Introduction
Mitochondrial diseases are a group of rare disorders
associated with defects (mutations or deletions) in mitochondrial
DNA (mtDNA) or nuclear DNA (1),
with an estimated birth prevalence of 20 cases per 100,000
individuals (2). Due to the fact
that mitochondria are major sources of energy in cells and are
present in all tissues (except for red blood cells), clinical
characteristics are depicted in tissues with high energy demand,
including the brain, skeletal muscle, cardiac muscle and endocrine
system (2). The clinical
manifestations, severity and prognosis of the different
mitochondrial diseases vary. The diseases usually exhibit a series
of symptoms and are correspondingly grouped into several syndromes
(1). Mitochondrial
encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS)
is one of the most common syndromes, with a prevalence as frequent
as 1 in 6,000(3). MELAS is
typically characterised by mitochondrial myopathy, encephalopathy
with stroke-like episodes, seizures and/or dementia, and lactic
acidosis. The clinical manifestations of MELAS syndrome often occur
before the age of 40 years (4).
The current study reports a case of MELAS with a
mutation in the adenine to guanine conversion at mitochondrial
genome 3243 (m.3243A>G), in which two stroke-like episodes
damaged different cerebral hemispheres.
Case report
A 48-year-old, right-handed female presented to the
Affiliated Hospital of North Sichuan Medical College (Nanchong,
China) with a sudden dysarthria in September 2021.
At 46 days before this hospital admission, the
patient had suddenly developed weakness in the left limb and was
unable to flexibly move the body. The condition of the patient had
continued to worsen over the next 2 days, after which the patient
experienced walking difficulties. There were no fevers, headaches
or limb convulsions. The patient underwent magnetic resonance
imaging (MRI) due to the symptoms and was suspected of having an
ischemic stroke, for which treatment was administered.
At the age of 35, the patient had developed a
hearing impairment, which resulted in the loss of hearing in the
left ear and very poor hearing in the right ear. At the age of 43,
the patient was diagnosed with type 2 diabetes, which was treated
with acarbose. At the age of 45, a diagnosis of cardiomyopathy was
made. The patient did not have any other ailments, such as
headaches, seizures, dementia or mental illness. The mother of the
patient, who was also diagnosed with diabetes in her 30s, died in
her mid-50s due to unknown reasons. The brother of the patient, who
was weak and suffered from diabetes, had died suddenly 3 years
prior to the current admission and the cause of death was unknown.
The father of the patient was still alive.
On admission (day 1), a temperature of 36.5˚C, a
blood pressure of 112/70 mmHg, a pulse rate of 76 beats per min and
a breathing rate of 20 breaths per min were recorded. The patient
had a height of 148 cm and weighed 40 kg. The physical examination
did not detect any other obvious abnormalities. The neurological
examination revealed a loss of hearing in the left ear and very
poor hearing in the right ear. Comprehension and reading ability
were normal, whereas left limb movement and sensation were
impaired, with a muscle strength level of 3.
Blood test results on admission were unremarkable,
except for the fact that the serum lactic acid level was 3.42
mmol/l (normal range, 0.5-2.2 mmol/l), the blood glucose was 8.6
mmol/l (normal range, 3.85-6.11 mmol/l) and HbA1c was elevated at
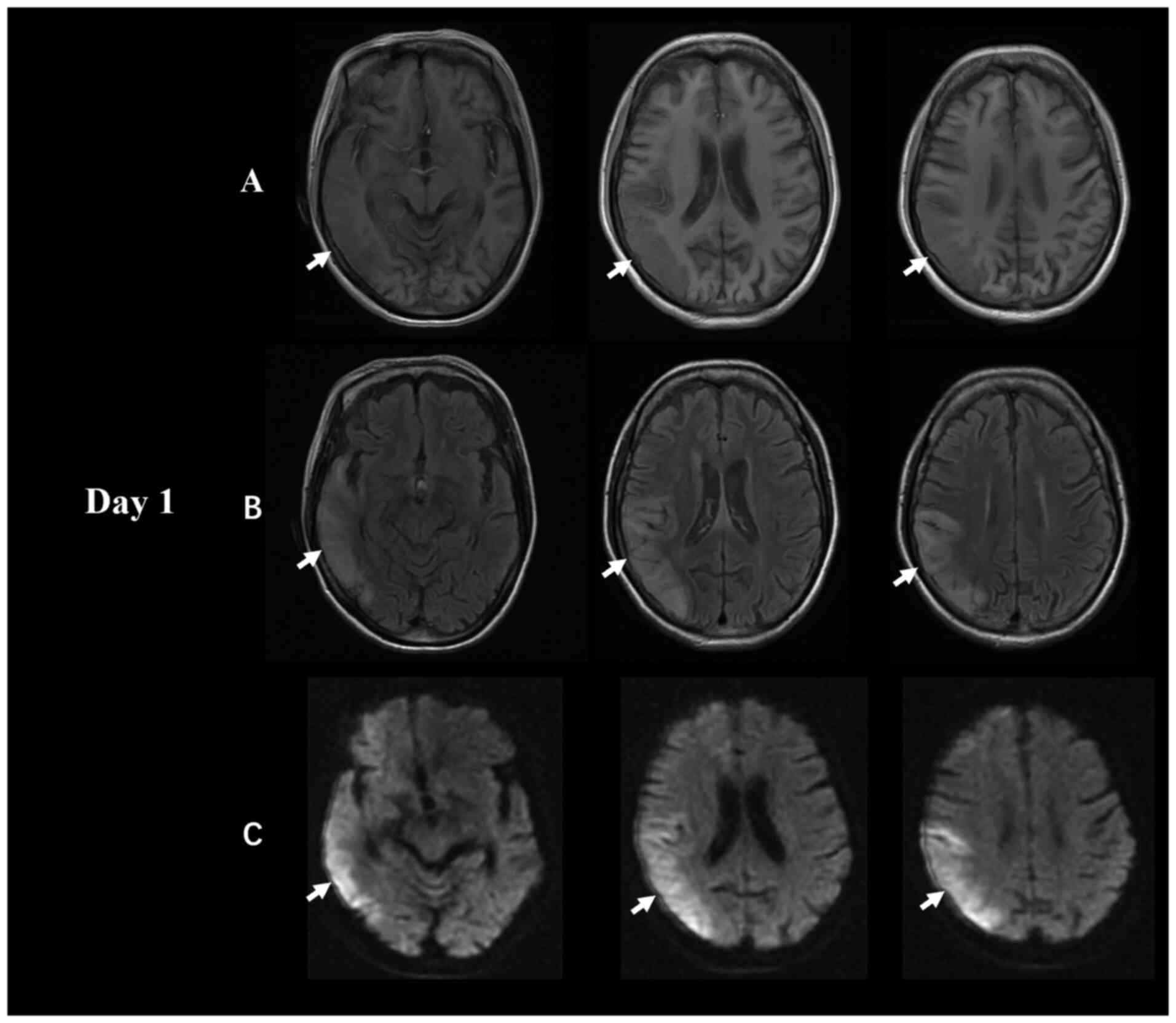
7.2% (normal range, 4-6%). The brain MRI scan obtained on admission
showed a decreased T1-weighted imaging signal and an increased
diffusion-weighted imaging (DWI)/fluid-attenuated inversion
recovery (FLAIR) signal in the right temporal and parietal lobes,
which mainly affected the cerebral cortex with underlying
subcortical oedema (Fig. 1).
However, no significant abnormality was observed on magnetic
resonance angiography (MRA) (Fig.
S1).
The patient was empirically treated with intravenous
butylphthalide (dose of 200 mg per time, three times a day) and
oral aspirin (dose of 100 mg, once a day) to address a possible
cardiogenic cerebral embolism. Dynamic electrocardiogram and
echocardiography did not show any evidence of a cardiogenic
embolism. The condition of the patient was improved after ~1 week
and they were discharged from the hospital.
At 2 days before the current admission, the family
members had observed that the speech of the patient lacked clarity,
with a slow response. Due to the fact that the patient had been
discharged from the hospital only 1 month earlier due to ischemic
stroke, the family members were worried about a recurrence. The
patient was admitted to hospital for emergency treatment due to
recurrent ischemic strokes.
On readmission (46 days after the initial stroke),
the physical examination resembled the previous examination. The
neurological examination demonstrated that the patient exhibited a
clear mind, poor higher neurological function (orientation, memory
and calculation functions), dysarthria and other obvious abnormal
neurological signs.
The blood tests did not reveal any significant
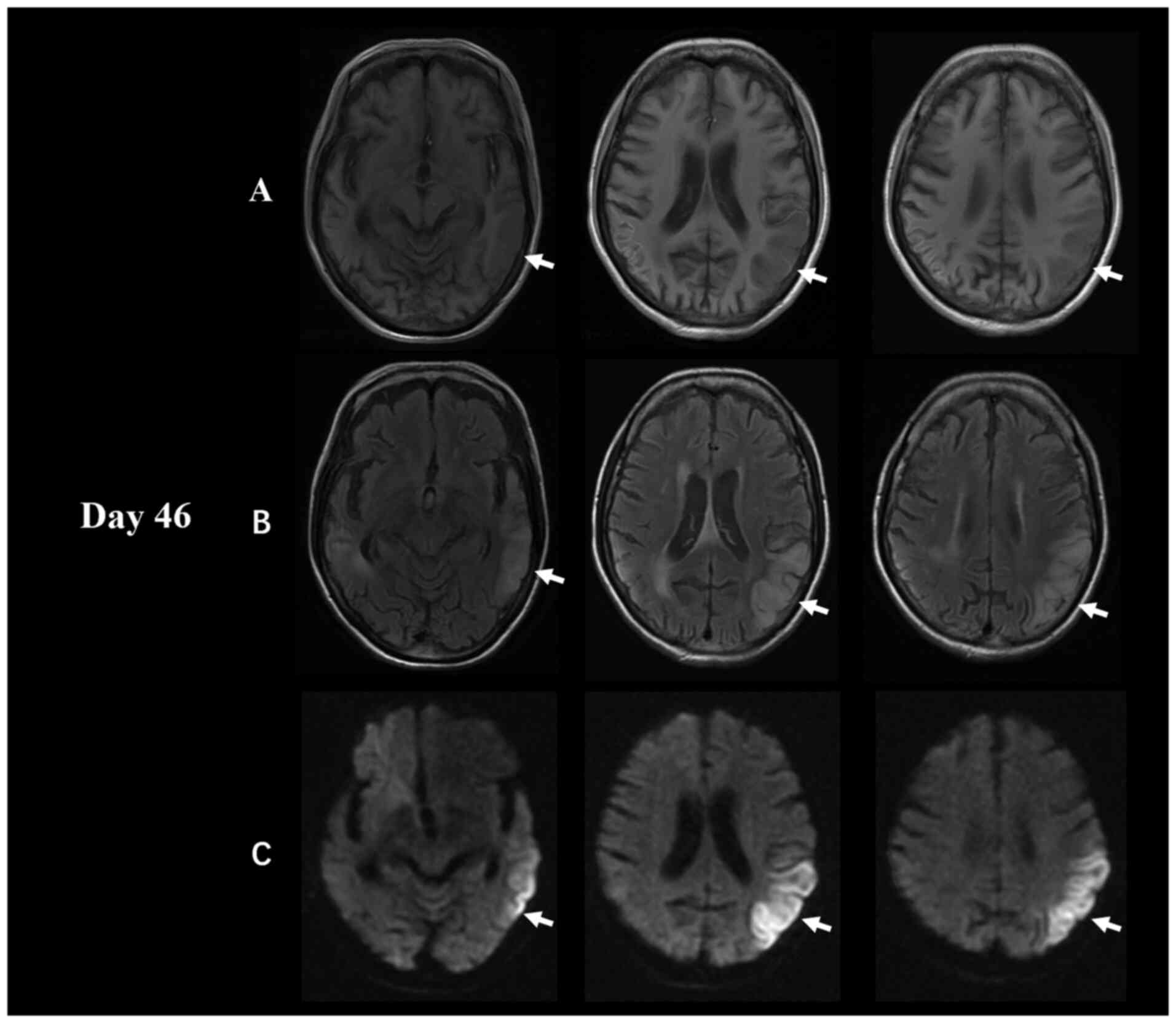
changes from the first admission. However, the MRI analysis showed
a new lesion, a decreased T1-weighted imaging signal and an
increased DWI/FLAIR signal in the left temporal, parietal and
occipital lobes (Fig. 2). In
addition, there was a reduction in the area covered by the old
lesion in the right temporal and parietal lobes compared with the
previous month. The MRA analysis was still normal (no significant
change from 1 month ago).
Therefore, the presence of the combination of
diabetes, hearing impairment, short stature, good cerebrovascular
status and lack of evidence of cardiogenic embolism indicated the
requirement for a genome analysis (mitochondrial gene sequencing,
positive genetic testing for the mitochondrial DNA mutation)
performed by Kindstar Global (Beijing) Technology, Inc., which
demonstrated a m.3243A>G mutation (Fig. S2), thus confirming the presence of
MELAS syndrome.
The patient was treated with L-carnitine (1,000
mg/day), vitamin B1 (100 mg/day) and vitamin B12 (100 mg/day), and
the diabetes treatment regimen was adjusted. The symptoms gradually
improved, after which the patient was discharged from the hospital.
The medication was well tolerated, and no adverse events occurred.
At the last follow-up (6 months after discharge), the Modified
Rankin Scale (5) was scored as 3,
which indicated residual cognitive deficits. Follow-up was
performed once every 3 months for 1 year. Electromyography, nerve
conduction and muscle biopsy tests were not performed during the
disease course, as these studies could not be conducted due to a
lack of consent from the patient and their family. After the
genetic analysis, the daughter of the patient was also determined
to have an m.3243A>G mutation, whereas no mutation was present
in the father of the patient.
Discussion
This case highlights the importance of considering
MELAS as a potential cause of recurrent stroke-like events when
imaging findings are atypical for ischemic stroke, even among
middle-aged patients with vascular risk factors. The patient in the
present study was middle-aged, had diabetes and had two stroke-like
episodes; however, the imaging findings were not consistent with
the symptoms of ischemic stroke, and recurrent stroke was not
considered past this point. This inconsistency triggered the
completion of a mitochondrial gene analysis.
Although MELAS syndrome is a matrilineal inherited
disorder, some sporadic cases also exist. The incidence of MELAS
syndrome is slightly higher in males than in females. Moreover, the
onset of MELAS syndrome is usually between 2 and 31 years, whereas
the onset after 40-years-old is extremely rare (4,6). In
the present study, the patient was 48-years-old before MELAS onset,
which is a relatively rare occurrence. The existence of a family
history, as well as the early deaths of the mother and brother of
the patient (likely from the same disease), and the presence of an
m.3243A>G mutation in the daughter, were demonstrated via the
mitochondrial gene analysis. Although there are specific mutations
that are typically associated with MELAS (m.3243A>G and
m.3271T>C), MELAS is a polygenetic disorder associated with at
least 29 specific point mutations. A number of mutations,
especially those involving protein subunits, have been implicated
in other mitochondrial syndromes [such as Leber's hereditary optic
neuropathy, Leigh Disease and myoclonic epilepsy with ragged-red
fibers (MERRF)] (5,7). This combination of different diseases
caused by a single mutation and polygenic mutations coexist in
polygenic syndromes, thus leading to a bewildering variety of
features of mitochondrial disease (5).
The clinical manifestations of MELAS syndrome are
diverse. However, stroke-like episodes are one of the cardinal
features of MELAS syndrome that occur in 84-99% of affected
individuals (8). Researchers
analysed the clinical characteristics of MELAS, and found that the
main neurological symptoms were epileptic seizures, hemiplegia or
partial numbness, cortical blindness or hemianopia, headaches,
mental retardation or dementia, exercise intolerance and
sensorineural deafness. Non-neurological symptoms included a short
stature, hirsutism, fever, vomiting and kidney damage (9). The clinical manifestations of the
present patient included motor intolerance, hemiplegia,
sensorineural deafness and a short stature. In addition, the
patient case was complicated with diabetes, which is not uncommon
in patients with MELAS. This is due to the fact that the
m.3243A>G mutation can not only cause neurological and muscular
system lesions, but is also one of the most common pathogenic point
mutations of mtDNA mutation in diabetes, and diabetes often
precedes the neurological symptoms (9). In addition, the present patient had
cardiomyopathy, and an m.3243A>G mutation is also a cause of
cardiomyopathy. In total, >30% of MELAS patients have heart
conditions (10). Thus, the
clinical manifestations of MELAS are often diverse and are often
misdiagnosed.
The preliminary screening of MELAS usually requires
a neuroimaging examination and laboratory biochemical tests.
Pathological biopsy is an important diagnostic tool, whereas
genetic testing is the gold standard for the diagnosis of MELAS
(11,12). Furthermore, lactic acid levels in
the blood and cerebrospinal fluid are often analysed during
biochemical tests, as mitochondrial dysfunction in patients with
MELAS syndrome results in enhanced anaerobic fermentation and
increased lactic acid production. The present case analysis
revealed that the patient had increased lactic acid levels.
However, a variety of factors, such as exercise, hypoxia and
improper sample collection and storage, can affect the blood lactic
acid level, which can lead to an increase in the lactic acid
reading and a corresponding incidence of false-positives. Although
lactic acid and pyruvate minimum exercise tests are often used in
clinical practice to understand mitochondrial function, there is
still no test standard. Other diseases with mitochondrial
involvement may also have similar positive results, thus leading to
false-positive results. It has been indicated that the
lactate/pyruvate ratio is meaningful only when the lactic acid
level is elevated to a certain level (>2.5 mmol/l) (12).
The characteristic neuroimaging areas involved in
MELAS do not conform to the classical vascular distribution and are
asymmetric; they mainly involve the temporal, parietal and
occipital lobes. Furthermore, they are limited to the cortical
areas or involve subcortical white matter and can migrate,
fluctuate or even disappear over time (13). Acute cranial MRI shows a decreased
T1 signal and increased T2, FLAIR, and DWI signals in cerebral
cortical and/or subcortical white matter lesions, whereas the
corresponding ADC shows equal or decreased signals (14). In the present patient, brain MRI
showed a large lesion in the right temporal and parietal lobes at
first onset (Fig. 1). However, the
second brain MRI showed a large area of fresh lesions in the left
temporal, parietal and occipital lobes 1 month later, and the old
lesion on the right side was smaller than before (Fig. 2). These lesion fluctuations at
different times were also consistent with the imaging
characteristics of MELAS.
Muscle pathological biopsy is an important basis for
the diagnosis of patients with suspected MELAS. Gomori staining of
frozen sections of muscle biopsy shows ragged-red fibres (RRF) and
a number of degenerated mitochondria. Furthermore, glycogen
staining also shows an accumulation of fat and glycogen. The
positive staining of the vascular wall of the muscle tissue with
subrinic acid dehydrogenase and the crystalloid inclusion bodies
under an electron microscope are helpful in the diagnosis of MELAS
(15). However, these pathological
manifestations are not solely present in MELAS, but can also appear
in MERRF and Kearns-Sayre syndrome, especially in early childhood.
In addition, due to the fact that it is an invasive examination,
the acceptance rate is low. The patient and the family in the
present case study were not willing to approve a muscle biopsy.
Nonetheless, muscle biopsy is useful in cases where there are no
mutation bases or during the investigation of other mitochondrial
disorders.
Mitochondrial DNA analysis is a diagnostic assay of
decisive significance, and urine-derived DNA testing is now
available in clinical practice, which is replacing the use of
muscle-derived DNA, due to the fact that the critical test in
investigating mitochondrial disorders is to establish the genetic
mutation, rather than necessarily demonstrating muscle pathology
(15). Currently, the detection of
gene mutation sites is the gold standard for the diagnosis of MELAS
syndrome. Although the most common cause of MELAS is m.3243A>G
mutation, MELAS can also be caused by a variety of other mtDNA and
nuclear gene mutations. In recent years, nearly 20 types of
MELAS-related mtDNA point mutations have been found, including
m.3243A>G, m.3252A>G, m.3291T>C, m.3271T>C,
m.3995A>G and m.3959G>A. Approximately 80% of patients with
MELAS syndrome have the m.3243A>G mutation of the mtDNA tRNA
leucine gene site (16). Similar
to these results, the patient and daughter in the present study
also had the same point mutation.
To date, there is no specific treatment for MELAS
syndrome, and symptomatic treatment is often applied in clinical
practice. Diet changes can reduce the production of endogenous
toxic metabolites. Specifically, a high-protein, high-carbohydrate
and low-fat diet compensates for impaired gluconeogenesis and
reduces fat breakdown. Valproic acid should be avoided in the
treatment of seizures due to its deleterious effects on
mitochondrial function. Moreover, metformin should be avoided in
individuals with MELAS syndrome due to its propensity to cause
lactic acidosis (16,17). The current patient had diabetes but
was fortunate to have been treated with acarbose. In addition, ATP,
coenzyme Q10 and large doses of B vitamins can reduce blood lactate
and pyruvate levels. L-carnitine can improve energy metabolism and
promote lipid metabolism. Furthermore, symptomatic treatments are
used for heart disease, seizures, cranial hypertension and
diabetes. Although a number of limitations exist for the current
treatment of MELAS, gene therapy remains an attractive research
direction. Stem cell-derived mitochondrial transplantation has been
shown to play an important role in metabolic rescue (18), which offers hope for the treatment
of MELAS. An earlier onset and increased incidence of clinical
symptoms corresponds to a worsened prognosis (19). Therefore, early diagnosis and early
intervention are particularly important, which may improve the
patient prognosis.
In conclusion, in patients with recurrent stroke,
there is a need for careful analysis of the occurrence and
development of the disease, and the past medical history and family
history of the patient, and a careful interpretation of the imaging
characteristics. Indeed, the aforementioned clinical manifestations
and imaging features are not specific to MELAS. Other potential
conditions to consider include subacute ischemic stroke,
progressive multifocal leukoencephalopathy, herpetic encephalitis,
posterior reversible encephalopathy syndrome and vasculitis.
Therefore, mitochondrial DNA testing may be important in
determining whether a patient has MELAS. At present, the treatment
of MELAS is still symptomatic. If the ultimate outcome is to be
improved, future therapeutic research should consider an
improvement in the potential of cell therapy-based mitochondrial
restoration, especially stem cell therapy for MELAS.
Supplementary Material
Brain MRA scans obtained after (A) the
first admission (day 1) and (B) the second admission (day 46).
Brain MRA did not reveal any evidence of intracranial arterial
stenosis or occlusion. MRA, magnetic resonance angiography.
Genome analysis. Genome analysis
demonstrated an adenine to guanine conversion at mitochondrial
genome 3243.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the Project of Nanchong
Science and Technology Bureau (grant no. 19SXHZ0051).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FY and SP were equal contributors in writing the
manuscript. FY, QP and SP analysed and interpreted the patient data
regarding the series of MRI data. FY and SP confirm the
authenticity of all the raw data. All of the authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written consent was obtained for publication from
the patient's legal representative (husband).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang S, Miao J and Feng J: Case report:
Mitochondrial encephalomyopathy presents as epilepsy, ataxia, and
dystonia with a rare mutation in MT-TW. Front Neurol.
12(679302)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gorman GS, Chinnery PF, DiMauro S, Hirano
M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M and
Turnbull DM: Mitochondrial diseases. Nat Rev Dis Primers.
2(16080)2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Koenig MK, Emrick L, Karaa A, Korson M,
Scaglia F, Parikh S and Goldstein A: Recommendations for the
management of strokelike episodes in patients with mitochondrial
encephalomyopathy, lactic acidosis, and strokelike episodes. JAMA
Neurol. 73:591–594. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sinnecker T, Andelova M, Mayr M, Rüegg S,
Sinnreich M, Hench J, Frank S, Schaller A, Stippich C, Wuerfel J
and Bonati LH: Diagnosis of adult-onset MELAS syndrome in a
63-year-old patient with suspected recurrent strokes-a case report.
BMC Neurol. 19(91)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sproule DM and Kaufmann P: Mitochondrial
encephalopathy, lactic acidosis, and strokelike episodes: Basic
concepts, clinical phenotype, and therapeutic management of MELAS
syndrome. Ann N Y Acad Sci. 1142:133–158. 2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Oyama M, Iizuka T, Nakahara J and Izawa Y:
Neuroimaging pattern and pathophysiology of cerebellar stroke-like
lesions in MELAS with m.3243A>G mutation: A case report. BMC
Neurol. 20(167)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wong LJ: Pathogenic mitochondrial DNA
mutations in protein-coding genes. Muscle Nerve. 36:279–293.
2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yatsuga S, Povalko N, Nishioka J, Katayama
K, Kakimoto N, Matsuishi T, Kakuma T and Koga Y: Taro Matsuoka for
MELAS Study Group in Japan. MELAS: A nationwide prospective cohort
study of 96 patients in Japan. Biochim Biophys Acta. 1820:619–624.
2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chen H, Hu Q, Raza HK, Chansysouphanthong
T, Singh S, Rai P, Cui G, Zhang Z, Ye X, Xu C, et al: An analysis
of the clinical and imaging features of mitochondrial
encephalopathy, lactic acidosis, and stroke-like episodes (MELAS).
Somatosens Mot Res. 37:45–49. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Brambilla A, Favilli S, Olivotto I,
Calabri GB, Porcedda G, De Simone L, Procopiow Pasquini E and
Donati MA: Clinical profile and outcome of cardiac involvement in
MELAS syndrome. Int J Cardiol. 276:14–19. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Finsterer J: Muscle biopsy is not
diagnostic for MELAS. J Neurol Sci. 410(116670)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Parikh S, Goldstein A, Koenig MK, Scaglia
F, Enns GM, Saneto R, Anselm I, Cohen BH, Falk MJ, Greene C, et al:
Diagnosis and management of mitochondrial disease: A consensus
statement from the mitochondrial medicine society. Genet Med.
17:689–701. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Doppler CEJ, Kabbasch C, Fink GR, Lehmann
HC and Wunderlich G: Rapid alterations in MR imaging in MELAS
syndrome. Pract Neurol. 19:447–448. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ito H, Mori K and Kagami S: Neuroimaging
of stroke-like episodes in MELAS. Brain Dev. 33:283–288.
2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Goodfellow JA, Dani K, Stewart W, Santosh
C, McLean J, Mulhern S and Razvi S: Mitochondrial myopathy,
encephalopathy, lactic acidosis and stroke-like episodes: An
important cause of stroke in young people. Postgrad Med J.
88:326–334. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
El-Hattab AW, Adesina AM, Jones J and
Scaglia F: MELAS syndrome: Clinical manifestations, pathogenesis,
and treatment options. Mol Genet Metab. 116:4–12. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Finsterer J: Optimising therapeutic
strategies for acute stroke-like lesions in MELAS.
eNeurologicalSci. 21(100278)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Liu K, Zhou Z, Pan M and Zhang L: Stem
cell-derived mitochondria transplantation: A promising therapy for
mitochondrial encephalomyopathy. CNS Neurosci Ther. 27:733–742.
2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Finsterer J and Wakil SM: Stroke-like
episodes, peri-episodic seizures, and MELAS mutations. Eur J
Paediatr Neurol. 20:824–829. 2016.PubMed/NCBI View Article : Google Scholar
|