Introduction
Acute myocardial infarction (AMI) is characterized
by myocardial necrosis caused by acute and persistent ischemia and
hypoxia of cardiac cells (1).
Clinically, AMI presents as sharp and persistent substernal pain
that cannot be fully relieved by resting or nitrate medications,
and is accompanied by elevated levels of serum myocardial enzymes,
including creatine kinase and lactate dehydrogenase (LDH), as well
as abnormal progression on an electrocardiogram (2-4).
AMI can occur at the same time as arrhythmias, shock or heart
failure, and is often life threatening. This disease is most common
in North American and European countries, where ~1.5 million new
cases of AMI are reported in the United States annually (5,6). In
China, the incidence of AMI has continued to increase in the last
decade, with ≥500,000 new cases annually and ~2 million patients
currently diagnosed (7).
Myocardial ischemia/reperfusion (I/R) injury is often the result of
different types of coronary artery disease, including myocardial
infarction, ischemic cardiomyopathy and sudden coronary death
(8). Reperfusion is a standard
therapy for acute cases of coronary artery diseases, including MI,
and is also an inevitable inducer of I/R injury of coronary
microvessels and myocardium (9-11).
Phosphodiesterase 4B (PDE4B) is one of the 11
members of the PDE enzyme family that are known for their exclusive
ability to decompose cyclic nucleotides, and it can regulate
several biological processes (12). A recent microarray study identified
PDE4B as a metabolism-related gene that was strongly associated
with the onset and recurrence of AMI (13). Previous studies have reported that
activation of PDE4B induced cognitive impairment and neuronal
apoptosis, and aggravated neuroinflammation and oxidative stress in
the central nervous system (14,15).
Jun dimerization protein 2 (JDP2) is a bZip-type
transcription factor that belongs to the activator protein-1 (AP-1)
family (16). JDP2 upregulation in
mice has been reported to have a direct association with impaired
cardiac function manifested as cardiomyocyte apoptosis, cardiac
hypertrophy, fibrosis and inflammation (17).
Therefore, the present study aimed to investigate
how PDE4B expression affected myocardial hypoxia/reoxygenation
(H/R) injury, and to determine the function of JDP2 in the
process.
Materials and methods
Bioinformatics tools
JASPAR database (jaspar.genereg.net) which is a regularly maintained
open-access database storing manually curated transcription factors
(TF) binding profiles as position frequency matrices (PFMs) was
applied to predict the interaction between the transcription factor
JDP2 and PDE4B promoter.
Cell culture and treatment
H9c2 rat embryo heart-derived cardiomyocytes
(American Type Culture Collection) were cultured in low-glucose
DMEM supplemented with 10% FBS (Gibco, Thermo Fisher Scientific,
Inc.) in a 5% CO2 incubator at 37˚C. To create a hypoxic
condition, H9c2 cells were cultured at 37˚C for 6 h in serum-free
and glucose-free DMEM in a tri-gas incubator with 94%
N2, 5% CO2 and 1% O2.
Subsequently, cells were re-oxygenated for 2 h in DMEM supplemented
with 10% FBS in 95% air and 5% CO2 at 37˚C, as
previously described (18). Cells
incubated in a humidified atmosphere of 5% CO2 incubator
at 37˚C for 20 h were used as a control throughout the
experiments.
Cell transfection
Short hairpin (sh)RNA plasmids specific for PDE4B
which were ligated into the plasmid of U6/GFP/Neo [sh-PDE4B-1
forward,
5'-CCGGTACTCCTTCTCTGTGAAATTTCTCGAGAAATTTCACAGAGAAGGAGTATTTTTG-3'
and reverse,
5'-AATTCAAAAATACTCCTTCTCTGTGAAATTTCTCGAGAAATTTCACAGAGAAGGAGTA-3';
sh-PDE4B-2 forward,
5'-CCGGTCCATCTACAGTCTGAGATTACTCGAGTAATCTCAGACTGTAGATGGATTTTTG-3'
and reverse,
5'-AATTCAAAAATCCATCTACAGTCTGAGATTACTCGAGTAATCTCAGACTGTAGATGGA-3',
shRNA with empty vectors which was regarded as negative control
(NC), pcDNA3.1(+) JDP2 overexpression vector (Oe-JDP2) and Oe-NC
were purchased from Shanghai GenePharma Co., Ltd. H9c2 cells
(4x105 cells/well) were transfected with 50 nM
sh-PDE4B-1/2, sh-NC, Oe-JDP2 or Oe-NC using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. After
incubation at 37˚C for 48 h, cells were harvested for subsequent
experiments.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from H9c2 cells using the
RNAeasy™ Viral RNA Isolation kit with Spin Column
(Beyotime Institute of Biotechnology). Total RNA was reverse
transcribed into cDNA using the Revert Aid™ cDNA
Synthesis kit (Takara Biotechnology co., Ltd.) according to the
manufacturer's protocol. Subsequently, qPCR was performed using
BeyoFast™ Probe qPCR mix (Beyotime Institute of
Biotechnology). The thermocycling conditions were as follows:
Pre-denaturation at 95˚C for 30 sec, followed by denaturation at
95˚C for 10 sec and annealing at 55˚C for 30 sec for 40 cycles. The
following primers were used for qPCR: PDE4B forward,
5'-ACGGTGGCTCATACATGCT-3' and reverse, 5'-GTACCAGTCCCGACGAAGAG-3';
JDP2 forward, 5'-CCCAGGGACCTCGAGCTTT-3' and reverse,
5'-GGTCTTCAGCTCTGCGTTCA-3'; and GADPH forward,
5'-GAATGGGCAGCCGTTAGGAA-3' and reverse, 5'-AAAAGCATCACCCGGAGGAG-3'.
Relative expression levels were calculated using the
2-ΔΔCq method (19) and
normalized to the internal reference gene GAPDH.
Western blotting
Total protein was extracted from H9c2 cells using
RIPA lysis buffer (Shanghai Yeasen Biotechnology Co., Ltd.) and
quantified using a BCA kit (Shanghai Enzyme-linked Biotechnology
Co., Ltd.). Proteins (20 µg per lane) were separated via 10%
SDS-PAGE (Beyotime Institute of Biotechnology) and transferred onto
PVDF membranes. The membranes were incubated at 4˚C overnight with
primary antibodies targeted against: PDE4B (1:1,000; cat. no.
ab14628; Abcam), Bcl-2 (1:1,000; cat. no. ab196495; Abcam), Bax
(1:1,000; cat. no. ab32503; Abcam), cleaved-caspase3 (1:1,000; cat.
no. 9661; Cell Signaling Technology, Inc.), cleaved-caspase9
(1:1,000; cat. no. 10380-1-AP; ProteinTech Group, Inc.), JDP2
(1:1,000; cat. no. orb336272; Biorbyt Ltd.) and GAPDH (1:10,000;
cat. no. ab181602; Abcam). Following the primary incubation,
membranes were incubated with a HRP-labeled Goat Anti-Rabbit IgG
(H+L) secondary antibody (1:1,000; cat. no. A0208; Beyotime
Institute of Biotechnology) at 37˚C for 2 h. Protein bands were
visualized using ECL reagents (PerkinElmer, Inc.) and analyzed by
Image J software (version 1.48v; National Institutes of Health).
GAPDH was used as the loading control.
Cell viability and cytotoxicity
The Cell Counting Kit-8 (CCK-8; Beijing Solarbio
Science & Technology Co., Ltd.) assay was performed was to
assess cell viability. Briefly, H9c2 cells (2x103
cells/100 µl/well) were incubated with 10 µl/well CCK-8 solution in
a 96-well plate for 2 h. Subsequently, optical density was measured
at a wavelength of 450 nm using a microplate reader (Bio-Rad
Laboratories, Inc.).
The LDH Cytotoxicity Assay Kit (Beyotime Institute
of Biotechnology) was used to detect H/R-induced cytotoxicity
according to the manufacturer's instructions. LDH activity was
measured at a wavelength of 490 nm using a microplate reader
(Bio-Rad Laboratories, Inc.).
Detection of oxidative stress
levels
The levels of malondialdehyde (MDA) and superoxide
dismutase (SOD) in H9c2 cells were detected using MDA (A003-1-2)
and SOD (A001-3-2) assay kits (both Nanjing Jiancheng
Bioengineering Institute), respectively. The glutathione (GSH) to
glutathione oxidized (GSSG) ratio was measured using the GSH/GSSG
Ratio Assay kit (cat. no. KA6046; Abnova). Briefly, the cell
precipitate was collected via centrifugation at 1,600 x g for 10
min at 4˚C and freeze-thawed twice with liquid nitrogen and 37˚C
water baths prior to centrifugation at 1,000 x g for 10 min at room
temperature. Subsequently, working solution and NADPH solution from
the kits were prepared and added successively into the supernatant.
Following incubation, the absorbance at 412 nm was determined using
a microplate reader (BioTek instruments, inc.).
Measurement of apoptotic rate and
apoptosis-related proteins
H9c2 cell apoptosis was detected using the TUNEL
Apoptosis Assay kit (Beyotime Institute of Biotechnology). After
washing with PBS, cells were fixed with 4% paraformaldehyde at 4˚C
for 10 min, permeabilized with 0.3% PBS in Triton X-100 at room
temperature for 30 min and then incubated with 0.3%
H2O2 in PBS on ice for 2 min prior to biotin
labeling. Cells were incubated with TUNEL detection solution at
37˚C for 60 min in the dark and stained with 10 µg/ml DAPI
(Shanghai Haoyang Bio Technology Co., Ltd.) at 37˚C for 2-3 min. In
total, three fields of view were selected at random and the images
were captured using a fluorescence microscope (magnification, x200;
Olympus Corporation). Apoptosis-related protein expression levels
were detected via western blotting according to the aforementioned
protocol.
Verification of interaction between
JDP2 and PDE4B
For the dual luciferase reporter assay, H9c2 cells
(5x105 cells/well) were seeded (1x104
cells/well) into 96-well plates. Subsequently, H9c2 cells were
co-transfected with PDE4B-WT and PDE4B-MUT, Oe-JDP2 or Oe-NC (0.5
µg) and Renilla luciferase reporter vector (Promega
Corporation) using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) for 48 h at 37˚C. At 48 h
post-transfection, firefly luciferase activities was measured using
a Dual Luciferase Reporter Assay system (Promega Corporation) and
normalized to Renilla luciferase activities.
For the chromatin immunoprecipitation (ChIP) assay,
ultrasound-treated samples were centrifuged at 12,000-14,000 x g
for 5 min at 4˚C. A total of 300 µl SDS lysis buffer (Active Motif,
Inc.) was then used to lyse the cells, which were subsequently
sonicated at 150 Hz and sheared with four sets of 10 sec pulses on
wet ice using a high intensity ultrasonic processor. ChIP Dilution
Buffer containing 1 mM PMSF was prepared using the ChIP Assay kit
(Beyotime Institute of Biotechnology) and added to part of the
samples to serve as the input. A total of 40 µl protein A/G agarose
beads (Santa Cruz Biotechnology, Inc.) was added to the rest of the
samples. Following centrifugation at 16,000 x g for 10 min at 4˚C,
supernatant (100 µl) was incubated with 5 µg anti-IgG (cat. no.
sc-2025; Santa Cruz Biotechnology, Inc.) or anti-JDP2 (cat. no.
sc-517133; Santa Cruz Biotechnology, Inc.) primary antibodies and
protein A/G agarose at 4˚C overnight. The precipitated chromatin
was purified by phenol/chloroform/isoamyl extraction and analyzed
via RT-qPCR according to the aforementioned protocol.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism (version 8; GraphPad Software, Inc.). All experiments were
performed in triplicate. Data are presented as the mean ± SD from
at least three independent experiments. Comparisons between two
groups were analyzed using an unpaired Student's t-test, whereas
comparisons among multiple groups were analyzed using one-way ANOVA
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
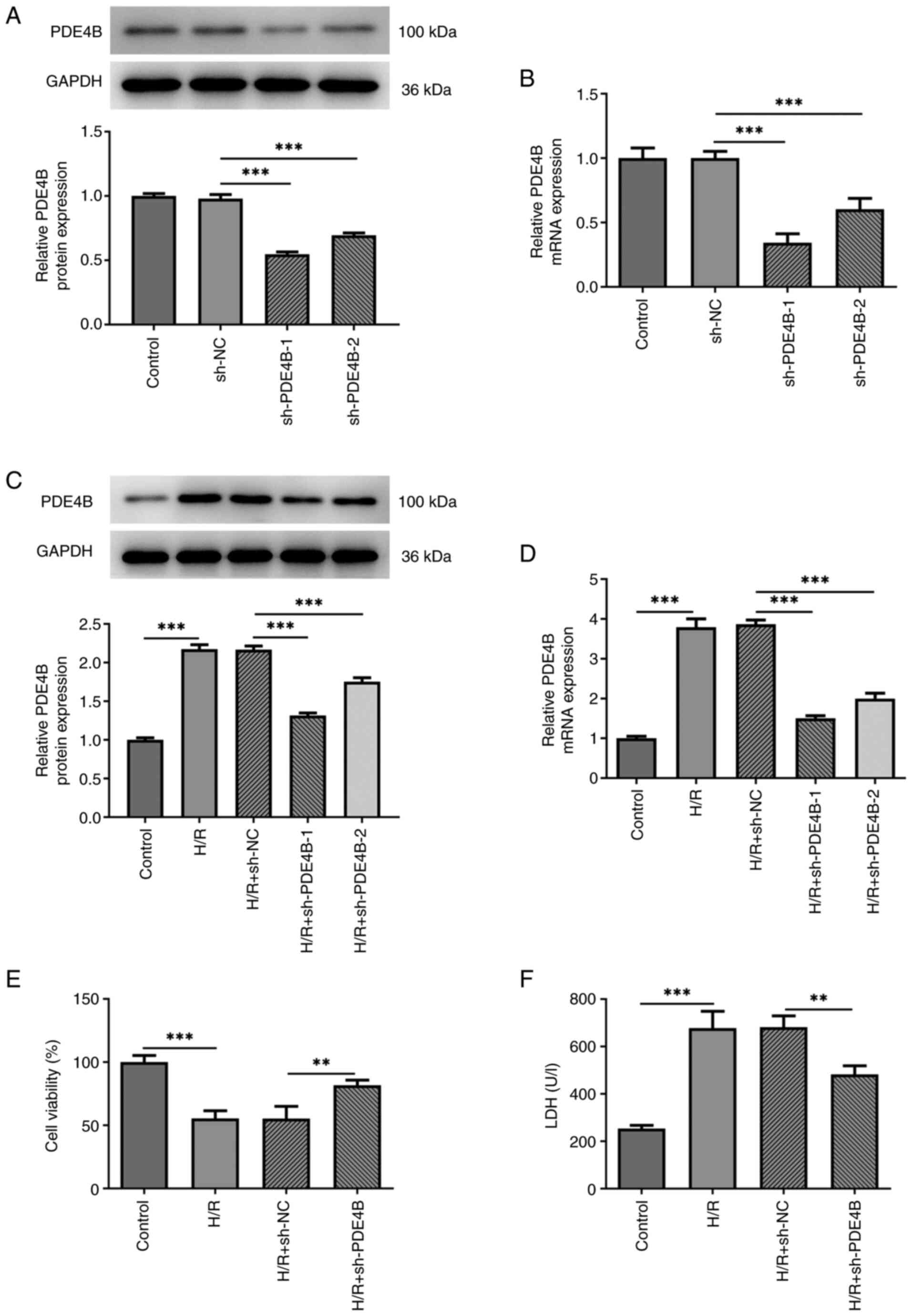
Effect of PDE4B knockdown on H9c2 cell
viability and cytotoxicity following H/R
Firstly, H9c2 cells were transfected with
sh-PDE4B-1/2 or sh-NC, and then PDE4B expression was detected via
RT-qPCR and western blotting (Fig.
1A and B). sh-PDE4B-1 and
sh-PDE4B-2 significantly decreased PDE4B expression levels compared
with sh-NC. Moreover, PDE4B was expressed at significantly higher
levels in H9c2 cells exposed to H/R compared with the control group
(Fig. 1C and D). In addition, transfection with
sh-PDE4B-1/2 significantly decreased the expression of PDE4B under
H/R conditions, with sh-PDE4B-1 displaying a higher knockdown
effect compared with sh-PDE4B-2 (Fig.
1C and D). Thus, sh-PDE4B-1
was selected for use in subsequent experiments. Compared with the
control group, cells exposed to H/R displayed a significant decline
in viability, whereas PDE4B interference significantly increased
cell viability under H/R conditions (Fig. 1E). Cytotoxicity detection in H9c2
cells demonstrated that PDE4B knockdown significantly decreased
H/R-induced upregulated LDH levels (Fig. 1F). Taken together, these results
suggested that PDE4B knockdown prevented decreases in cell
viability and alleviated the cytotoxicity of H/R-stimulated H9c2
cells.
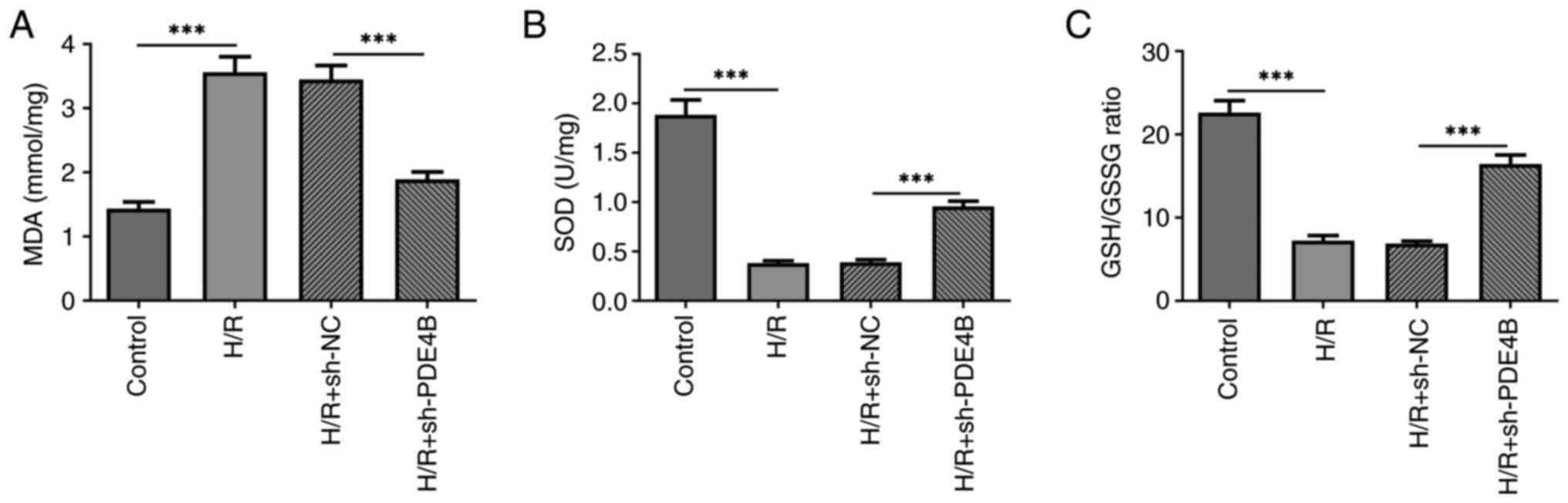
Effect of PDE4B knockdown on
H/R-induced oxidative stress and apoptosis in H9c2 cells
The present study assessed the degrees of oxidative
stress and apoptosis of H9c2 cells following H/R, before and after
transfection with sh-PDE4B. The results demonstrated that the
concentration of MDA was significantly higher (Fig. 2A), whereas the concentration of SOD
and the GSH/GSSG ratio were significantly lower (Fig. 2B and C) in cells exposed to H/R compared with
those in the control group. However, PDE4B knockdown significantly
decreased the concentration of MDA, and significantly elevated the
concentration of SOD and the ratio of GSH to GSSG under H/R
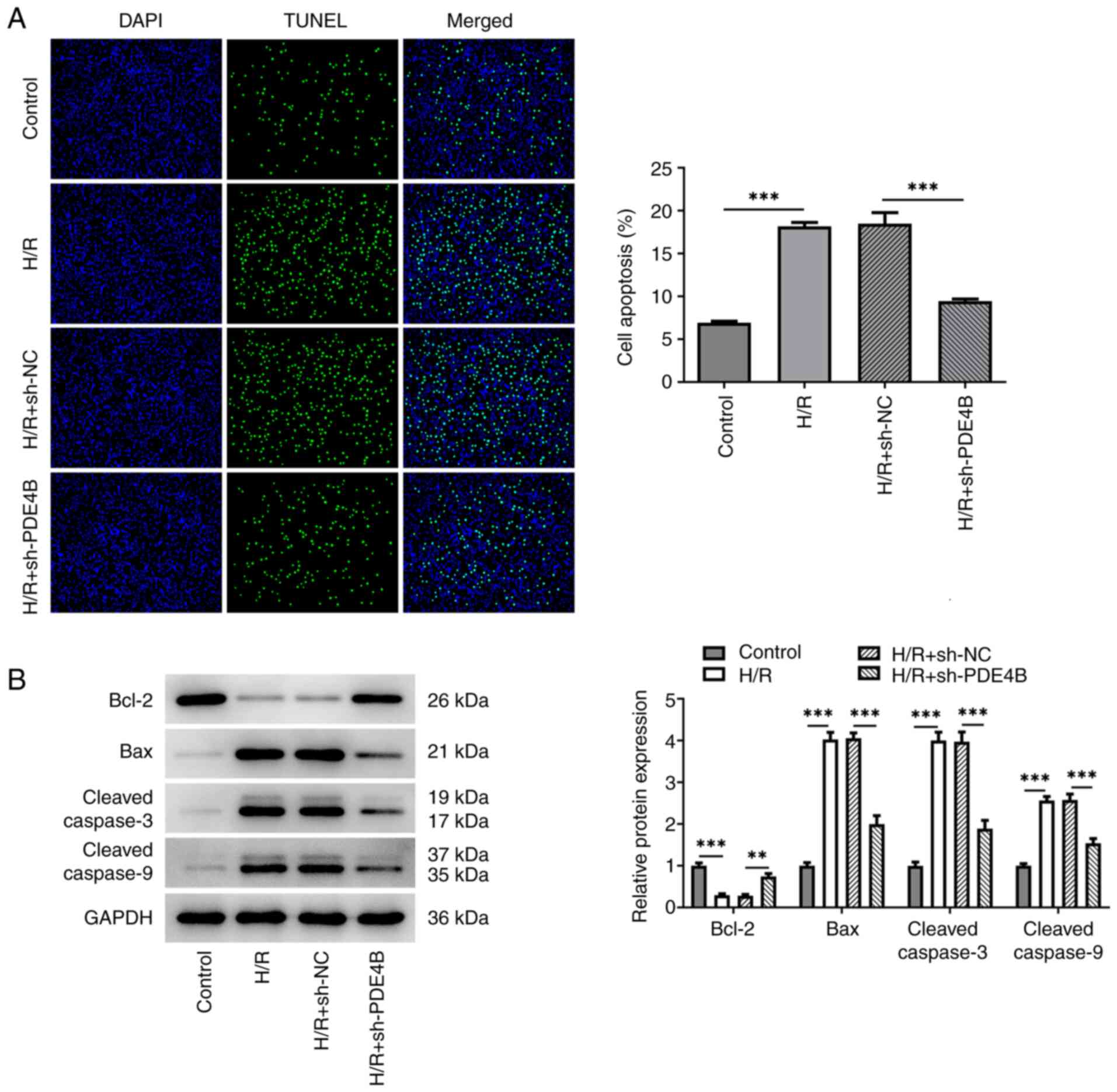
conditions. In addition, the number of TUNEL+ cells was
significantly increased in the H/R group compared with the control
group, which was significantly reversed following PDE4B knockdown
(Fig. 3A). Consistently, in
H/R-stimulated H9c2 cells, anti-apoptotic Bcl-2 expression was
significantly decreased, whereas proapoptotic Bax expression was
significantly increased, and the expression levels of
cleaved-caspase3 and cleaved-caspase9 were significantly increased
compared with those in the control group. Notably, these effects
were significantly reversed following PDE4B knockdown (Fig. 3B). Collectively, these results
suggested that PDE4B knockdown ameliorated H/R-induced oxidative
stress and apoptosis in H9c2 cells.
Binding interaction between PDE4B and
transcription factor JDP2
RT-qPCR and western blotting confirmed that JDP2
expression was upregulated in H9c2 cells following H/R (Fig. 4A and B). JASPAR was used to predict the binding
sites between JDP2 and PDE4B (Fig.
4C). This interaction was confirmed by performing dual
luciferase reporter and ChIP assays, which demonstrated
significantly upregulated luciferase activities in cells
co-transfected with PDE4B-wild-type (WT) and Oe-JDP2 compared with
those co-transfected with PDE4B-WT and Oe-NC. No apparent changes
were observed in the luciferase activity of PDE4B-MUT after
co-transfection of Oe-NC or Oe-JDP2 (Fig. 4D). Compared with cells treated with
IgG, significantly increased PDE4B expression was observed in cells
treated with anti-JDP2 (Fig. 4E).
The RT-qPCR results confirmed the successful transfection of
Oe-JDP2 in H9c2 cells (Fig. 4F).
mRNA level of PDE4B was also enhanced after transfection of Oe-JDP2
(Fig. 4G). Additionally, the
results demonstrated that JDP2 overexpression significantly
increased PDE4B expression in H9c2 cells exposed to H/R (Fig. 4H). Taken together, these results
suggested that the transcription factor JDP2 activated PDE4B in
H9c2 cells exposed to H/R.
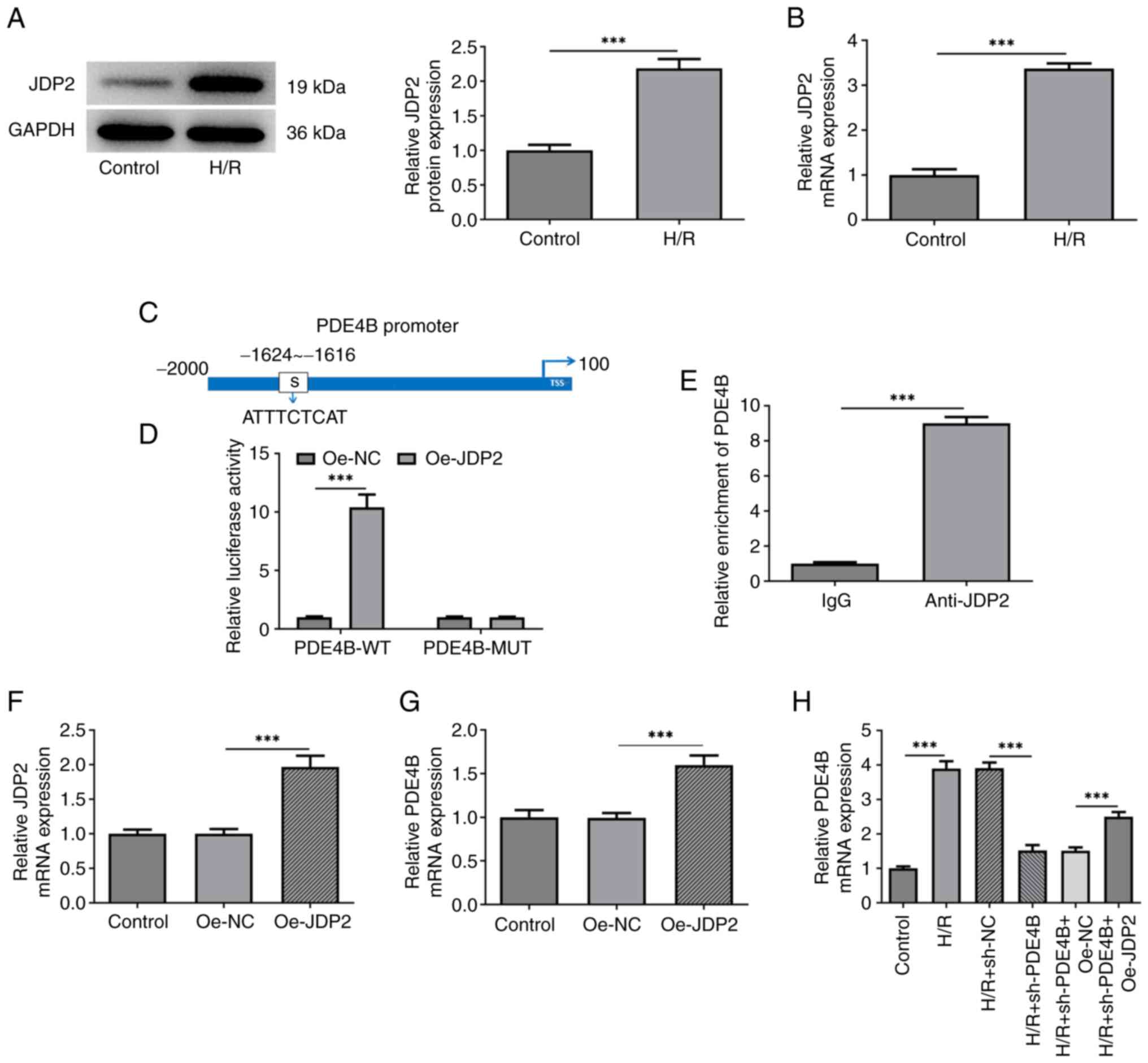 | Figure 4JDP2 expression is upregulated in
H/R-stimulated H9c2 cells, which activates PDE4B expression. (A)
Western blotting and (B) RT-qPCR were performed to detect JDP2
expression in H/R-stimulated H9c2 cells. (C) JASPAR database was
used to predict the binding sites between JDP2 and PDE4B promoter
regions. (D) Dual luciferase reporter assays were performed to
detect relative luciferase activities in cells co-transfected with
PDE4B-WT or MUT and Oe-NC or Oe-JDP2. (E) Chromatin
immunoprecipitation assays were performed to detect PDE4B
enrichment in IgG and anti-JDP2 groups. (F) RT-qPCR was performed
to assess the transfection efficiency of Oe-JDP2 in H9c2 cells. (G)
RT-qPCR was performed to detect PDE4B mRNA expression levels after
JDP2 overexpression in H9c2 cells. (H) RT-qPCR was performed to
detect PDE4B mRNA expression in H/R-stimulated H9c2 cells following
transfection with sh-PDE4B or co-transfection with Oe-JDP2.
***P<0.001. JDP2, c-Jun dimerization protein 2; H/R,
hypoxia/reoxygenation; PDE4B, phosphodiesterase 4B; RT-qPCR,
reverse transcription-quantitative PCR; WT, wild-type; MUT, mutant;
Oe, overexpression; NC, negative control; sh, short hairpin
RNA. |
Role of JDP2 in PDE4B
knockdown-mediated alleviation of H9c2 cell injury following
H/R
The present study investigated how the interaction
between JDP2 and PDE4B affected H/R-induced H9c2 cell injury.
Analysis of cell viability and cytotoxicity under H/R conditions
demonstrated a significant decrease in H9c2 cell viability
(Fig. 5A) and a significant
increase in the level of LDH (Fig.
5B) following co-transfection with sh-PDE4B and Oe-JDP2
compared with that observed in sh-PDE4B + Oe-NC group. Therefore,
the results indicated that JDP2 overexpression reversed the effect
of PDE4B knockdown on H/R-induced H9c2 cell injury.
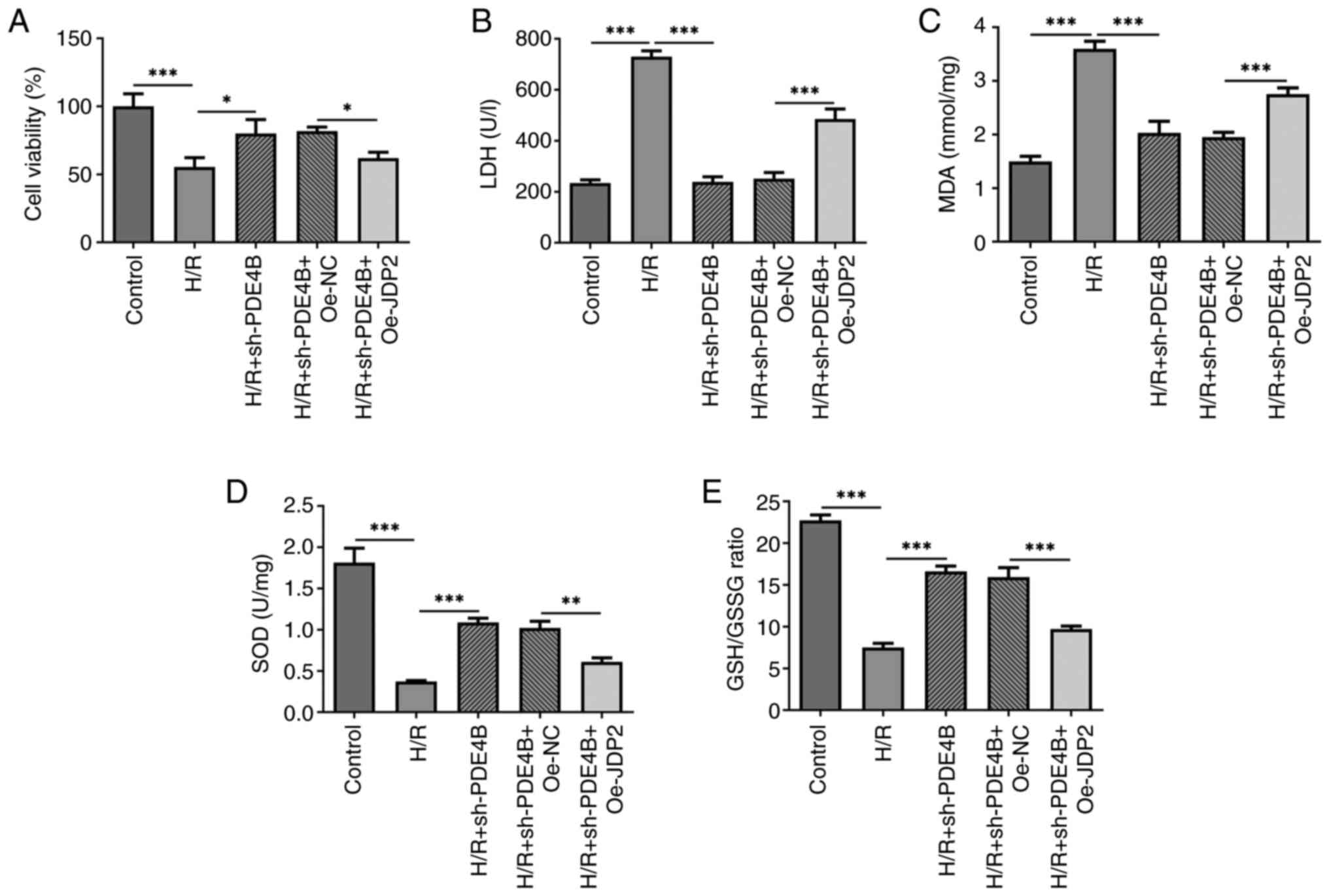 | Figure 5JDP2 overexpression partly reverses
the effects of PDE4B knockdown on cell viability, cytotoxicity and
oxidative stress in H/R-stimulated H9c2 cells. Analysis of (A) cell
viability, (B) cytotoxicity, (C) MDA concentration, (D) SOD
concentration and (E) GSH/GSSG ratio in H/R-stimulated H9c2 cells
following transfection with sh-PDE4B or co-transfection with
Oe-JDP2. *P<0.05, **P<0.01 and
***P<0.001. JDP2, c-Jun dimerization protein 2;
PDE4B, phosphodiesterase 4B; H/R, hypoxia/reoxygenation; MDA,
malondialdehyde; SOD, superoxide dismutase; GSH, glutathione; GSSG,
glutathione oxidized; sh, short hairpin RNA; Oe, overexpression;
NC, negative control; LDH, lactate dehydrogenase. |
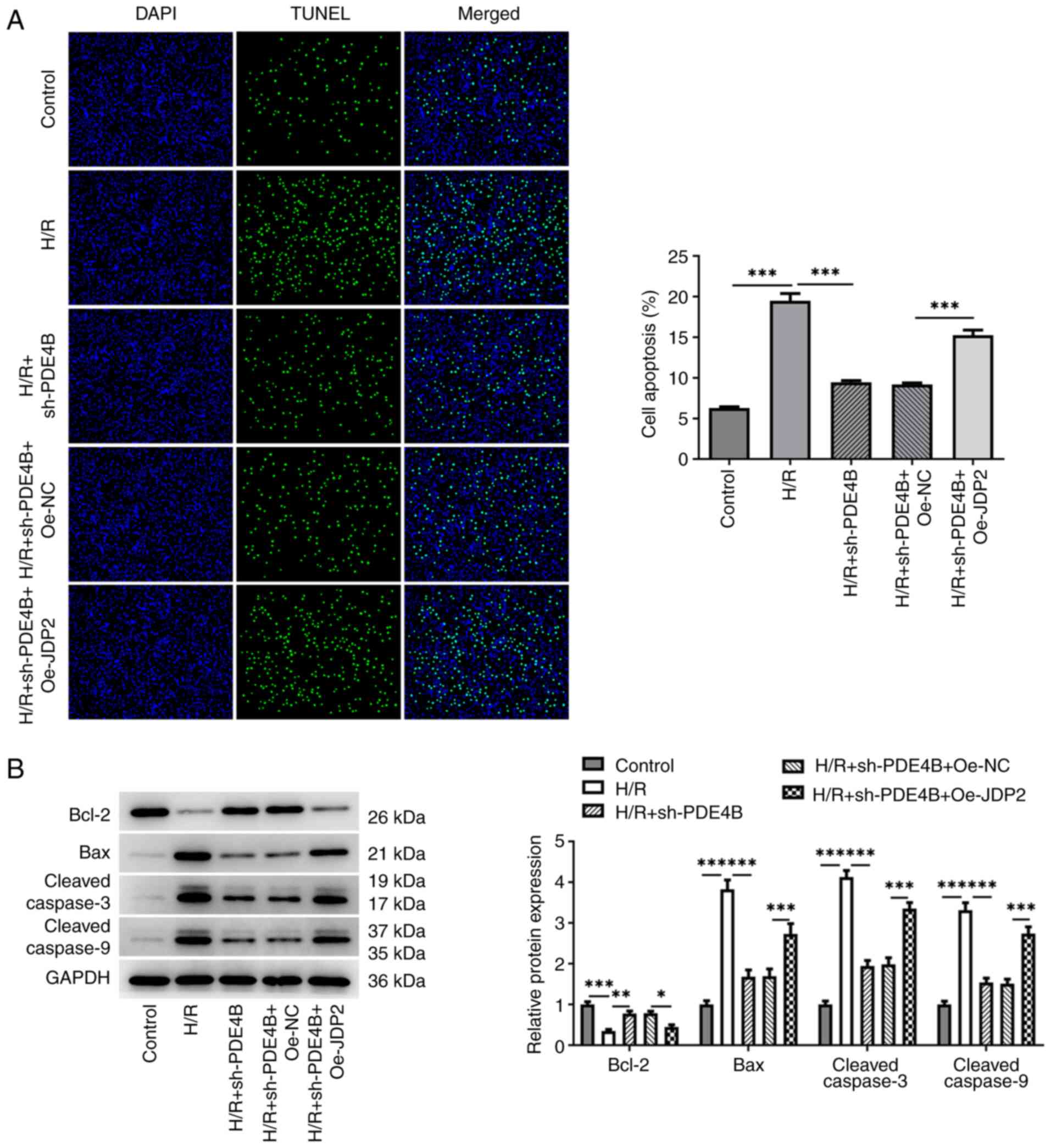
Role of JDP2 in PDE4B
interference-alleviated oxidative stress and apoptosis of H9c2
cells following H/R
The results demonstrated significantly increased MDA
concentration (Fig. 5C), and
significantly decreased SOD concentration (Fig. 5D) and GSH/GSSG ratio (Fig. 5E) in H/R-stimulated H9c2 cells
co-transfected with sh-PDE4B and Oe-JDP2 compared with those
co-transfected with sh-PDE4B and Oe-NC. Notably, JDP overexpression
significantly increased the number of TUNEL+ cells
(Fig. 6A), decreased Bcl-2
expression, increased Bax expression, and increased the expression
levels of cleaved-caspase3 and cleaved-caspase9 following H/R in
PDE4B-knockdown H9c2 cells (Fig.
6B). Collectively, these results suggested that JDP2
overexpression reversed the ameliorative effect of PDE4B knockdown
on H/R-induced oxidative stress and apoptosis in H9c2 cells.
Discussion
AMI is a serious consequence of atherosclerotic
cardiovascular disease and coronary artery disease. Rapid
development of cardiac interventional therapy and bypass surgery
has improved the treatment of coronary heart disease (20). However, several issues associated
with AMI treatment remain, particularly cardiomyocyte necrosis or
apoptosis from prolonged myocardial ischemia (21,22).
Myocardial I/R injury is the primary pathological manifestation of
coronary artery disease, which continues to increase the global
morbidity and mortality of AMI despite therapeutic intervention
(23). Cytotoxic oxygen species
production, inflammatory response, oxidative stress, and interplay
between apoptosis and autophagy are critical participants in
I/R-induced cardiomyocyte injury (24-26).
Previous studies have reported the roles of certain
genes in the process of AMI (27-29).
In the present study, a high expression level of PDE4B expression
was observed in H9c2 cardiomyocytes exposed to H/R, indicating the
involvement of PDE4B in H/R-induced AMI. Aberrantly high PDE4B
expression has been identified as a contributing factor in the
pathogenesis of several diseases, including colorectal cancer,
neurological conditions and injury, allergy and heart failure
(30-33).
In the setting of oxygen-glucose deprivation-induced HT22 mouse
hippocampal neuronal cell injury, PDE4B overexpression promotes
endoplasmic reticulum stress and the release of reactive oxygen
species (34). Furthermore, PDE4B
knockdown in primary microglia increases LC-3II expression and
decreases inflammasome activity (35). The results of the present study
demonstrated that PDE4B knockdown increased H9c2 viability and
decreased cytotoxicity under H/R conditions. According to a study
on acute lung injury, PDE4B knockout could effectively inhibit
lipopolysaccharide (LPS)-induced activation of the NF-κB-related
inflammatory response and generation of reactive oxygen species in
multiple pulmonary cell lines, including epithelial cells (A549),
microvascular endothelial cells (pulmonary microvascular
endothelial cells) and vascular smooth muscle cells (36). PDE4B has also been reported to be
upregulated in patients with acute kidney injury, whereas treatment
with rutaecarpine derivative compound-6c exerts an anti-oxidative
effect by downregulating PDE4B to protect kidney tubular epithelial
cells against oxidative stress and programmed cell death (37). The results of the present study
demonstrated that PDE4B knockdown alleviated H/R-induced oxidative
stress and apoptosis in H9c2 cardiomyocytes, which was consistent
with previous findings.
In the present study, the JASPAR database was used
to predict the binding sites between PDE4B promoter and JDP2, a
transcription factor that is differentially expressed in patients
with MI (38). In view of the
critical regulatory role of JDP2 in impaired cardiac function
(17), the present study also
confirmed that JDP2 was upregulated in H9c2 cells exposed to H/R,
and verified the interaction between JDP2 and the PDE4B promoter
region. In addition, JDP2 expression is activated by hypoxia in a
murine model of MI and is regulated by testis-specific transcript,
Y-linked 15-targeted microRNA-455-5p to prevent cardiomyocyte
apoptosis and rescue cell migration and invasion (39). In mice with traumatic brain injury,
LPS stimulates the expression of JDP2, and JDP2 overexpression
aggravates LPS-induced inflammation and apoptosis of mouse
astrocytes (40). Consistent with
these findings, the results of the present study demonstrated that
JDP2 overexpression attenuated the ameliorative effect of PDE4B
knockdown on cell viability, cytotoxicity, oxidative stress and
apoptosis in H/R-stimulated H9c2 cardiomyocytes.
In conclusion, the results of the present study
demonstrated that the transcription factor JDP2 activated PDE4B
expression to participate in H/R-induced oxidative and apoptotic
injuries of H9c2 cardiomyocytes, partially explaining the
pathogenic mechanism underlying myocardial I/R injury. Taken
together, the results of the present study suggested that PDE4B and
JDP2 may serve as promising biomarkers and therapeutic targets in
AMI diagnosis and treatment. However, further in vivo
studies are required to confirm the results of the present
study.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SL and ZZ were responsible for the conception of the
study and the drafting of the manuscript. SL, YC, YJ, TX and XH
assisted in obtaining experimental data and analyzed the data. ZZ
revised the manuscript. SL and ZZ confirm the authenticity of all
the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pollard TJ: The acute myocardial
infarction. Prim Care. 27:631–649;vi. 2000.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Beltrame JF: Nitrate therapy in acute
myocardial infarction: Potion or poison? Cardiovasc Drugs Ther.
22:165–168. 2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jensen AE, Reikvam A and Asberg A:
Diagnostic efficiency of lactate dehydrogenase isoenzymes in serum
after acute myocardial infarction. Scand J Clin Lab Invest.
50:285–289. 1990.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hurst JW: Thoughts about the abnormalities
in the electrocardiogram of patients with acute myocardial
infarction with emphasis on a more accurate method of interpreting
ST-segment displacement: Part I. Clin Cardiol. 30:381–390.
2007.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sattar Y and Alraies MC: Ventricular
aneurysm. In: StatPearls. StatPearls Publishing, Treasure Island,
FL, 2021.
|
|
6
|
Lindower P, Embrey R and Vandenberg B:
Echocardiographic diagnosis of mechanical complications in acute
myocardial infarction. Clin Intensive Care. 4:276–283.
1993.PubMed/NCBI
|
|
7
|
Li J, Li X, Wang Q, Hu S, Wang Y, Masoudi
FA, Spertus JA, Krumholz HM and Jiang L: China PEACE Collaborative
Group. ST-segment elevation myocardial infarction in China from
2001 to 2011 (the China PEACE-retrospective acute myocardial
infarction study): A retrospective analysis of hospital data.
Lancet. 385:441–451. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Neri M, Riezzo I, Pascale N, Pomara C and
Turillazzi E: Ischemia/reperfusion injury following acute
myocardial infarction: A critical issue for clinicians and forensic
pathologists. Mediators Inflamm. 2017(7018393)2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bagai A, Dangas GD, Stone GW and Granger
CB: Reperfusion strategies in acute coronary syndromes. Circ Res.
114:1918–1928. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Borer JS and Lewis BS: Therapeutic
coronary reperfusion and reperfusion injury: An introduction.
Cardiology. 135(13)2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhou H and Toan S: Pathological roles of
mitochondrial oxidative stress and mitochondrial dynamics in
cardiac microvascular ischemia/reperfusion injury. Biomolecules.
10(85)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Tibbo AJ and Baillie GS: Phosphodiesterase
4B: Master regulator of brain signaling. Cells.
9(1254)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Xie H, Zha E and Zhang Y: Identification
of featured metabolism-related genes in patients with acute
myocardial infarction. Dis Markers. 2020(8880004)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Guo H, Cheng Y, Wang C, Wu J, Zou Z, Niu
B, Yu H, Wang H and Xu J: FFPM, a PDE4 inhibitor, reverses learning
and memory deficits in APP/PS1 transgenic mice via cAMP/PKA/CREB
signaling and anti-inflammatory effects. Neuropharmacology.
116:260–269. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhong J, Yu H, Huang C, Zhong Q, Chen Y,
Xie J, Zhou Z, Xu J and Wang H: Inhibition of phosphodiesterase 4
by FCPR16 protects SH-SY5Y cells against MPP+-induced
decline of mitochondrial membrane potential and oxidative stress.
Redox Biol. 16:47–58. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tsai MH, Wuputra K, Lin YC, Lin CS and
Yokoyama KK: Multiple functions of the histone chaperone Jun
dimerization protein 2. Gene. 590:193–200. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Heger J, Bornbaum J, Würfel A, Hill C,
Brockmann N, Gáspár R, Pálóczi J, Varga ZV, Sárközy M, Bencsik P,
et al: JDP2 overexpression provokes cardiac dysfunction in mice.
Sci Rep. 8(7647)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Duan Y, Cheng S, Jia L, Zhang Z and Chen
L: PDRPS7 protects cardiac cells from hypoxia/reoxygenation injury
through inactivation of JNKs. FEBS Open Bio. 10:593–606.
2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Rocha EAV: Fifty years of coronary artery
bypass graft surgery. Braz J Cardiovasc Surg. 32:2–3.
2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang MX, Liu X, Li JM, Liu L, Lu W and
Chen GC: Inhibition of CACNA1H can alleviate endoplasmic reticulum
stress and reduce myocardial cell apoptosis caused by myocardial
infarction. Eur Rev Med Pharmacol Sci. 24:12887–12895.
2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chen YQ, Yang X, Xu W, Yan Y, Chen XM and
Huang ZQ: Knockdown of lncRNA TTTY15 alleviates myocardial
ischemia-reperfusion injury through the miR-374a-5p/FOXO1 axis.
IUBMB Life. 73:273–285. 2021.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Tibaut M, Mekis D and Petrovic D:
Pathophysiology of myocardial infarction and acute management
strategies. Cardiovasc Hematol Agents Med Chem. 14:150–159.
2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Dong Y, Chen H, Gao J, Liu Y, Li J and
Wang J: Molecular machinery and interplay of apoptosis and
autophagy in coronary heart disease. J Mol Cell Cardiol. 136:27–41.
2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Shen Y, Liu X, Shi J and Wu X: Involvement
of Nrf2 in myocardial ischemia and reperfusion injury. Int J Biol
Macromol. 125:496–502. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Granger DN and Kvietys PR: Reperfusion
injury and reactive oxygen species: The evolution of a concept.
Redox Biol. 6:524–551. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lee SJ, Lee CK, Kang S, Park I, Kim YH,
Kim SK, Hong SP, Bae H, He Y, Kubota Y and Koh GY: Angiopoietin-2
exacerbates cardiac hypoxia and inflammation after myocardial
infarction. J Clin Invest. 128:5018–5033. 2018.PubMed/NCBI View
Article : Google Scholar
|
|
28
|
Liu Z, Ma C, Gu J and Yu M: Potential
biomarkers of acute myocardial infarction based on weighted gene
co-expression network analysis. Biomed Eng Online.
18(9)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sheng X, Fan T and Jin X: Identification
of key genes involved in acute myocardial infarction by comparative
transcriptome analysis. Biomed Res Int.
2020(1470867)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Pearse DD and Hughes ZA: PDE4B as a
microglia target to reduce neuroinflammation. Glia. 64:1698–1709.
2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zheng XY, Chen JC, Xie QM, Chen JQ and
Tang HF: Anti-inflammatory effect of ciclamilast in an allergic
model involving the expression of PDE4B. Mol Med Rep. 19:1728–1738.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Pleiman JK, Irving AA, Wang Z, Toraason E,
Clipson L, Dove WF, Deming DA and Newton MA: The conserved
protective cyclic AMP-phosphodiesterase function PDE4B is expressed
in the adenoma and adjacent normal colonic epithelium of mammals
and silenced in colorectal cancer. PLoS Genet.
14(e1007611)2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Karam S, Margaria JP, Bourcier A, Mika D,
Varin A, Bedioune I, Lindner M, Bouadjel K, Dessillons M, Gaudin F,
et al: Cardiac overexpression of PDE4B blunts β-adrenergic response
and maladaptive remodeling in heart failure. Circulation.
142:161–174. 2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Xu B, Qin Y, Li D, Cai N, Wu J, Jiang L,
Jie L, Zhou Z, Xu J and Wang H: Inhibition of PDE4 protects neurons
against oxygen-glucose deprivation-induced endoplasmic reticulum
stress through activation of the Nrf-2/HO-1 pathway. Redox Biol.
28(101342)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
You T, Cheng Y, Zhong J, Bi B, Zeng B,
Zheng W, Wang H and Xu J: Roflupram, a phosphodiesterase 4
inhibitior, suppresses inflammasome activation through autophagy in
microglial cells. ACS Chem Neurosci. 8:2381–2392. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ma H, Shi J, Wang C, Guo L, Gong Y, Li J,
Gong Y, Yun F, Zhao H and Li E: Blockade of PDE4B limits lung
vascular permeability and lung inflammation in LPS-induced acute
lung injury. Biochem Biophys Res Commun. 450:1560–1567.
2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Liu XQ, Jin J, Li Z, Jiang L, Dong YH, Cai
YT, Wu MF, Wang JN, Ma TT, Wen JG, et al: Rutaecarpine derivative
Cpd-6c alleviates acute kidney injury by targeting PDE4B, a key
enzyme mediating inflammation in cisplatin nephropathy. Biochem
Pharmacol. 180(114132)2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Wang S and Cao N: Uncovering potential
differentially expressed miRNAs and targeted mRNAs in myocardial
infarction based on integrating analysis. Mol Med Rep.
22:4383–4395. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Huang S, Tao W, Guo Z, Cao J and Huang X:
Suppression of long noncoding RNA TTTY15 attenuates hypoxia-induced
cardiomyocytes injury by targeting miR-455-5p. Gene. 701:1–8.
2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wang XH, Liu Q and Shao ZT: Deletion of
JDP2 improves neurological outcomes of traumatic brain injury (TBI)
in mice: Inactivation of caspase-3. Biochem Biophys Res Commun.
504:805–811. 2018.PubMed/NCBI View Article : Google Scholar
|