Introduction
Glutamate excitotoxicity is a condition in which
excessive glutamate accumulates in the central nervous system (CNS)
causing acute neuronal injury and long-term neurodegeneration
(1). Abrupt increases in glutamate
are known to accompany traumatic brain injuries and cerebral
ischemia, as well as to contribute to neurodegenerative diseases,
such as amyotrophic lateral sclerosis, fibromyalgia, multiple
sclerosis, Alzheimer's, Parkinson's, and Huntington's diseases.
Glutamate-induced injury is primarily the result of increased
intracellular calcium levels facilitated by the engagement of
glutamate receptors (2). In
addition to Ca2+ overload, receptor stimulation also
leads to collapse of the electrochemical gradient, and activation
of protein kinases and endonucleases. These alterations accelerate
cell death through multiple pathways and through degradation of
important substances (3). Work in
our own lab has shown that HT22 mouse hippocampal cells exposed to
glutamate experience mitochondrial dysfunction causing formation of
the mitochondrial permeability transition pore (mPTP). This
occurred with increased calcium retention, alteration of the
mitochondrial membrane potential (MMP), fragmentation of the
mitochondria, release of apoptosis inducing factor (AIF), and
breakdown of DNA (4,5).
Much work has been done on how to best limit the
effects of glutamate to prevent overstimulation of its receptors.
We have found that supplementation with coenzyme Q10 (CoQ10) can
prevent many of the glutamate- or rotenone-induced changes within
the mitochondria, and improve viability of neurons (4,6-9).
One of the principal defenses against mitochondrial
dysfunction is the detoxification of reactive oxygen species (ROS)
whose stimulation is both triggered and enhanced when the
mitochondrial membrane potential is disrupted. CoQ10 is a major
cofactor of the electron transport chain (ETC) where majority ROS
are produced. Within the ETC, CoQ10 plays a key role in
transporting electrons between complexes I, II and III. It is also
recognized as having antioxidant functions. Thus, when ETC function
is disrupted by a mitochondrial insult, CoQ10's location positions
itself as a key antioxidant to reduce oxidative damage (10). However, continuous, high levels of
oxidative stress can lead to CoQ10 depletion and prevent adequate
detoxification of ROS.
Therefore, exogenous antioxidant supplementation of
CoQ10, can improve outcomes after mitochondrial damage (6-8),
including damage caused by glutamate (4). CoQ10 is produced naturally within
cells as part of the cholesterol pathway, but the cellular content
can be further increased through consumption of food sources high
in CoQ10, such as meat, nuts and green leafy vegetables. Exogenous
ubiquinone (oxidized form of CoQ10), or ubiquinol [a reduced and
more bioavailable form of CoQ10(11)], can also be administered. This
additional supplementation may particularly benefit aging adults as
CoQ10 levels naturally decline with age (12).
The benefits from CoQ10 supplementation have been
documented in a number of clinical trials. For example, in chronic
heart failure patients, CoQ10 decreased mortality, reduced the
incidence of hospitalization, and improved patients' functional
classification by the New York Heart Association (13). CoQ10 increases total capacity of
antioxidant enzymes and reduces inflammatory biomarkers in diabetic
hemodialysis patients (14,15). CoQ10 benefits have also been
reported in a clinical trial of multiple sclerosis (16). More clinical trials are needed to
establish the efficacy of CoQ10 in preventing neurodegeneration and
in preserving mitochondrial function. Our lab has been interested
in the neuroprotective benefits of CoQ10 supplementation. We
believe this protection can be attributed to the effects of CoQ10
on the mitochondria, beyond antioxidant functions. In this study,
our goal was to determine whether CoQ10 could increase
mitochondrial biogenesis to improve the outcome against glutamate
toxicity.
Mitochondrial biogenesis is a process by which new
mitochondria are formed through the growth and division of
pre-existing mitochondria (17).
Activation of this process often occurs during an insult to the
cell. This is an attempt to counteract the damage process.
Essentially, increasing numbers of mitochondria serves to boost ATP
production and to increase detoxification of ROS. Unfortunately,
this response is usually not sufficient to counteract the damage
process (18). However, because of
its protective effect, enhancing the mitochondrial biogenesis could
be used as a strategy to protect cells against various damages
including neurological diseases (19).
Changes in mitochondrial biogenesis can be
determined by examining expression of key proteins mediating
mitochondrial biogenesis pathways. Peroxisome
proliferator-activated receptor gamma coactivator-1α (PGC-1α)
appears to be the master regulator of mitochondrial biogenesis
based on current knowledge (17).
Once being activated, either through phosphorylation or
deacetylation, PGC-1α activates the mitochondrial transcription
factors, nuclear respiratory factor 1 and 2 (NRF1,2). The NRF 1,2
could activate the mitochondrial NDA and protein synthesis either
directly or through activating mitochondrial transcription factor A
(TFAM) (20). Activation of these
transcription factors by PGC-1α, leads to the synthesis of
mitochondrial genes encoded in nuclear and mitochondrial DNA,
promoting generation of new mitochondria.
The overall objectives of this work were to
determine whether glutamate exposure itself affected mitochondrial
biogenesis in hippocampal cells and whether CoQ10 supplementation
conferred protection against glutamate-induced toxicity by
promoting mitochondrial biogenesis. To this end, we measured
mitochondrial biogenesis in hippocampal cell exposed to glutamate,
with and without CoQ10 pre-treatment. We further analyzed
mitochondrial biogenesis protein expression patterns to determine
the molecular mechanisms involved. Given the active role
mitochondria play in many disease models, including neuronal injury
from glutamate excitotoxicity, we believe elucidating mitochondrial
changes, and targeting them for correction, may improve therapeutic
outcomes in patients.
Materials and methods
Cell culture
HT22 is a mouse hippocampal cell line kindly
provided by Dr Panee at the University of Hawaii. HT22 cells were
cultured as previously described (21). Stock preparations of glutamate
(Sigma-Aldrich; Merck KGaA) and CoQ10 supplement, Ubisol-Q10 (Zymes
LLC), dissolved in water, were diluted with cell culture media
before being added to the cells. Glutamate concentrations (1-8 mM)
were tested in pilot study and 4 mM was selected for subsequent
studies because this concentration resulted in a 30% reduction of
cell viability assessed by resazurin assay after 24 h exposure.
Treatment of Ubisol-Q10 (25 µg/µl) was initiated 24 h prior to
glutamate addition.
Reproductive potential assay
The reproductive potential of HT22 cells after
exposure to glutamate and/or CoQ10 treatment was assessed using
colony formation assay. Cells were treated as described above for
untreated, glutamate, CoQ10, and glutamate plus CoQ10 experimental
groups. After treatment, the cells were harvested and seeded at a
density of 100 cells per 10 cm dish, except in the case of the
glutamate only group, which were seeded at a higher density of
5,000 cells per dish. The cells were incubated under standard
culture conditions for 14 days to allow visible colonies to form
from individual cells seeded. The colonies were stained with a
0.05% crystal violet solution. Numbers of colonies were counted
using ImageJ software version 1.49(22). Plating efficiencies were calculated
by taking the actual numbers of colonies formed in each plate and
dividing by the number of cells originally seeded and then
multiplying by 100 to obtain a percent. The percent of cells
surviving was calculated by taking the individual plates' plating
efficiencies and dividing by the average plating efficiency of the
untreated, control group and again multiplying by 100. The average
percent survival for each group was quantified and analyzed for
statistical significance using one-way ANOVA and Bonferroni's
multiple comparisons test.
Western blotting
After undergoing experimental treatments for 24 h
[untreated, glutamate alone (4 mM), CoQ10 alone (25 µg/µl), and
glutamate plus CoQ10 24 h pre-treatment], cells were harvested and
washed with phosphate-buffered saline (PBS) before lysing to obtain
either total protein extracts, or protein fractions from cytosolic,
mitochondrial, and nuclear compartments as previously described
(21,23). Protein concentrations were measured
using standard Bradford assays (Bio-Rad Laboratories). Western blot
analysis was performed essentially as previously described
(21). Equal amount of protein (20
µg) samples were loaded and separated on 4-12% Bis-Tris NuPAGE gels
(Invitrogen; Thermo Fisher Scientific, Inc.) and then transferred
to Polyvinylidene fluoride membranes. The following primary
antibodies from Cell Signaling Technology were used at 1:1,000
dilutions: Anti-AKT, anti-p-AKT, anti-CREB, anti-p-CREB,
anti-COX-IV, and anti-Histone 3. Anti-PGC-1α (1:1,000) was obtained
from Abcam. Anti-NRF2 (1:200) was from Santa Cruz Biotechnology and
anti-TFAM (1:1,000) was from Calbiochem. Both anti-Histone 3 and
β-actin (1:5,000) were used as protein loading controls. IRDye
680LT goat anti-rabbit, IRDye 800CW goat anti-mouse, and donkey
anti-goat secondary antibodies from Li-COR, Inc. were used at
1:10,000 dilutions for visualization using the Li-COR Odyssey
Classic Imaging System scanner. Protein bands were analyzed with
the Li-COR Image Studio Software version 5.2.5. as previously
described (21). Statistical
significance was measured using one-way ANOVA and Bonferroni's
multiple comparisons test.
Measurement of mitochondrial
biogenesis
Different levels of mitochondrial biogenesis among
experimental groups were assessed using a MitoBiogenesis™ In-Cell
ELISA Kit (Abcam) according to the manufacturer's protocol.
Briefly, 20,000 cells per well were seeded in 96-well plates and
allowed to adhere overnight. The cells were then fixed with 4%
paraformaldehyde, briefly permeabilized with Triton X-100, blocked,
and incubated overnight at 4˚C with primary antibodies. Primary
antibodies were specific against mitochondrial DNA-encoded protein,
COX-I, and nuclear DNA-encoded protein, SDH-A. The cells were
washed and incubated for 1 h at room temperature with a solution of
secondary antibodies containing an AP-labeled antibody specific for
SDH-A and an HRP-labeled antibody specific for COX-I. The reactions
were sequentially developed; first with an AP development solution,
and then an HRP development solution. Fifteen minute kinetic
reactions with 1 min intervals were recorded using a PHERAstar
microplate reader (BMG Labtech) to measure optical density at 405
nm for AP development and 600 nm for HRP development. Whole cell
staining with Janus Green was also done to compensate for
variations due to cell loss during the procedure. The AP and HRP
data were normalized to the Janus Green optical density at 595 nm.
The signal ratio of COX-I/SDH-A was calculated to determine
increased production of mitochondrial DNA-encoded protein without a
matching increase in nuclear DNA-encoded protein. Such an increase
was interpreted as an increase in mitochondrial biogenesis.
Statistical significance was measured using one-way ANOVA and
Bonferroni's multiple comparisons test.
Statistical analysis
Each experiment was repeated at least three times.
Data is presented as either mean values ± standard deviation (SD),
or as a percentage of the control. Statistical analysis was carried
out using ordinary one- or two-way ANOVA with Bonferroni's multiple
comparisons test where indicated. P-values ≤0.05 were considered
statistically significant.
Results
CoQ10 protects against cellular damage
caused by glutamate exposure
Because the toxic effect of glutamate exposure can
vary between cell lines, we first looked at glutamate-induced loss
of viability in HT22 cells using a range of glutamate
concentrations from 1 to 8 mM. The cells were exposed to these
concentrations for 24 h before adding the resazurin dye as
described previously (21). We
observed a dose-dependent decrease in cell viability as glutamate
concentration rises. At 4 mM, glutamate decreased viability by
roughly 30% (P<0.001) and at the highest concentration of 8 mM,
>80% of cells were died (data not shown). As a result, 4 mM
glutamate was selected for subsequent study. CoQ10 (25 µg/µl)
treatment alone did not affect cell viability. However, CoQ10
pretreatment to glutamate-exposed cells effectively prevented the
decline of cell viability caused by glutamate (data not shown).
In addition to reducing cell viability directly,
cells surviving glutamate exposure may lose their proliferative
capability even after glutamate has been removed. We assessed the
the reproductive potential of HT22 cells after exposure to
glutamate and/or CoQ10 treatment using clonogenic colony forming
assay, an indication that they have retained their reproductive
ability and are still capable of cellular division. Our experiments
assessed this ability in cells that survived the initial 24 h
glutamate exposure, CoQ10 treatment alone, or glutamate plus CoQ10
pretreatment. Surviving cells were harvested, washed and then
seeded at low densities to allow individual cells to form visible
colonies.
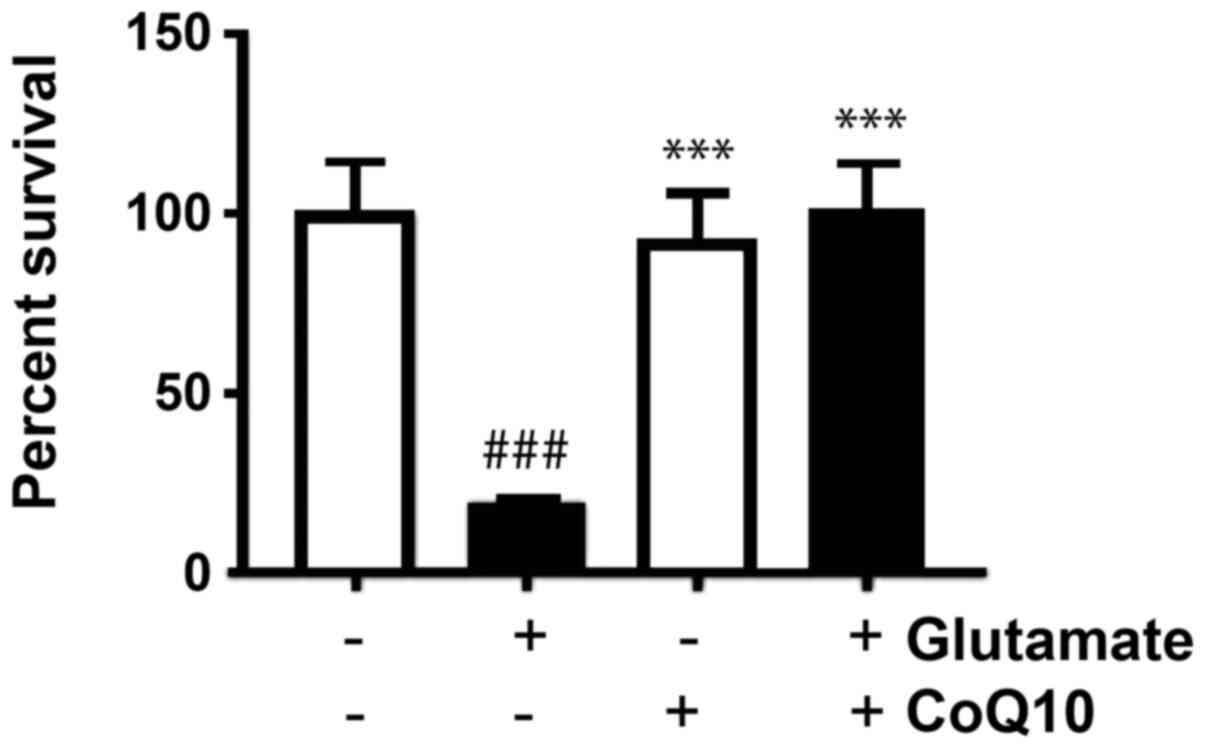
After staining colonies with crystal violet, the
plating efficiency (PE) for each group was calculated by taking the
number of colonies formed and dividing it by the number of cells
seeded and then multiplying by 100 to obtain a percent. We found
that in untreated cells, the average PE was 68%. Our results showed
the PE was reduced in glutamate-exposed cells dropping to 13%, but
was not significantly altered from control in the CoQ10 alone, or
the glutamate plus CoQ10 group. To find the surviving fraction, we
divided the PE of the treated sample by the PE of the control cells
and multiplied by 100. Value in untreated control group was set at
100% survival and our experimental groups were reported relative to
this value. Glutamate exposure dropped survival to 19%
(P<0.001). CoQ10 alone and CoQ10 plus glutamate groups were not
significantly different from untreated cells with average survival
fractions of 93 and 101% respectively (Fig. 1). In another word, CoQ10 reinstalled
the cell survival.
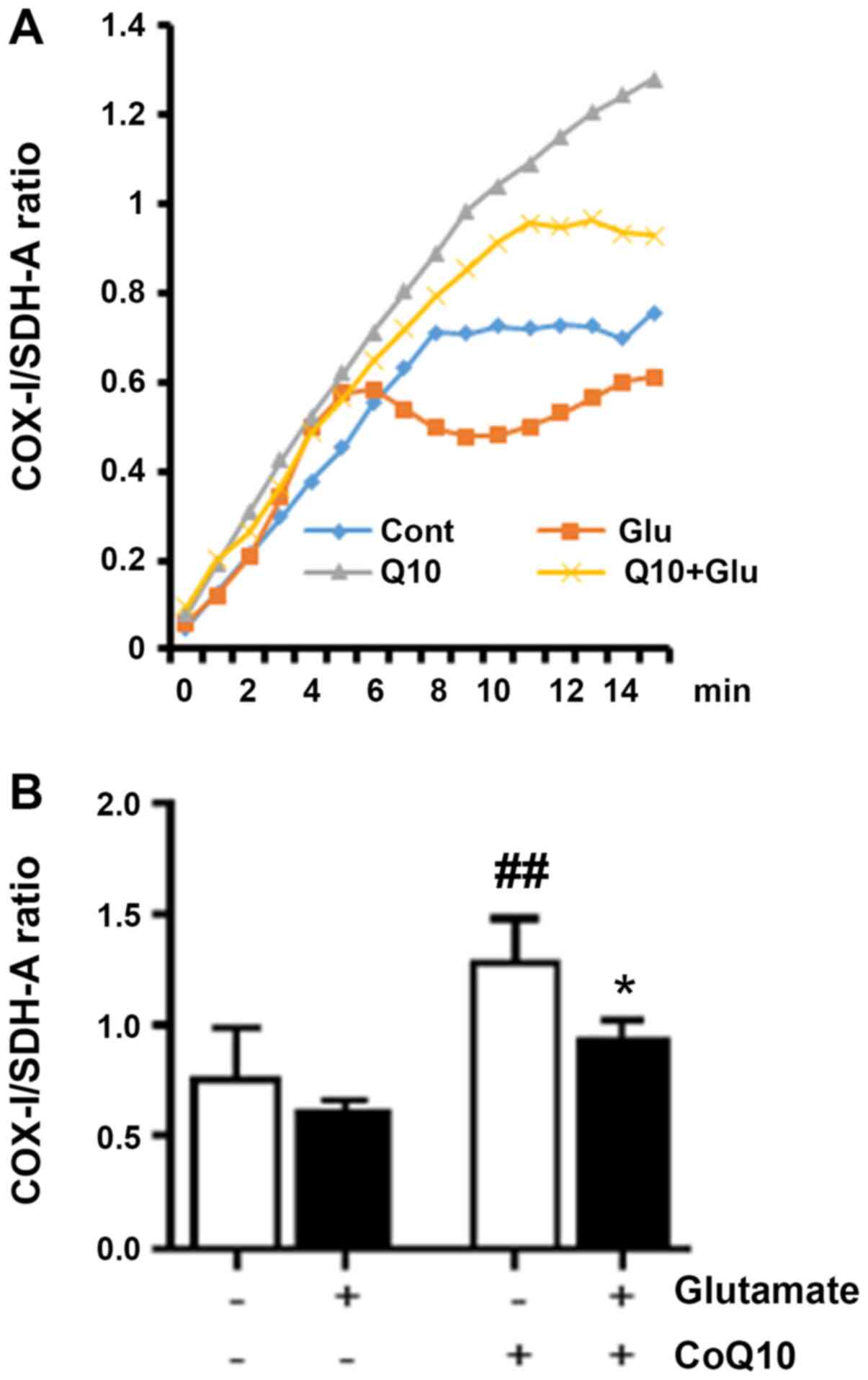
CoQ10 promotes mitochondrial
biogenesis
Labeling both a mitochondrial DNA-encoded protein
(COX-I) and a nuclear DNA-encoded protein (SDH-A), we measured
increases, or decreases, in COX-I expression over SDH-A that were
interpreted as corresponding to increases, and decreases, in
mitochondrial biogenesis, respectively. AP and HRP enzyme-catalyzed
reactions (as described in methods) were developed for 15 min with
optical density measured at 1 min intervals. Fig. 2A shows the ratio of the COX-I signal
to the SDH-A signal (normalized to Janus Green) at each interval
reading. The bar graph (Fig. 2B)
shows the average ratio for each experimental group at the 15 min
endpoint. We observed that the COX-I/SDH-A ratio from glutamate
exposure was moderately depressed indicating reduced mitochondrial
biogenesis. CoQ10 treatment alone significantly increased
mitochondrial biogenesis by roughly 60% compared to control
(P<0.01). Combination of CoQ10 plus glutamate significantly
increased the mitochondrial biogenesis compared to glutamate
exposure alone (P<0.05).
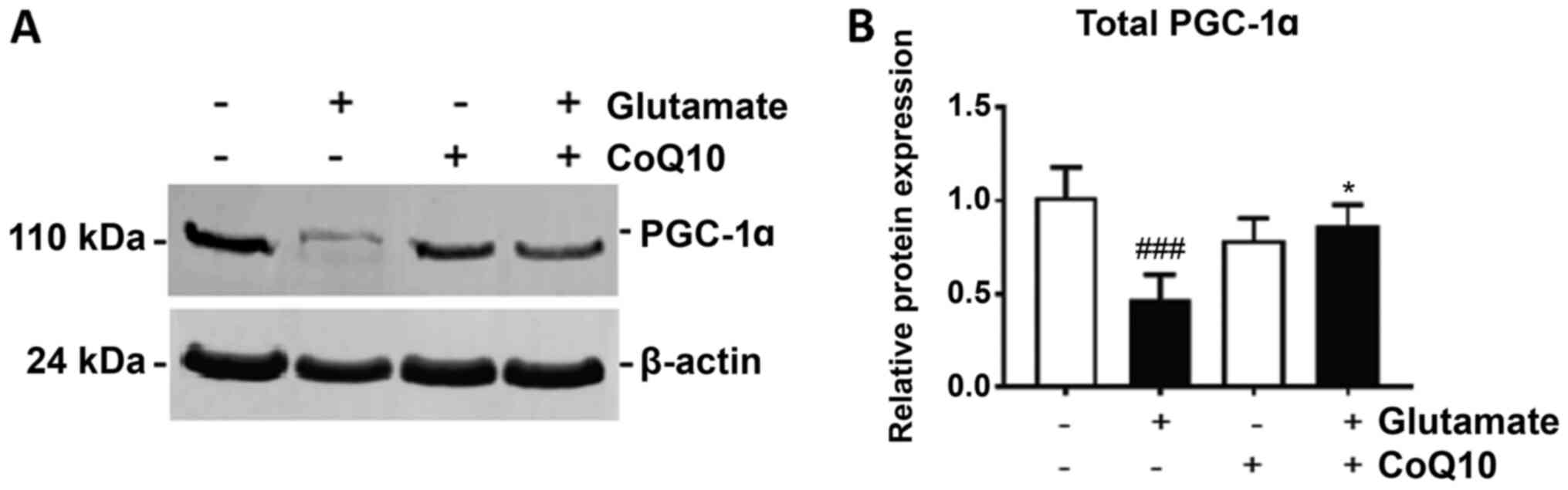
CoQ10 reseved glutamate-suppressed
PGC-1α, p-Akt and p-CREB
PGC-1α is considered the master regulator of
mitochondrial biogenesis through its action as a transcription
factor for genes central in promoting mitochondrial growth. We show
in Fig. 3 that glutamate exposure
significantly reduced PGC-1α expression by 40% in HT22 cells after
24 h (P<0.01). Treatment with CoQ10 alone had no effect on
PGC-1α expression, but when added prior to glutamate addition, it
significantly improved PGC-1α expression, increasing it by 35% from
glutamate exposure alone (P<0.05).
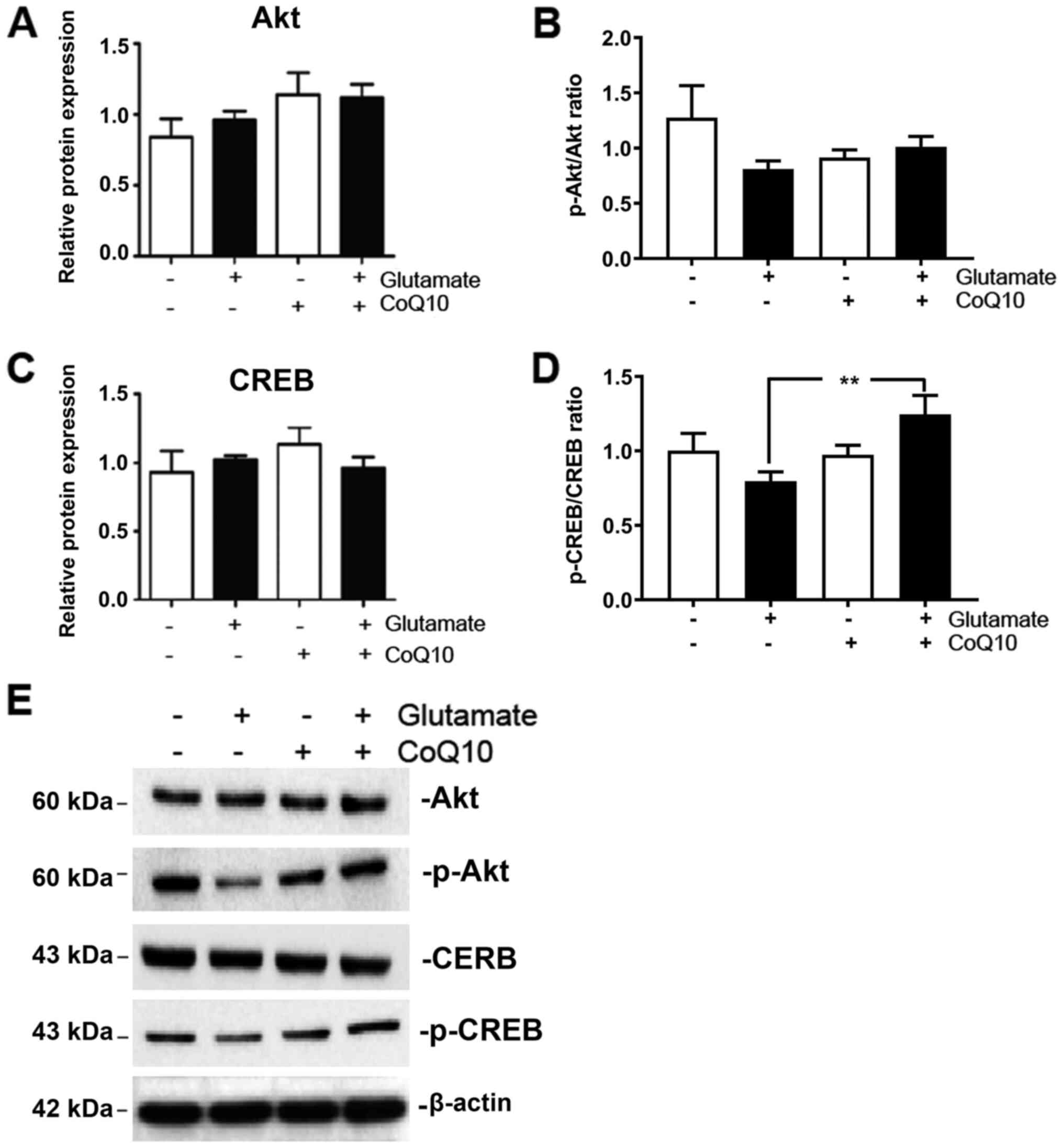
PGC-1α is most commonly activated by either
phosphorylation, or deacetylation, of its promoter region. A number
of endogenous and exogenous actors can facilitate these reactions.
One of noted inducers of PGC-1α phosphorylation is the AKT/CREB
pathway (20). Therefore, we
measured expression of total and phosphorylated AKT and CREB in our
experimental groups. The results showed that both phosphorylated
AKT (p-AKT) and phosphorylated CREB (p-CREB) were moderately
reduced from glutamate exposure albeit did not reach statistical
significant. They were both upregulated when CoQ10 was added
(Fig. 4; P<0.01 for p-CREB).
CoQ10 prevented glutamate-caused
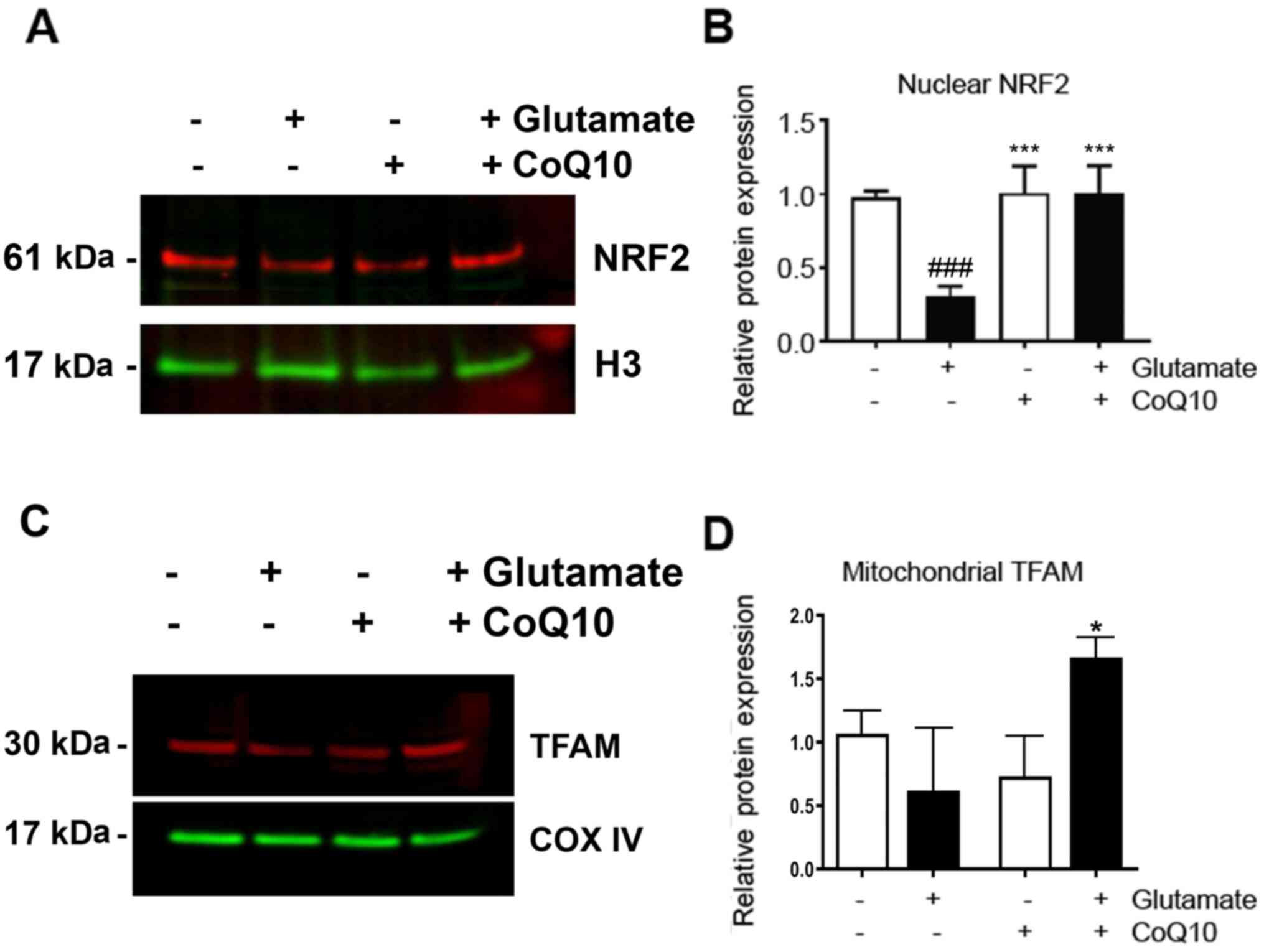
suppressions of NRF2 and TFAM
The downstream effectors, NRF2 and TFAM, were also
measured (Fig. 5). We observed a
nearly 70% decrease in NRF2 expression in the nuclear fraction
after glutamate exposure compared to the control (P<0.001).
Addition of CoQ10 resulted in a complete restoration of NRF2 from
this glutamate-inhibited level (P<0.001 vs. Glutamate). TFAM
protein level was reduced by glutamate exposure. However, due to
relative large variation, this reduction did not reach statistical
significance. CoQ10 pretreatment to glutamate exposed cells
resulted in a slight, but significantly increase in TFAM expression
(20%) compared to control (P<0.05).
Discussion
The present study showed that glutamate
significantly reduced cell viability and reproductivity (cell
proliferation) after glutamate exposure. Pretreatment with CoQ10 24
h prior to glutamate exposure significant limited the damaging
effects of glutamate on cell viability and cell proliferation
capacity after glutamate exposure. These results are consistent
with our previous published results (4,5,24,25).
The glutamate toxicity is time-dependent, with mild damage observed
after 10 h, decreased to about 40-60% after 24 h, and further
reduced to less than 20% of the control value after 36 and 48 h
(5,25). It is likely that glutamate caused
reduction in cell viability is caused by cell death. We have
previously shown that glutamate exposure increases ROS production,
causes mitochondrial membrane hyperpolarization, suppressed
mitochondrial respiration and resulted in mitochondrial
fragmentation. Furthermore, glutamate increases the release of AIF
to nuclei, activates caspases 9 and 3, and resulted in TUNEL
positive cell death (4,5,24,25).
Damage from glutamate excitotoxicity is exacerbated
by oxidative stress (5) and
increasing the number of mitochondria is a way to boost the
antioxidant, detoxification functions of cells against this stress.
A study by Wang et al, showed that NAD+ protected
against glutamate-induced apoptosis through increasing
mitochondrial biogenesis (26). We
believe CoQ10 similarly protects against cell death by boosting
mitochondrial biogenesis. Protecting PGC-1α expression may be
crucial to this effect. Our results demonstrated that glutamate
markedly suppressed PGC-1α protein level and CoQ10 treatment
provided restoration of glutamate-diminished PGC-1α protein
expression, and at the same time increased cell viability and
proliferative potential. It is interesting that CoQ10 treatment
alone significantly increased mitochondrial biogenesis without much
increase in PGC-1α. The reason for this phenomenon is not quite
clear but could be because there were other unknown factors except
PGC-1a regulating mitochondrial biogenesis, PGC-1α was upregulated
at transcription level, or a mild upregulation of PGC-1α was
sufficient to increase the synthesis of mitochondrial DNA-encoded
proteins. We expanded our understanding of this mechanism by
exploring alterations to PGC-1α signaling pathways both upstream
and downstream this central regulator.
Among the many upstream facts that regulate PGC-1α,
we measured AKT and CREB. While the suppressive effect of glutamate
on p-Akt and pCREB was moderate, treatment with CoQ10 in
combination of glutamate increased p-Akt and p-CREB levels,
suggesting upregulation of PGC-1α and mitochondrial biogenesis by
CoQ10 may involve phosphorylation of Akt and CREB.
NRF1,2 and TFAM are the known downstream targets of
PGC-1α. While we did not observe a change in NRF1 in our model, we
did observe significant degradation of NRF2 from glutamate
exposure. NRF2 is a transcription factor, activated under stress
conditions, to increase survival through enhancing transcription of
genes involved in anti-inflammatory, antioxidant, and general
cytoprotective pathways (27).
Stimulated by PGC-1α, NRF2 has been shown to be a key contributor
to mitochondrial biogenesis pathways (28,29).
NRF2's upregulation after CoQ10 treatment may thus contribute to
our observed increase in mitochondrial biogenesis. This effect
could be achieved either directly activating mitochondria DNA and
protein synthesis or indirectly through activating TFAM (30,31).
TFAM is an important mitochondrial transcription factor integral to
maintaining the mitochondrial genome by regulating its packaging,
stability, and replication (32).
While we did not observe a significant decrease in TFAM after
glutamate exposure, it was significantly elevated by CoQ10
treatment. This suggest that TFAM may or may not involve
glutamate-caused suppression in mitochondrial biogenesis signaling,
but it participated surely in CoQ10 stimulated mitochondrial
biogenesis.
In summary, our results show that glutamate, while
causing severe cellular damage, affects mitochondrial biogenesis
pathways, particularly through inhibition of PGC-1α, to decrease
mitochondrial biogenesis. CoQ10 is an effective option for
mitigating this damage. In addition to preventing decreases in
PGC-1α, CoQ10 stimulates p-AKT, p-CREB, NRF2, and TFAM. Glutamate
toxicity is involved in the pathogenesis of many neurodegenerative
disorders and CoQ10 blocks the glutamate toxicity through
stimulating mitochondrial biogenesis. Therefore, CoQ10 could be
considered as complementary treatment for neurological diseases.
Further exploration of mechanisms of CoQ10-mediated mitochondrial
biogenesis may improve and broaden the clinical application of
CoQ10.
Acknowledgements
Not applicable.
Funding
Funding: The current study was supported by the National Natural
Science Foundation of China (grant no. 8156050251 to GC).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GC and PAL conceived and designed the experiments of
the current study. MAZ, MH, QQ and SLM performed the experiments.
MAZ, GC, PAL wrote the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Olivares-Banuelos TN, Chí-Castañeda D and
Ortega A: Glutamate transporters: Gene expression regulation and
signaling properties. Neuropharmacology. 161(107550)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Amantea D and Bagetta G: Excitatory and
inhibitory amino acid neurotransmitters in stroke: From
neurotoxicity to ischemic tolerance. Curr Opin Pharmacol.
35:111–119. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
You J, Feng L, Xin M, Ma D and Feng J:
Cerebral ischemic postconditioning plays a neuroprotective role
through regulation of central and peripheral glutamate. Biomed Res
Int. 2018(6316059)2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kumari S, Mehta SL, Milledge GZ, Huang X,
Li H and Li PA: Ubisol-Q10 prevents glutamate-induced cell death by
blocking mitochondrial fragmentation and permeability transition
pore opening. Int J Biol Sci. 12:688–700. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kumari S, Mehta SL and Li PA: Glutamate
induces mitochondrial dynamic imbalance and autophagy activation:
Preventive effects of selenium. PLoS One. 7(e39382)2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jing L, Kumari S, Mendelev N and Li PA:
Coenzyme Q10 ameliorates ultraviolet B irradiation induced cell
death through inhibition of mitochondrial intrinsic cell death
pathway. Int J Mol Sci. 12:8302–8315. 2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Li H, Chen G, Ma W and Li PA:
Water-soluble coenzyme Q10 inhibits nuclear translocation of
apoptosis inducing factor and cell death caused by mitochondrial
complex I inhibition. Int J Mol Sci. 15:13388–13340.
2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Jing L, He MT, Chang Y, Mehta SL, He QP,
Zhang JZ and Li PA: Coenzyme Q10 protects astrocytes from
ROS-induced damage through inhibition of mitochondrial-mediated
cell death pathways. Int J Biol Sci. 11:59–66. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Li HN, Zimmerman M, Milledge GZ, Hou XL,
Cheng J, Wang ZH and Li PA: Water soluble coenzyme Q10 reduced
rotenone-induced mitochondrial fission. Neurochem Res.
42:1096–1103. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Niyazov DM, Kahler SG and Frye RE: Primary
mitochondrial disease and secondary mitochondrial dysfunction:
Importance of distinction for diagnosis and treatment. Mol
Syndromol. 7:122–137. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Failla ML, Chitchumroonchokchai C and Aoki
F: Increased bioavailability of ubiquinol compared to that of
ubiquinone is due to more efficient micellarization during
digestion and greater GSH-dependent uptake and basolateral
secretion by Caco-2 cells. J Agric Food Chem. 62:7174–7182.
2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kalen A, Appelkvist EL and Dallner G:
Age-related changes in the lipid compositions of rat and human
tissues. Lipids. 24:579–584. 1989.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Mortenson SA, Rosenfeldt F, Kumar A,
Dolliner P, Filipiak KJ, Pella D, Alehagen U, Steurer G and
Littarru GP: Q-SYMBIO study investigators: The effect of coenzyme
Q10 on morbidity and mortality in chronic heart failure: Results
from Q-SYMBIO: A randomized double-blind trial. JACC Heart Fail.
2:641–649. 2014.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Folkers K and Simonsen R: Two successful
double-blind trials with coenzyme Q10 (vitamin Q10) on muscular
dystrophies and neurogenic atrophies. Biochim Biophys Acta.
1271:281–286. 1995.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fallah M, Askari G, Soleimani A, Feizi A
and Asemi Z: Clinical trial of the effects of coenzyme Q10
supplementation on biomarkers of inflammation and oxidative stress
in diabetic hemodialysis patients. Int J Prev Med.
10(12)2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sanoobar M, Dehghan P, Khalili M, Azimi A
and Seifar F: Coenzyme Q10 as a treatment for fatigue and
depression in multiple sclerosis patients: A double blind
randomized clinical trial. Nutr Neurosci. 19:138–143.
2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Valero T: Mitochondrial biogenesis:
Pharmacological approaches. Curr Pharm Des. 20:5507–5509.
2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Uittenbogaard M and Chiaramello A:
Mitochondrial biogenesis: A therapeutic target for
neurodevelopmental disorders and neurodegenerative diseases. Curr
Pharm Des. 20:5574–5593. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Procaccio V, Bris C, Chao de la Barca JM,
Oca F, Chevrollier A, Amati-Bonneau P, Bonneau D and Reynier P:
Perspectives of drug-based neuroprotection targeting mitochondria.
Rev Neurol (Paris). 170:390–400. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li PA, Hou X and Hao S: Mitochondrial
biogenesis in neurodegeneration. J Neurosci Res. 95:2025–2029.
2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zimmerman MA, Biggers CD and Li PA:
Rapamycin treatment increases hippocampal cell viability in an
mTOR-independent manner during exposure to hypoxia mimetic, cobalt
chloride. BMC Neurosci. 19(82)2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Guzman C, Bagga M, Kaur A, Westermarck J
and Abankwa D: ColonyArea: an ImageJ plugin to automatically
quantify colony formation in clonogenic assays. PLoS ONE.
9(e92444)2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Andrews NC and Faller DV: A rapid
micro-preparation technique for extraction of DNA-binding proteins
from limiting numbers of mammalian cells. Nucleic Acids Res.
19(2499)1991.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mehta SL, Kumari S, Mendelev N and Li PA:
Selenium preserves mitochondrial function, stimulates mitochondrial
biogenesis, and reduces infarct volume after focal cerebral
ischemia. BMC Neurosci. 13(79)2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ma YM, Ibeanu G, Wang LY, Zhang JZ, Chang
C, Dong JD, Li PA and Jing L: Selenium suppresses glutamate-induced
cell death and prevents mitochondrial morphological dynamic
alterations in hippocampal HT22 neuronal cells. BMC Neurosci.
18(15)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang X, Li H and Ding S: The effects of
NAD+ on apoptotic neuronal death and mitochondrial biogenesis and
function after glutamate excitotoxicity. Int J Mol Sci.
15:20449–20468. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dinkova-Kostova AT and Abramov AY: The
emerging role of Nrf2 in mitochondrial function. Free Radic Biol
Med. 88:179–188. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hayashi G, Jasoliya M, Sahdeo S, Saccà F,
Pane C, Filla A, Marsili A, Puorro G, Lanzillo R, Brescia Morra V
and Cortopassi G: Dimethyl fumarate mediates Nrf2-dependent
mitochondrial biogenesis in mice and humans. Hum Mol Genet.
26:2864–2873. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Bernard K, Logsdon NJ, Miguel V, Benavides
GA, Zhang J, Carter AB, Darley-Usmar VM and Thannickal VJ: NADPH
oxidase 4 (Nox4) suppresses mitochondrial biogenesis and
bioenergetics in lung fibroblasts via a nuclear factor
erythroid-derived 2-like 2 (Nrf2)-dependent pathway. J Biol Chem.
292:3029–3038. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chaturvedi RK and Beal MF: Mitochondrial
diseases of the brain. Free Radic Biol Med. 63:1–29.
2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Khatua TN, Dinda AK, Putcha UK and
Banerjee SK: Diallyl disulfide ameliorates isoproterenol induced
cardiac hypertrophy acting mitochondrial biogenesis via
eNOS-Nrf2-Tfam pathway in rats. Biochem Biophys Rep. 5:77–88.
2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kang I, Chu CT and Kaufman BA: The
mitochondrial transcription factor TFAM in neurodegeneration:
Emerging evidence and mechanisms. FEBS Lett. 592:793–811.
2018.PubMed/NCBI View Article : Google Scholar
|