Introduction
Apigenin (APG), a flavonoid widely distributed in a
variety of fruits, vegetables and herbs, including oranges, onions
and parsley (1), has been
demonstrated to be potent against cancer and effective for
chemoprophylaxis (2). Its
anticancer effects have been examined in multiple cancer types,
including breast (3), prostate
(4), colon (5) and cervical cancer (6). The anticancer mechanisms of APG are
complex. Previous studies demonstrated that APG inhibited the
growth of numerous types of cancer cell via inhibition of epidermal
growth factor receptor (7-9).
APG is also able to induce cell-cycle arrest in G2/M
phase in multiple cell lines (8,10) and
trigger apoptosis in leukemia cells (6). APG in combination with tumor necrosis
factor-α was able to reduce the cell viability and proliferation of
hepatocellular carcinoma cells (11). APG was indicated to target inhibitor
proteins of apoptosis in ovarian cancer to induce apoptotic cell
death (12). In addition, APG was
demonstrated to induce reactive oxygen species and p38, which are
essential substances in the process of cell death (7,13,14).
Furthermore, it was observed that APG induced DNA lesions involving
DNA breaks (15), micronuclei,
chromosomal aberrations (CAs) and chromatid exchanges (16-18).
APG was further demonstrated to have significant topoisomerase I
(Top1) inhibitor activities (19).
A previous in vitro study demonstrated that APG inhibited
the DNA binding or the DNA re-ligation step of Top1(20).
Induction of DNA lesions is an important mechanism
of action of numerous clinical anticancer agents. DNA double-strand
breaks (DSBs), the most severe type of DNA lesion, induces cell
death if not repaired (21). DSBs
were induced by numerous exogenous factors, including ionizing
radiation (IR) (22) and endogenous
factors, including topoisomerase (23,24).
Camptothecin (CPT), a DNA Top1 inhibitor, has been widely used in
the clinic as an anticancer drug. Regarding the mechanism of
action, CPT has been demonstrated to selectively bind to and
stabilize the 3'phospho-tyrosyl bond formed between Top1 and DNA
[Top1 covalent protein DNA complex (Top1cc)] to trap Top1 in
Top1cc, which inhibits DNA and RNA synthesis when colliding with
the replication fork or transcription machinery to cause DSBs and
cell death (25-28).
To avoid CPT-induced cell death, two major DNA repair pathways are
in place: One is the removal of covalent 3'-DNA adducts by
tyrosyl-DNA phosphodiesterase I (TDP1), which hydrolyzes the
drug-stabilized 3'phospho-tyrosyl bond of Top1cc (29,30);
the other is DSB repair by homologous recombination (HR) (31). Cells deficient of either TDP1 or HR
factors, including Rad54, were demonstrated to enhance DNA damage
and cell death (32,33).
The DT40 cell line is derived from bursal
B-lymphocyte cells (34). It has a
stable karyotype and exhibits extraordinarily high gene-targeting
efficiency; thus, it has been widely used in genetic studies,
including comprehensive research on the mechanisms of DNA damage
and repair (35-38).
In the present study, the sensitivity to APG, induction of DNA
damage and formation of Top1cc were investigated using wild-type
(WT) DT40 cells, as well as DT40 cells with deletions in
several DNA repair genes. The TK6 cell line, a human lymphoblastoid
cell line, has numerous advantages, including stable spontaneous
mutation frequencies, ability to grow in suspension and simplicity
of culture. As a human-derived cell line, TK6 has been widely used
for genotoxicity testing (39).
In the present study, it was identified that Rad54,
an HR factor, has a critical role in the tolerance to APG-induced
DNA damage in DT40 cells. The
Rad54-/- TK6 cell line was
also used to verify these results and it was demonstrated that
human Rad54 has the same function in response to APG as that in the
chicken DT40 cell line.
Materials and methods
Chemicals
APG and CPT were purchased from MedChemExpress. APG
(50 mM) and CPT (100 µM) were prepared and stored at -20˚C. The
chemicals were dissolved with DMSO to produce stock solutions.
Cell culture
DT40 cells and human lymphoblast TK6 cells were
provided by Dr Shunichi Takeda (Department of Radiation Genetics,
Graduate School of Medicine, Kyoto University, Kyoto, Japan;
Table I). DT40 cells were
maintained in RPMI-1640 medium (Wisent, Inc.) containing 10%
heat-inactivated FBS (Wisent, Inc.), 1% chicken serum (Gibco;
Thermo Fisher Scientific, Inc.), 1% penicillin-streptomycin
(Wisent, Inc.) and 50 µM β-mercaptoethanol (Gibco; Thermo Fisher
Scientific, Inc.). The medium for TK6 cells was RPMI-1640,
supplemented with 10% heat-inactivated horse serum (HyClone;
Cytiva) and 1% penicillin-streptomycin. All cell lines were
maintained at 37˚C in a humidified atmosphere with 5%
CO2.
 | Table IDNA repair genes mutated in the DT40
and TK6 clones analyzed. |
Table I
DNA repair genes mutated in the DT40
and TK6 clones analyzed.
| Gene | Name of cell
line | Function | Reference |
|---|
| Fen1 | Chicken DT40 | Base excision
repair, processing 5'flap in long-patch and lagging strand DNA | (48) |
| Rad18 | Chicken DT40 | TLS | (49) |
| Parp1 | Chicken DT40 | Poly(ADP)
ribosylation, related to single-strand break and BER | (50) |
| Rad54 | Chicken DT40 | DSB repair by
HR | (51) |
| Rad54 | Human TK6 | DSB repair by
HR | (53) |
| TDP1 | Human TK6 | Tyrosyl-DNA
phosphodiesterase I | (54) |
Cell viability assay
The cells were exposed to APG (0, 10, 20 and 30 µM)
and CPT (0, 10, 20, 30 and 40 nM) for 72 h, and subsequently, 20 µl
Cell Counting Kit-8 (CCK-8; MedChemExpress) reagent was added to
each well. The absorbance of each well was measured at 450 nm using
a microplate reader (SynergyMx; BioTek Instruments, Inc.). Cell
survival curves were constructed from the CCK-8 assay as previously
described (40). SPSS software
version 20.0 (IBM Corp.) was used to calculate the survival
percentage compared with the control. Relative IC50
value = (The IC50 of deletion cells/The IC50
of WT cells) x100%.
Cell-cycle assays
Flow cytometry was used to determine cell-cycle
arrest (41). In total,
4x105 cells were cultured with 20 µΜ APG or 6 nM CPT for
16 h. Subsequently, the cells were fixed with 70% ethanol for 4˚C
overnight prior to staining with propidium iodide (PI) in the
presence of RNase A (cat. no. ST579; Beyotime Institute of
Biotechnology) (42). The results
were determined using a flow cytometer (CytoFLEX; Beckman Coulter,
Inc.).
Colony formation assay
The colony formation assay was performed as
previously described (39,43). The medium for DT40 cells included
various amounts of APG, mixed with DMEM-F12 (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 15% heat-inactivated FBS
(Wisent, Inc.), 1.5% chicken serum (Wisent, Inc.), 1.5% (w/v)
methylcellulose, 200 mΜ L-glutamine (Gibco; Thermo Fisher
Scientific, Inc.) and 50 µΜ β-mercaptoethanol (Gibco; Thermo Fisher
Scientific, Inc.) using a slowly rotating shaker overnight at 4˚C.
The medium for TK6 cells contained various amounts of drugs, which
were mixed with 1.5% (w/v) methylcellulose medium containing 10%
heat-inactivated horse serum using slowly rotating tubes overnight
at 4˚C. Cells (100 cells/well) were seeded into six-well plates
containing 3 ml methylcellulose medium per well and after 14 days,
visible white colonies were counted in each well.
Immunofluorescence staining and image
analysis
Immuno-fluorescence staining was performed as
previously described (43). In
total, 5x105 cells were treated with 30 µM APG or 100 nM
CPT for 3, 6 and 9 h. The cells were then fixed with 4%
paraformaldehyde for 10 min at room temperature. After washing with
PBS, the cells were permeabilized with 0.1% Nonidet P-40. The cells
were then blocked with PBS containing 3% BSA (cat. no. A8020;
Beijing Solarbio Science & Technology Co., Ltd.) at room
temperature for 30 min and incubated with a mouse monoclonal
γ-phosphorylated H2A.X variant histone (γ-H2AX; Ser139) antibody
(1:1,000; cat. no. 05-636; EMD Millipore) at 4˚C overnight.
Anti-mouse Alexa Fluor 488-conjugated antibody (1:1,000; cat. no.
A0216; Beyotime Institute of Biotechnology) was used as the
secondary antibody at 37˚C for 1 h. The γ-H2AX foci were visualized
under a fluorescent microscope (AX10 imager A2/AX10 Cam HRC;
Zeiss). The nuclei were stained with DAPI for 10 min at room
temperature. The foci of 100 nuclei were counted per well as
described in previous reports using Photoshop (version 12.0.3;
Adobe Systems, Inc.) (39,43). Experiments were performed three
times independently.
Detection of CAs
Karyotype analysis was performed according to a
previous study (44). In brief,
cells were treated with APG or CPT for different durations. At 3 h
prior to harvesting, 0.2 µg/ml colchicine solution (Gibco; Thermo
Fisher Scientific, Inc.) was added. The suspended cells were
incubated in 75 mM KCl solution for 25 min and then fixed in
Carnoy's solution. The cell suspension was dripped onto
ethanol-soaked glass slides and dried using a flame. The dried
slides were stained with 5% Giemsa solution and rinsed with
water.
In vitro complex of enzyme (ICE)
assay
Top1-DNA adducts were isolated using the ICE
Bio-assay as described in a previous study (36). In total, 1.5x107 cells
per sample were treated with APG or CPT and then suspended in 2 ml
buffer A (10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 0.34 M
sucrose, 10% glycerol, 0.1% Triton X-100; 1 mM phenylmethylsulfonyl
fluoride) and put on ice for 10 min. The precipitate was then
resuspended in 2 ml buffer B (2 mM EDTA, 0.3 mM egtazic acid, 0.1%
Triton X-100 and protease inhibitor) and incubated for 30 min on
ice. The chromatin sediment was washed with buffer C (25 mM
Tris-Cl, 300 mM NaCl, 10% glycerol and protease inhibitor) five
times. After being resuspended in 2 ml 6 M guanidinium chloride,
DNA solutions were loaded onto the top of a cushion of CsCl
(densities of CsCl solutions: 1.82, 1.7, 1.5 and 1.45 g/ml) and
samples were centrifuged at 100,000 x g for 16 h at room
temperature. Subsequently, 1 ml supernatant of each part was
collected. For dot blot analysis, 100 µl of each fraction was
loaded onto a Bio-Dot apparatus (cat. no. 1706545; Bio-Rad
Laboratories, Inc.). After washing with Tris-buffered saline with
Tween-20, the membrane was incubated with anti-Top1 antibody at 4˚C
for 24 h (1:10,000; cat. no. ab109374; Abcam) for 24 h and
subsequently incubated with an anti-rabbit antibody conjugated with
horseradish peroxidase at 37˚C for 1 h (1:1,000; cat. no. A0208;
Beyotime Institute of Biotechnology).
Statistical analysis
Statistical significance of differences was examined
using Student's t-test or two-way analysis of variance with
Bonferroni's test. Statistical analysis was performed with SPSS
25.0 software (IBM Corp.). P<0.05 was considered to indicate a
statistically significant difference.
Results
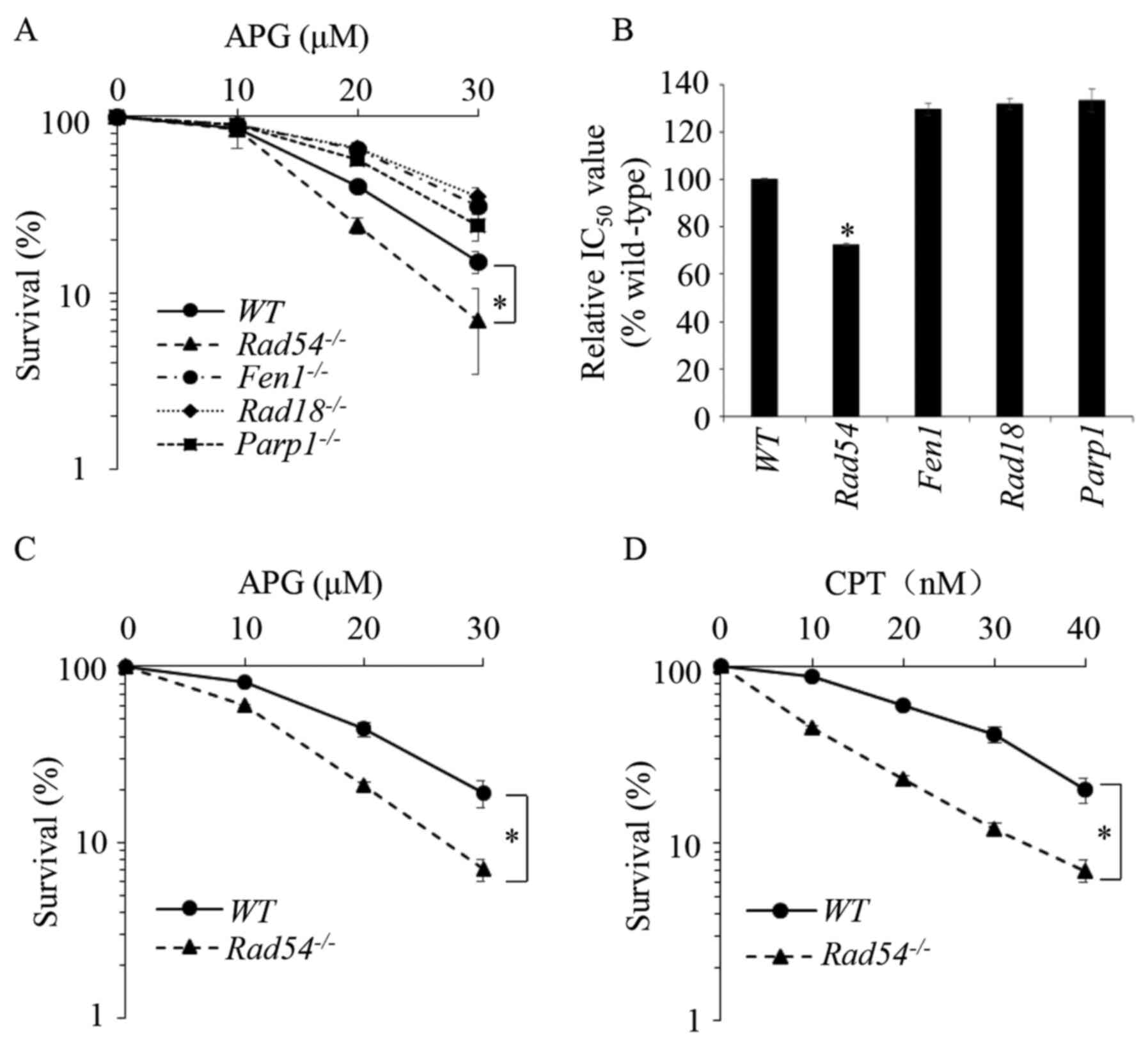
Rad54-/- DT40 cells are sensitized
to APG
DT40 chicken B-lymphocyte cells (45) have been widely used in research on
DNA lesions and repair (46,47).
To examine the molecular mechanisms of APG-induced DNA lesions and
their repair, the effects of APG on the viability of WT DT40
cells, as well as DT40 cells deletions in several DNA repair genes,
flap structure-specific endonuclease 1
(FEN1)-/- (48),
Rad18-/- (49), poly(ADP-ribose) polymerase 1
(PARP1)-/- (50) and
Rad54-/- (51), were studied and compared. Those
cells were separately defective in several DNA repair pathways
(Table I). Cells were treated with
different concentrations of APG for 72 h and a CCK-8 assay was used
to determine their viability. The 50% inhibitory concentration was
calculated using SPSS 20.0 software (IBM Corp.). As presented in
Fig. 1A and B,
Rad54-/- cells deficient of
HR repair demonstrated significant sensitivity to APG, while the
cells deficient in other DNA repair pathways, were not sensitive to
APG. The sensitivity of
Rad54-/- cells to APG was
also demonstrated in the colony formation assay, as presented in
Figs. 1C and S1A. CPT, a Top1 inhibitor, which has been
previously demonstrated to sensitize
Rad54-/- DT40 cells
(52), was used as the positive
control, as presented in Fig.
1D.
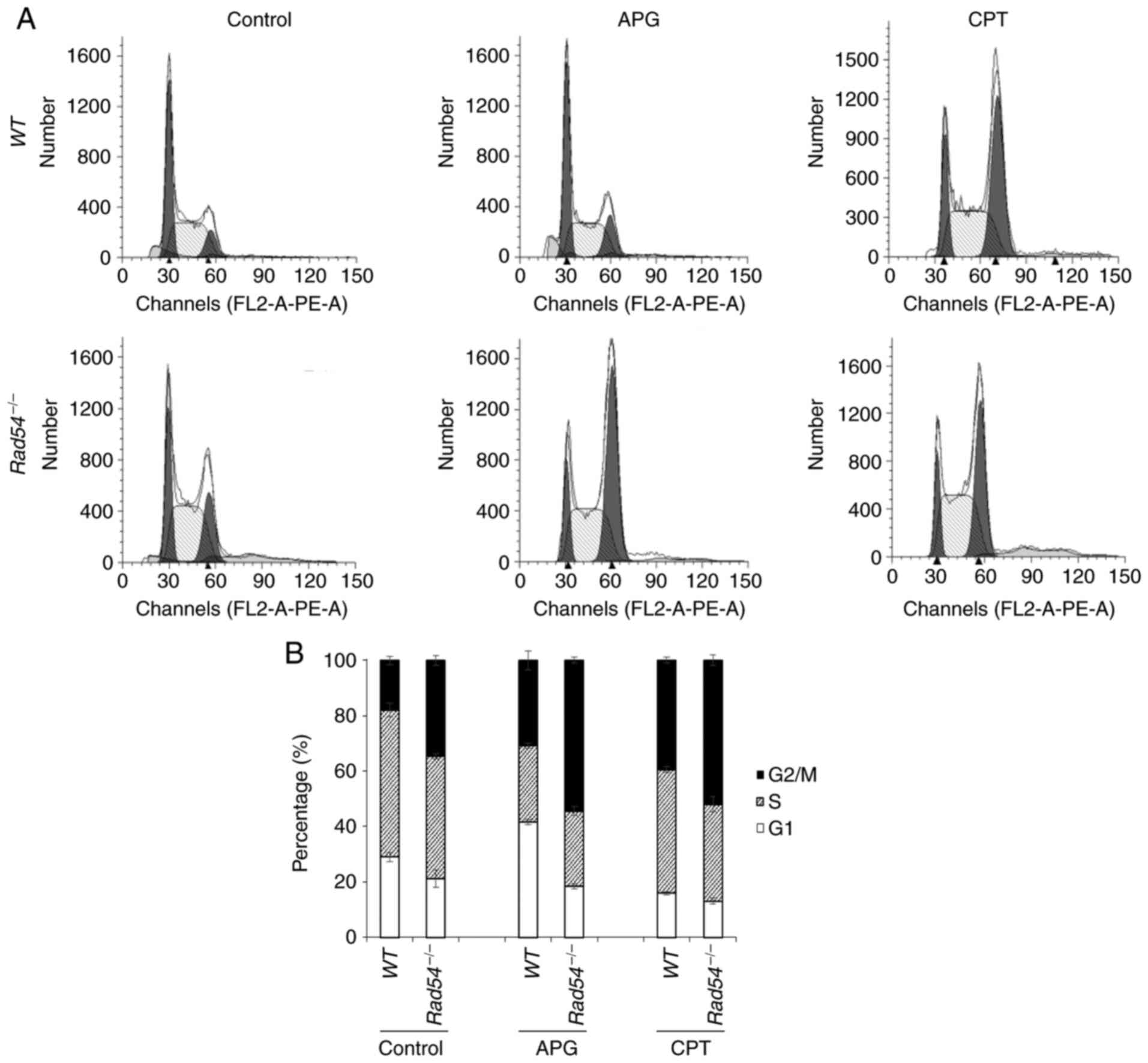
APG treatment leads to
G2/M-phase cell-cycle arrest of
Rad54-/- DT40 cells
Changes in the cell cycle of
Rad54-/- DT40 cells
compared with WT cells were determined. After 16 h of APG
treatment, flow cytometric analysis of PI-stained cells
demonstrated that the WT and
Rad54-/- cells were
arrested at the G2/M phase of the cell cycle.
Furthermore, APG led to a higher degree of accumulation of in the
G2/M phase for
Rad54-/- cells compared
with that in WT cells. CPT also promoted
G2/M-phase arrest of
Rad54-/- cells (Fig. 2).
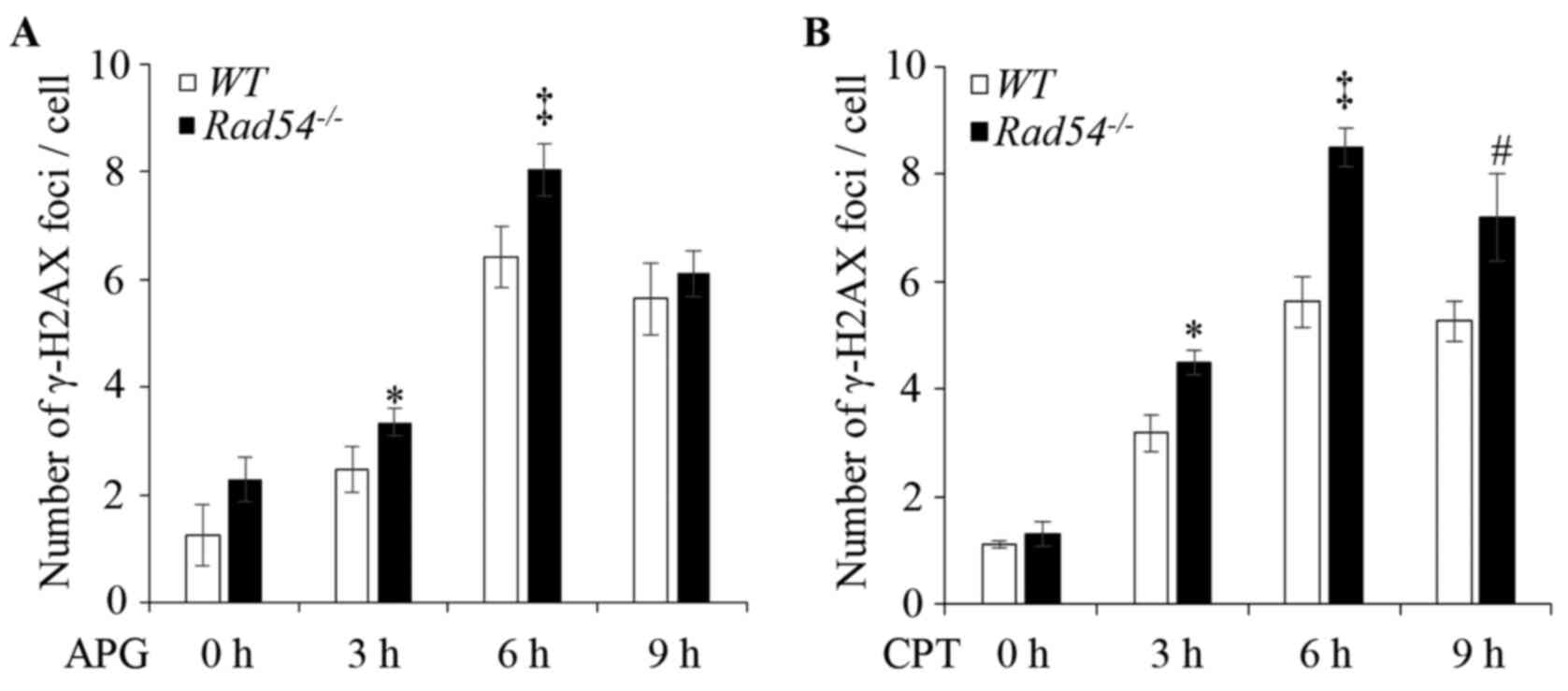
APG treatment produces more DSBs in
Rad54-/- DT40 cells
To verify the effect of APG to induce DNA damage in
DT40 cells, the APG-induced γ-H2AX foci in DT40 cells were
quantified. DT40 WT and
Rad54-/- cells were treated
with 30 µM APG for 0, 3, 6 and 9 h. As presented in Figs. 3A and S2A, the numbers of γ-H2AX foci were
significantly increased in the nuclei and peaked at 6 h after APG
exposure in both WT and
Rad54-/- cells. However, in
the Rad54-/- cells, γ-H2AX
foci increased more significantly in response to APG (Figs. 3A and S2A) or CPT treatment (Figs. 3B and S2A). The patterns of γ-H2AX foci induced
by APG and CPT in Rad54-/-
cells were similar. However, compared with the WT, the
increase of CPT-induced γ-H2AX foci in
Rad54-/- cells was
significantly greater than that induced by APG at 9 h.
APG-induced DSBs in
Rad54-/- cells were also
compared with the WT by measuring CAs in DT40 cells. Cells
were incubated with 20 µM APG or 10 nM CPT and CAs were confirmed
dynamically at 8, 16 and 24 h. The largest number of CAs was
counted after 16 h of treatment with APG (Fig. 4A and B) or CPT (Fig.
4C). As presented in Fig. 4,
Rad54-/- cells had
significantly increased numbers of CAs than WT cells in
response to APG or CPT treatment. These results suggested that
Rad54 participated in repairing APG-induced DNA damage.
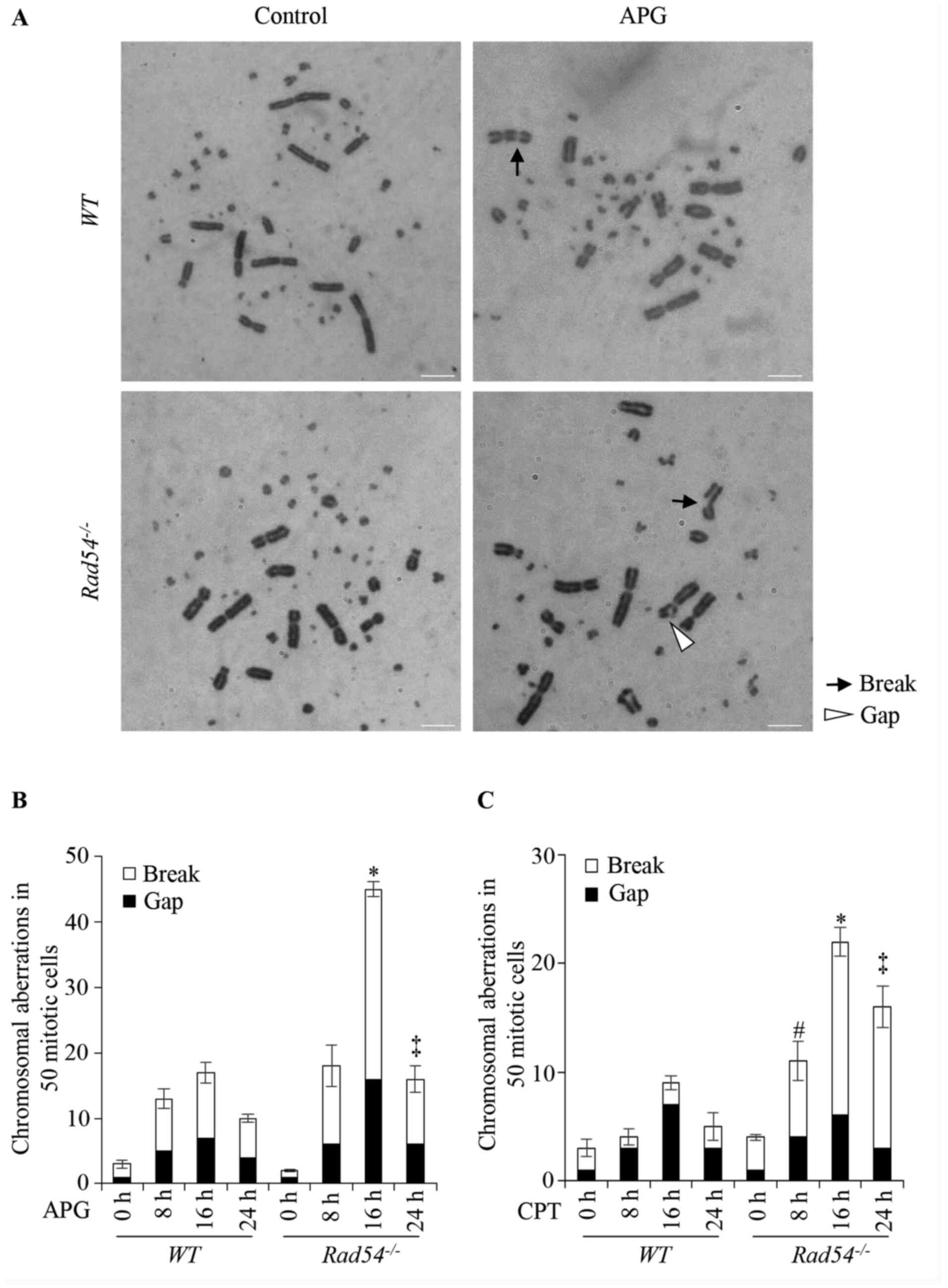 | Figure 4APG induces double-strand breaks in
DT40 cells. (A) Representative karyotype analysis of untreated DT40
cells and cells treated with 20 µM APG. Magnification, x1,000.
Black arrows indicate breaks whereas white arrowheads indicate
gaps. (B) CAs in WT and Rad54-/- cells
after treatment with APG (20 µM) for 8-24 h. (C) CAs in WT
and Rad54-/- cells after treatment with CPT (10
nM) for 8-24 h. In total, 50 metaphase cells per each experiment
were analyzed under a light microscope (magnification, x1,000).
Values are expressed as the mean ± standard deviation. Experiments
were performed three times independently. #P<0.05,
total number of breaks and gaps in Rad54-/- cells
compared with WT DT40 cells after treatment for 8 h;
*P<0.05, total number of breaks and gaps in
Rad54-/- cells compared with WT DT40 cells
after treatment for 16 h; ‡P<0.05, total number of
breaks and gaps compared with WT DT40 cells after treatment
for 24 h. APG, apigenin; CAs, chromosomal aberrations; CPT,
camptothecin; WT, wild-type. |
APG traps Top1cc in
Rad54-/- DT40 cells
It was then was further assessed whether APG induced
Top1cc in DT40 cells. Cells were lysed and Top1cc was separated
from free Top1 in the cellular lysates by subjecting cellular
extract to CsCl gradient ultracentrifugation. Free Top1 remained in
the top two fractions, while Top1cc moved to lower fractions of the
CsCl gradient corresponding to the migration of chromosomal DNA.
DT40 cells were cultivated with 100 µM APG or CPT for 2 h and an
ICE assay was used to detect Top1cc and free Top1. As presented in
Fig. 5, fractions 1-2 were mainly
free protein and Top1cc peaked at fraction 3. The results in
Fig. 5 demonstrated that both APG
and CPT induced more accumulation of Top1cc in
Rad54-/- cells than in
WT cells (Fig. 5). This
observation suggested that APG is able to trap Top1 to remain in a
covalent protein-DNA complex in DT40 cells, suggesting that APG is
a Top1 inhibitor.
APG induces sensitivity and increases
DSBs and Top1cc in Rad54-/- and
TDP1-/- TK6 cells
In order to identify whether human Rad54 was also
involved in tolerance of DNA lesions induced by APG, the effects of
APG in Rad54-/- TK6 cells
were studied and compared (53). As
presented in Figs. 6A and B, and S1B,
Rad54-/- TK6 cells were
more sensitive to APG or CPT than WT cells in the colony
formation assay. Following treatment with 80 µM APG or 20 nM CPT
for 0, 3, 6 and 9 h, the number of γ-H2AX foci in
Rad54-/- TK6 cells was
significantly increased as compared with that in WT TK6
cells (Figs. 6C and D, and S2B). Furthermore, Top1cc was observed in
TK6 cells after treatment with 100 µM APG or 2 nM CPT for 2 h, and
the drugs induced increased Top1cc in
Rad54-/- compared with
WT cells (Fig. 6E). These
data were consistent with the results obtained with DT40 cells,
suggesting that APG may be a human Top1 inhibitor, which may induce
Rad54-dependent tolerance of DNA damage.
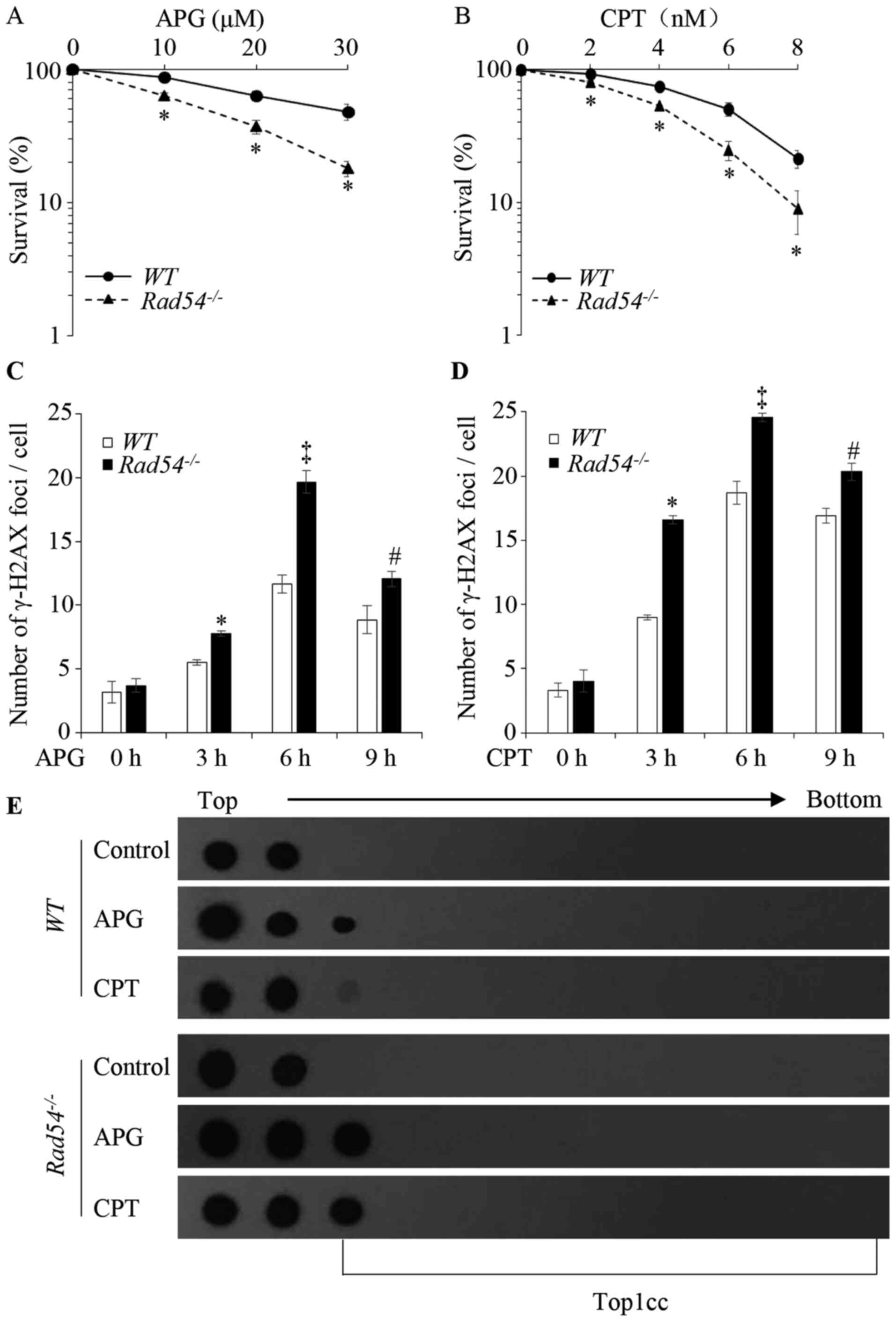 | Figure 6Effects of APG on the proliferative
ability and DNA damage in Rad54-/- TK6 cells.
Proliferative ability of (A) APG- and (B) CPT-treated TK6 cells
determined by clonogenic assays. The x-axis represents the
concentration of the drugs and the y-axis represents the fractions
of surviving colonies on a logarithmic scale. Survival data were
log-transformed to approximate normality. Two-way analysis of
variance was used to test for differences in the linear
dose-response curves between WT and
Rad54-/- cells. *P<0.05,
Rad54-/- cells compared with WT. (C)
Quantification of γ-H2AX foci in WT and
Rad54-/- cells after treatment with 80 µM APG.
(D) Quantification of γ-H2AX foci in WT and
Rad54-/- cells after treatment with 20 nM CPT.
TK6 cells were treated for 0, 3, 6 and 9 h, respectively; 100 cells
were analyzed for each data-point. (E) APG induced the formation of
Top1cc in TK6 cells. TK6 cells were treated with 100 µM APG and 2
µM CPT for 2 h to detect Top1cc. In the dot blot, top to bottom
indicates the order in which the fractions were taken out of the
sample after ultracentrifugation. Values are expressed as the mean
± standard deviation. Experiments were performed three times
independently. *P<0.05, compared with WT after
treatment for 3 h; ‡P<0.05, compared with WT
after treatment for 6 h; #P<0.05, compared with
WT after treatment for 9 h. APG, apigenin; CPT,
camptothecin; WT, wild-type; γ-H2AX, γ-phosphorylated H2A.X
variant histone; Top1cc, topoisomerase I covalent protein DNA
complex. |
Based on the function of TDP1 in Top1-associated DNA
damage repair by hydrolyzing the 3' phospho-tyrosyl bond of Top1cc
(54,55), it was hypothesized that cells
deficient in TDP1 should also be sensitive to APG. Therefore, the
cytotoxic effects in
TDP1-/- TK6 cells were
investigated after treating cells with different concentrations of
APG (0, 10, 20 and 30 µM) and CPT (0, 2, 4, 6 and 8 nM) for 72 h.
The results suggested that
TDP1-/- TK6 cells were more
sensitive to APG than WT cells with similar effects of CPT
in the colony formation assay (Figs.
S1B and S3A and B). Subsequently, the effects of
APG-induced DNA-damage on
TDP1-/- cells were
examined. WT and
TDP1-/- TK6 cells were
treated with 80 µM APG or 20 nM CPT for 0, 3, 6 and 9 h. The
results indicated that the numbers of γ-H2AX foci in the nuclei
were significantly increased and peaked at 6 h after APG exposure
in WT and TDP1-/-
TK6 cells. In TDP1-/- TK6
cells, γ-H2AX foci were increased more significantly in response to
APG or CPT treatment compared with those in WT cells
(Figs. S2B and S3C and D). These results suggested that APG or
CPT induced more DNA damage in
TDP1-/- TK6 cells.
Furthermore, Top1cc was observed in
TDP1-/- TK6 cells after
treatment with 100 µM APG or 2 nM CPT for 2 h, and the results
demonstrated that APG or CPT induced more Top1cc in
TDP1-/- TK6 cells than in
WT cells (Fig. S3E). These
results further suggested that APG is a Top1 inhibitor, which is
able to trap Top1 in Top1cc, and increased Top1cc was observed in
TDP1-/- vs. WT
cells.
Discussion
It has been demonstrated that APG possesses a
series of biological effects, including antiviral, antibacterial,
antioxidant, anti-inflammatory and anticancer effects (3,56-59).
APG was also indicated to cause DNA lesions (60,61).
In the present study, DT40 cells with deletions of various DNA
damage repair genes were used to examine the possible molecular
mechanisms of APG-induced DNA damage repair, and it was identified
that Rad54-/- DT40 cells
were specifically sensitive to APG. The cell-cycle assay
demonstrated that APG also induced more cell-cycle arrest in
G2/M phase in
Rad54-/- than in WT
cells.
Rad54 is the most highly conserved eukaryotic HR
protein, which is involved in various activities that contribute to
the progression of HR (62). The
Rad54 protein is part of a nucleoprotein filament to confer DNA
homology (63,64). It interacts with the Rad51
nucleoprotein filament and induces the activity of DNA pairing in
HR (51). HR is an important DNA
repair pathway for DSBs. It repairs DSBs in the S phase (62). It was demonstrated that lack of
Rad54 in DT40 cells leads to sensitivity to endogenous factors,
including topoisomerase (65), and
exogenous factors, such as IR (66), which is able to induce DSBs in the
S-phase (67,68). In the present study,
Rad54-/- DT40 cells were
sensitive to APG, demonstrating that APG may induce DSBs that are
tolerated in the presence of Rad54. Cell-cycle arrest is an
important cell response to DNA lesions that is initiated by
activating cell-cycle checkpoint proteins. It was previously
demonstrated that APG caused arrest of the cell cycle in
G0/G1-phase by reducing the level of cyclin
D1 in LNCaP and PC-3 prostate cancer cell lines (1). However, in other studies, APG caused
cell-cycle arrest at the G2-phase by decreasing cyclin B
in human colon carcinoma and pancreatic cancer cells (10), or regulated other cell-cycle phases
by affecting cyclin A and cell division cycle 25A and cell division
cycle 25C in different cell types (8,69-71).
The results of the cell-cycle assay of the present study suggested
that APG arrested Rad54-/-
cells in G2/M-phase. To identify whether APG induced
DSBs, the γ-H2AX foci were quantified as a marker for DSBs and CAs
representing stable DSBs were also evaluated. Analysis of both
γ-H2AX foci and CAs identified that APG induced increased DSBs in
Rad54-/- cells compared
with the WT cells. The results suggested that the enhanced
sensitivity and increased amount of cell-cycle arrest of
Rad54-/- cells in
G2/M-phase were associated with the induction of DSBs by
APG.
HR has been identified as the essential
Top1-associated repair pathway for DSBs (72). Numerous flavonoids, including APG,
quercetin, kaempferol and morin, have been identified as Top1
inhibitors (20,73). Previous studies (20,73)
reported that several flavonoids, including APG, were similar to
CPT and were able to stabilize Top1cc in vitro and in
vivo. Consistent with these previous studies, the present study
demonstrated that APG induced significant Top1cc formation in
Rad54-/- cells.
Furthermore, human Rad54-/-
TK6 cells exhibited the same sensitivities to APG and increased
DSBs and Top1cc formation as
Rad54-/- DT40 cells. These
results suggested that APG may be a Top1 inhibitor, which traps
Top1 in the form of Top1cc. When Top1cc encounters a replication
fork, it leads to cell-cycle arrest and even DSBs, which is mainly
repaired by HR. Lack of Rad54 affects the repair of DSBs caused by
the sustained existence of Top1cc.
Top1 is an important enzyme for DNA replication,
transcription, recombination and chromatin remodeling, which
relaxes the supercoil structure of DNA molecules by cutting one
strand of duplex DNA and generating Top1cc that causes supercoiled
DNA to untwist (74). Under
physiological conditions, Top1cc is resolved by TDP1, which
catalyzes hydrolysis of the Top1 tyrosine residue covalently linked
to the 3'-phosphate of DNA, removes Top1 from the DNA 3'-end so
that the broken DNA strand is religated (75). A previous study has demonstrated
that deficiency of TDP1 leads to accumulation of Top1cc and DSBs,
and a Top1 inhibitor, e.g. CTP, was able to enhance accumulation of
Top1cc and DSBs (76). In the
present study, it was identified that TK6 cells deficient of TDP1
were also sensitive to APG, which generated increased DSBs and
Top1cc. This result further suggested that APG is a Top1
inhibitor.
To the best of our knowledge, the present study was
the first to examine the role of Rad54 in the tolerance of
APG-induced Top1-mediated DNA damage by using DNA repair-deficient
DT40 and TK6 cells. The data suggested that inhibition of Rad54 may
enhance the toxicity of APG to tumor cells. The present results
also suggested that APG-induced toxicity was enhanced by Rad54
deletion, such that Rad54 may be included in the potential DNA
rearrangements caused by APG for the treatment of other diseases.
The present study provided insight for developing novel anticancer
medicines with higher therapeutic efficacy and less
genotoxicity.
Supplementary Material
Representative images of colony
formation assays in DT40 and TK6 cells. (A) DT40 cells treated with
APG. (B) TK6 cells treated with APG. The representative images were
taken with a camera. APG, apigenin; WT, wild-type; TDP1,
tyrosyl-DNA phosphodiesterase 1.
Representative images of
immunostaining in DT40 and TK6 cells. (A) Immunostaining of
WT and Rad54-/- DT40 cells using an anti-γ-H2AX
antibody and DAPI. Cells were treated with 30 μM APG or 100 nM CPT
for 6 h (scale bar, 10 μM; magnification, x1,000). (B)
Immunostaining of WT, Rad54-/- and TDP1-/- TK6
cells using an anti-γ-H2AX antibody and DAPI. Cells were treated
with 80 μM APG or 20 nM CPT for 6 h (scale bar, 10 μM;
magnification, x1,000). TDP1, tyrosyl-DNA phosphodiesterase 1;
WT, wild-type; γ-H2AX, γ-phosphorylated H2A.X variant
histone; APG, apigenin; CPT, camptothecin.
Effects of APG on proliferative
ability and DNA damage in TDP1-/- TK6 cells. Proliferative
ability of (A) APG- and (B) CPT-treated TK6 cells determined by
colony formation assays. The x-axis represents the concentration of
drugs and the y-axis represents the fractions of surviving colonies
on a logarithmic scale. Survival data were log-transformed to
approximate normality. Two-way analysis of variance was used to
test for differences in the linear dose-response curves between
WT and TDP1-/- cells after 72 h.
*P<0.05 (C) Quantification of γ-H2AX foci in
WT and TDP1-/- cells after treatment with 80 μM APG.
(D) Quantification of γ-H2AX foci in WT and TDP1-/-
cells after treatment with 20 nM CPT. TK6 cells were treated for 0,
3, 6 and 9 h; 100 cells were analyzed for each data-point. (E) APG
induced formation of Top1cc in TK6 cells. TK6 cells were treated
with 100 μM APG and 2 μM CPT for 2 h to detect Top1cc. Values are
expressed as the mean ± standard deviation. The experiments were
performed three times independently. *P<0.05,
compared with WT after treatment for 3 h; ‡P<0.05,
compared with WT after treatment for 6 h; #P<0.05,
compared with WT after treatment for 9 h. APG, apigenin;
CPT, camptothecin; WT, wild-type; Top1cc, topoisomerase I
covalent protein DNA complex; TDP1, tyrosyl-DNA phosphodiesterase
1; γ-H2AX, γ-phosphorylated H2A.X variant histone.
Acknowledgements
The authors would like to thank Dr Hiroyuki
Sasanuma (Department of Radiation Genetics, Graduate School of
Medicine, Kyoto University, Koyot, Japan) for providing the
complete protocol of in vitro complex of enzyme
analysis.
Funding
The present study was supported by the Fundamental Research
Funds for the Central Universities and The 111 Project (grant no.
B18035).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
YQ, XW, FH and ZZ conceived and designed the
experiments. ZZ and CX performed the experiments. ZZ, YQ, XW, FH,
CX, XF and XL analyzed the data. ZZ, YQ, XW, CX, JZ and XB
contributed reagents/materials/analysis tools. ZZ, XW and YQ wrote
the manuscript. ST was responsible for the generation of gene
knock-out cells. XB and JZ developed the method for the ICE assay,
verified the results and ensured that the results could be
reproduced. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shukla S, Bhaskaran N, Babcook MA, Fu P,
Maclennan GT and Gupta S: Apigenin inhibits prostate cancer
progression in TRAMP mice via targeting PI3K/Akt/FoxO pathway.
Carcinogenesis. 35:452–460. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tong X and Pelling JC: Targeting the
PI3K/Akt/mTOR axis by apigenin for cancer prevention. Anticancer
Agents Med Chem. 13:971–978. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Perrott KM, Wiley CD, Desprez PY and
Campisi J: Apigenin suppresses the senescence-associated secretory
phenotype and paracrine effects on breast cancer cells.
Geroscience. 39:161–173. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Shukla S, Kanwal R, Shankar E, Datt M,
Chance MR, Fu P, MacLennan GT and Gupta S: Apigenin blocks IKKα
activation and suppresses prostate cancer progression. Oncotarget.
6:31216–31232. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Shao H, Jing K, Mahmoud E, Huang H, Fang X
and Yu C: Apigenin sensitizes colon cancer cells to antitumor
activity of ABT-263. Mol Cancer Ther. 12:2640–2650. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Souza RP, Bonfim-Mendonça PS, Gimenes F,
Ratti BA, Kaplum V, Bruschi ML, Nakamura CV, Silva SO, Maria-Engler
SS and Consolaro ME: Oxidative stress triggered by apigenin induces
apoptosis in a comprehensive panel of human cervical cancer-derived
cell lines. Oxid Med Cell Longev. 2017(1512745)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yin F, Giuliano AE, Law RE and Van Herle
AJ: Apigenin inhibits growth and induces G2/M arrest by modulating
cyclin-CDK regulators and ERK MAP kinase activation in breast
carcinoma cells. Anticancer Res. 21:413–420. 2001.PubMed/NCBI
|
|
8
|
Wang W, Heideman L, Chung CS, Pelling JC,
Koehler KJ and Birt DF: Cell-cycle arrest at G2/M and growth
inhibition by apigenin in human colon carcinoma cell lines. Mol
Carcinog. 28:102–110. 2000.PubMed/NCBI
|
|
9
|
Caltagirone S, Rossi C, Poggi A,
Ranelletti FO, Natali PG, Brunetti M, Aiello FB and Piantelli M:
Flavonoids apigenin and quercetin inhibit melanoma growth and
metastatic potential. Int J Cancer. 87:595–600. 2000.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ujiki MB, Ding XZ, Salabat MR, Bentrem DJ,
Golkar L, Milam B, Talamonti MS, Bell RH Jr, Iwamura T and Adrian
TE: Apigenin inhibits pancreatic cancer cell proliferation through
G2/M cell cycle arrest. Mol Cancer. 5(76)2006.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Chen Z, Chen J, Liu H, Dong W, Huang X,
Yang D, Hou J and Zhang X: The SMAC mimetic APG-1387 sensitizes
immune-mediated cell apoptosis in hepatocellular carcinoma. Front
Pharmacol. 9(1298)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Li BX, Wang HB, Qiu MZ, Luo QY, Yi HJ, Yan
XL, Pan WT, Yuan LP, Zhang YX, Xu JH, et al: Novel smac mimetic
APG-1387 elicits ovarian cancer cell killing through TNF-alpha,
Ripoptosome and autophagy mediated cell death pathway. J Exp Clin
Cancer Res. 37(53)2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Vargo MA, Voss OH, Poustka F, Cardounel
AJ, Grotewold E and Doseff AI: Apigenin-induced-apoptosis is
mediated by the activation of PKCdelta and caspases in leukemia
cells. Biochem Pharmaco. 72:681–692. 2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Shukla S and Gupta S: Apigenin-induced
cell cycle arrest is mediated by modulation of MAPK, PI3K-Akt, and
loss of cyclin D1 associated retinoblastoma dephosphorylation in
human prostate cancer cells. Cell Cycle. 6:1102–1114.
2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Meng S, Zhu Y, Li JF, Wang X, Liang Z, Li
SQ, Xu X, Chen H, Liu B, Zheng XY, et al: Apigenin inhibits renal
cell carcinoma cell proliferation. Oncotarget. 8:19834–19842.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Farooqi AA, Wu SJ, Chang YT, Tang JY, Li
KT, Ismail M, Liaw CC, Li RN and Chang HW: Activation and
inhibition of ATM by phytochemicals: Awakening and sleeping the
guardian angel naturally. Arch Immunol Ther Exp (Warsz).
63:357–366. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Noel S, Kasinathan M and Rath SK:
Evaluation of apigenin using in vitro cytochalasin blocked
micronucleus assay. Toxicol In Vitro. 20:1168–1172. 2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Papachristou F, Chatzaki E, Petrou A,
Kougioumtzi I, Katsikogiannis N, Papalambros A, Tripsianis G,
Simopoulos C and Tsaroucha AK: Time course changes of anti- and
pro-apoptotic proteins in apigenin-induced genotoxicity. Chin Med.
8(9)2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Song J, Parker L, Hormozi L and Tanouye
MA: DNA topoisomerase I inhibitors ameliorate seizure-like
behaviors and paralysis in a Drosophila model of epilepsy.
Neuroscience. 156:722–728. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Boege F, Straub T, Kehr A, Boesenberg C,
Christiansen K, Andersen A, Jakob F and Köhrle J: Selected novel
flavones inhibit the DNA binding or the DNA religation step of
eukaryotic topoisomerase I. J Biol Chem. 271:2262–2270.
1996.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Adachi N, Suzuki H, Iiizumi S and Koyama
H: Hypersensitivity of nonhomologous DNA end-joining mutants to
VP-16 and ICRF-193: Implications for the repair of topoisomerase
II-mediated DNA damage. J Biol Chem. 278:35897–35902.
2003.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Plante I, Slaba T, Shavers Z and Hada M: A
bi-exponential repair algorithm for radiation-induced double-strand
breaks: Application to simulation of chromosome aberrations. Genes
(Basel). 10(10)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Morimoto S, Tsuda M, Bunch H, Sasanuma H,
Austin C and Takeda S: Type II DNA topoisomerases cause spontaneous
double-strand breaks in genomic DNA. Genes (Basel).
10(10)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Cho JE and Jinks-Robertson S: Deletions
associated with stabilization of the Top1 cleavage complex in yeast
are products of the nonhomologous end-joining pathway. Proc Natl
Acad Sci USA. 116:22683–22691. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li F, Jiang T, Li Q and Ling X:
Camptothecin (CPT) and its derivatives are known to target
topoisomerase I (Top1) as their mechanism of action: Did we miss
something in CPT analogue molecular targets for treating human
disease such as cancer? Am J Cancer Res. 7:2350–2394.
2017.PubMed/NCBI
|
|
26
|
Pommier Y: Drugging topoisomerases:
Lessons and challenges. ACS Chem Biol. 8:82–95. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ji Y, Dang X, Nguyen LN, Nguyen LN, Zhao
J, Cao D, Khanal S, Schank M, Wu XY, Morrison ZD, et al:
Topological DNA damage, telomere attrition and T cell senescence
during chronic viral infections. Immun Ageing.
16(12)2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Petermann E, Orta ML, Issaeva N, Schultz N
and Helleday T: Hydroxyurea-stalled replication forks become
progressively inactivated and require two different RAD51-mediated
pathways for restart and repair. Mol Cell. 37:492–502.
2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Borda MA, Palmitelli M, Verón G,
González-Cid M and de Campos Nebel M: Tyrosyl-DNA-phosphodiesterase
I (TDP1) participates in the removal and repair of stabilized-Top2α
cleavage complexes in human cells. Mutat Res. 781:37–48.
2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Cuya SM, Comeaux EQ, Wanzeck K, Yoon KJ
and van Waardenburg RC: Dysregulated human Tyrosyl-DNA
phosphodiesterase I acts as cellular toxin. Oncotarget.
7:86660–86674. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tripathi K, Mani C, Clark DW and Palle K:
Rad18 is required for functional interactions between FANCD2,
BRCA2, and Rad51 to repair DNA topoisomerase 1-poisons induced
lesions and promote fork recovery. Oncotarget. 7:12537–12553.
2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yonetani Y, Hochegger H, Sonoda E, Shinya
S, Yoshikawa H, Takeda S and Yamazoe M: Differential and
collaborative actions of Rad51 paralog proteins in cellular
response to DNA damage. Nucleic Acids Res. 33:4544–4552.
2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Vance JR and Wilson TE: Yeast Tdp1 and
Rad1-Rad10 function as redundant pathways for repairing Top1
replicative damage. Proc Natl Acad Sci USA. 99:13669–13674.
2002.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Buerstedde JM, Reynaud CA, Humphries EH,
Olson W, Ewert DL and Weill JC: Light chain gene conversion
continues at high rate in an ALV-induced cell line. EMBO J.
9:921–927. 1990.PubMed/NCBI
|
|
35
|
Buerstedde JM and Takeda S: Increased
ratio of targeted to random integration after transfection of
chicken B cell lines. Cell. 67:179–188. 1991.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Hoa NN, Shimizu T, Zhou ZW, Wang ZQ,
Deshpande RA, Paull TT, Akter S, Tsuda M, Furuta R, Tsutsui K, et
al: Mre11 is essential for the removal of lethal topoisomerase 2
covalent cleavage complexes. Mol Cell. 64:580–592. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Sasanuma H, Tsuda M, Morimoto S, Saha LK,
Rahman MM, Kiyooka Y, Fujiike H, Cherniack AD, Itou J, Callen Moreu
E, et al: BRCA1 ensures genome integrity by eliminating
estrogen-induced pathological topoisomerase II-DNA complexes. Proc
Natl Acad Sci USA. 115:E10642–E10651. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zong D, Adam S, Wang Y, Sasanuma H, Callén
E, Murga M, Day A, Kruhlak MJ, Wong N, Munro M, et al: BRCA1
haploinsufficiency is masked by RNF168-mediated chromatin
ubiquitylation. Mol Cell. 73:1267–1281.e7. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Liu H, Wu Y, He F, Cheng Z, Zhao Z, Xiang
C, Feng X, Bai X, Takeda S, Wu X, et al: Brca1 is involved in
tolerance to adefovir dipivoxil induced DNA damage. Int J Mol Med.
43:2491–2498. 2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zhang Z, Bu X, Chen H, Wang Q and Sha W:
Bmi-1 promotes the invasion and migration of colon cancer stem
cells through the downregulation of E-cadherin. Int J Mol Med.
38:1199–1207. 2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Thapa M, Bommakanti A, Shamsuzzaman M,
Gregory B, Samsel L, Zengel JM and Lindahl L: Repressed synthesis
of ribosomal proteins generates protein-specific cell cycle and
morphological phenotypes. Mol Biol Cell. 24:3620–3633.
2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Lecomte S, Demay F, Pham TH, Moulis S,
Efstathiou T, Chalmel F and Pakdel F: Deciphering the molecular
mechanisms sustaining the estrogenic activity of the two major
dietary compounds zearalenone and apigenin in ER-positive breast
cancer cell lines. Nutrients. 11(11)2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Hu X, Wu X, Liu H, Cheng Z, Zhao Z, Xiang
C, Feng X, Takeda S and Qing Y: Genistein-induced DNA damage is
repaired by nonhomologous end joining and homologous recombination
in TK6 cells. J Cell Physiol. 234:2683–2692. 2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Liu Y, Wu X, Hu X, Chen Z, Liu H, Takeda S
and Qing Y: Multiple repair pathways mediate cellular tolerance to
resveratrol-induced DNA damage. Toxicol In Vitro. 42:130–138.
2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Baba TW, Giroir BP and Humphries EH: Cell
lines derived from avian lymphomas exhibit two distinct phenotypes.
Virology. 144:139–151. 1985.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Sonoda E, Morrison C, Yamashita YM, Takata
M and Takeda S: Reverse genetic studies of homologous DNA
recombination using the chicken B-lymphocyte line, DT40. Philos
Trans R Soc Lond B Biol Sci. 356:111–117. 2001.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Dhar PK, Sonoda E, Fujimori A, Yamashita
YM and Takeda S: DNA repair studies: experimental evidence in
support of chicken DT40 cell line as a unique model. J Environ
Pathol Toxicol Oncol. 20:273–283. 2001.PubMed/NCBI
|
|
48
|
Asagoshi K, Tano K, Chastain PD II, Adachi
N, Sonoda E, Kikuchi K, Koyama H, Nagata K, Kaufman DG, Takeda S,
et al: FEN1 functions in long patch base excision repair under
conditions of oxidative stress in vertebrate cells. Mol Cancer Res.
8:204–215. 2010.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Yoshinaga N, Shindo K, Matsui Y, Takiuchi
Y, Fukuda H, Nagata K, Shirakawa K, Kobayashi M, Takeda S and
Takaori-Kondo A: A screening for DNA damage response molecules that
affect HIV-1 infection. Biochem Biophys Res Commun. 513:93–98.
2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Ooka M, Abe T, Cho K, Koike K, Takeda S
and Hirota K: Chromatin remodeler ALC1 prevents replication-fork
collapse by slowing fork progression. PLoS One.
13(e0192421)2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Petukhova G, Stratton S and Sung P:
Catalysis of homologous DNA pairing by yeast Rad51 and Rad54
proteins. Nature. 393:91–94. 1998.PubMed/NCBI View
Article : Google Scholar
|
|
52
|
Qing Y, Yamazoe M, Hirota K, Dejsuphong D,
Sakai W, Yamamoto KN, Bishop DK, Wu X and Takeda S: The epistatic
relationship between BRCA2 and the other RAD51 mediators in
homologous recombination. PLoS Genet. 7(e1002148)2011.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Takata M, Sasaki MS, Sonoda E, Morrison C,
Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A and Takeda S:
Homologous recombination and non-homologous end-joining pathways of
DNA double-strand break repair have overlapping roles in the
maintenance of chromosomal integrity in vertebrate cells. EMBO J.
17:5497–5508. 1998.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Al Abo M, Sasanuma H, Liu X, Rajapakse VN,
Huang SY, Kiselev E, Takeda S, Plunkett W and Pommier Y: TDP1 is
critical for the repair of DNA breaks induced by sapacitabine, a
nucleoside also targeting ATM- and BRCA-deficient tumors. Mol
Cancer Ther. 16:2543–2551. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Lountos GT, Zhao XZ, Kiselev E, Tropea JE,
Needle D, Pommier Y, Burke TR Jr and Waugh DS: Identification of a
ligand binding hot spot and structural motifs replicating aspects
of tyrosyl-DNA phosphodiesterase I (TDP1) phosphoryl recognition by
crystallographic fragment cocktail screening. Nucleic Acids Res.
47:10134–10150. 2019.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Sharma H, Kanwal R, Bhaskaran N and Gupta
S: Plant flavone apigenin binds to nucleic acid bases and reduces
oxidative DNA damage in prostate epithelial cells. PLoS One.
9(e91588)2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Jayasooriya RG, Kang SH, Kang CH, Choi YH,
Moon DO, Hyun JW, Chang WY and Kim GY: Apigenin decreases cell
viability and telomerase activity in human leukemia cell lines.
Food Chem Toxicol. 50:2605–2611. 2012.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Chung CS, Jiang Y, Cheng D and Birt DF:
Impact of adenomatous polyposis coli (APC) tumor supressor gene in
human colon cancer cell lines on cell cycle arrest by apigenin. Mol
Carcinog. 46:773–782. 2007.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Gupta S, Afaq F and Mukhtar H: Involvement
of nuclear factor-kappa B, Bax and Bcl-2 in induction of cell cycle
arrest and apoptosis by apigenin in human prostate carcinoma cells.
Oncogene. 21:3727–3738. 2002.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Subhasitanont P, Chokchaichamnankit D,
Chiablaem K, Keeratichamroen S, Ngiwsara L, Paricharttanakul NM,
Lirdprapamongkol K, Weeraphan C, Svasti J and Srisomsap C: Apigenin
inhibits growth and induces apoptosis in human cholangiocarcinoma
cells. Oncol Lett. 14:4361–4371. 2017.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Sharma NK: Modulation of radiation-induced
and mitomycin C-induced chromosome damage by apigenin in human
lymphocytes in vitro. J Radiat Res (Tokyo). 54:789–797.
2013.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Agarwal S, van Cappellen WA, Guénolé A,
Eppink B, Linsen SE, Meijering E, Houtsmuller A, Kanaar R and
Essers J: ATP-dependent and independent functions of Rad54 in
genome maintenance. J Cell Biol. 192:735–750. 2011.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Mazin AV, Alexeev AA and Kowalczykowski
SC: A novel function of Rad54 protein. Stabilization of the Rad51
nucleoprotein filament. J Biol Chem. 278:14029–14036.
2003.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Mazin AV, Bornarth CJ, Solinger JA, Heyer
WD and Kowalczykowski SC: Rad54 protein is targeted to pairing loci
by the Rad51 nucleoprotein filament. Mol Cell. 6:583–592.
2000.PubMed/NCBI View Article : Google Scholar
|
|
65
|
García-Luis J and Machín F: Fanconi
anaemia-like Mph1 helicase nacks up Rad54 and Rad5 to circumvent
replication stress-driven chromosome bridges. Genes (Basel).
9(9)2018.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Tang S, Liu B, Liu M, Li Z, Liu J, Wang H,
Wang J, Oh YT, Shen L and Wang Y: Ionizing radiation-induced growth
in soft agar is associated with miR-21 upregulation in wild-type
and DNA double strand break repair deficient cells. DNA Repair
(Amst). 78:37–44. 2019.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Crickard JB, Kaniecki K, Kwon Y, Sung P,
Lisby M and Greene EC: Regulation of Hed1 and Rad54 binding during
maturation of the meiosis-specific presynaptic complex. EMBO J.
37(37)2018.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Zhang XP, Janke R, Kingsley J, Luo J,
Fasching C, Ehmsen KT and Heyer WD: A conserved sequence extending
motif III of the motor domain in the Snf2-family DNA translocase
Rad54 is critical for ATPase activity. PLoS One.
8(e82184)2013.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Zhang L, Cheng X, Gao Y, Zheng J, Xu Q,
Sun Y, Guan H, Yu H and Sun Z: Apigenin induces autophagic cell
death in human papillary thyroid carcinoma BCPAP cells. Food Funct.
6:3464–3472. 2015.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Fang J, Bao YY, Zhou SH and Fan J:
Apigenin inhibits the proliferation of adenoid cystic carcinoma via
suppression of glucose transporter-1. Mol Med Rep. 12:6461–6466.
2015.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Lim W, Park S, Bazer FW and Song G:
Apigenin reduces survival of choriocarcinoma cells by inducing
apoptosis via the PI3K/AKT and ERK1/2 MAPK pathways. J Cell
Physiol. 231:2690–2699. 2016.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Nitiss J and Wang JC: DNA
topoisomerase-targeting antitumor drugs can be studied in yeast.
Proc Natl Acad Sci USA. 85:7501–7505. 1988.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Chen G and Guo M: Screening for natural
inhibitors of topoisomerases I from Rhamnus davurica by affinity
ultrafiltration and high-performance liquid chromatography-mass
spectrometry. Front Plant Sci. 8(1521)2017.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Champoux JJ: DNA topoisomerases:
Structure, function, and mechanism. Annu Rev Biochem. 70:369–413.
2001.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Interthal H, Pouliot JJ and Champoux JJ:
The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the
phospholipase D superfamily. Proc Natl Acad Sci USA.
98:12009–12014. 2001.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Krawczyk C, Dion V, Schär P and Fritsch O:
Reversible Top1 cleavage complexes are stabilized
strand-specifically at the ribosomal replication fork barrier and
contribute to ribosomal DNA stability. Nucleic Acids Res.
42:4985–4995. 2014.PubMed/NCBI View Article : Google Scholar
|