Introduction
As a prevalent disease, chronic obstructive
pulmonary disease (COPD) is characterized by incessant respiratory
disorders as well as airflow restriction, commonly associated with
airway and alveolar abnormalities (1). Dyspnea, cough and phlegm are the most
frequent clinical manifestations of COPD, which usually results
from constant exposure to detrimental particles or gases (2). In the majority of patients, COPD is
often accompanied by other chronic diseases, which can
significantly increase the incidence and mortality rate, seriously
threatening human public health (3). It is widely accepted that COPD is
closely associated with the abnormal reaction of airways and lung
tissue to harmful gases, such as cigarette smoke or toxic particles
(4). Chronic exposure to cigarette
smoke often results in COPD and it has been reported that >50%
of smokers eventually develop the disease (5). Therefore, smoking cessation either at
home or in a hospital setting is of significant importance for
preventing the development of COPD.
As one of the most naturally occurring carotenoids,
fucoxanthin (FX), chiefly found in sea zones (brown algae algae)
and microalgae (diatoms), serves a critical role in alga
photosynthesis (6). It has been
reported that FX has several biological properties, since it can
protect against oxidative stress, tumors, bacteria, viruses,
obesity and neuron injury, with minimal toxicity and side effects
(7). At present, FX can be used as
a weight loss health care product (8). Clinically, as a drug, FX can be used
in the treatment of skin cancer, colon cancer, prostate cancer,
liver cancer and other cancers (9). A previous study showed that FX
promoted lipopolysaccharide (LPS)-induced acute lung injury (ALI)
via inhibiting Toll-like receptor 4/major myeloid differentiation
response gene 88 signaling (10).
FX was also demonstrated to inhibit fibrogenesis to alleviate
bleomycin-induced pulmonary fibrosis (11). Another study in asthmatic mice
revealed that FX improved oxidative stress and airway inflammation
in tracheal epithelial cells (12). Additionally, the antitumor activity
of FX has been previously reported in lung cancer (13). However, the effects of FX on COPD
have not been previously investigated.
A previous study demonstrated that rosiglitazone
inhibited the polarization of M1 macrophages via triggering
peroxisome proliferator-activated receptor γ (PPARγ) and retinoid X
receptor-α, thereby improving cigarette smoke-induced airway
inflammation (14). Additionally,
another study revealed that erythromycin exhibited suppressive
effects on cigarette smoke-induced inflammation via triggering the
PPARγ/NF-κB signaling pathway in macrophages (15). It was therefore hypothesized that
PPARγ may be a promising target in terms of pathophysiology and
pharmacology for the improvement of COPD. PPARγ activation possibly
results in NF-κB-dependent, CSE-induced and chemokine-modulated
inflammatory responses (16).
Bioinformatics analysis using the search tool for interactions of
chemicals (STITCH) database predicted that PPARγ could be a target
of FX. In addition, it has been shown that FX regulates the
expression of PPARγ in 3T3-L1 cells and inhibits the uptake of
glucose and mature adipocytes (17). Moreover, FX was revealed to inhibit
proinflammatory cytokines by regulating NF-κB and NLRP3
inflammasome activation (18).
Thus, it was hypothesized that FX may regulate COPD via targeting
the PPARγ/NF-κB signaling pathway.
Therefore, the present study aimed to investigate
the role of FX in oxidative injury and inflammation in cigarette
smoke-induced human bronchial epithelial cells of patients with
COPD, as well as its underlying mechanism.
Materials and methods
Preparation of aqueous cigarette smoke
extract (CSE)
Firstly, three cigarettes from commercial Da Qianmen
cigarettes (containing 2.5 mg of nicotine and 12 mg of tar per
cigarette) were burned and the smoke was then collected in a
container supplemented with 10 ml PBS using a vacuum pump. The pH
of 100% CSE solution was adjusted to 7.4, followed by filtering
through a 0.22-µm sterile filter. Prior to use, the well-prepared
100% CSE was diluted in RPMI 1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) supplemented without FBS (19). Prior to treatment with FX,
GSE-exposed cells were co-treated with 10 µM PPARγ inhibitor,
T0070907 (Beijing BioLab Technology Co., Ltd.).
Cell culture
Human bronchial BEAS-2B cells (cat. no. BS-C1281173;
Shanghai Binsui Biotechnology Co., Ltd.) were cultured in DMEM with
10% FBS (both from Gibco; Thermo Fisher Scientific, Inc.). BEAS-2B
cells were cultured in medium supplemented with 1, 2 or 5% CSE for
24 h. Additionally, BEAS-2B cells were pre-treated with 5, 10, 20
and 40 µM FX followed by treatment with 5% CSE (10). FX concentrations of 0, 5, 10, 20
and 40 µM were used at first, and Cell Counting Kit-8 (CCK8) was
used to detect cell viability. It was determined that a
concentration of 40 µM FX damaged the cells, thus concentrations of
0, 5, 10 and 20 µM FX were selected for the experiments.
CCK-8 assay
Cells were seeded into 96-well culture plates at a
density of 1x104 cells/well and were then treated as
aforementioned. Subsequently, the cells were incubated with CCK-8
solution (Millipore Sigma) at 37˚C for 3 h and the absorbance at
450 nm was decided using a microplate reader (Thermo Fisher
Scientific, Inc.).
ELISA
The levels of tumor necrosis factor (TNF)-α (cat.
no. H052-1), interleukin (IL)-1β (cat. no. H002) and IL-6 cat. no.
(H007-1-1) in culture supernatants were measured using the
corresponding ELISA kits. Briefly, 100 µl cell supernatant was
supplemented into each well and ELISA was carried out according to
the manufacturer's instructions. Additionally, a lactate
dehydrogenase (LDH; cat. no. A020-2-2) assay was used to detect LDH
levels according to the manufacturer's instructions. All kits were
purchased from Nanjing Jiancheng Bioengineering Institute.
TUNEL assay
BEAS-2B cells (2x104 cells/well) were
seeded into 6-well plates and were then treated as aforementioned.
Subsequently, the cells were fixed with 4% paraformaldehyde at 37˚C
for 15 min followed by the cultivation with TUNEL solution for 1 h
at 37˚C. The cells were then stained with 3,3-diaminobenzidine
(Sigma-Aldrich; Merck KGaA) for 10 min at room temperature
according to the manufacturer's protocol. Cell nuclei were stained
with 0.1 µg/ml DAPI for 5 min at room temperature and nuclear DNA
fragmentation was assessed using the DeadEnd™ Fluorometric TUNEL
system (Promega Corporation). Finally, the cells were observed in
five randomly selected fields under an Olympus IX71 fluorescence
microscope (Olympus Corporation). TUNEL-positive cells and total
cells were analyzed using ImageJ 1.8.0 software (National
Institutes of Health).
Western blot analysis
Total proteins were extracted from BEAS-2B cells
following treatment with RIPA lysis buffer (Shanghai Absin
Biotechnology Co., Ltd.) on ice. The cells were centrifuged at
16,000 x g for 10 min at 4˚C, and the supernatant was collected for
western blotting. A bicinchoninic acid protein assay kit (Thermo
Fisher Scientific, Inc.) was used to detect the protein
concentrations. Subsequently, protein extracts (30 µg) were
separated by 10% SDS-PAGE and were then transferred onto PVDF
membranes (Sigma-Aldrich; Merck KGaA). Following the block with 5%
non-fat milk for 60 min at room temperature, PVDF membranes were
incubated with primary antibodies Bcl-2 (1:1,000; product code
ab32124), Bax (1:1,000; product code ab32503), PPARγ (1:1,000;
product code ab272718), phosphorylated (p)-NF-κB p65 (1:1,000;
product code ab32536), NF-κB p65 (1:1,000; product code ab207297)
or GAPDH (1:1,000; product code ab9485) at 4˚C overnight.
Subsequently, the membranes were cultivated with goat anti-rabbit
IgG H&L (HRP) preadsorbed antibody (1:5,000; product code
ab7090; all from Abcam) at room temperature for 1 h. An ECL system
(Beyotime Institute of Biotechnology) was used to develop the
membranes and blots were analyzed using ImageJ 1.8.0 (National
Institutes of Health).
Reactive oxygen species (ROS)
detection
Briefly, to measure ROS levels, cells
(1x104 cells/well) seeded into a 96-well plate were
treated with 2,7-dichlorodi-hydrofluorescein diacetate (DCFH-DA;
Beyotime Institute of Biotechnology) solution (dilution, 1:1,000)
at a concentration of 2 µM at 37˚C for 20 min. ROS content was
assessed by measuring the fluorescence intensity of each well at a
wavelength of 488 and 525 nm, separately, using a microplate
reader. The control group served as the baseline absorbance value
to calculate the average fluorescence intensity of each group.
Detection of malondialdehyde (MDA),
superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px)
The activities of SOD (cat. no. A001-3-1; WST-1
method), MDA (cat. no. A003-1-1; colorimetric method) and GSH-Px
(cat. no. A005-1-2; colorimetric method) were assessed using the
corresponding kits according to the manufacturer's protocol
(Nanjing Jiancheng Bioengineering Institute) by measuring the
absorbance at 450, 532 and 412 nm, respectively, with a microplate
reader.
Molecular docking
ChemDraw software 21 (ChemDraw; PerkinElmerk, Inc.)
was used to visualize the structure of FX, which was subsequently
hydrogenated using OpenBabel (v2.2.1) software and converted into a
mol2 format file (20). The
structure of PPARγ was downloaded from the RCSB Protein Data Bank
(PDB) webpage (https://www.rcsb.org/). The removal
of surplus water molecules, the deletion of irrelevant small
ligands originally carried and protein structure analysis were
performed by opening the PDB file in PyMOL (v2.2.0) software
(21). Since the predicted protein
structure carried a ligand, this ligand was removed and the
original ligand position was considered as the docking site.
Following analysis in AutoDock (v4.2), the specific docking energy
values were displayed (22). The
results were analyzed using Protein-Ligand Interaction Profiler
(https://plip-tool.biotec.tu-dresden.de/plip-web)
database.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA that was isolated with a RNeasy Mini
Kit (Qiagen China Co., Ltd) was then reversely transcribed into
cDNA using M-MLV RTase and random primer (GeneCopoeia, Inc.). qPCR
was performed on an ABI PRISM 7900HT (Applied Biosystems; Life
Technologies; Thermo Fisher Scientific, Inc.) using SYBR Premix Ex
Taq™ (Takara Bio, Inc.) or Taqman probes (Applied Biosystems;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The thermocycling conditions were as follows: 95˚C for 5
min, followed by 40 cycles at 94˚C for 15 sec, 60˚C for 20 sec and
72˚C for 40 sec. Relative expression levels were calculated using
2-ΔΔCq method (23).
The primers were as follows: PPARγ forward,
5'-CCAGAAGCCTGCATTTCTGC-3' and reverse, 5'-CACGGAGCTGATCCCAAAGT-3';
GAPDH forward, 5'-AATGGGCAGCCGTTAGGAAA-3' and reverse,
5'-GCGCCCAATACGACCAAATC-3'.
Database
The search tool for interactions of chemicals
(STITCH) database was used to predict the targets of FX (http://stitch.embl.de/) .
Statistical analysis
All data were analyzed using SPSS 19.0 software (IBM
Corp.). All results are expressed as the mean ± SD. The differences
between multiple groups were compared using one-way ANOVA followed
by Tukey's post hoc test with GraphPad Prism 5 software (GraphPad
Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference. All experiments were repeated
at least three times.
Results
FX enhances CSE-induced BEAS-2B cell
activity
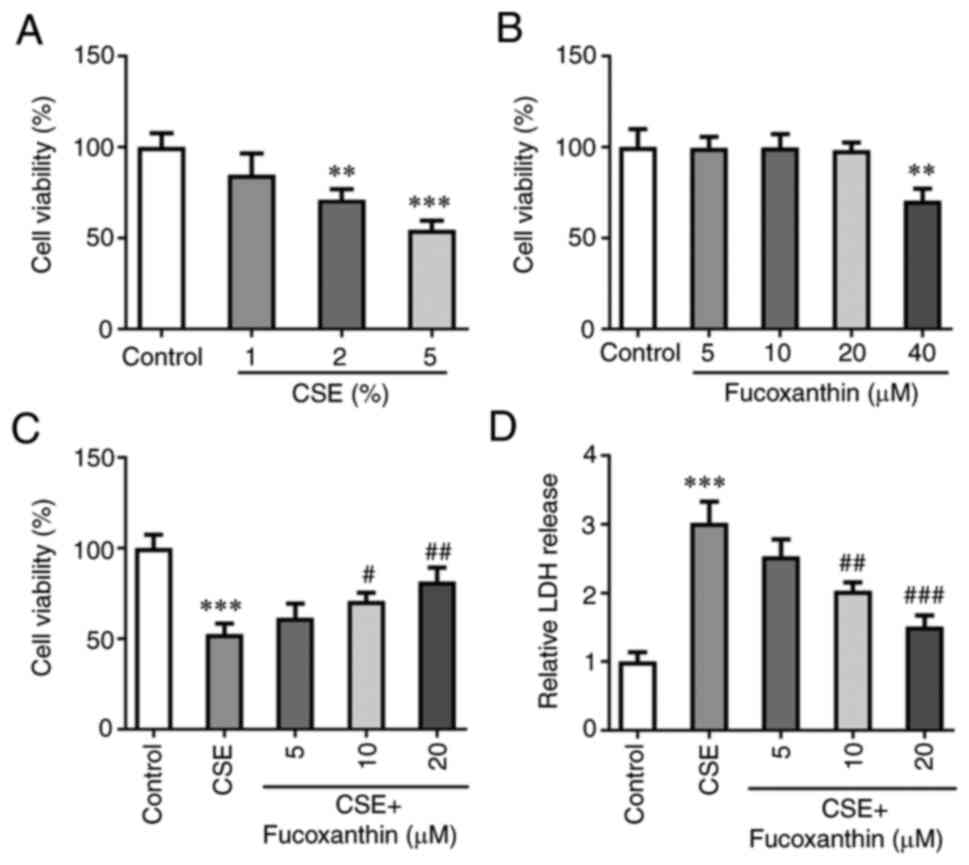
Following exposure of BEAS-2B cells to 1, 2 or 5%
CSE, cell viability was evaluated. The results of the CCK-8 assay
demonstrated that cell viability of BEAS-2B cells was markedly and
gradually decreased with the increasing concentrations of CSE
(Fig. 1A). A concentration of 5%
CSE was selected for the follow-up experiments. Subsequently,
BEAS-2B cells were induced with 5, 10, 20 or 40 µM FX, and the
results demonstrated that FX at concentrations of 5, 10 and 20 µM
did not affect the cell viability of BEAS-2B cells, and FX at a
concentration of 40 µM significantly decreased cell viability
(Fig. 1B). To avoid FX-induced
cell injury, concentrations of 5, 10 and 20 µM FX were selected for
the subsequent experiments. Cells were then divided into the
following five groups: The control group; the CSE group; the CSE +
5 µM FX group; the CSE + 10 µM FX group; and the CSE + 20 µM FX
group. The results of the CCK-8 assay demonstrated that the
CSE-induced decrease of cell viability was reversed with increasing
concentrations of FX (Fig. 1C).
Additionally, LDH levels were assessed. The results revealed that
LDH production was significantly enhanced in the CSE group compared
with the control group. However, compared with the CSE group, the
levels of LDH were significantly reduced in the CSE + FX groups
(Fig. 1D).
FX attenuates CSE-induced inflammation
and oxidative damage in BEAS-2B cells
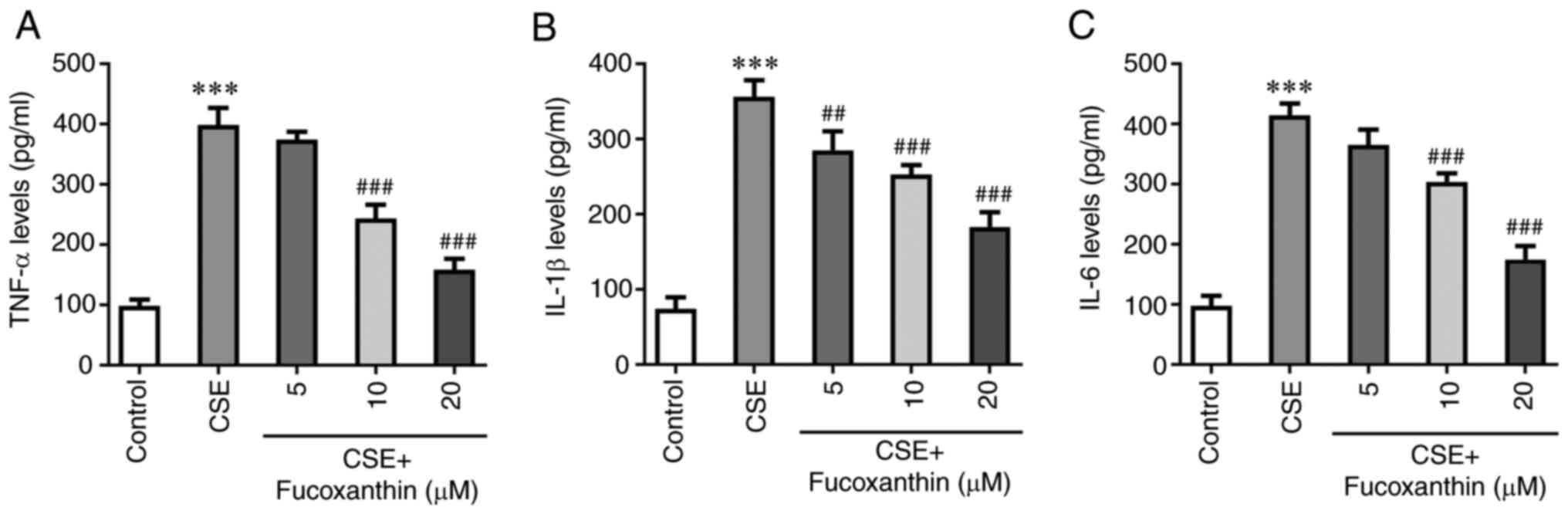
The results of ELISA demonstrated that the levels of
TNF-α, IL-1β and IL-6 were significantly increased in the CSE group
compared with the control group. However, the levels of the
aforementioned cytokines were decreased with increasing
concentrations of FX in the CSE + FX groups compared with the CSE
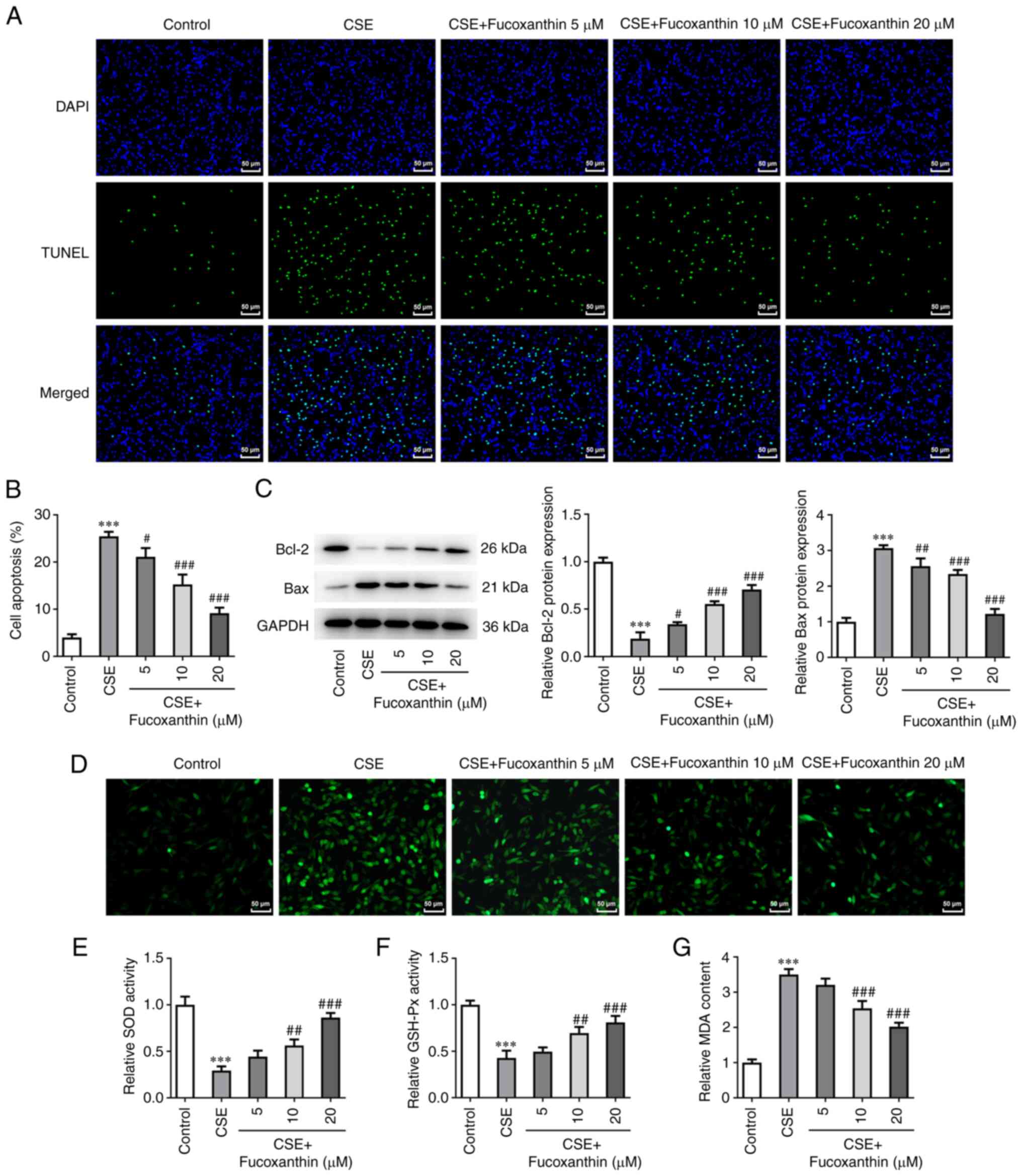
group (Fig. 2). TUNEL assay and
western blot analysis revealed that compared with the control
group, cell apoptosis was evidently enhanced in the CSE group,
accompanied by Bcl-2 downregulation and Bax upregulation. Compared
with the CSE group, cell apoptosis was decreased in the CSE + FX
groups in a dose-dependent manner, as verified by the increased
Bcl-2 expression and decreased Bax expression (Fig. 3A-C). Additionally, ROS levels were
increased following cell induction with CSE compared with the
control group. However, ROS production was dose-dependently reduced
in CSE-induced cells after cell exposure to FX (Fig. 3D). Furthermore, MDA activity was
significantly elevated, while that of SOD and GSH-Px was attenuated
in the CSE-induced cells. Following cell treatment with increasing
concentrations of FX, MDA activity was dose-dependently reduced,
while SOD and GSH-Px activities were dose-dependently enhanced in
the CSE-induced cells (Fig.
3D).
FX regulates PPARγ/NF-κB
signaling
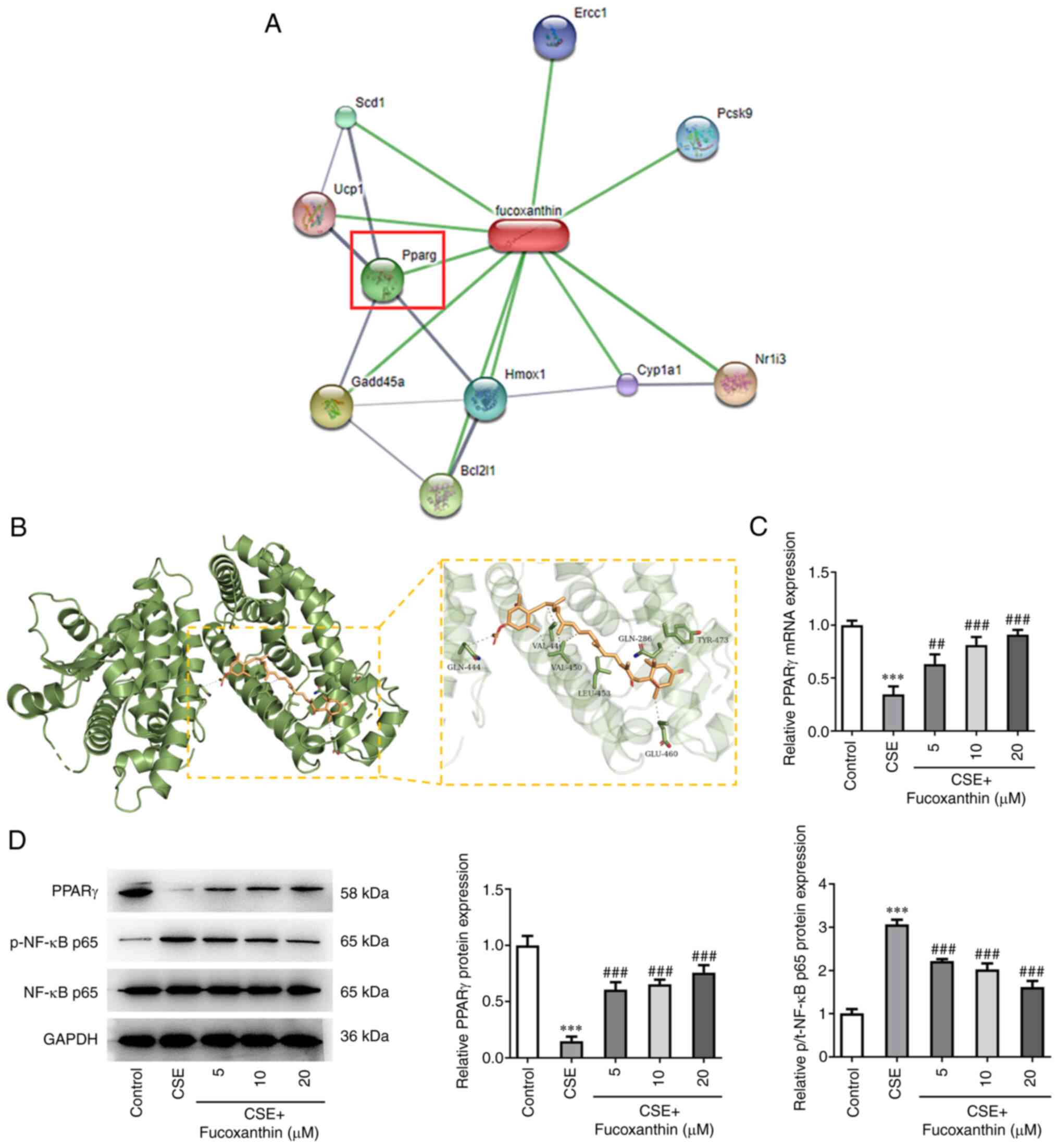
Bioinformatics analysis using the STITCH database
predicted that PPARγ could be a target of FX (Fig. 4A). The three-dimensional structure
of PPARγ protein was obtained from PDB database (PDB ID: 1KNU) and
the Autodock (version 4.2) database was used for molecular docking
(Fig. 4B). The analysis indicated
that FX could regulate PPARγ expression. In addition, western blot
and RT-qPCR analyses showed that PPARγ was downregulated and
p-NF-κB p65 was upregulated following cell treatment with CSE.
However, compared with the CSE group, the expression levels of
PPARγ were gradually increased, while those of p-NF-κB p65 were
gradually reduced in the CSE + FX groups, in a dose-dependent
manner (Fig. 4C and D).
Pretreatment with the PPARγ inhibitor,
T0070907, reverses the protective effects of FX on CSE-induced
BEAS-2B cells
Subsequent assays were carried out using 20 µM FX.
Cells were divided into the following four groups: The control
group; the CSE group; the CSE + FX group; and the CSE + FX +
T0070907 group. Cells in the CSE + FX + T0070907 group were
co-treated with a PPARγ inhibitor, namely T0070907. The results
revealed that the levels of inflammatory cytokines were
significantly increased in the CSE + FX + T0070907 group compared
with the CSE + FX group (Fig.
5A-C). TUNEL assay and western blot analysis revealed that
compared with the CSE + FX group, cell apoptosis was enhanced in
the CSE + FX + T0070907 group, as indicated by Bcl-2 downregulation
and Bax upregulation (Fig. 5D-G).
In addition, DCFH-DA assay showed that T0070907 significantly
reversed the effects of FX on ROS, MDA, SOD and GSH-Px levels in
CSE-induced BEAS-2B cells (Fig.
5H-K).
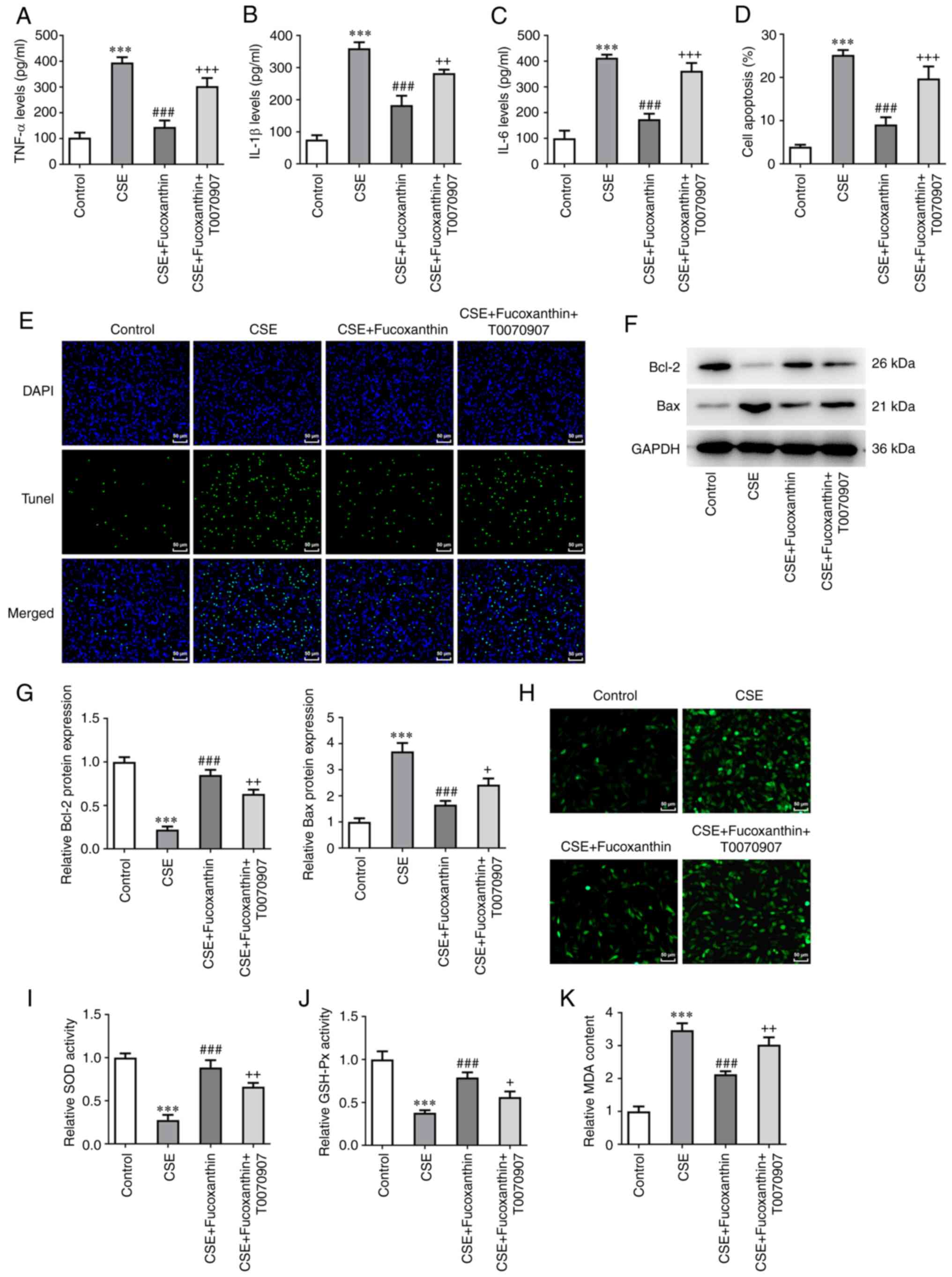 | Figure 5Pretreatment with the PPARγ
inhibitor, T0070907, reverses the protective effects of FX on
CSE-induced BEAS-2B cells. Contents of (A) TNF-α, (B) IL-1β and (C)
IL-6 were examined using ELISA. (D and E) A TUNEL assay was used to
determine the apoptotic rate of cells. (F and G) Western blotting
was used to examine Bcl-2 and Bax expression. (H) DCFH-DA was used
to assess ROS activity. Contents of (I) SOD, (J) GSH-Px and (K) MDA
were examined using relative kits. ***P<0.001 vs.
Control; ###P<0.001 vs. CSE; +P<0.05,
++P<0.01 and +++P<0.001 vs. CSE +
Fucoxanthin. PPARγ, peroxisome proliferator-activated receptor γ;
FX, fucoxanthin; CSE, cigarette smoke extract; TNF-α, tumor
necrosis factor-α; IL-, interleukin; ROS, reactive oxygen species;
SOD, superoxide dismutase; GSH-Px, glutathione peroxidase; MDA,
malondialdehyde. |
Discussion
Epidemiological and genetic risk factors for COPD
include long-term smoking, persistent exposure to air pollution,
respiratory infections, inhalation of biofuel smoke and
occupational dust (24). Among
them, smoking is a pivotal risk factor, significantly contributing
to the development of COPD, thus increasing the rate of impaired
lung function (25,26). This could mainly be due to the
harmful gases and particulate matter produced by tobacco, causing
lung inflammation and oxidative stress response. In turn, lung
inflammation and oxidative stress response could further lead to
lung tissue injury and small airway fibrosis, eventually resulting
in irreversible airflow restriction and various respiratory
symptoms in individuals (27). In
addition, persistent chronic inflammation induces the recurrence of
vascular wall damage and repair processes, thus leading to airway
remodeling, which is the primary cause of irreversible COPD
progression (28). In the present
study, human bronchial BEAS-2B cells were induced with CSE, thus
resulting in oxidative damage and inflammatory response, simulating
COPD in vitro. Moreover, the model of CSE-induced human
bronchial epithelial cells is a recognized COPD model (14,15,19)
When patients with COPD are affected by air pollutants and other
inducements, the epithelial cells are stimulated to release
oxidative stress-related factors SOD, GSH-Px, MDA, which lead to
the accumulation of inflammation-related factors TNF-α, IL-1β and
IL-6 in the airway giving rise to cell damage, and thus causing the
acute exacerbation of COPD (29).
Therefore, oxidative stress and inflammatory response were detected
to be activated after CSE induction in the experiments of the
present study.
FX, a natural carotenoid, is extensively found in
various algae, marine phytoplankton and aquatic shellfish (7). This compound can protect against
inflammation, obesity, diabetes and cancer (6). It has been also reported that FX
exhibits particular therapeutic effects on airway inflammatory or
traumatic diseases. Therefore, a study demonstrated that FX exerted
anti-inflammatory and antiapoptotic effects on lung cancer in
benzo(A) pyrene-induced mice (30). In asthma, FX could effectively
diminish ROS secretion and protect against oxidative stress and
inflammation in bronchoalveolar lavage fluid (31). Another study suggested that FX
could attenuate LPS-induced ALI via suppressing RhoA activation
together with the NF-κB pathway (32). However, the therapeutic effect of
FX on COPD and its underlying mechanism have not been previously
reported. The results of the present study demonstrated that FX
significantly inhibited CSE-induced BEAS-2B cell inflammation and
oxidative damage, thus supporting its effect on improving COPD.
Subsequently, the mechanism underlying the effect of
FX on improving COPD was investigated. Analysis in the STITCH
database predicted that PPARγ could be a target of FX. Furthermore,
the three-dimensional structure of PPARγ was obtained from the PDB
database. Autodock (version 4.2) database was used for molecular
docking. A previous study on type 2 diabetic mice revealed that FX
could distinctly increase PPARγ expression in adipose tissue, thus
improving the metabolism of sugar and fat (33). Herein, FX promoted the expression
of PPARγ and downregulated p-NF-κB p65. It was therefore
hypothesized that PPARγ may be a promising target in terms of
pathophysiology and pharmacology for improving COPD. PPARγ
activation could possibly result in NF-κB-dependent, GSE-induced
and chemokine-mediated regulation of inflammatory responses
(16). The expression levels of
PPARγ were reduced in primary human bronchial cells of patients
with COPD compared with those in healthy smokers (34). Additionally, another study
demonstrated that CSE-induced airway remodeling could be
significantly improved via activating the PPARγ/TGF-β1/Smad
signaling pathway (35). Curcumin
suppressed cigarette-induced inflammation via modulating the
PPARγ/NF-κB signaling pathway, while PPARγ inhibitor T0070907
inhibited this protective effect (36). Furthermore, a previous study
revealed that PPARγ inhibitor T0070907 downregulated the tight
junction barrier function of human nasal epithelial cells through
the PKC signaling pathway (37).
FX was also demonstrated to downregulate the expression of PPARγ
and inhibit adipogenesis in adipocytes, thereby exerting an
anti-obesity effect (38).
Therefore, it was hypothesized that FX may serve an important
regulatory role in improving COPD via regulating the PPARγ/NF-κB
signaling pathway. Herein, the protective effects of FX on
CSE-induced cells were reversed following cell treatment with the
PPARγ inhibitor, T0070907.
FX was revealed to inhibit the inflammatory response
by suppressing the activation of NF-κB and MAPKs in
lipopolysaccharide-induced RAW 264.7 macrophages (39). FX was also demonstrated to have
anti-inflammatory activity in high-fat diet-induced obesity in mice
and an antioxidant function in PC12 cells (40). FX suppressed lipid accumulation and
ROS production during differentiation in 3T3-L1 adipocytes
(41). FX and its metabolite,
fucoxanthinol, suppressed adipocyte differentiation in 3T3-L1 cells
(42). However, the regulation of
FX on PPARγ was not involved, and the regulation of FX on PPARγ in
COPD has not been reported to date, to the best of our knowledge,
which is also the novelty of the present study.
The present study has certain limitations.
Conclusions were drawn from cell experiments only, and have yet to
be verified in animal experiments or clinical samples. In
vivo experiments will be performed in future studies. In the
present study, whether PPARγ protein is a post-transcriptional
regulator or stable protein was not determined, and to establish
this, further experiments are required in the future. Moreover, the
experiments were only carried out in one cell line, BEAS-2B cells,
which is also a limitation of the present study.
In conclusion, the present study indicated that FX
ameliorated oxidative damage and inflammation in CSE-induced human
bronchial epithelial cells in patients with COPD via modulating the
PPARγ/NF-κB signaling pathway.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SC and LZ conceived and designed the study. SC, JL
and LZ performed the experiments. JL and LZ were major contributors
to writing the manuscript. SC and LZ collected the clinical data
and analyzed the data. All authors have read and approved the final
manuscript. SC, LZ and JL confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rabe KF and Watz H: Chronic obstructive
pulmonary disease. Lancet. 389:1931–1940. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Labaki WW and Rosenberg SR: Chronic
obstructive pulmonary disease. Ann Intern Med. 173:ITC17–ITC32.
2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Vogelmeier CF, Criner GJ, Martinez FJ,
Anzueto A, Barnes PJ, Bourbeau J, Celli BR, Chen R, Decramer M,
Fabbri LM, et al: Global strategy for the diagnosis, management,
and prevention of chronic obstructive lung disease 2017 report.
GOLD executive summary. Am J Respir Crit Care Med. 195:557–582.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Brandsma CA, Van den Berge M, Hackett TL,
Brusselle G and Timens W: Recent advances in chronic obstructive
pulmonary disease pathogenesis: From disease mechanisms to
precision medicine. J Pathol. 250:624–635. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Tashkin DP: Smoking cessation in chronic
obstructive pulmonary disease. Semin Respir Crit Care Med.
36:491–507. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bae M, Kim MB, Park YK and Lee JY: Health
benefits of fucoxanthin in the prevention of chronic diseases.
Biochim Biophys Acta Mol Cell Biol Lipids.
1865(158618)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Liu M, Li W, Chen Y, Wan X and Wang J:
Fucoxanthin: A promising compound for human inflammation-related
diseases. Life Sci. 255(117850)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Gammone MA and D'Orazio N: Anti-obesity
activity of the marine carotenoid fucoxanthin. Mar Drugs.
13:2196–2214. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Meresse S, Fodil M, Fleury F and Chenais
B: Fucoxanthin, a marine-derived carotenoid from brown seaweeds and
microalgae: A promising bioactive compound for cancer therapy. Int
J Mol Sci. 21(9273)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Li X, Huang R, Liu K, Li M, Luo H, Cui L,
Huang L and Luo L: Fucoxanthin attenuates LPS-induced acute lung
injury via inhibition of the TLR4/MyD88 signaling axis. Aging
(Albany NY). 13:2655–2667. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ma SY, Park WS, Lee DS, Choi G, Yim MJ,
Lee JM, Jung WK, Park SG, Seo SK, Park SJ, et al: Fucoxanthin
inhibits profibrotic protein expression in vitro and attenuates
bleomycin-induced lung fibrosis in vivo. Eur J Pharmacol.
811:199–207. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wu SJ, Liou CJ, Chen YL, Cheng SC and
Huang WC: Fucoxanthin ameliorates oxidative stress and airway
inflammation in tracheal epithelial cells and asthmatic mice.
Cells. 10(1311)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Mei C, Zhou S, Zhu L, Ming J, Zeng F and
Xu R: Antitumor effects of laminaria extract fucoxanthin on lung
cancer. Mar Drugs. 15(39)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Feng H, Yin Y, Zheng R and Kang J:
Rosiglitazone ameliorated airway inflammation induced by cigarette
smoke via inhibiting the M1 macrophage polarization by activating
PPARgamma and RXRalpha. Int Immunopharmacol.
97(107809)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Qiu JF, Ma N, He ZY, Zhong XN, Zhang JQ,
Bai J, Deng JM, Tang XJ, Luo ZL, Huang M, et al: Erythromycin
inhibits cigarette smoke-induced inflammation through regulating
the PPARgamma/NF-kappaB signaling pathway in macrophages. Int
Immunopharmacol. 96(107775)2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Solleti SK, Simon DM, Srisuma S, Arikan
MC, Bhattacharya S, Rangasamy T, Bijli KM, Rahman A, Crossno JT Jr,
Shapiro ST and Mariani TJ: Airway epithelial cell PPARgamma
modulates cigarette smoke-induced chemokine expression and
emphysema susceptibility in mice. Am J Physiol Lung Cell Mol
Physiol. 309:L293–L304. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kang SI, Ko HC, Shin HS, Kim HM, Hong YS,
Lee NH and Kim SJ: Fucoxanthin exerts differing effects on 3T3-L1
cells according to differentiation stage and inhibits glucose
uptake in mature adipocytes. Biochem Biophys Res Commun.
409:769–774. 2011.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lee AH, Shin HY, Park JH, Koo SY, Kim SM
and Yang SH: Fucoxanthin from microalgae Phaeodactylum tricornutum
inhibits pro-inflammatory cytokines by regulating both NF-kappaB
and NLRP3 inflammasome activation. Sci Rep. 11(543)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Dang X, He B, Ning Q, Liu Y, Guo J, Niu G
and Chen M: Alantolactone suppresses inflammation, apoptosis and
oxidative stress in cigarette smoke-induced human bronchial
epithelial cells through activation of Nrf2/HO-1 and inhibition of
the NF-κB pathways. Respir Res. 21(95)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
O'Boyle NM, Banck M, James CA, Morley C,
Vandermeersch T and Hutchison GR: Open babel: An open chemical
toolbox. J Cheminform. 3(33)2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Berman H, Henrick K and Nakamura H:
Announcing the worldwide protein data bank. Nat Struct Biol.
10(980)2003.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Morris GM, Huey R, Lindstrom W, Sanner MF,
Belew RK, Goodsell DS and Olson AJ: AutoDock4 and AutoDockTools4:
Automated docking with selective receptor flexibility. J Comput
Chem. 30:2785–2791. 2009.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chen L, Yuan X, Zou L, Peng J and Hu X:
Effects of 1,25-Dihydroxyvitamin D3 on the prevention of chronic
obstructive pulmonary disease (COPD) in rats exposed to air
pollutant particles less than 2.5 micrometers in diameter (PM2.5).
Med Sci Monit. 24:356–362. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kohansal R, Martinez-Camblor P, Agusti A,
Buist AS, Mannino DM and Soriano JB: The natural history of chronic
airflow obstruction revisited: An analysis of the Framingham
offspring cohort. Am J Respir Crit Care Med. 180:3–10.
2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kankaanranta H, Harju T, Kilpelainen M,
Mazur W, Lehto JT, Katajisto M, Peisa T, Meinander T and Lehtimäki
L: Diagnosis and pharmacotherapy of stable chronic obstructive
pulmonary disease: The finnish guidelines. Basic Clin Pharmacol
Toxicol. 116:291–307. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Vestbo J, Hurd SS, Agusti AG, Jones PW,
Vogelmeier C, Anzueto A, Barnes PJ, Fabbri LM, Martinez FJ,
Nishimura M, et al: Global strategy for the diagnosis, management,
and prevention of chronic obstructive pulmonary disease: GOLD
executive summary. Am J Respir Crit Care Med. 187:347–365.
2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Lopez-Campos JL, Tan W and Soriano JB:
Global burden of COPD. Respirology. 21:14–23. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Barnes PJ: Oxidative stress-based
therapeutics in COPD. Redox Biol. 33(101544)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chen W, Zhang H and Liu Y:
Anti-inflammatory and apoptotic signaling effect of fucoxanthin on
benzo(A)pyrene-induced lung cancer in mice. J Environ Pathol
Toxicol Oncol. 38:239–251. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Yang X, Guo G, Dang M, Yan L, Kang X, Jia
K and Ren H: Assessment of the therapeutic effects of fucoxanthin
by attenuating inflammation in ovalbumin-induced asthma in an
experimental animal model. J Environ Pathol Toxicol Oncol.
38:229–238. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lee CY, Chen SP, Huang-Liu R, Gau SY, Li
YC, Chen CJ, Chen WY, Wu CN and Kuan YH: Fucoxanthin decreases
lipopolysaccharide-induced acute lung injury through the inhibition
of RhoA activation and the NF-κB pathway. Environ Toxicol.
37:2214–2222. 2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lin HV, Tsou YC, Chen YT, Lu WJ and Hwang
PA: Effects of low-molecular-weight fucoidan and high stability
fucoxanthin on glucose homeostasis, lipid metabolism, and liver
function in a mouse model of type II diabetes. Mar Drugs.
15(113)2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kwak N, Lee KH, Woo J, Kim J, Lee CH and
Yoo CG: Synergistic cycles of protease activity and inflammation
via PPARγ degradation in chronic obstructive pulmonary disease. Exp
Mol Med. 53:947–955. 2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Pan K, Lu J and Song Y: Artesunate
ameliorates cigarette smoke-induced airway remodelling via
PPAR-γ/TGF-β1/Smad2/3 signalling pathway. Respir Res.
22(91)2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Li Q, Sun J, Mohammadtursun N, Wu J, Dong
J and Li L: Curcumin inhibits cigarette smoke-induced inflammation
via modulating the PPARγ-NF-κB signaling pathway. Food Funct.
10:7983–7994. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ogasawara N, Kojima T, Go M, Ohkuni T,
Koizumi J, Kamekura R, Masaki T, Murata M, Tanaka S, Fuchimoto J,
et al: PPARgamma agonists upregulate the barrier function of tight
junctions via a PKC pathway in human nasal epithelial cells.
Pharmacol Res. 61:489–498. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Koo SY, Hwang JH, Yang SH, Um JI, Hong KW,
Kang K, Pan CH, Hwang KT and Kim SM: Anti-obesity effect of
standardized extract of microalga phaeodactylum tricornutum
containing fucoxanthin. Mar Drugs. 17(311)2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kim KN, Heo SJ, Yoon WJ, Kang SM, Ahn G,
Yi TH and Jeon YJ: Fucoxanthin inhibits the inflammatory response
by suppressing the activation of NF-κB and MAPKs in
lipopolysaccharide-induced RAW 264.7 macrophages. Eur J Pharmacol.
649:369–375. 2010.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Tan CP and Hou YH: First evidence for the
anti-inflammatory activity of fucoxanthin in high-fat-diet-induced
obesity in mice and the antioxidant functions in PC12 cells.
Inflammation. 37:443–450. 2014.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Seo MJ, Seo YJ, Pan CH, Lee OH, Kim KJ and
Lee BY: Fucoxanthin suppresses lipid accumulation and ROS
production during differentiation in 3T3-L1 adipocytes. Phytother
Res. 30:1802–1808. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
42
|
Maeda H, Hosokawa M, Sashima T, Takahashi
N, Kawada T and Miyashita K: Fucoxanthin and its metabolite,
fucoxanthinol, suppress adipocyte differentiation in 3T3-L1 cells.
Int J Mol Med. 18:147–152. 2006.PubMed/NCBI
|