Introduction
Atherosclerosis (AS) is a chronic inflammatory
disease detrimental to human health and a common cause of mortality
among the elderly. Inflammation occupies a pivotal position during
the process of AS (1). The
symptoms of AS are mainly manifested as inadequate blood supply,
dizziness, memory loss and even myocardial infarction and sudden
death in severe cases of coronary AS. Previous therapies focus on
the prevention of risk factors, such as high cholesterol and high
blood pressure (2). However, the
incidence rate of AS complications remains high. Numerous reports
about epidemiological and clinical studies have shown that
abnormally high concentrations of oxidized low density lipoprotein
(ox-LDL) play a central role in the development of AS (3-5).
As a risk factor of AS, ox-LDL has been found to
damage vascular endothelial cells, bring about endothelial
dysfunction and contribute to the production of inflammatory
cytokines, all of which are engaged in and drive the process of AS
(6). Ox-LDL downregulates
insulin-like growth factor-1 receptor in human smooth muscle cells,
but overexpression of dominant-negative NEDD4 prevents
ox-LDL-induced downregulation of insulin-like growth factor-1
receptor, which suggests that neuronally expressed developmentally
downregulated 4 (NEDD4) is a ubiquitin ligase that mediated
receptor downregulation (7).
NEDD4, serves as an essential member of HECT domain
E3 ligase family in eukaryotes (8)
and has been proved to play a prominent part in various cellular
processes through the ubiquitination-mediated degradation of
multiple substrates (9). Various
cardiovascular diseases are strongly dependent on the ubiquitin
proteasome system (10). Previous
studies found that aberrant activity of the TGF-β signaling pathway
may contribute to the process of the calcification of vascular
smooth muscle cells (VSMCs) (11,12).
In addition, the phosphorylated Smad1 in BMP/TGF-β signaling could
be regulated by NEDD4 E3 ligase which serves as a key inhibitor
(13). As a result, the role of
NEDD4 in the progression of vascular calcification has been
confirmed. Furthermore, the methylation levels of the NEDD4 gene
promoter are significantly increased in patients with AS and NEDD4
deficiency enhance vascular calcification (14). However, there is no direct evidence
for a specific link between NEDD4 and AS. On the basis of BioGRID
database (https://thebiogrid.org), NEDD4 could
interact with APEX1.
Apurinic-apyrimidinic endonuclease-1 (APEX1) is a
multifunctional protein involved in the DNA damage response and is
expressed in a variety of human tissues (15). The abnormal expression of APEX1 can
mediate several physiological processes (16). Meanwhile, APEX1 has been reported
to attenuate oxidative stress in ox-LDL-induced endothelial cells
(17). Recent study has shown that
APEX1 is able to inhibit AS-induced foam cell formation (18). However, whether APEX1 can bind to
NEDD4 and participate in ox-LDL-induced endothelial cell
inflammation and endothelial dysfunction remains to be
elucidated.
Hence, the present study sought to investigate
whether NEDD4 can act on ox-LDL-induced inflammation and
endothelial dysfunction in vascular endothelial cells by regulating
APEX1.
Materials and methods
Bioinformatics tools
The relationship between NEDD4 and APEX1 was
predicted by BioGRID database (https://thebiogrid.org).
Cell culture and treatment
DMEM (Thermo Fisher Scientific, Inc.) with 10% FBS
in an incubator at 37˚C with 5% CO2 was used to culture
human umbilical vein endothelial cells (HUVECs) from Fudan IBS Cell
Center。HUVECs between the third and fifth passages were
selected for experiments. Increasing doses of ox-LDL (25, 50, 100
and 200 µg/ml) were to treat cells for 24 h at 37˚C.
RPMI 1640 medium (Thermo Fisher Scientific, Inc.)
with 20% FBS at 37˚C in a 5% CO2 incubator with 90%
humidity was to culture U937 monocytes (CRL.1593.2) procured from
American Type Culture Collection.
Cell transfection
The overexpression plasmid of NEDD4 (4 µg) and
negative control plasmid (empty vector; 4 µg) were purchased from
Addgene, Inc. and transfected into HUVECs. The short interfering
(si)RNA-APEX1-1, siRNA-APEX1-1 and negative control plasmid were
obtained from Nanjing Cobioer Gene Technology Co., Ltd. Target
sequence of siRNA-APEX1-1 was AGGGTACAAGGCACTATGAAATG, target
sequence of siRNA-APEX1-2 was GGCACTATGAAATGATCTAGTTT and target
sequence of siRNA-NC was UAGCGACUAAACACAUCAA. HUVECs were seeded
into 6-well plates (2x105 cells/ml). HUVECs were
transfected with 20 µM siRNA-APEX1-1 and siRNA APEX1-2 and negative
control siRNA using Lipofectamine® RNAiMAX (Invitrogen;
Thermo Fisher Scientific, Inc.) for 24 h at 37˚C following the
directions of the manufacturer. Subsequent experimentation was
performed at 24 h after cell transfection.
Cell counting Kit-8 assay
CCK-8 solution (10 µl; GlpBio) was added to HUVECs
grown in 96-well plates at 37˚C for 2 h. OD450 nm value was
examined with a microplate reader (BioTek Instruments, Inc.).
Reverse transcription-quantitative
(RT-q) PCR
HUVECs suspension was placed in a 6-well plate
(2x105 cells/ml). By means of reverse
transcription kit (Roche Diagnostics), cDNA was generated from the
extracted RNA from HUVECs dependent on TRIzol® reagent
(Thermo Fisher Scientific, Inc.) in accordance with the guidance
from the manufacturer. The expression levels of target mRNAs in the
HUVECs were subsequently analyzed through RT-qPCR using SYBR-Green
I dye (Vazyme Biotech Co., Ltd.) according to the manufacturer's
protocols. The thermocycling conditions were: Initial denaturation
at 95˚C for 5 min; 38 cycles of denaturation at 95˚C for 10 sec,
annealing at 60˚C for 20 sec and extension at 72˚C for 30 sec. The
data were analyzed using 2-ΔΔCq method (19) and normalized to GAPDH gene
expression. The reactions were performed in duplicate for each
sample. The primer sequences used were as follows: TNF-α forward,
5'-CTGGGCAGGTCTACTTTGGG-3' and reverse, 5'-CTGGAGGCCCCAGTTTGAAT-3';
IL-1β forward, 5'-AGCCATGGCAGAAGTACCTG-3' and reverse,
5'-TGAAGCCCTTGCTGTAGTGG-3'; IL-6 forward,
5'-GTCCAGTTGCCTTCTCCCTGG-3' and reverse,
5'-CCCATGCTACATTTGCCGAAG-3'; NEDD4 forward,
5'-GAGCTCAGCTTAAAGGTCGC-3' and reverse, 5'-TCTCTGTCCGTAGACAGGCT-3';
APEX1 forward, 5'-GCAGATACGGGGTTGCTCTT-3' and reverse,
5'-ATTTTTACCGCGTTGCTCGC-3'; APEX1 forward,
5'-GCAGATACGGGGTTGCTCTT-3' and reverse, 5'-ATTTTTACCGCGTTGCTCGC-3';
GAPDH forward, 5'-AATGGGCAGCCGTTAGGAAA-3' and reverse,
5'-GCGCCCAATACGACCAAATC-3'.
Western blotting assay
After washing twice with cold PBS, total protein was
extracted from HUVECs using RIPA lysis buffer. The protein
concentration was measured with a bicinchoninic acid protein (BCA)
assay kit (Beyotime Institute of Biotechnology). PVDF membranes
(MilliporeSigma) were used to move the extracted proteins (50
µg/lane) separated by 10% SDS-PAGE. Skimmed milk (5%) was to impede
the membranes for 2 h at room temperature following washing with
TBST containing 0.1% Tween-20. Treatment with primary antibodies
for one night and secondary antibody for 1.5 h was performed at 4˚C
and at room temperature respectively. Finally, protein bands were
visualized using enhanced chemiluminescence reagent (Thermo Fisher
Scientific, Inc.) and ImageJ software (version 1.43; National
Institutes of Health) was used for protein band analysis. NEDD4
(cat. no. 2740; 1:1,000; Cell Signaling Technology), eNOS (cat. no.
ab300071; 1:1,000; Abcam), iNOS (cat. no. ab178945; 1:1,000;
Abcam), APEX1 (cat. no. ab189474; 1:1,000; Abcam) and GAPDH (cat.
no. ab9485; 1:2,500; Abcam) antibodies were used in this study.
Lactate dehydrogenase (LDH) assay
HUVECs were treated with different concentrations of
ox-LDL (25, 50, 100 and 200 µg/ml) for 24 h at 37˚C and then
transferred into 96-well flat bottom plate. An LDH assay kit
(Shanghai Coibo Biotechnology Co., Ltd.) was used to test the
released LDH in the media based on the instructions of the
manufacturer. The maximum amount of LDH release was determined and
the absorbance was measured at 490 nm with the aid of a Synergy HT
Microplate Reader (BioTek Instruments, Inc.).
Measurement of reactive oxygen species
(ROS)
ROS generation was detected using a ROS ELISA kit
(F06100; Shanghai Yanjin Biotechnology Co., Ltd.). Briefly, after
the cells were treated, the supernatant was taken and the level of
ROS was detected according to the ROS kit instructions. A
microplate reader was used to measure the OD value of each well at
the wavelength of 450 nm and the ROS concentration calculated
according to the standard curve. The ratio of ROS concentration in
each group to that in the control group was the relative ROS
level.
ELISA
The antigens were solubilized by 50 mM of carbonate
buffer (pH 9.6). The solution at a concentration of 10-20 µg/ml was
added to a 96-well enzyme labeling plate overnight at 4˚C. Finally,
the levels of vascular cell adhesion molecule (VCAM)-1 (cat. no.
E-EL-H5587c) and intercellular adhesion molecule (ICAM)-1 (cat. no.
E-EL-H6114) were determined by an ELISA kit (Elabscience
Biotechnology, Inc.).
Monocyte adhesion assay
TNF-α (10 ng/ml) was used to pretreat HUVECs for 12
h at 37˚C. U937 monocytes were labelled with calcein-acetoxymethyl
ester (Abcam) and cultured for 20 min at 37˚C. Then, the labelled
U937 cells (5x105) were added to the culture media
containing 1x105 HUVECS for 2 h at 37˚C followed by
washing 3 times with PBS. The estimation of attached green cells
was conducted using an Olympus fluorescence microscope
(magnification, x100; Olympus Corporation). Cells from five random
high power fields for each well were counted to assess the average
number of adherent cells.
Co-immunoprecipitation (Co-IP)
assay
Antibodies of NEDD4 (cat. no. 5344; 1:50; Cell
Signaling Technology, Inc.) and APEX1 (cat. no. 10203-1-AP; 1:100;
ProteinTech Group, Inc.) were used for the co-immunoprecipitation
assay and anti-rabbit IgG antibody (cat. no. ab205718, 1:500;
Abcam) served as a negative control. The cells were separated,
washed twice with PBS and proteins were extracted with RIPA lysis
buffer and the supernatant was collected after centrifugation at
13,000 x g for 10 min at 4˚C. The supernatant of cell lysate (500
µg) was incubated with anti-NEDD4 and anti-APEX1 at 4˚C for 24 h.
Then, 50 µg protein A magnetic beads were added for capturing the
complexes of NEDD4 and APEX1. After the IP reaction, agarose beads
were centrifuged at 1,000 x g for 3 min at 4˚C to the bottom of the
tube. The supernatant was then carefully absorbed and the agarose
beads were washed three times with 1 ml lysis buffer. A total of 15
µl 2X SDS sample buffer was finally added for boiling at 100˚C for
5 min. Afterwards, the collected complexes were subjected to
western blotting. Finally, an ECL reagent (Vazyme Biotech Co.,
Ltd.) was adopted to observe the immunoreactive bands.
Statistical analysis
Data are given as the mean ± SD and were analyzed by
IBM SPSS Statistics 25 (IBM Corp.). Student t-test along with
one-way ANOVA followed by Tukey's post hoc test was for comparisons
among two or more means. P<0.05 was considered to indicate a
statistically significant difference.
Results
NEDD4 expression is declined in
ox-LDL-induced endothelial cells
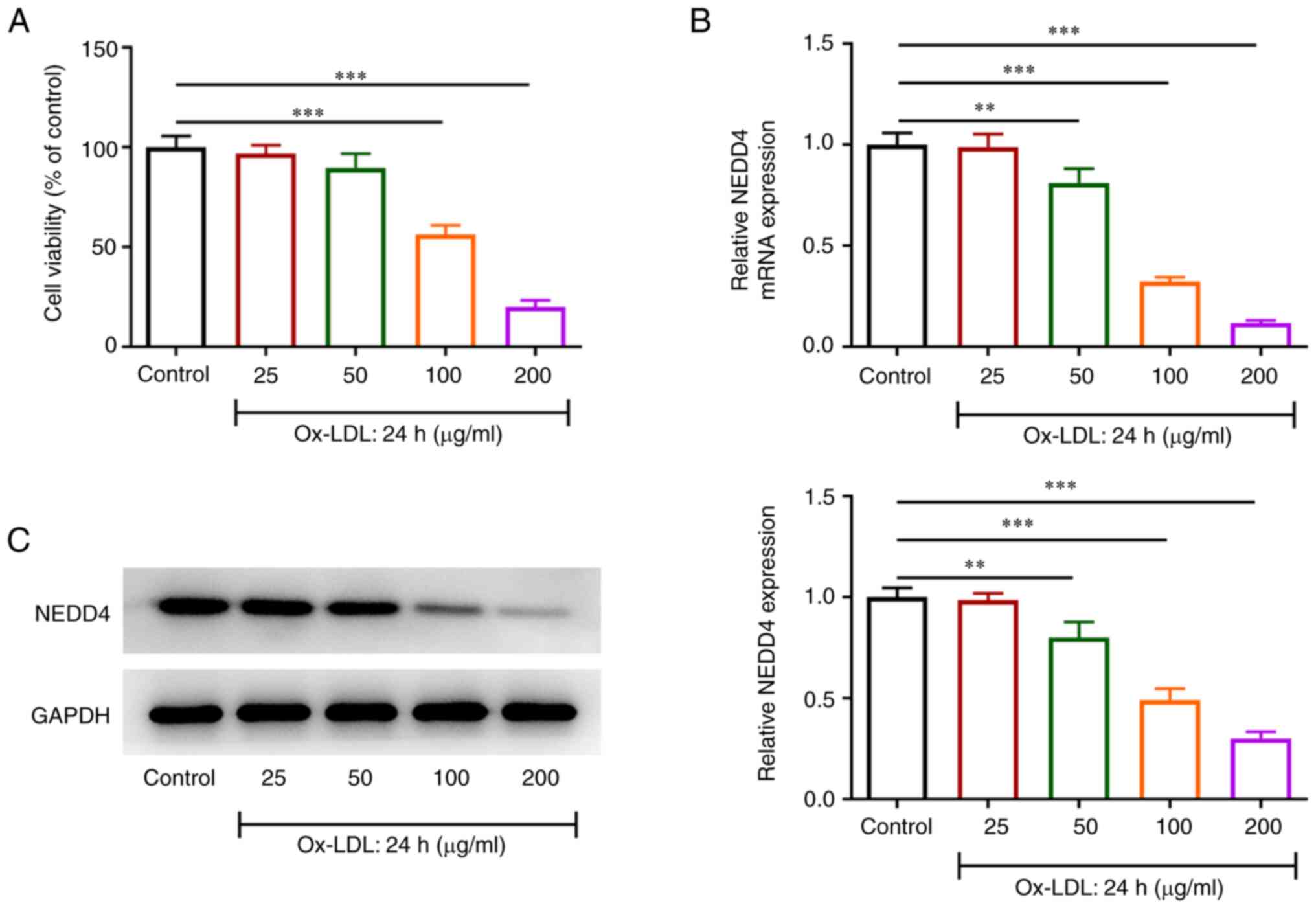
To validate the effects of NEDD4, CCK-8 was used to
judge cell activity. RT-qPCR and western blotting were performed to
verify NEDD4 expression in ox-LDL-exposed HUVECs. Fig. 1A shows a sharp fall in the
viability of HUVECs with increasing concentrations of ox-LDL.
Additionally, Fig. 1B and C showed that there was a steep drop in
NEDD4 expression relative to the control group. The above results
suggested that ox-LDL exerted a greater effect on the viability of
HUVECs and that NEDD4 expression was remarkably reduced by ox-LDL
induction.
NEDD4 overexpression attenuates
ox-LDL-induced cellular damage and release of inflammatory
factors
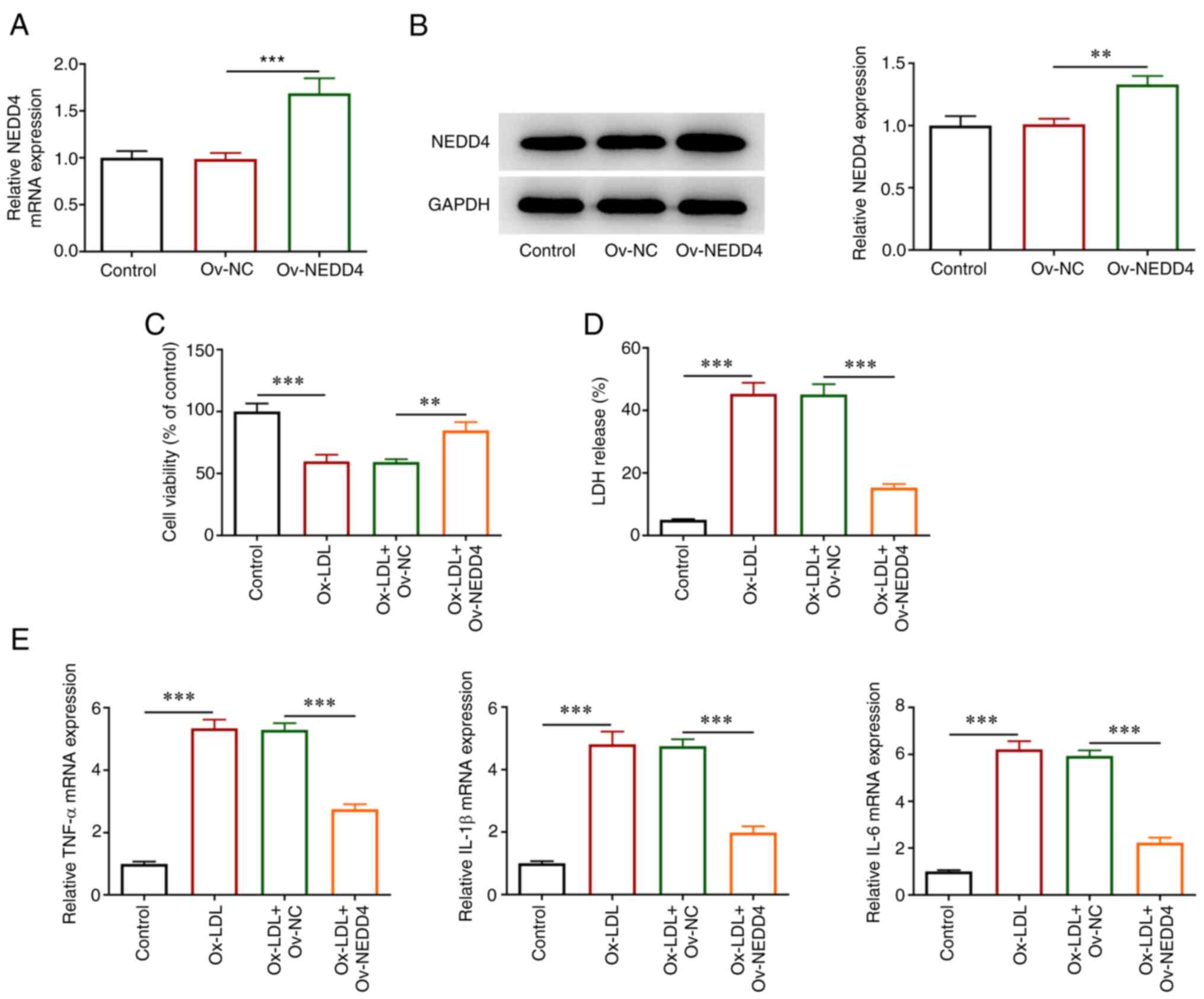
To understand the state of HUVECs induced by ox-LDL,
overexpression plasmids of NEDD4 were transfected. RT-qPCR, western
blotting, CCK-8 and LDH kits were applied for the evaluation of
cellular damage and the release of inflammatory factors. As shown
in Fig. 2A and B, NEDD4 expression was markedly elevated
in contrast to the negative control group. In addition, it can be
observed in Fig. 2C that the
reduced viability of HUVECs imposed by ox-LDL was conspicuously
restored following the overexpression of NEDD4. Cell cytotoxicity
was decreased (Fig. 2D). Moreover,
Fig. 2E shows that TNF-α, IL-1β
andIL-6 levels were decreased by contrast with the negative control
group. Overall, these results indicated that overexpression of
NEDD4 was an efficient way to attenuate cellular damage and
inflammatory effects of HUVECs induced by ox-LDL.
NEDD4 overexpression ameliorates
endothelial cell dysfunction of HUVECs induced by ox-LDL
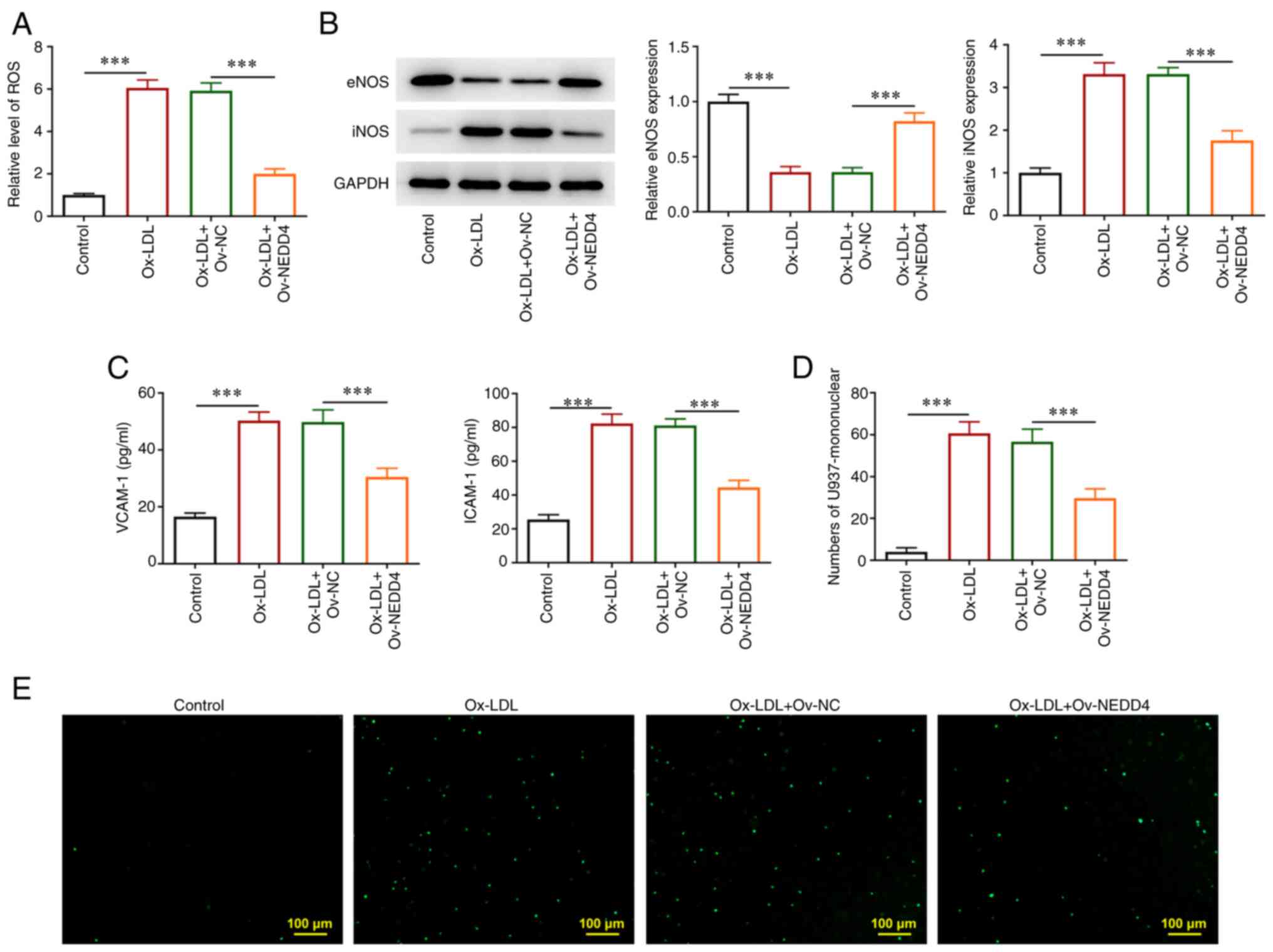
ROS content was estimated by corresponding kit. The
levels of endothelial nitric oxide synthase (eNOS) and inducible
nitric oxide synthase (iNOS), VCAM-1 and ICAM-1 in addition to the
number of endothelial cells of U937 monocytes were detected
respectively. As seen in Fig. 3A,
overexpression of NEDD4 was found to lead to a marked drop in ROS
generation. Conversely, Fig. 3B
revealed that NEDD4 overexpression promoted eNOS generation but
reduced iNOS production. Additionally, the rapid decrease in the
levels of adhesion molecules VCAM-1 and ICAM-1 in atherosclerotic
lesions in response to NEDD4 overexpression (Fig. 3C). Fig. 3D and E show that the stained cells
and the number of endothelial cells that adhered to U937 monocytes
were reduced. Together, these results provided important insights
that NEDD4 overexpression alleviated inflammatory response and
endothelial cell dysfunction of HUVECs induced by ox-LDL.
NEDD4 binds to APEX1 and its
overexpression promotes the expression of APEX1
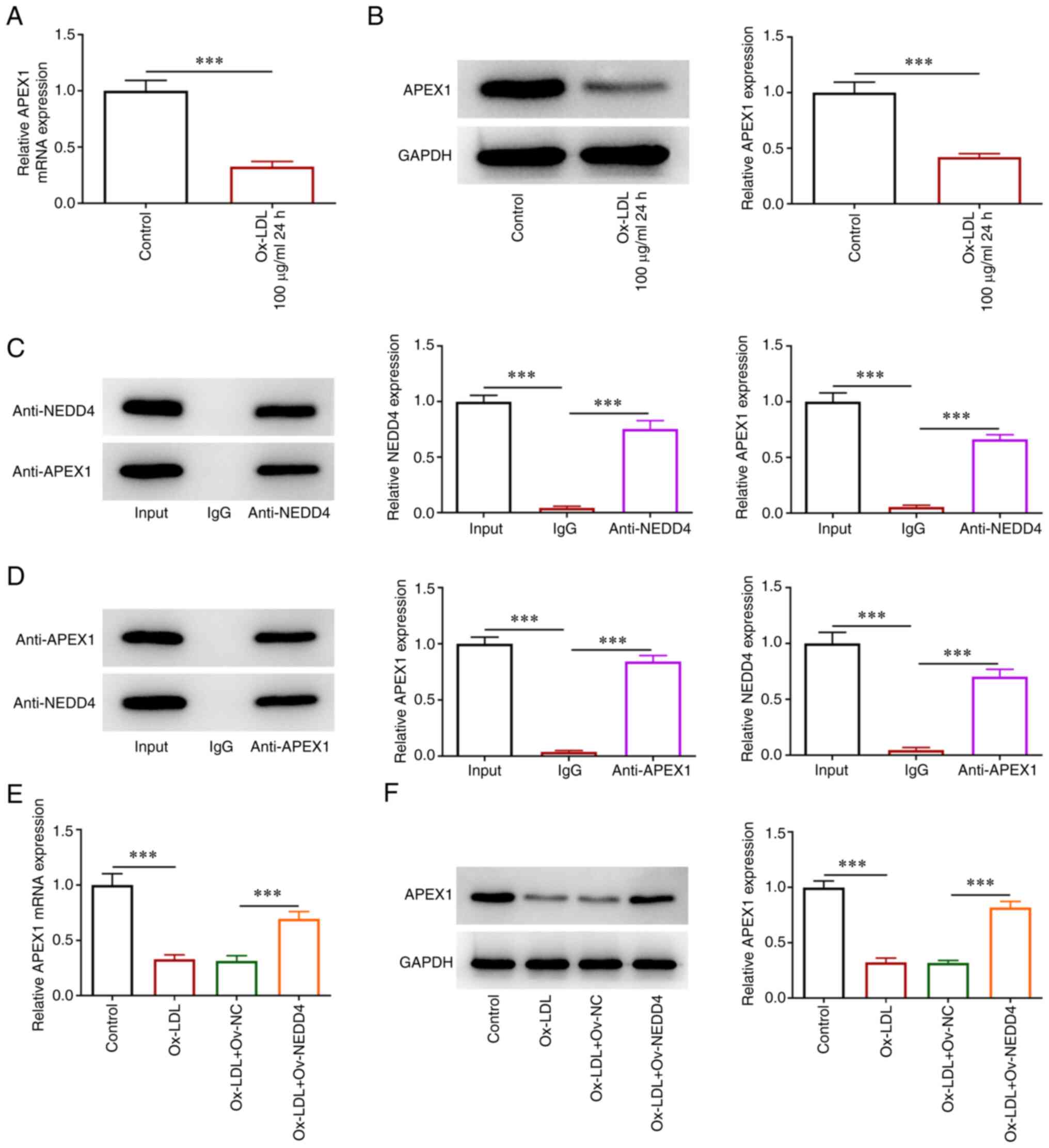
To determine the affinity of NEDD4 with APEX1,
RT-qPCR and western blotting were used to examine the expression of
APEX1 with or without NEDD4 overexpression. Co-IP assay was used to
test whether NEDD4 and APEX1 were combined in HUVECs. There was a
steep fall in the expression of APEX1 in ox-LDL-induced HUVECs
(Fig. 4A and B). As shown in Fig. 4C and D, NEDD4 protein could directly bind with
APEX1 protein in HUVECs. The expression of APEX1 clearly declined
in HUVECs following ox-LDL induction, which was increased after
NEDD4 overexpression (Fig. 4E and
F). The above data showed that
NEDD4 could bind to APEX1 and its overexpression was able to
promote APEX1 expression.
NEDD4 attenuates cellular damage and
release of inflammatory factors in ox-LDL-induced HUVECs via
regulating APEX1 expression
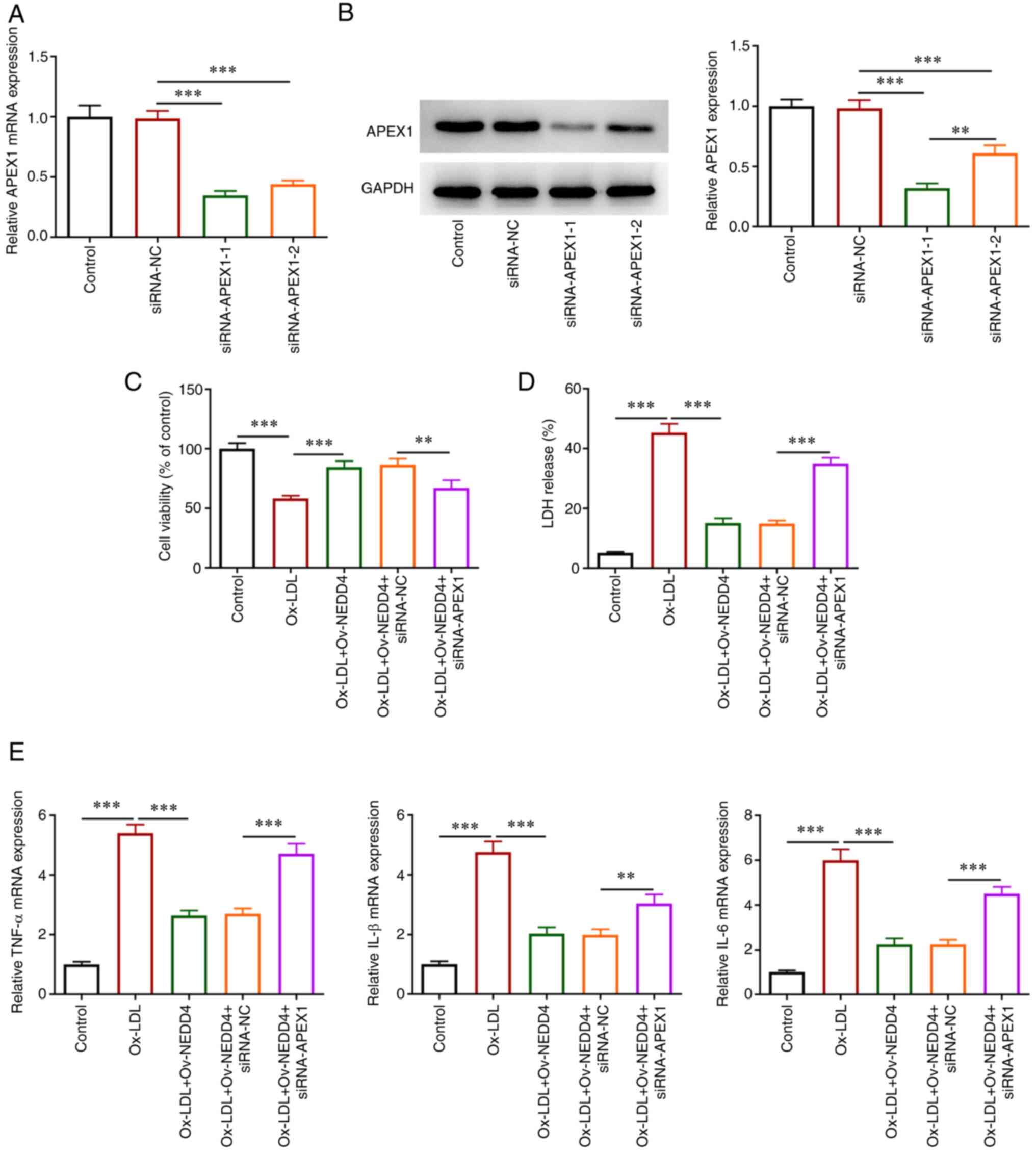
To understand the role of NEDD4 and APEX1 in the
viability injury of HUVECs, the interference plasmids siRNA-APEX1-1
and siRNA-APEX1-2 for APEX1 were constructed. The expression of
APEX1 in the siRNA-APEX1-1 group was the lowest among all groups
and siRNA-APEX1-1 was selected to perform subsequent experiments
(Fig. 5A and B). CCK-8 assay (Fig. 5C) showed that simultaneous NEDD4
elevation and APEX1 absence suppressed the viability of HUVECs
following ox-LDL exposure. Using an LDH kit to assess cytotoxic
injury, an apparent increase in the cell cytotoxic injury was found
(Fig. 5D). TNF-α, IL-1β, IL-6
expression were also fortified relative to the negative group
(Fig. 5E). In brief, the results
revealed that NEDD4 mitigated cell viability damage and the release
of inflammatory factors under the induction of ox-LDL by regulating
APEX1 expression.
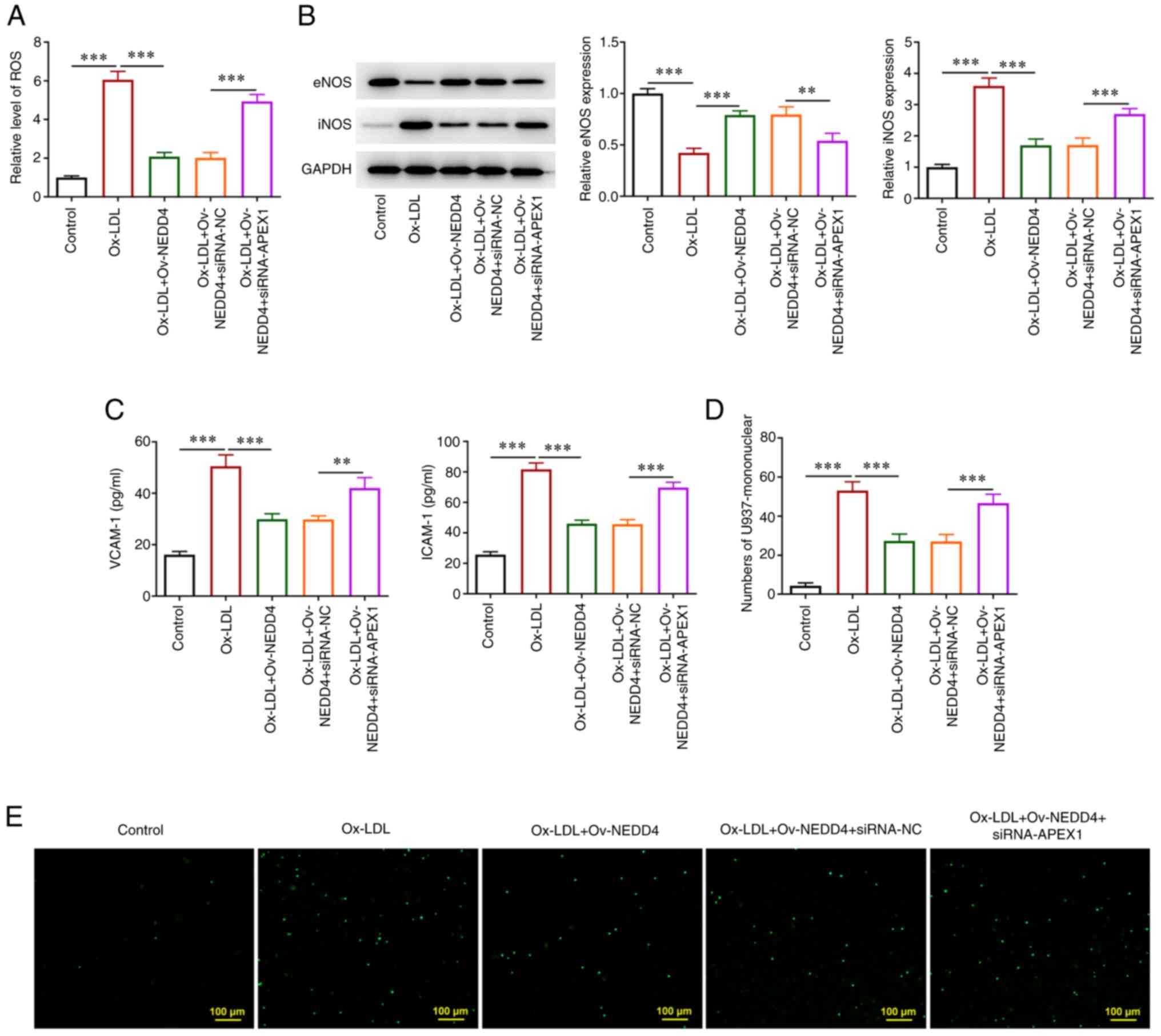
NEDD4 ameliorates ox-LDL-induced
endothelial dysfunction by regulating APEX1 expression
To understand the effect of NEDD4 on the endothelial
dysfunction of HUVECs induced by ox-LDL through the regulation of
APEX1 expression, the level of ROS was first examined using the
corresponding kit and it was increased after overexpression of
NEDD4 and interference of APEX1 (Fig.
6A). Then, under the aforementioned circumstances, a drop of
eNOS protein level and a rise in the protein level of iNOS were
observed (Fig. 6B). In addition,
the results of ELISA showed that NEDD4 overexpression led to a
significant upregulation of both VCAM-1 and ICAM-1 expression
(Fig. 6C). Finally, as shown in
Fig. 6D and E, there was an increasing number of
stained HUVECs and the number of endothelial cells that adhered to
U937 monocytes in the cell adhesion experiment following
overexpression of NEDD4 and interference of APEX1. From the above,
it can be seen that NEDD4 had a certain effect on migrating
ox-LDL-induced endothelial dysfunction by regulating APEX1
expression.
Discussion
AS is a threatening and slowly progressive disease.
One of the risk factors for AS is ox-LDL, which can contribute to
the production of inflammatory cytokines and result in endothelial
dysfunction (3). NEDD4 is a
ubiquitin ligase that mediates receptor downregulation, elicits
vital activities in AS (14) and
has been found to bind to APEX1 in the database. The present study
found that NEDD4 overexpression attenuated ox-LDL-induced
endothelial cell inflammation and dysfunction and ameliorated these
AS-related symptoms by regulating APEX1 expression.
A number of studies about NEDD4 are related to the
development of cancers. For example, NEDD4 participates in cell
migration in lung cancer through mediating EGFR signaling (20). NEDD4 mediates the reduction of CX43
regulated by fulvestrant in breast cancer cells (21). There are also studies demonstrating
the regulatory role of NEDD4 in some of the symptoms of AS. NEDD4
negatively modulates the activation of non-classical inflammasomes
(22). NEDD4 promotes p38α
ubiquitination which plays a crucial regulatory role in
inflammatory cells (23). A
previous study has suggested that NEDD4 can reduce AngII-induced
apoptosis in endothelial cells (24). To investigate the role of NEDD4 in
endothelial cell dysfunction and inflammation, the present study
examined the expression of NEDD4 in ox-LDL-induced HUVECs and found
that the overexpression of NEDD4 was capable of enhancing the cell
activity, diminishing HUVECs toxicity and cutting down TNF-α, IL-1β
and IL-6 expression. These results were consistent with the fact
that NEDD4 poses an important role in reducing endothelial
dysfunction and inflammation.
The imbalance between reactive ROS and the
antioxidant defense system is a major cause of endothelial
dysfunction, leading to vascular damage in metabolic and
atherosclerotic diseases (25). NO
is an anti-atherosclerotic molecule that reduces the inflammatory
response of tissues (26). iNOS
and eNOS are abundant isoforms expressed in endothelium. eNOS
uncoupling is one of the important mechanisms of AS. eNOS
decoupling reduces the production of NO and its protective effect
on blood vessels (27). Previous
experiments have shown that eNOS gene knockout or its inhibitor can
accelerate the formation of AS in experimental animals (27). In addition, eNOS is uncoupled to
produce superoxide, which increases the oxidative pressure and
promotes the formation of AS (28). ROS levels are reduced by eNOS
inhibition (29). Under normal
circumstances, iNOS is not expressed. However, under some stimuli,
iNOS can be regulated through overexpression, transcription and
translation and then regulates the synthesis of NO and finally
participates in inflammation and injury repair (30). ROS is abundantly produced by iNOS
(31). Moreover, macrophages which
can be differentiated from U973 monocytes can produce ROS during
phagocytosis of foreign particles (32). The inhibition of ROS results in the
reduction of U937-HUVECs adhesion (33). In addition, adhesion molecules
VCAM-1 and ICAM-2 mediate the migration of cells between the
bloodstream and inflamed tissues (34). Moreover, when atherosclerotic
injury occurs, endothelial cells release adhesion factors and
attract monocytes to accumulate, which can reflect cell adhesion
(35), and U937 is a human
monocyte. U937 cells were used to detect the changes of HUVEC
adhesion ability in the present study (36). It was found that NEDD4
overexpression led to a decrease in the levels of ROS, iNOS,
VCAM-1, ICAM-2 and the number of U937-HUVEC adhesion cells, but an
increase in the level of eNOS. Consequently, NEDD4 elevation eased
ox-LDL-evoked endothelial cell dysfunction.
APEX1 is a multifunctional protein which is related
to cancers and cardiovascular diseases (37). APEX1 mediates redox function
against vascular calcification which plays a role in the
pathogenesis of AS and chronic kidney disease (38). In the present study, the
combination of NEDD4 and APEX1 was confirmed, which corresponded to
BioGRID database. Subsequently, an interference plasmid for APEX1
was constructed and it was found that NEDD4 promoted APEX1
expression and attenuated ox-LDL-induced inflammation and
endothelial dysfunction by regulating APEX1 expression.
Taken together, in the present study, increasing
doses of ox-LDL were used to treat HUVECs to simulate the
inflammatory environment. NEDD4 and APEX1 expression as well as the
influence on ox-LDL-induced endothelial cell inflammation and
dysfunction were examined under different conditions. Experimental
results showed that NEDD4 could reduce ox-LDL-induced endothelial
cell dysfunction and inflammation by promoting the expression of
APEX1. These findings are of great value for understanding the
underlying mechanism of AS.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by Scientific Research
Incentive Fund of Shanxi Cardiovascular Hospital (grant no.
XYS20170307) and Scientific research project of Shanxi Provincial
Health and Family Planning Commission (grant no. 2017105).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HX and WZ conceived and designed the study. LT and
QQ performed the experiments. HX and WZ analyzed the experimental
data. HX and LT wrote and revised the manuscript. HX and WZ confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Li B, Li W, Li X and Zhou H: Inflammation:
A novel therapeutic target/direction in atherosclerosis. Curr Pharm
Des. 23:1216–1227. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kattoor AJ, Kanuri SH and Mehta JL: Role
of Ox-LDL and LOX-1 in atherogenesis. Curr Med Chem. 26:1693–1700.
2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Khatana C, Saini NK, Chakrabarti S, Saini
V, Sharma A, Saini RV and Saini AK: Mechanistic insights into the
oxidized low-density lipoprotein-induced atherosclerosis. Oxid Med
Cell Longev. 2020(5245308)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Pirillo A, Norata GD and Catapano AL:
LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm.
2013(152786)2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bian W, Jing X, Yang Z, Shi Z, Chen R, Xu
A, Wang N, Jiang J, Yang C, Zhang D, et al: Downregulation of
LncRNA NORAD promotes Ox-LDL-induced vascular endothelial cell
injury and atherosclerosis. Aging (Albany NY). 12:6385–6400.
2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Higashi Y, Sukhanov S, Parthasarathy S and
Delafontaine P: The ubiquitin ligase Nedd4 mediates oxidized
low-density lipoprotein-induced downregulation of insulin-like
growth factor-1 receptor. Am J Physiol Heart Circ Physiol.
295:H1684–H1689. 2008.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yang Y, Luo M, Zhang K, Zhang J, Gao T,
Connell DO, Yao F, Mu C, Cai B, Shang Y and Chen W: Nedd4
ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in
melanoma. Nat Commun. 11(433)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wang ZW, Hu X, Ye M, Lin M, Chu M and Shen
X: NEDD4 E3 ligase: Functions and mechanism in human cancer. Semin
Cancer Biol. 67:92–101. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zhang Y, Qian H, Wu B, You S, Wu S, Lu S,
Wang P, Cao L, Zhang N and Sun Y: E3 Ubiquitin ligase NEDD4
family-regulatory network in cardiovascular disease. Int J Biol
Sci. 16:2727–2740. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
He F, Li L, Li PP, Deng Y, Yang YY, Deng
YX, Luo HH, Yao XT, Su YX, Gan H and He BC:
Cyclooxygenase-2/sclerostin mediates TGF-β1-induced calcification
in vascular smooth muscle cells and rats undergoing renal failure.
Aging (Albany NY). 12:21220–21235. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Luong TTD, Estepa M, Boehme B, Pieske B,
Lang F, Eckardt KU, Voelkl J and Alesutan I: Inhibition of vascular
smooth muscle cell calcification by vasorin through interference
with TGFβ1 signaling. Cell Signal. 64(109414)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kim BG, Lee JH, Yasuda J, Ryoo HM and Cho
JY: Phospho-Smad1 modulation by nedd4 E3 ligase in BMP/TGF-β
signaling. J Bone Miner Res. 26:1411–1424. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Lee JH, Jeon SA, Kim BG, Takeda M, Cho JJ,
Kim DI, Kawabe H and Cho JY: Nedd4 deficiency in vascular smooth
muscle promotes vascular calcification by stabilizing pSmad1. J
Bone Miner Res. 32:927–938. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kim MH, Kim HB, Yoon SP, Lim SC, Cha MJ,
Jeon YJ, Park SG, Chang IY and You HJ: Colon cancer progression is
driven by APEX1-mediated upregulation of Jagged. J Clin Invest.
123:3211–3230. 2013.PubMed/NCBI View
Article : Google Scholar : (Epub ahead of
print).
|
|
16
|
Pei DS, Jia PP, Luo JJ, Liu W and Strauss
PR: AP endonuclease 1 (Apex1) influences brain development linking
oxidative stress and DNA repair. Cell Death Dis.
10(348)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lee SK, Chung JI, Park MS, Joo HK, Lee EJ,
Cho EJ, Park JB, Ryoo S, Irani K and Jeon BH: Apurinic/apyrimidinic
endonuclease 1 inhibits protein kinase C-mediated p66shc
phosphorylation and vasoconstriction. Cardiovasc Res. 91:502–509.
2011.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hu Z, Hui B, Hou X, Liu R, Sukhanov S and
Liu X: APE1 inhibits foam cell formation from macrophages via LOX1
suppression. Am J Transl Res. 12:6559–6568. 2020.PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Shao G, Wang R, Sun A, Wei J, Peng K, Dai
Q, Yang W and Lin Q: The E3 ubiquitin ligase NEDD4 mediates cell
migration signaling of EGFR in lung cancer cells. Mol Cancer.
17(24)2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Butt G, Yaylim I, Attar R, Aras A, Romero
MA, Qureshi MZ, Purenovic J and Farooqi AA: NEDD4 family of E3
ubiquitin ligases in breast cancer: Spotlight on SMURFs, WWPs and
NEDD4. Adv Exp Med Biol. 1152:365–375. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Xiang F, Fan Y, Ni Z, Liu Q, Zhu Z, Chen
Z, Hao W, Yue H, Wu R and Kang X: Ursolic acid reverses the
chemoresistance of breast cancer cells to paclitaxel by targeting
MiRNA-149-5p/MyD88. Front Oncol. 9(501)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liu Q, Zhang S, Chen G and Zhou H: E3
ubiquitin ligase Nedd4 inhibits AP-1 activity and TNF-α production
through targeting p38α for polyubiquitination and subsequent
degradation. Sci Rep. 7(4521)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Xu J, Sheng Z, Li F, Wang S, Yuan Y, Wang
M and Yu Z: NEDD4 protects vascular endothelial cells against
Angiotensin II-induced cell death via enhancement of XPO1-mediated
nuclear export. Exp Cell Res. 383(111505)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Incalza MA, D'Oria R, Natalicchio A,
Perrini S, Laviola L and Giorgino F: Oxidative stress and reactive
oxygen species in endothelial dysfunction associated with
cardiovascular and metabolic diseases. Vascul Pharmacol. 100:1–19.
2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
de Meirelles LR, Mendes-Ribeiro AC, Mendes
MA, da Silva MN, Ellory JC, Mann GE and Brunini TM: Chronic
exercise reduces platelet activation in hypertension: Upregulation
of the L-arginine-nitric oxide pathway. Scand J Med Sci Sports.
19:67–74. 2009.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hong FF, Liang XY, Liu W, Lv S, He SJ,
Kuang HB and Yang SL: Roles of eNOS in atherosclerosis treatment.
Inflamm Res. 68:429–441. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Li H and Förstermann U: Uncoupling of
endothelial NO synthase in atherosclerosis and vascular disease.
Curr Opin Pharmacol. 13:161–167. 2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kumar S, Kang DW, Rezvan A and Jo H:
Accelerated atherosclerosis development in C57Bl6 mice by
overexpressing AAV-mediated PCSK9 and partial carotid ligation. Lab
Invest. 97:935–945. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Collins JL, Vodovotz Y, Hierholzer C,
Villavicencio RT, Liu S, Alber S, Gallo D, Stolz DB, Watkins SC,
Godfrey A, et al: Characterization of the expression of inducible
nitric oxide synthase in rat and human liver during hemorrhagic
shock. Shock. 19:117–122. 2003.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Matziouridou C, Rocha SDC, Haabeth OA,
Rudi K, Carlsen H and Kielland A: iNOS- and NOX1-dependent ROS
production maintains bacterial homeostasis in the ileum of mice.
Mucosal Immunol. 11:774–784. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Prasad A, Sedlářová M, Balukova A, Ovsii
A, Rác M, Křupka M, Kasai S and Pospíšil P: Reactive oxygen species
imaging in U937 cells. Front Physiol. 11(552569)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chai Y, Cao Z, Yu R, Liu Y, Yuan D and Lei
L: Dexmedetomidine attenuates LPS-induced monocyte-endothelial
adherence via inhibiting Cx43/PKC-α/NOX2/ROS signaling pathway in
monocytes. Oxid Med Cell Longev. 2020(2930463)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
de Sousa J, Sousa Aarão TL, Rodrigues de
Sousa J, Hirai KE, Silva LM, Dias LB Jr, Oliveira Carneiro FR,
Fuzii HT and Quaresma JA: Endothelium adhesion molecules ICAM-1,
ICAM-2, VCAM-1 and VLA-4 expression in leprosy. Microb Pathog.
104:116–124. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xu K, Saaoud F, Yu S, Drummer C IV, Shao
Y, Sun Y, Lu Y, Sun J, Yu J, Jiang X, et al: Monocyte adhesion
assays for detecting endothelial cell activation in vascular
inflammation and atherosclerosis. Methods Mol Biol. 2419:169–182.
2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kang H, Yu H, Fan J and Cao G: Rotigotine
protects against oxidized low-density lipoprotein(ox-LDL)-induced
damages in human umbilical vein endothelial cells(HUVECs).
Bioengineered. 12:10568–10579. 2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kim JM, Yeo MK, Lim JS, Song IS, Chun K
and Kim KH: APEX1 expression as a potential diagnostic biomarker of
clear cell renal cell carcinoma and hepatobiliary carcinomas. J
Clin Med. 8(1151)2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lee KM, Lee EO, Lee YR, Joo HK, Park MS,
Kim CS, Choi S, Jeong JO and Jeon BH: APE1/Ref-1 inhibits
phosphate-induced calcification and osteoblastic phenotype changes
in vascular smooth muscle cells. Int J Mol Sci.
18(2053)2017.PubMed/NCBI View Article : Google Scholar
|