle cells. Biochem Biophys Res
Commun. 353:1074–1079. 2007.PubMed/NCBI View Article : Google Scholar
|
|
53
|
He J, Qi D, Wang DX, Deng W, Ye Y, Feng
LH, Zhu T, Zhao Y and Zhang CR: Insulin upregulates the expression
of epithelial sodium channel in vitro and in a mouse model of acute
lung injury: Role of mTORC2/SGK1 pathway. Exp Cell Res.
331:164–175. 2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Deng W, Li CY, Tong J, He J, Zhao Y and
Wang DX: Insulin ameliorates pulmonary edema through the
upregulation of epithelial sodium channel via the PI3K/SGK1 pathway
in mice with lipopolysaccharide-induced lung injury. Mol Med Rep.
19:1665–1677. 2019.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Bashir SO: Concomitant administration of
resveratrol and insulin protects against diabetes mellitus
type-1-induced renal damage and impaired function via an
antioxidant-mediated mechanism and up-regulation of
Na+/K+-ATPase. Arch Physiol Biochem.
125:104–113. 2019.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Liu H, Chen Z, Wu D, Huang X and Wan Q:
Insulin attenuates the inhibition effect of lipopolysaccharide on
Na+ -K+ -ATPase α1 via PI3 K/AKT and PI3
K/ERK pathway. Chin J Clin Pharm Therap. 24:896–902. 2019.
|
|
57
|
Obradovic M, Bjelogrlic P, Rizzo M,
Katsiki N, Haidara M, Stewart AJ, Jovanovic A and Isenovic ER:
Effects of obesity and estradiol on
Na+/K+-ATPase and their relevance to
cardiovascular diseases. J Endocrinol. 218:R13–R23. 2013.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Siddiqui MR, Moorthy K, Taha A, Hussain ME
and Baquer NZ: Low doses of vanadate and trigonella synergistically
regulate Na+/K+ -ATPase activity and GLUT4
translocation in alloxan-diabetic rats. Mol Cell Biochem.
285:17–27. 2006.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Sun HJ, Cao L, Zhu MY, Wu ZY, Shen CY, Nie
XW and Bian JS: DR-region of Na+/K+-ATPase is
a target to ameliorate hepatic insulin resistance in obese diabetic
mice. Theranostics. 10:6149–6166. 2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Pirkmajer S and Chibalin AV: Na,K-ATPase
regulation in skeletal muscle. Am J Physiol Endocrinol Metab.
311:E1–E31. 2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Pirkmajer S and Chibalin AV: Hormonal
regulation of Na+-K+-ATPase from the
evolutionary perspective. Curr Top Membr. 83:315–351.
2019.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Catalán RE, Martínez AM, Aragonés MD,
Fernández I and Miguel BG: Inhibitory effect of insulin and
cytoplasmic factor(s) on brain (Na(+) + K+) ATPase. Neurosci Res.
13:139–145. 1992.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Banday AA, Asghar M, Hussain T and
Lokhandwala MF: Dopamine-mediated inhibition of renal Na,K-ATPase
is reduced by insulin. Hypertension. 41:1353–1358. 2003.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Sowers JR: Effects of insulin and IGF-I on
vascular smooth muscle glucose and cation metabolism. Diabetes. 45
(Suppl 3):S47–S51. 1996.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Silva CID, Gonçalves-De-Albuquerque CF,
Moraes BPT, Garcia DG and Burth P: Na/K-ATPase: Their role in cell
adhesion and migration in cancer. Biochimie. 185:1–8.
2021.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Lu S, Cai S, Peng X, Cheng R and Zhang Y:
Integrative transcriptomic, proteomic and functional analysis
reveals ATP1B3 as a diagnostic and potential therapeutic target in
hepatocellular carcinoma. Front Immunol. 12(636614)2021.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Vigneri R, Goldfine ID and Frittitta L:
Insulin, insulin receptors, and cancer. J Endocrinol Invest.
39:1365–1376. 2016.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Vigneri R, Sciacca L and Vigneri P:
Rethinking the relationship between insulin and cancer. Trends
Endocrinol Metab. 31:551–560. 2020.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Belfiore A, Frasca F, Pandini G, Sciacca L
and Vigneri R: Insulin receptor isoforms and insulin
receptor/insulin-like growth factor receptor hybrids in physiology
and disease. Endocr Rev. 30:586–623. 2009.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Vella V, Sciacca L, Pandini G, Mineo R,
Squatrito S, Vigneri R and Belfiore A: The IGF system in thyroid
cancer: New concepts. Mol Pathol. 54:121–124. 2001.PubMed/NCBI View Article : Google Scholar
|