Introduction
Chronic glomerulonephritis (CGN) is an autoimmune
glomerulopathy, characterized by the excessive proliferation of
mesangial cells, accumulation of extracellular matrix and
infiltration of circulating inflammatory cells (1). Glomerular mesangial cells (GMCs),
distributed in the mesangial matrix of the glomerulus, constitute
the mesangial region of the glomerulus together with the mesangial
matrix. GMCs have multiple physiological functions, including
stabilizing the structure of glomerular capillaries, maintaining
mesangial matrix homeostasis, regulating filtration surface area,
phagocytosis of apoptotic cells and immune complexes (2). Under the influence of certain
pathological factors such as hyperglycemia and inflammation, GMCs
can proliferate abnormally, increase intracellular protein
synthesis and, increase the secretion of the extracellular matrix
(3-4). Therefore, excessive proliferation of
GMCs is an important pathological feature of certain human kidney
diseases, including chronic glomerulonephritis and diabetic
nephropathies. Knowledge of the responses of GMCs to pathological
stimuli is crucial to the understanding of the pathogenesis of
chronic glomerulonephritis. Thus, a deeper understanding of the
excessive proliferation of GMCs is required to devise more
effective prevention and therapies for CGN.
Long non-coding RNAs (lncRNAs) are generally
considered as non-coding transcripts >200 nucleotides that are
important genetic regulators. LncRNAs have been previously reported
to be crucial determinants of epigenetic regulation and serve a key
role in regulation of chromatin structure, scaffolding or the decoy
function of mRNAs, and post-transcriptional regulation of mRNAs
(5). The regulatory role of lncRNA
can be summarized as cis-action on neighboring genes or as
trans-action through effects on mRNA stability, mRNA translation or
the regulation of microRNA-mRNA interactions and RNA binding
proteins (6-7). For example, Gao et al
(8) reported that lncRNA
NONRATG001910.2 promoted CTNNB1 expression by targeting miR-339-3p
in lipopolysaccharide (LPS)-stimulated rat mesangial cells, which
suggested that NONRATG001910.2 may be a potential biomarker for
CGN. Moreover, Zhou et al (9) suggested that lncRNA NORAD directly
regulated RUNX2 transcription and promoted the proliferation,
migration and invasion of breast cancer cells. Competitive
endogenous (ce)RNA is the most frequently reported mechanism of
lncRNAs.
Materials and methods
GMC culture conditions
Mouse SV40-MES-13 GMCs were purchased from BNCC
Biological Technology and cultured in DMEM (Beijing Solarbio
Science & Technology Co., Ltd.) supplemented with 10% FBS
(Biological Industries Sartorius AG) and penicillin/streptomycin at
37˚C in a humidified, 5% CO2 atmosphere. GMCs stimulated
with 3.0 µg/ml LPS (MilliporeSigma) (10-11)
were defined as the model group (n=3) and unstimulated GMCs were
defined as the control group (n=3).
Cell proliferation assay
Cell proliferation rates were assessed using the
Cell Counting Kit-8 (CCK-8) assay (cat. no. BB19071X; Shanghai
Besto Biological Technology Co., Ltd.). GMCs were seeded
(5.0x104 cells/well) into 96-well tissue culture plates
with DMEM medium and treated with CCK-8 solution (10 µl/well) for 1
h. The optical density (absorbance at 450 nm) was assessed using an
RT-6100 ELISA reader (Rayto Life and Analytical Sciences Co.,
Ltd.).
LncRNA sequence analysis
To ensure the quality of paired-end sequencing
reads, FastQC software (version 0.10.1) was used to evaluate the
quality of the original sequencing data. The input reads were
considered good data quality when the Q20 base percentage in
‘Reads’ was ≥90% and the Q30 base percentage in ‘Reads’ was
≥80%.
High-throughput RNA-seq was performed by Genesky
Bio-tech Co., Ltd. RNA fragmentation was performed using Bioruptor
Pico (cat. no. B01060001; Diagenode SA) sonication in RNase-free
water. RNA integrity was detected by denaturing gel electrophoresis
and quantified using a NanoDrop 2000 spectrophotometer (Thermo
Fisher Scientific, Inc.). The purified RNA fragments were then used
to construct libraries using the TruSeq RNA Sample Prep Kit (cat.
no. RS-122-2002; Illumina, Inc.). Libraries underwent quality
control and were quantified using an Agilent 2100 bioanalyzer
system (Agilent Technologies, Inc.). Paired-end 150 bp sequencing
was performed using an Illumina HiSeq 2500 (Illumina, Inc.).
Isolation of RNA and reverse
transcription-quantitative (RT-qPCR)
The expression levels of NONMMUG036949.2,
NONMMUG089165.1, NONMMUG030447.2, NONMMUG028702.2, NONMMUG039651.2
and NONMMUG032587.2 were assessed using RT-qPCR. Total RNA was
extracted from the cells using TRIzol® (Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Primers (Table I) were designed
based on the cDNA sequences, which were accessed from the National
Center for Biotechnology Information (NCBI) and tested using NCBI
Primer BLAST, using Primer Premier 5 (Premier Biosoft
International), and synthesized by Sangon Biotech Co., Ltd. The
concentration and purity of the isolated RNA were determined using
an OD1000+ Ultra Micro Spectrophotometer (WuYi Technologies), and
reverse transcription was performed using the PrimeScript™ RT
Reagent Kit with gDNA Eraser (cat. no. RR047A; Takara Biotechnology
Co., Ltd.). qPCR was performed using a StepOne Plus fluorescence
quantitative PCR instrument (Applied Biosystems; Thermo Fisher
Scientific, Inc.) with SYBR Green qPCR Master Mix (cat. no.
G3322-05; Wuhan Servicebio Technology Co., Ltd.). β-actin was used
as the internal control; for quantitative results, the expression
of lncRNA was expressed as fold change using the 2-ΔΔCq
method (12).
 | Table IPrimer sequences used for reverse
transcription-quantitative PCR. |
Table I
Primer sequences used for reverse
transcription-quantitative PCR.
| Gene | Amplicon size,
bp | Sequence
(5'-3') |
|---|
|
NONMMUG036949.2 | 185 | F:
TTCTCCAGGACACTCACCAC |
| | | R:
AGAGAGCCAGGCATAGTGTG |
|
NONMMUG089165.1 | 140 | F:
AGACCACGACGCCTTAAGTA |
| | | R:
GAATGGGAGTCCGAATGCAG |
|
NONMMUG030447.2 | 73 | F:
GCGTCGCCTAACGGTCC |
| | | R:
GGGACAAGAAGGTCATCGGT |
|
NONMMUG028702.2 | 181 | F:
ACTGATACCATGCACCTCTCA |
| | | R:
GCATGTCACTTCAGCCTCTG |
|
NONMMUG039651.2 | 160 | F:
TCCCTGCTGCAGTGTCATAA |
| | | R:
AGCAAAGCTCCCTTGTCTCT |
|
NONMMUG032587.2 | 175 | F:
CAAGCCTGGATGTTCCATCG |
| | | R:
AGGGCACACCCTTCAAAGAT |
Construction of the lncRNA-mRNA
regulatory network
LncTar (http://www.cuilab.cn/lnctar) was used to predict
lncRNA-mRNA interactions utilizing free energy minimization
(13-14). LncTar utilizes a variation on the
standard ‘sliding’ algorithm approach to calculate the normalized
binding free energy (ndG) and predicts the minimum free energy
joint structure. ndG ≤0.1 was regarded as the cutoff to determine
the paired RNAs as interacting, ndG >0.1 was considered to
indicate that paired RNAs did not interact. Subsequently, Cytoscape
3.8.1 was used to visualize the lncRNA-mRNA network after screening
the target mRNAs of differentially expressed lncRNAs.
Construction of the lncRNA-associated
ceRNA network
Interactions between miRNAs and lncRNAs were
predicted using the miRNA target prediction software, miRanda
(http://www.miranda.org) (15), and the three miRNAs of the highest
confidence that can bind to lncRNAs were chosen. The target mRNAs
of miRNAs were predicted using TargetScan 8.0 (https://www.targetscan.org/vert_80/) (16), and mRNAs with high confidence
(cumulative weighted context score cutoff level <-0.8) bound to
the miRNAs were screened out (17). Cytoscape 3.8.1 was then used to
delineate the lncRNA-miRNA-mRNA ceRNA network.
Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) analysis
To evaluate the role of differentially expressed
lncRNAs in LPS-induced GMCs, GO biological process (BP) term
enrichment and KEGG pathway enrichment were assessed (18). GO terms with P<0.05 were
considered statistically significant. KEGG pathway enrichment
analysis was used to identify significantly enriched signal
transduction pathways or metabolic pathways (P<0.05). GO and
KEGG enrichment analysis were performed using the free online data
analysis platform OmicShare tools (https://www.omicshare.com/tools).
Protein-protein interaction (PPI)
network construction and core gene screening
The PPI network of the mRNA involved in the ceRNA
network was generated using the STRING online database tool
(Version 11.5, https://string-db.org) (19); interaction pairs with a confidence
value >0.4 were deemed significant and were retained. The top 10
genes in the PPI network were evaluated using the MCC, MNC and
Degree algorithms through the CytoHubba plug-in for Cytoscape
(20). The final hub genes were
identified as those that were identified by all three of the
algorithms.
GEO public dataset analysis
The public datasets (GSE104066) utilized in this
study were obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo) and were initially
screened based on disease (CGN), organism (Homo sapiens) and
experiment type (expression profiling by array). Ultimately, the
GSE104066 dataset was selected for use in CGN analysis.
Statistical analysis
Statistical analysis was performed using SPSS 22.0
software (IBM Corp.), and data are reported as the mean + SD. The
data were subjected to one-way ANOVA with Tukey's post hoc multiple
comparison tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
Optimal concentration and treatment
time of LPS were determined using the CCK-8 assay
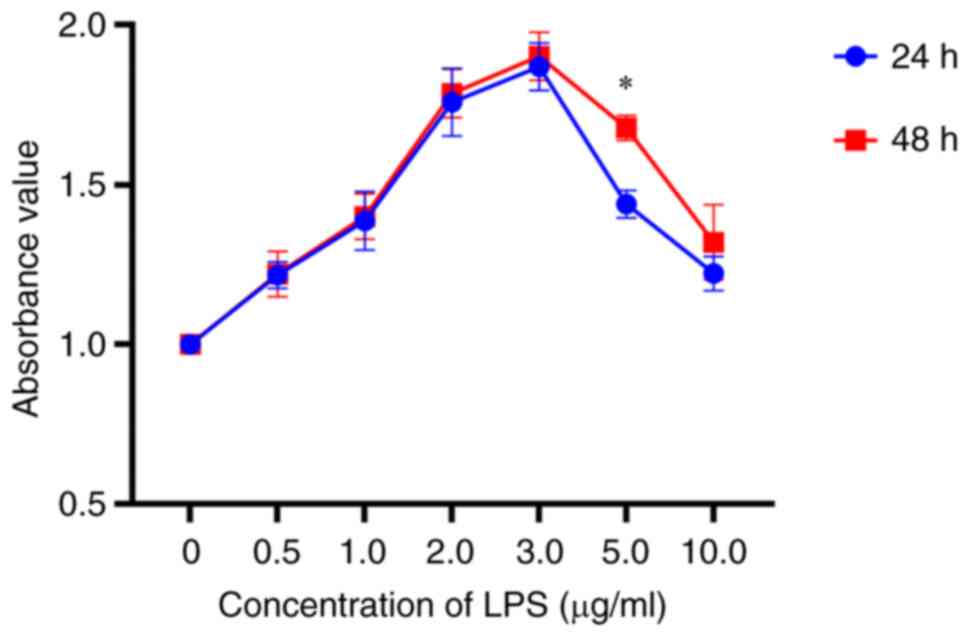
The present study evaluated the proliferative
ability of GMCs induced by different concentrations of LPS (0.5,
1.0, 3.0, 5.0 and 10.0 µg/ml) using the CCK-8 assay at 24 and 48 h
following treatment. The relationship between cell proliferation,
LPS concentration and intervention time were assessed (Fig. 1). The results of the CCK-8
proliferation assay demonstrated that LPS could effectively induce
cell proliferation in GMCs, with the highest absorbance (optical
density 450 nm) observed at a concentration of 3.0 µg/ml (Table SI), which indicated that 3.0 µg/ml
was the optimal concentration of LPS for the induction of cell
proliferation in GMCs. Furthermore, there was no significant
difference demonstrated between the treatment times of 24 and 48 h
(P=0.62) at a concentration of 3.0 µg/ml LPS. Consequently, the
concentration of 3.0 µg/ml and the treatment time of 24 h were
selected for use in subsequent experiments.
Characteristics of differentially
expressed lncRNAs
In the present study, six groups of cells were used
in subsequent experiments, including three groups of LPS-induced
GMCs as the model group (LPS1-3) and three groups of normal GMCs as
the control group (CON1-3).
After deduplication, quality trimming and quality
filtering, the sequencing data at both ends of the R1 and R2 paired
reads were assessed as being of good quality (Q20 base and Q30 base
were both >95%). The proportion of ‘clean reads’ retained after
cleaning was >95% for each of the six samples which met the
pre-determined quality requirements for sequencing. The quality
control results of the sequencing data were presented (Table II; Table SII).
| Table IIThe quality control results for the
sequencing data. |
Table II
The quality control results for the
sequencing data.
| | R1 | R2 | |
|---|
| Sample | Q20, % | Q30, % | Q20, % | Q30, % | Clean reads, % of
total |
|---|
| CON-1 | 98.1 | 95.3 | 98.5 | 95.9 | 97.7 |
| CON-2 | 98.1 | 95.2 | 98.5 | 97.5 | 97.5 |
| CON-3 | 98.1 | 95.3 | 98.5 | 97.5 | 97.5 |
| LPS-1 | 98.1 | 95.2 | 98.2 | 97.7 | 97.7 |
| LPS-2 | 98.2 | 95.4 | 98.3 | 96.9 | 96.9 |
| LPS-3 | 98.1 | 95.3 | 98.4 | 97.3 | 97.3 |
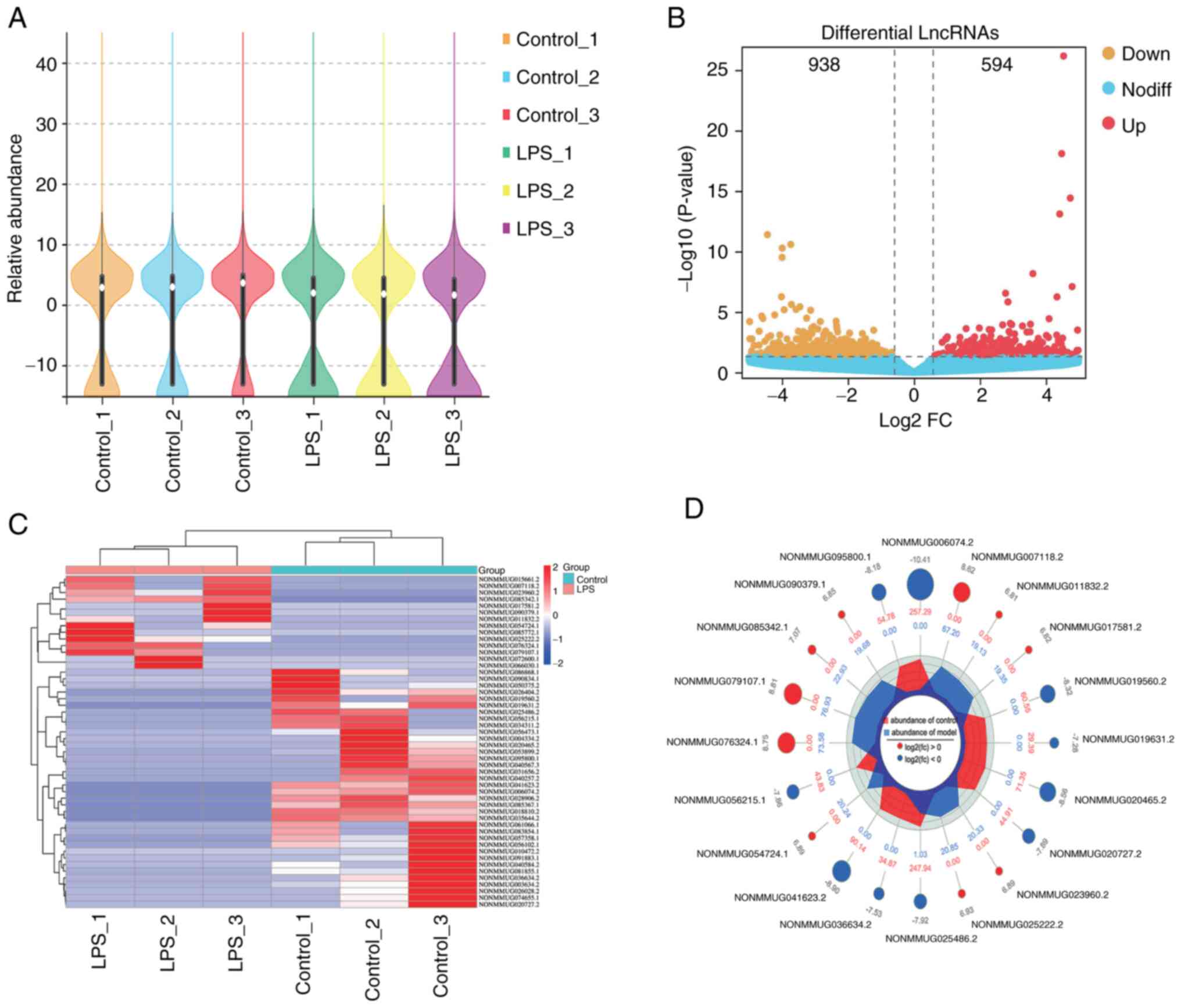
The violin plot demonstrated the relative abundance
of lncRNAs in each sample (Fig.
2A). The violin plot demonstrated that there were significant
differences in the expression of lncRNAs in LPS-induced GMCs
compared with control GMCs. The threshold for differentially
expressed lncRNAs was set at absolute fold change |(FC)|≥1.5 and
P<0.05 (21-22). A total of 1,532 differentially
expressed lncRNAs, including 594 upregulated lncRNAs and 938
downregulated lncRNAs, were identified from a total of 46,879
lncRNAs (Fig. 2B; Table SIII). A heat map of the top 50
differentially expressed lncRNAs was presented (Fig. 2C). The top 10 upregulated and
downregulated lncRNAs are presented in Table III, and the radar map in Fig. 2D presents the top 10 upregulated
and downregulated lncRNA expression levels in LPS-induced GMCs
compared with control GMCs.
| Table IIITop 10 upregulated and downregulated
expressed log non-coding RNAs. |
Table III
Top 10 upregulated and downregulated
expressed log non-coding RNAs.
| Gene ID |
Log2(FC) | P-value | Regulation |
|---|
|
NONMMUG011832.2 | 6.8068 | 0.0013 | Up |
|
NONMMUG017581.2 | 6.8226 | 0.0052 | Up |
|
NONMMUG090379.1 | 6.8473 | 0.0049 | Up |
|
NONMMUG054724.1 | 6.8894 | 0.001 | Up |
|
NONMMUG023960.2 | 6.895 | 0.0001 | Up |
|
NONMMUG025222.2 | 6.9314 | 0.0001 | Up |
|
NONMMUG085342.1 | 7.0671 | <0.0001 | Up |
|
NONMMUG007118.2 | 8.6184 | 0.0274 | Up |
|
NONMMUG076324.1 | 8.7489 | 0.0252 | Up |
|
NONMMUG079107.1 | 8.8134 | 0.0241 | Up |
|
NONMMUG006074.2 | -10.4113 | <0.0001 | Down |
|
NONMMUG041623.2 | -8.8981 | <0.0001 | Down |
|
NONMMUG020465.2 | -8.5609 | 0.0285 | Down |
|
NONMMUG019560.2 | -8.3242 | 0.0332 | Down |
|
NONMMUG095800.1 | -8.1796 | 0.0364 | Down |
|
NONMMUG025486.2 | -7.9163 | <0.0001 | Down |
|
NONMMUG020727.2 | -7.8928 | 0.0435 | Down |
|
NONMMUG056215.1 | -7.8581 | 0.0444 | Down |
|
NONMMUG036634.2 | -7.5271 | <0.0001 | Down |
|
NONMMUG019631.2 | -7.2817 | <0.0001 | Down |
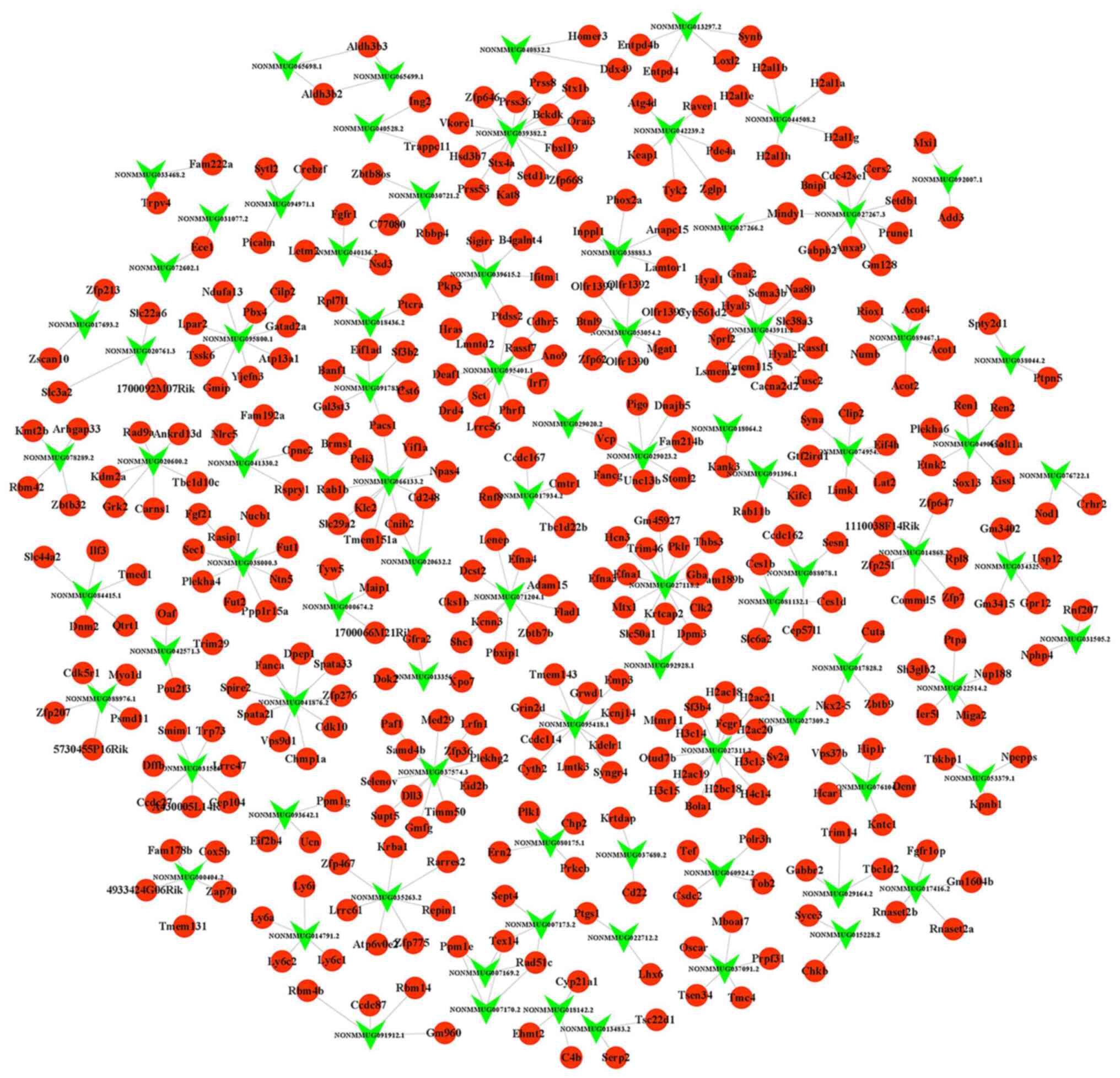
Construction of the lncRNA-mRNA
regulatory network
To evaluate the functions of differentially
expressed lncRNAs, 556 target mRNAs of the 236 lncRNAs were
predicted using LncTar (Table
SIV). The highly coordinated expression between lncRNAs and
target mRNAs may be due to complementary base pairing between
lncRNA and mRNA (23). The
lncRNA-mRNA regulatory network presented in Fig. 3 shows the interaction relationship
between lncRNA and mRNA. For example, lncRNA NONMMUG029023.2 may
regulate Stoml2 mRNA expression. Ptdss2 may be affected by both
lncRNA NONMMUG039651.2 and lncRNA NONMMUG095401.1.
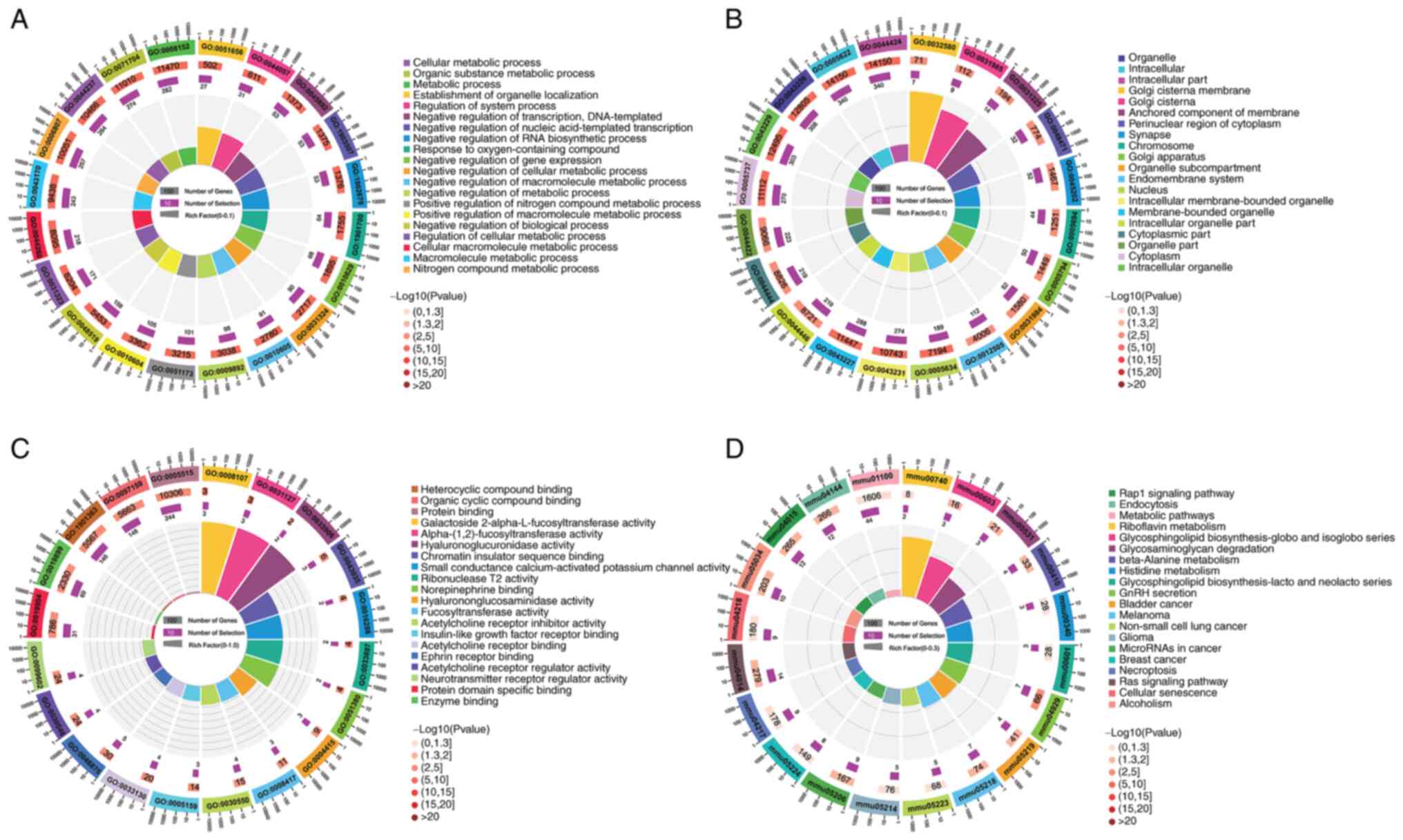
To elucidate the biological functions of
differentially expressed lncRNAs in LPS-induced GMCs, GO BP term
enrichment and KEGG pathway analyses were performed on all mRNAs in
the lncRNA-mRNA regulatory network (Fig. 4). Within the GO BP classification,
‘cellular macromolecule metabolic process’, ‘negative regulation of
biological process’ and ‘nitrogen compound metabolic process’ were
the top three over-represented terms (Fig. 4A). Within the GO cellular
components (CC) classification, ‘intracellular’, ‘intracellular
part’ and ‘membrane-bounded organelle’ were the top three
over-represented terms (Fig. 4B).
Within the GO molecular function (MF) classification, ‘galactoside
2-α-L-fucosyltransferase activity’, ‘α-(1,2)-fucosyltransferase activity’ and
‘chromatin insulator sequence’ binding were the top three
over-represented terms (Fig.
4C).
The KEGG metabolic pathway enrichment analysis
demonstrated that ‘GnRH secretion’, ‘Melanoma’, ‘Ras signaling
pathway’, ‘Glycosphingolipid biosynthesis-globo and isoglobo
series’ and ‘β-Alanine metabolism’ were the top five most
significantly enriched KEGG pathways (Fig. 4D). In addition,
inflammation-related signaling pathways, such as the ‘Rap1
signaling pathway’ and ‘Ras signaling pathway’, and substance
metabolism pathways, such as ‘Ribofiavin metabolism’, ‘β-Alanine
metabolism’ and ‘Histidine metabolism’ were also significantly
enriched. These results suggested that inflammation and substance
metabolism disorder may be the underlying CGN pathogenesis.
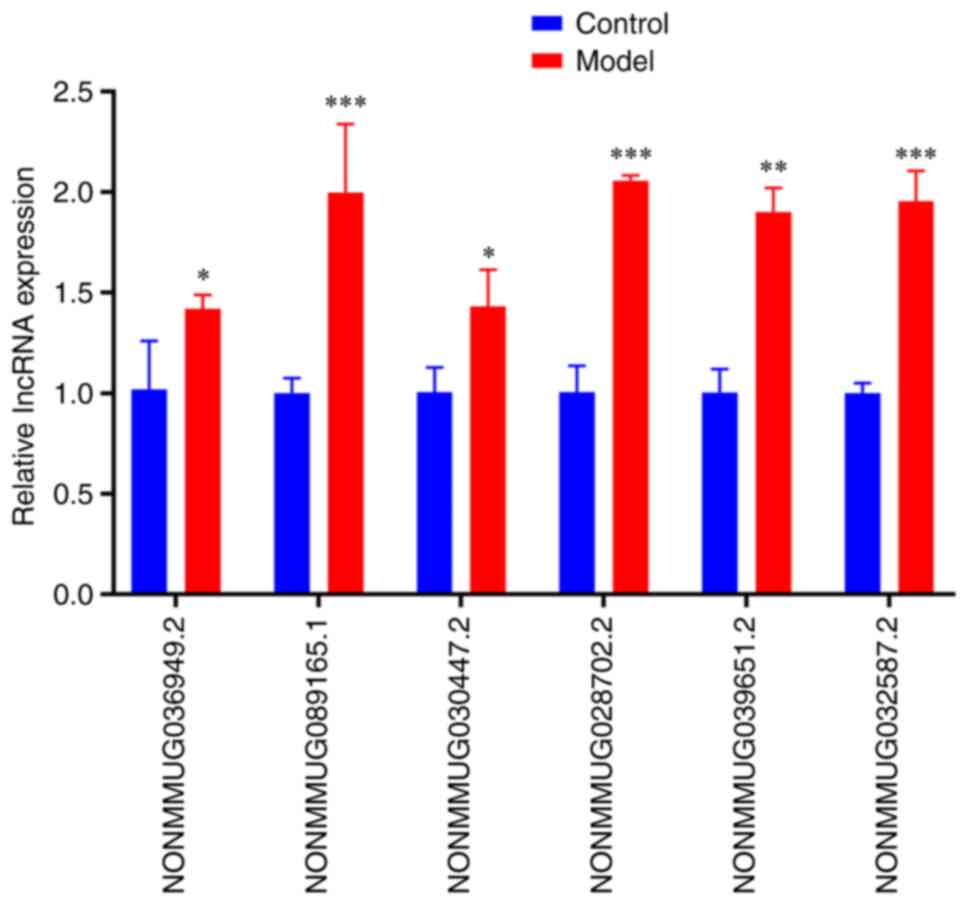
RT-qPCR validation
To verify the reliability of the sequencing data and
to support more clinically meaningful follow-up research, RT-qPCR
validation of certain highly conserved lncRNAs between humans and
mice was performed. The conservation information of lncRNAs was
obtained from the NONCODE database (http://www.noncode.org). The conservation analysis of
lncRNAs was assessed using the E-value, where a threshold of
E-value <1x10-5 was used (24). After conservation analysis among
species (Table IV),
NONMMUG036949.2, NONMMUG089165.1, NONMMUG030447.2, NONMMUG028702.2,
NONMMUG039651.2 and NONMMUG032587.2 were selected for RT-qPCR
validation (25). The RT-qPCR
results demonstrated the same expression trend as the RNA-Seq
results, with the six selected lncRNAs all significantly
upregulated in LPS-induced GMCs compared with the control group
(Fig. 5; Table SV).
| Table IVConservative information for the six
selected long non-coding RNAs. |
Table IV
Conservative information for the six
selected long non-coding RNAs.
| Gene | Chromosome |
log2(FC) | P-value | Alignment | Score, bits | E-value |
|---|
|
NONMMUG036949.2 | 6 | 1.2899 | 0.0476 |
NONHSAG058656.2 | 60 |
6.00x10-7 |
|
NONMMUG089165.1 | 12 | 1.9608 | 0.0054 |
NONHSAG048570.2 | 283 |
3.00x10-74 |
|
NONMMUG030447.2 | 4 | 1.4135 | 0.0250 |
NONHSAG056378.1 | 232 |
7.00x10-59 |
|
NONMMUG028702.2 | 4 | 1.2152 | 0.0287 |
NONHSAG038858.2 | 56 |
2.00x10-5 |
|
NONMMUG039651.2 | 7 | 1.0544 | 0.0353 |
NONHSAG063259.1 | 168 |
3.00x10-39 |
|
NONMMUG032587.2 | 5 | 1.3938 | 0.0013 |
NONHSAG088273.1 | 66 |
2.00x10-8 |
Construction of the lncRNA-associated
ceRNA network
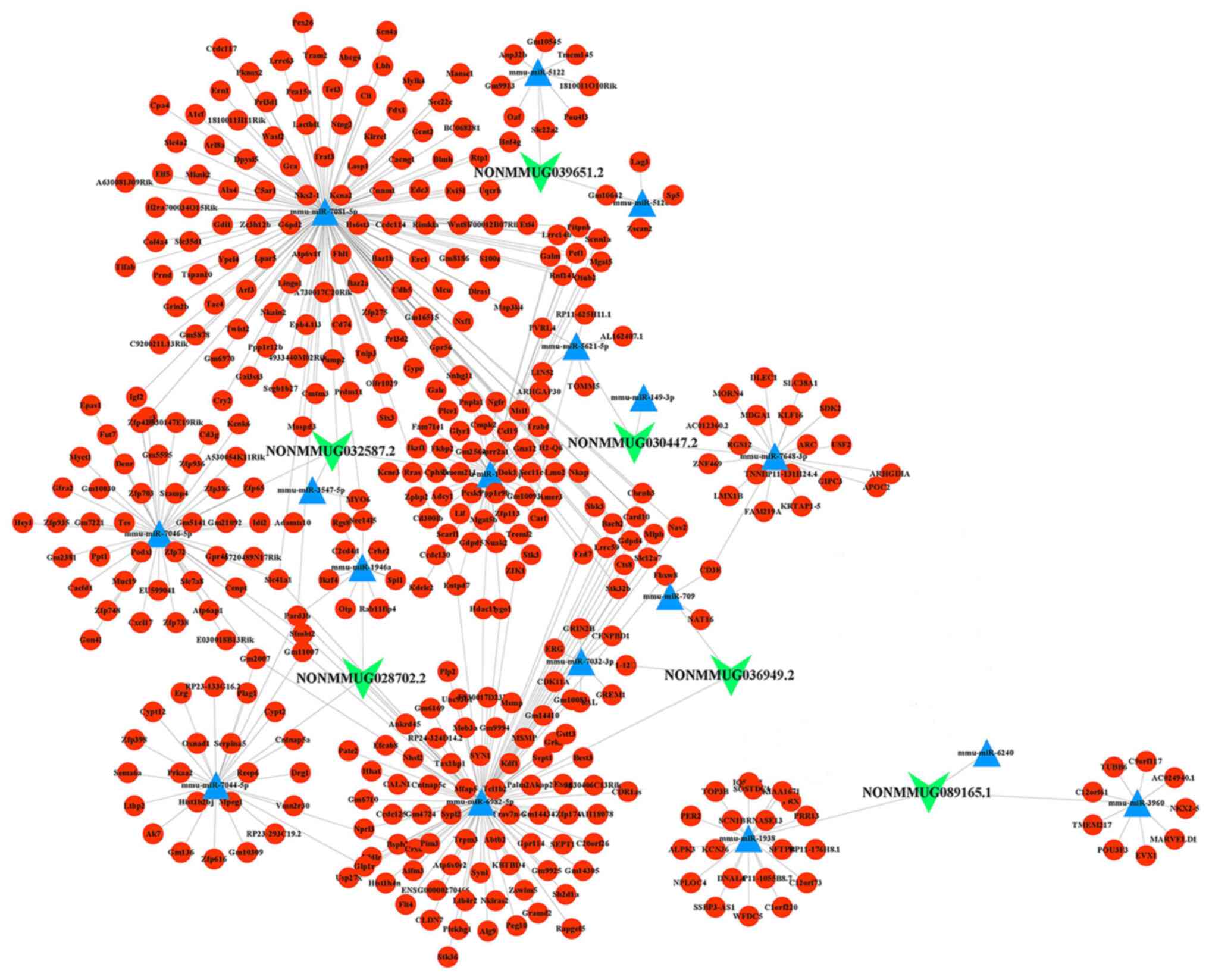
In addition to the lncRNA-mRNA regulatory network,
lncRNAs can also serve a role in other gene networks that regulate
diverse biological processes, such as the ceRNA network (26). To construct an lncRNA-associated
ceRNA network, the aforementioned six highly conserved and
RT-qPCR-validated lncRNAs were used to construct ceRNA networks.
The ceRNA network consisted of miRNAs of the highest confidence
with lncRNAs, and mRNAs with a high confidence (cumulative weighted
context score cutoff level <-0.8) (27) bound to the miRNAs, which included 6
lncRNAs, 18 miRNAs and 419 mRNAs (Fig.
6; Tables SVI and SVII). lncRNAs can be competing targets
of shared miRNAs with other mRNAs and form a complex regulatory
ceRNA network. For example, the lncRNA NONMMUG089165.1 may act as a
sponge for mmu-miR-3960 to affect EVX1 expression. PARD3B could be
affected by both the lncRNA NONMMUG039651.2/mmu-miR-7081-5P axis
and the lncRNA NONMMUG028702.2/mmu-miR-7044-5P axis at the same
time.
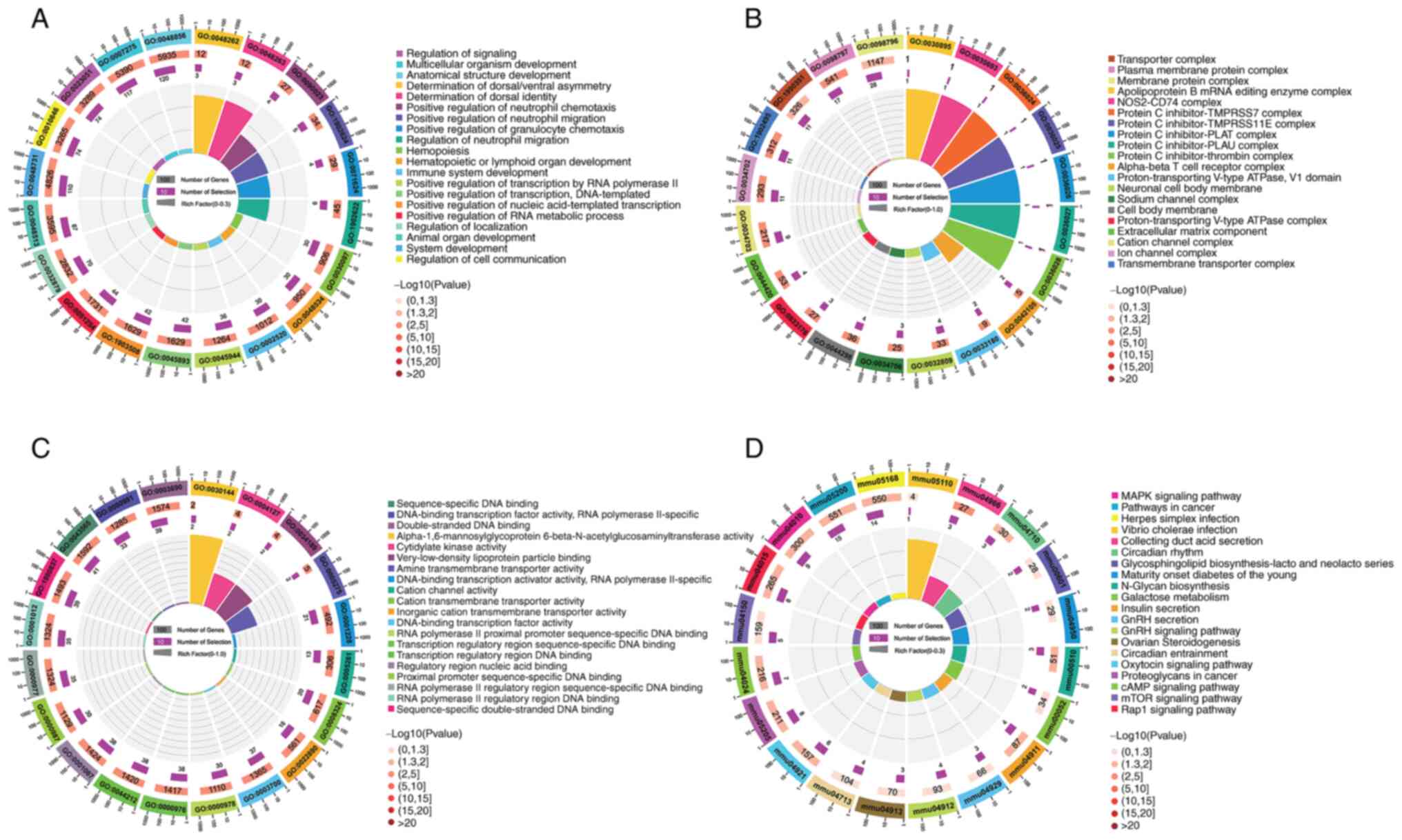
To evaluate the biological functions of the lncRNAs
in the ceRNA network, GO BP and KEGG pathway analysis was performed
on all mRNAs involved in the ceRNA network (Fig. 7). Within the GO BP classification,
‘system development’, ‘animal organ development’ and ‘multicellular
organism development’ were the top three over-represented terms
(Fig. 7A). Within the GO CC
classification, ‘neuronal cell body membrane’, ‘cell body membrane’
and ‘α-β T cell receptor complex’ were the top three
over-represented terms (Fig. 7B).
Within the GO MF classification, ‘DNA-binding transcription
activator activity, RNA polymerase II-specific’,
‘α-1,6-mannosylglycoprotein 6-β-N-acetylglucosaminyltransferase
activity’ and ‘DNA-binding transcription factor activity’ were the
top three over-represented terms (Fig.
7C). The KEGG metabolic pathway enrichment analysis
demonstrated that ‘Collecting duct acid secretion’, ‘Circadian
rhythm’, ‘Proteoglycans in cancer’, ‘Pathways in cancer’ and
‘Oxytocin signaling pathway’ were the top five most significantly
enriched KEGG pathways (Fig. 7D).
Furthermore, inflammation-related signaling pathways, such as ‘MAPK
signaling pathway’, ‘mTOR signaling pathway’ and ‘Rap1 signaling
pathway’ were significantly enriched KEGG pathways. These results
suggested that inflammation may be the underlying CGN
pathogenesis.
Screening of PPI core genes and
validation on GEO dataset
The PPI network of all the mRNAs involved in ceRNA
was successfully constructed, including 396 nodes and 260 edges
(Fig. 8A). The top 10 genes were
evaluated in the PPI network using the MCC, MNC and Degree
algorithms; the results of the three algorithms were cross-compared
to identify five core genes, including IKAROS family zinc finger 1
(IKZF1), CD3 ε subunit of T-cell receptor complex (CD3E), synapsin
1 (SYN1), glutamate ionotropic receptor NMDA subtype 2B (GRIN2B)
and SPI1) (Fig. 8B). According to
the ceRNA hypothesis, lncRNAs compete with miRNAs, to bind to the
target genes, which increases their mRNA expression. Public dataset
GSE104066 from the GEO database was used to validate the expression
of IKZF1, CD3E, SYN1, GRIN2B and SPI1 in CGN. The data demonstrated
that IKZF1, CD3E, SYN1 and GRIN2B were significantly upregulated;
however, no significant difference was demonstrated for SPI1
(Fig. 8C; Table SVIII).
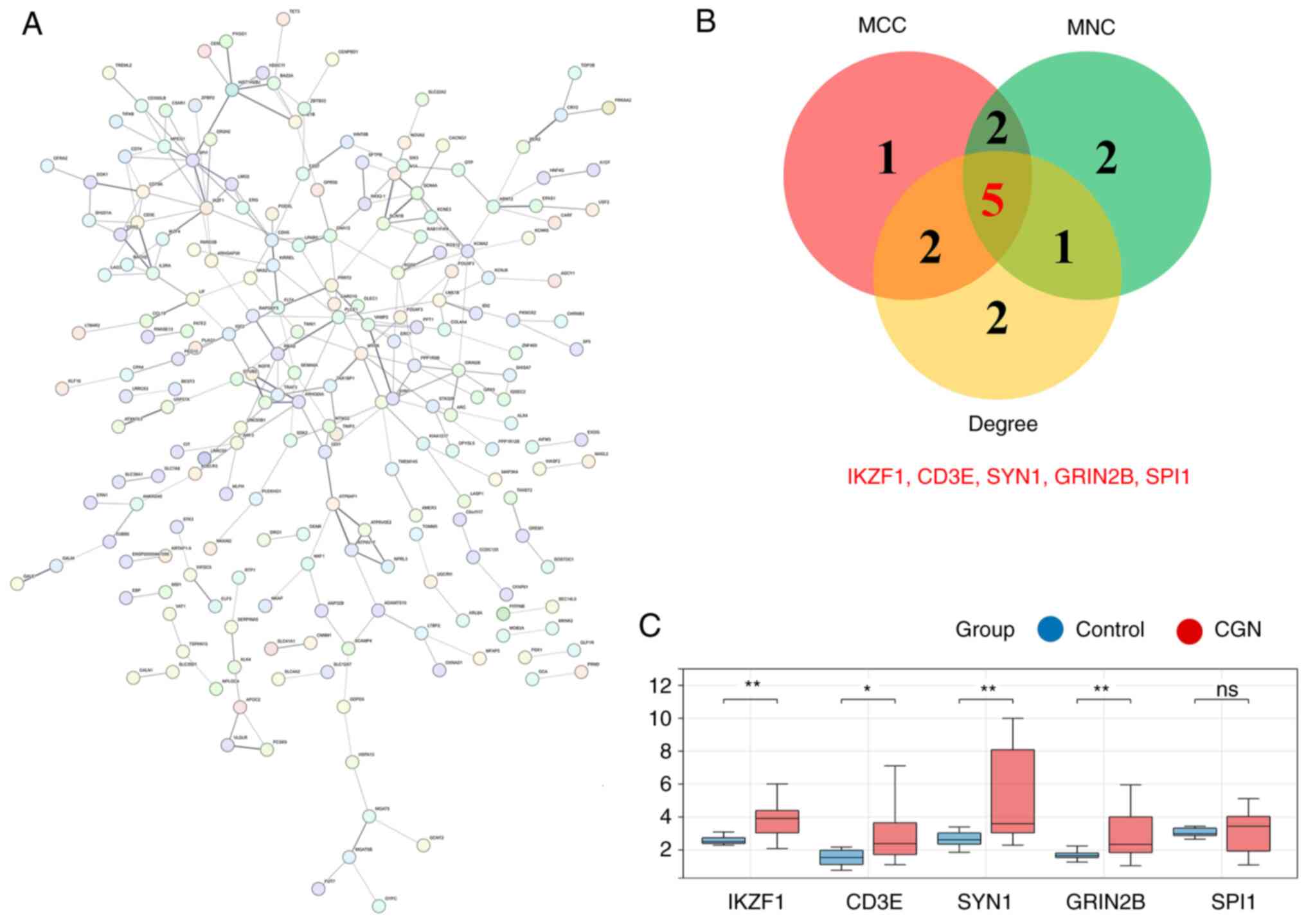 | Figure 8Screening of PPI core genes and
validation on GEO dataset. (A) PPI network diagram. (B) Screened
core genes identified using the MCC, MNC and Degree algorithms. (C)
The expression levels of IKZF1, CD3E, SYN1, GRIN2B and SPI1 in the
GEO GSE104066 dataset. *P<0.05 and
**P<0.01. CD3E, CD3 ε subunit of T-cell receptor
complex; GRIN2B, glutamate ionotropic receptor NMDA subtype 2B;
IKZF1, IKAROS family zinc finger 1; ns, not significant; PPI,
protein-protein interaction; SYN1, synapsin 1. |
Discussion
Chronic inflammation is an essential factor in the
occurrence and development of CGN, which may activate cell
proliferation and induce the deregulation of cells (28-29).
Excessive proliferation of GMCs, commonly observed in
glomerulonephritis, is an important pathological basis of kidney
disease (30-31). LPS is a major constituent of the
outer membrane of gram-negative bacteria that can stimulate the
activation of inflammatory factors in cells, leading to systemic
inflammatory response and activation of the immune system. LPS has
frequently been used as an inducer of cell proliferation in
numerous previous studies. For example, it has been previously
reported that LPS can induce THP-1 cell (32) and naive B cell (33) proliferation. Our research group has
previously used LPS-induced GMCs as an in vitro model of CGN
(34); therefore, in the present
study, RNA-seq was used to evaluate CGN-related lncRNAs in
LPS-induced GMCs, and an lncRNA-mRNA regulatory network and an
lncRNA-miRNA-mRNA ceRNA network were constructed to elucidate the
possible molecular mechanism of CGN.
The ceRNA theory was first proposed by Salmena
(35) in 2011; it provided a new
perspective for studying the role of RNA biological behavior in the
occurrence and development of disease. According to the ceRNA
theory, RNA transcripts, including non-coding RNAs, circular RNAs
and pseudogene transcripts, could function as miRNA sponges and so
regulate miRNA expression. Previous studies have reported that
lncRNAs could affect mRNA stability through a ceRNA theory. For
example, it has been previously reported that lncRNA linc00673 acts
as a ceRNA by sponging miR-150-5p and thus regulating ZEB1
expression; as such, it was suggested that linc00673 served an
essential role in the regulation of non-small cell lung cancer
proliferation, migration and invasion (36). Previous studies have reported on
the ceRNA theory and the link between lncRNAs and miRNAs (37). Therefore, the construction of a
lncRNA-associated ceRNA network in CGN could have important
research significance.
The enrichment analysis of the GO terms and KEGG
pathways of lncRNA-targeted genes presented a preliminary depiction
of the lncRNA function under investigation. The present study
performed a comprehensive analysis of the GO terms and KEGG
pathways for all mRNAs involved in the lncRNA-mRNA regulatory
network and lncRNA-associated ceRNA network. The results of the two
KEGG pathway analyses indicated significant enrichment of classical
inflammatory signaling pathways, including MAPK, Rap1, Ras and
mTOR. In instances of renal dysfunction, the timely clearance of
pro-inflammatory cytokines and small to medium-sized molecular
toxins is hindered, which leads to an elevation in inflammatory
cytokines. Furthermore, the accumulation of these toxins within the
body can stimulate the production of additional inflammatory
cytokines (38), as evidenced by
the enrichment of numerous inflammatory signaling pathways in the
present study.
Furthermore, the metabolism of certain substances
appeared to be affected, with pathways such as ‘N-Glycan
biosynthesis’, ‘Glycosphingolipid biosynthesis’, ‘Galactose
metabolism’, ‘Glycosaminoglycan degradation’, ‘β-Alanine
metabolism’, ‘Riboflavin metabolism’ and ‘Histidine metabolism’
being significantly enriched in the KEGG enrichment analysis of the
lncRNA-mRNA regulatory network. The kidneys are vital organs
responsible for the elimination of metabolic waste products from
the bloodstream and the regulation of the homeostatic levels of
electrolytes and metabolites, while simultaneously eliminating
harmful toxins from the body. In instances where kidney function is
compromised, the metabolic equilibrium of specific substances
within the body is disrupted (39). Previous studies have reported
notable variances in the plasma amino acid profile of individuals
with chronic kidney disease in comparison with those of healthy
individuals, which is typically characterized by alterations in the
levels of endogenous and essential amino acids (40,41).
There were certain limitations in the present study.
Firstly, the number of samples tested in RNA-seq was small and
should be increased in future studies to reduce possible bias in
the sequencing results. Secondly, both the lncRNA-miRNA and
miRNA-mRNA regulatory relationships were only predicted using
bioinformatics analysis software, and all hypotheses and relevant
mechanisms need to be verified by further experimental molecular
studies.
In conclusion, 1,532 differentially expressed
lncRNAs, including 594 upregulated lncRNAs and 938 downregulated
lncRNAs, were identified using RNA-seq in LPS-induced GMCs.
Furthermore, the lncRNA-mRNA regulatory network including 236
lncRNAs and 556 mRNAs, and the lncRNA-miRNA-mRNA ceRNA network
including 6 lncRNAs, 18 miRNAs and 419 mRNAs were constructed. KEGG
pathway analysis demonstrated that certain classical inflammatory
signaling pathways and substances metabolism were significantly
enriched. The present study demonstrated a global view of the
lncRNA-associated ceRNA network, and may offer novel insights into
the roles of lncRNAs in the pathogenesis of CGN as well as in
identifying promising diagnostic biomarkers.
Supplementary Material
Raw data of CCK-8 proliferation
assay.
The quality control result of
sequencing data
All long non-coding RNAs detected by
RNA sequencing
The target mRNAs of 236 differentially
expressed lncRNAs
Raw reverse transcription-quantitative
PCR data
The target miRNAs of long non-coding
RNAs
The target mRNAs of miRNAs
The expression of IKZF1, CD3E, SYN1,
GRIN2B, and SPI1 in dataset GSE104066
Acknowledgements
Not applicable.
Funding
Funding: This study was financially supported by The National
Natural Science Foundation of China (grant no. 81973546) and The
Key Scientific Research Projects of Natural Science in Colleges and
Universities in Anhui Province (grant no. 2022AH050747).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The raw RNA sequencing data were deposited in the National
Genomics Data Center (accession number PRJCA017179; https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA017179.
Authors' contributions
XXZ, TL, LBW and JRG wrote the manuscript. JRG
conceived and designed the experiment. XXZ performed the cell
experiment and acquisition of data. TL and LBW interpreted and
analyzed the sequencing data. LBW and JRG revised the manuscript
critically for important intellectual content. XXZ, TL, LBW and JRG
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
References
|
1
|
Sethi S and Fervenza FC: Standardized
classification and reporting of glomerulonephritis. Nephrol Dial
Transplant. 34:193–199. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Avraham S, Korin B, Chung JJ, Oxburgh L
and Shaw AS: The Mesangial cell-the glomerular stromal cell. Nat
Rev Nephrol. 17:855–864. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Chao S, Xu Q, Dong S, Guo M, Liu X and
Cheng X: Polygala fallax Hemsl combined with compound Sanqi
granules relieves glomerulonephritis by regulating proliferation
and apoptosis of glomerular mesangial cells. J Int Med Res.
48(300060519894124)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Shen J, Wu Q, Liang T, Zhang J, Bai J,
Yuan M and Shen P: TRIM40 inhibits IgA1-induced proliferation of
glomerular mesangial cells by inactivating NLRP3 inflammasome
through ubiquitination. Mol Immunol. 140:225–232. 2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bridges MC, Daulagala AC and Kourtidis A:
LNCcation: lncRNA localization and function. J Cell Biol.
220(e202009045)2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhu P, He F, Hou Y, Tu G, Li Q, Jin T,
Zeng H, Qin Y, Wan X, Qiao Y, et al: A novel hypoxic long noncoding
RNA KB-1980E6.3 maintains breast cancer stem cell stemness via
interacting with IGF2BP1 to facilitate c-Myc mRNA stability.
Oncogene. 40:1609–1627. 2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Tan YT, Lin JF, Li T, Li JJ, Xu RH and Ju
HQ: LncRNA-mediated posttranslational modifications and
reprogramming of energy metabolism in cancer. Cancer Commun (Lond).
41:109–120. 2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Gao J, Zhu X, Chen H, Jiang H, Shi M, Wei
L and Qin X: Long non-coding NONRATG001910.2 promotes the
proliferation of rat mesangial cell line HBZY-1 through the
miR-339-3p/CTNNB1 axis. Front Genet. 28(834144)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zhou K, Ou Q, Wang G, Zhang W, Hao Y and
Li W: High long non-coding RNA NORAD expression predicts poor
prognosis and promotes breast cancer progression by regulating
TGF-β pathway. Cancer Cell Int. 19(63)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Gao JR, Shi MM, Jiang H, Zhu XL, Wei LB
and Qin XJ: MicroRNA-339-5p inhibits lipopolysaccharide-induced rat
mesangial cells by regulating the Syk/Ras/c-Fos pathway. Naunyn
Schmiedebergs Arch Pharmacol. 395:1075–1085. 2022.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Liu T, Zhuang XX, Qin XJ, Wei LB and Gao
JR: The potential role of N6-methyladenosine modification of
LncRNAs in contributing to the pathogenesis of chronic
glomerulonephritis. Inflamm Res. 72:623–638. 2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-(Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Khoei MA, Karimi M, Karamian R, Amini S
and Soorni A: Identification of the complex interplay between
nematode-related lncRNAs and their target genes in glycine max L.
Front Plant Sci. 12(779597)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li J, Ma W, Zeng P, Wang J, Geng B, Yang J
and Cui Q: LncTar: A tool for predicting the RNA targets of long
noncoding RNAs. Brief Bioinform. 16:806–812. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
John B, Enright AJ, Aravin A, Tuschl T,
Sander C and Marks DS: Human MicroRNA targets. PLoS Biol.
2(e363)2004.PubMed/NCBI View Article : Google Scholar
|
|
16
|
McGeary SE, Lin KS, Shi CY, Pham TM,
Bisaria N, Kelley GM and Bartel DP: The biochemical basis of
microRNA targeting efficacy. Science. 366(eaav1741)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Su H, Tao T, Yang Z, Kang X, Zhang X, Kang
D, Wu S and Li C: Circular RNA cTFRC acts as the sponge of
MicroRNA-107 to promote bladder carcinoma progression. Mol Cancer.
18(27)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen L, Zhang YH, Lu G, Huang T and Cai
YD: Analysis of cancer-related lncRNAs using gene ontology and KEGG
pathways. Artif Intell Med. 76:27–36. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Szklarczyk D, Kirsch R, Koutrouli M,
Nastou K, Mehryary F, Hachilif R, Gable AL, Fang T, Doncheva NT,
Pyysalo S, et al: The STRING database in 2023: Protein-protein
association networks and functional enrichment analyses for any
sequenced genome of interest. Nucleic Acids Res. 51:D638–D646.
2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Doncheva NT, Morris JH, Gorodkin J and
Jensen LJ: Cytoscape StringApp: Network analysis and visualization
of proteomics data. J Proteome Res. 18:623–632. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Gao R, Liang X, Cheedipudi S, Cordero J,
Jiang X, Zhang Q, Caputo L, Günther S, Kuenne C, Ren Y, et al:
Pioneering function of Isl1 in the epigenetic control of
cardiomyocyte cell fate. Cell Res. 29:486–501. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Weber MD, McKim DB, Niraula A, Witcher KG,
Yin W, Sobol CG, Wang Y, Sawicki CM, Sheridan JF and Godbout JP:
The influence of microglial elimination and repopulation on stress
sensitization induced by repeated social defeat. Biol Psychiatry.
85:667–678. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yang KC, Yamada KA, Patel AY, Topkara VK,
George I, Cheema FH, Ewald GA, Mann DL and Nerbonne JM: Deep RNA
sequencing reveals dynamic regulation of myocardial noncoding RNAs
in failing human heart and remodeling with mechanical circulatory
support. Circulation. 129:1009–1021. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Liang F, Zhang Y, Wang X, Yang S, Fang T,
Zheng S and Zeng L: Integrative mRNA and long noncoding RNA
analysis reveals the regulatory network of floral bud induction in
longan (Dimocarpus longan Lour.). Front Plant Sci.
13(923183)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang Y, Zhu P, Luo J, Wang J, Liu Z, Wu W,
Du Y, Ye B, Wang D, He L, et al: LncRNA HAND2-AS1 promotes liver
cancer stem cell self-renewal via BMP signaling. EMBO J.
38(e101110)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Li S, Cao Y, Zhang H, Lu X, Wang T, Xu S,
Kong T, Bo C, Li L, Ning S, et al: Construction of lncRNA-Mediated
ceRNA network for investigating immune pathogenesis of ischemic
stroke. Mol Neurobiol. 58:4758–4769. 2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Mulari S, Eskin A, Lampinen M, Nummi A,
Nieminen T, Teittinen K, Ojala T, Kankainen M, Vento A, Laurikka J,
et al: Ischemic heart disease selectively modifies the right atrial
appendage transcriptome. Front Cardiovasc Med.
8(728198)2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Chung H, Komada T, Lau A, Chappellaz M,
Platnich JM, de Koning HD, Petri B, Luque Y, Walker S, Benediktsson
H, et al: AIM2 Suppresses inflammation and epithelial cell
proliferation during glomerulonephritis. J Immunol. 207:2799–2812.
2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Eaton JE, Fritcher EG, Gores GJ, Atkinson
EJ, Tabibian JH, Topazian MD, Gossard AA, Halling KC, Kipp BR and
Lazaridis KN: Biliary multifocal chromosomal polysomy and
cholangiocarcinoma in primary sclerosing cholangitis. Am J
Gastroenterol. 110:299–309. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Harendza S, Schneider A, Helmchen U and
Stahl RA: Extracellular matrix deposition and cell proliferation in
a model of chronic glomerulonephritis in the rat. Nephrol Dial
Transplant. 14:2873–2879. 1999.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Harendza S, Behrens U, Zahner G, Schneider
A and Stahl RA: In vitro characterization of the mesangial
phenotype in a proliferative glomerulonephritis of the rat. Nephrol
Dial Transplant. 12:2537–2541. 1997.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Sun J, Shigemi H, Cao M, Qin E, Tang J,
Shen J and Iwasaki H: Minocycline induces autophagy and inhibits
cell proliferation in LPS-Stimulated THP-1 cells. Biomed Res Int.
2020(5459209)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ribeiro de Almeida C, Dhir S, Dhir A,
Moghaddam AE, Sattentau Q, Meinhart A and Proudfoot NJ: RNA
helicase DDX1 converts RNA G-quadruplex structures into R-loops to
promote IgH class switch recombination. Mol Cell. 70:650–662.
2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Liu T, Zhuang XX, Qin XJ, Wei LB and Gao
JR: Alteration of N6-methyladenosine epitranscriptome profile in
lipopolysaccharide-induced mouse mesangial cells. Naunyn
Schmiedebergs Arch Pharmacol. 395:445–458. 2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lu W, Zhang H, Niu Y, Wu Y, Sun W, Li H,
Kong J, Ding K, Shen HM, Wu H, et al: Long non-coding RNA linc00673
regulated non-small cell lung cancer proliferation, migration,
invasion and epithelial mesenchymal transition by sponging
miR-150-5p. Mol Cancer. 16(118)2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wang L, Cho KB, Li Y, Tao G, Xie Z and Guo
B: Long noncoding RNA (lncRNA)-mediated competing endogenous RNA
networks provide novel potential biomarkers and therapeutic targets
for colorectal cancer. Int J Mol Sci. 20(5758)2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Suliman ME, Qureshi AR, Stenvinkel P,
Pecoits-Filho R, Bárány P, Heimbürger O, Anderstam B, Ayala ER,
Filho JCD, Alvestrand A and Lindholm B: Inflammation contributes to
low plasma amino acid concentrations in patients with chronic
kidney disease. Am J Clin Nutr. 82:342–349. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yan LJ: Redox imbalance and mitochondrial
abnormalities in kidney disease. Biomolecules.
12(476)2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Taherkhani A, Kalantari S, Oskouie AA,
Nafar M, Taghizadeh M and Tabar K: Network analysis of membranous
glomerulonephritis based on metabolomics data. Mol Med Rep.
18:4197–4212. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Carter JL, Parker CT, Stevens PE,
Eaglestone G, Knight S, Farmer CK and Lamb EJ: Biological variation
of plasma and urinary markers of acute kidney injury in patients
with chronic kidney disease. Clin Chem. 62:876–883. 2016.PubMed/NCBI View Article : Google Scholar
|