The tumor microenvironment (TME), which consists
primarily of the extracellular matrix (ECM), is crucial to various
aspects of tumor progression, such as tumorigenesis, metastasis,
relapse and treatment resistance, making it a potential target for
cancer therapies (1,2). The TME is a complex system comprising
a range of cellular and noncellular elements, and cancer-associated
fibroblasts (CAFs), which are highly abundant in the tumor stroma
and exhibit potent regulatory effects on tumor growth (3,4). The
induction of cell senescence through traditional cancer treatments
is one of the most notable mechanisms of tumor suppression.
However, in cancer, the process of therapy-induced senescence (TIS)
is a ‘double-edged sword’ (5).
Despite its indispensable role in combating tumor growth by halting
cancer cell division, the chronic accumulation of senescent cells
can conceivably promote tumor development (6). Specifically, secretion of the
senescence-associated secretory phenotype (SASP) (7), which is induced by TIS, can
significantly affect the TME through autocrine and paracrine
effects (8).
The aim of the present review was to provide an
overview of the most recent research regarding the biological
tumor-promoting functions of CAFs. Additionally, this review aimed
to emphasize the antitumorigenic properties of senescence-like CAFs
within the TME, which are induced by anticancer therapies.
Furthermore, a comprehensive overview of the existing senolytic
therapies, and their potential advantages in the context of cancer
treatment, is provided.
Previous studies have highlighted the complexity and
diversity of CAFs, one of the diverse components of the tumor
stroma, which could be due to their multiple cellular origins and
the emergence of various CAF subpopulations (9,10).
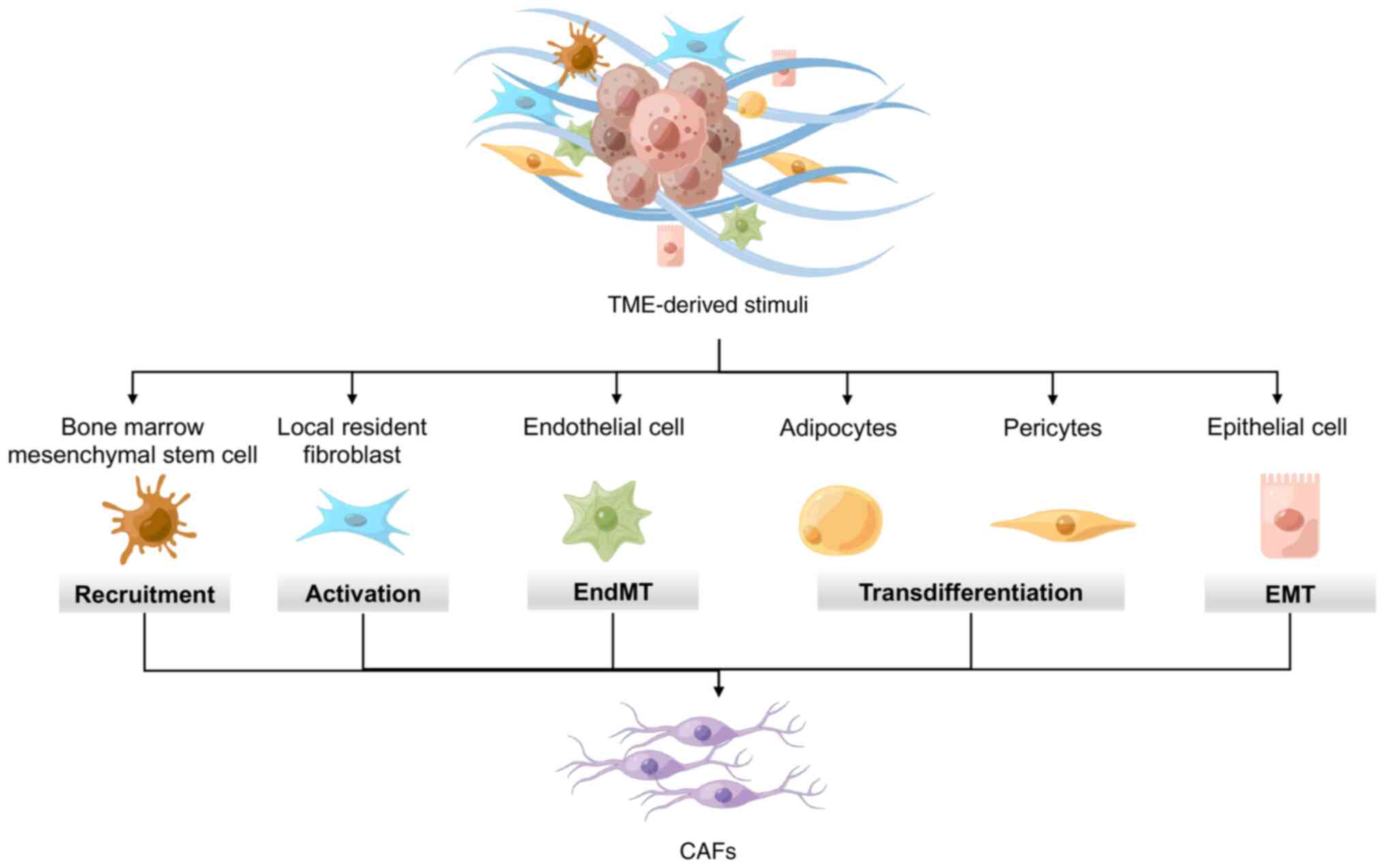
While the exact origin of CAFs remains unclear, they can be
produced through various means. During tumorigenesis, remnant local
fibroblasts in the surrounding tissues can undergo gene expression
and phenotype changes in response to tumor driver stimuli. For
example, ‘quiescent’ stellate cells in the liver and pancreas,
which are the resident fibroblasts in these tissues, can be
activated by inflammatory stimuli released by tumors to acquire a
myofibroblast-like CAF phenotype (11-13).
In addition, CAFs can be recruited from remotely circulating cell
populations. One such precursor of CAFs is bone marrow mesenchymal
stem cells, which is the most well-studied cell population among
the known sources of CAFs (14,15).
Moreover, nonfibroblastic lineages that are in close proximity to
tumor cells, such as endothelial or epithelial cells, can undergo
transdifferentiation into CAFs through endothelial-mesenchymal or
epithelial-mesenchymal transition (EMT), respectively. These cells
exhibit CAF-like gene expression and represent another source of
heterogeneity for the population of CAFs (16,17).
Furthermore, transdifferentiation from other uncommon CAF precursor
cells, such as adipocytes and pericytes, has been observed under
limited conditions (18,19) (Fig.
1).
Cancer progression relies heavily on tumor stromal
support to maintain continuous tumor growth and metastasis
(20,21). Despite the presence of normal
fibroblasts in the matrix surrounding the tumor, they do not hinder
cancer cell invasion and metastasis (22). Hence, it becomes essential for
cancer cells to transform normal fibroblasts into tumor-promoting
CAFs, which are recruited and activated by various tumor-derived
signals and specific stimuli in the TME, such as hypoxia and
oxidative stress damage (23,24).
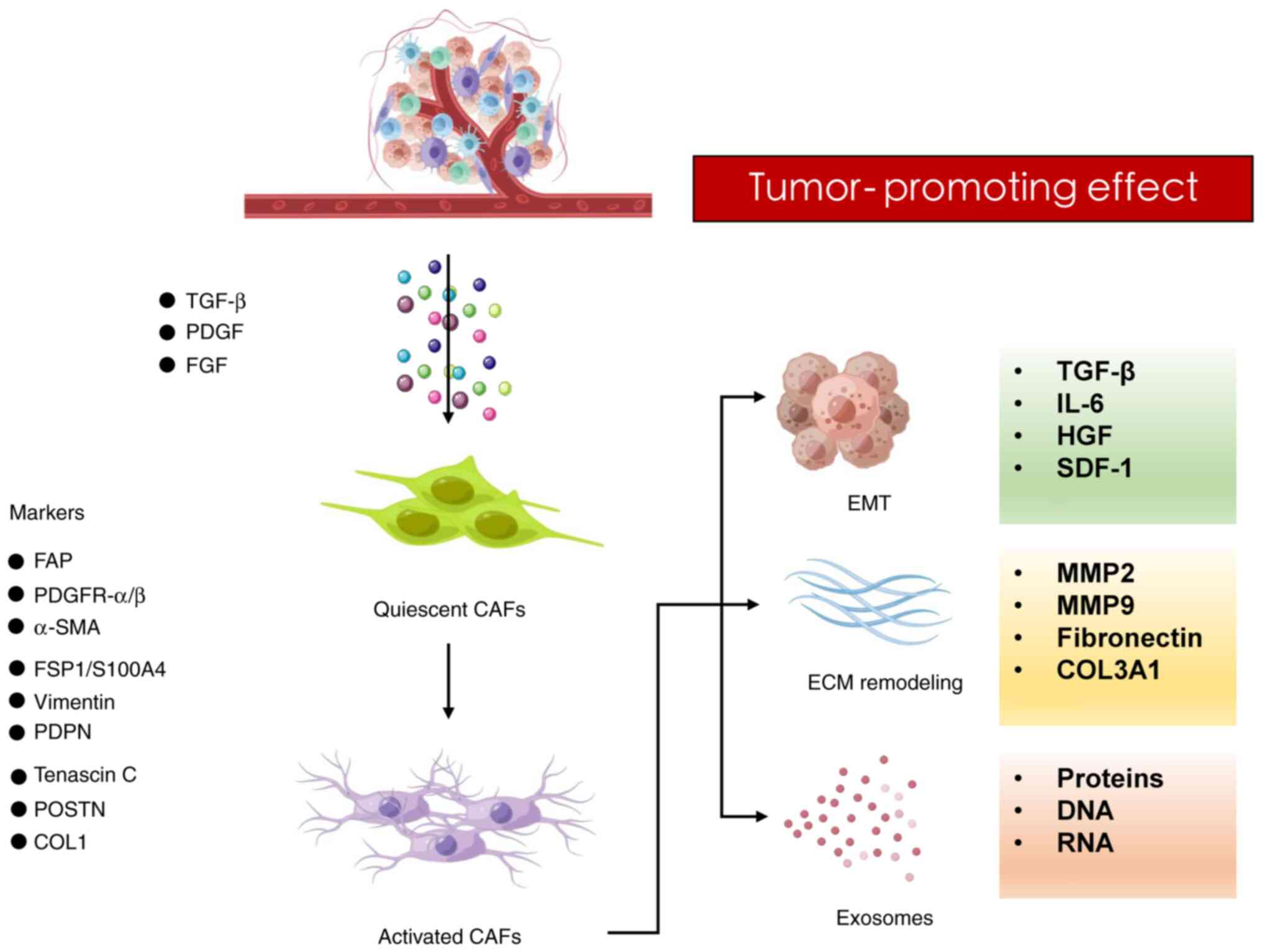
Among these, transforming growth factor β (TGF-β), platelet-derived
growth factor (PDGF) and fibroblast growth factor (FGF) play
crucial roles as biochemical mediators in CAFs (25). It has been indicated that TGF-β has
a strong interaction with ECM components in tissues and is an
essential cytokine that activates the cancer stroma (26). TGF-β is typically activated
locally, and as a result, CAFs produce TGF-β locally in response to
its secretion by cancer cells in the surrounding area. This
triggers a response in CAFs, enhancing their potential to produce
tumor-promoting factors (27,28).
PDGF is another growth factor secreted by tumor cells that is
closely linked to cancer progression (29). Cellular processes, such as
chemotaxis, cell proliferation, division and angiogenesis, are some
of the numerous biological processes affected by PDGF, and
overexpression of PDGF and its receptors in tumors is a common
occurrence. However, most tumor cells do not express PDGF
receptors. Rather, PDGF levels are upregulated, implying that PDGF
mainly promotes tumor progression through paracrine signaling from
other cells, such as fibroblasts and endothelial cells (30,31).
Unlike the TGF-β-induced ECM over-deposition phenotype, PDGF mainly
enhances fibroblast recruitment and proliferation (32). FGF2 was initially known as a ‘basic
fibroblast growth factor’ that promotes cell differentiation and
proliferation between epithelial and mesenchymal cells through
autocrine and paracrine processes. Further research has revealed
that FGF2 stimulates the activation of normal fibroblasts in
vivo, conferring their ability to promote metastasis (33). Furthermore, FGF2 can act
synergistically with PDGF to promote tumor angiogenesis and
metastasis (34).
CAF populations are highly heterogeneous, with
differences across various tumor types and cell sources. However,
unlike other cell lineages, CAFs do not express specific
biomarkers, posing a challenge to their comprehensive
characterization. Representative CAF markers have been established,
including fibroblast activation protein, PDGF receptor-α/β,
α-smooth muscle actin, fibroblast-specific protein 1/S100A4,
vimentin, podoplanin, periostin and type-I collagen (35,36).
Nevertheless, while these markers are commonly used to identify CAF
phenotypic features, none of them offer specificity to CAFs, and
the expression of these markers can also be observed in healthy
tissues and other cell types. Table
I (37-54)
lists some typical CAF markers used in cancer studies.
Tumor metastasis is frequently associated with an
unfavorable prognosis for patients (55). CAFs are known to be important in
tumor development and resistance to cancer therapy; therefore, the
functions of CAFs should be closely examined in relation to tumor
metastasis. The modulatory effects of CAFs on tumor progression
have been well documented in the TME, where they promote tumor
progression and metastasis through various mechanisms, including
ECM modification, EMT regulation in cancer cells and the release of
cytokines that support tumor growth.
ECM remodeling is a vital physiological process for
maintaining tissue homeostasis that occurs throughout body
development, regeneration and wound healing (56,57).
Fibroblasts, found in both healthy and tumor tissues, make a
critical difference in producing ECM components and remodeling
enzymes in the interstitial space. Tumor-derived factors activate
fibroblasts, transforming them into CAFs, which facilitate
communication between the tumor and its surrounding stroma. CAFs
are responsible for ECM remodeling in distant organs, promoting
tumor invasion and metastasis, and can create pathways within the
ECM by degrading matrix proteins, allowing cancer cells to
penetrate the basement membrane and enter the surrounding tissues,
ultimately metastasizing to distant locations (58). This ECM remodeling process,
facilitated by ECM-degrading enzymes such as rho-associated kinase
and matrix metalloproteinases, serves an important role in driving
malignant cell invasion and metastasis (59-61).
In both embryogenesis and tumor metastasis, the
involvement of fibroblasts in promoting EMT is crucial. Generally,
cells with a mesenchymal phenotype are more prone to invasion
(62). However, carcinomas
typically retain the epithelial characteristics that limit their
invasive potential. Therefore, cancer cells rely on nonmalignant
stromal cell types, with CAFs being the most ideal matrix partners,
to facilitate invasion and metastasis. Hybrid EMT, in which
malignant cells exhibit a combination of mesenchymal and epithelial
characteristics, strongly promotes metastasis (17,63-65).
The interaction between malignant cells and CAFs is mediated by
cytokines, which is vital for EMT. In recent years, extensive
research has been conducted on the CAF-secreted factors involved in
EMT in various types of cancer. Among these factors, TGF-β,
released by CAFs, has been extensively studied. In advanced cancer,
TGF-β enhances the migratory and invasive capabilities of cancer
cells by inducing a mesenchymal phenotype. For example, CAFs can
activate homeobox transcript antisense intergenic RNA (HOTAIR)
transcription through the secretion of TGF-β1, and SMAD2/3/4 can
directly bind to the HOTAIR promoter site to increase the ability
of breast cancer cells to metastasize (66). Other signaling pathways that drive
cancer cells to acquire mesenchymal phenotypes, such as Janus
kinase/signal transducer and activator of transcription proteins
(JAK/STAT), Wnt/β-catenin, MAPK and PI3K/Akt, can be triggered by
CAF-released cytokines, growth factors and chemokines, such as
interleukin-6(67), hepatocyte
growth factor (68) and stromal
cell-derived factor-1(69).
As previously mentioned, activated CAFs secrete
various messengers that can induce remodeling of the ECM or EMT in
malignant cells to facilitate tumor development and progression. In
the present section, the ways in which CAF-derived exosomes promote
metastasis was focused on. Exosomes, a type of extracellular
vesicle, have emerged as novel messengers in intercellular
communication; notably, they deliver proteins, DNA and RNA
(70). Several studies have
identified exosomes secreted by CAFs as key players in the
crosstalk between tumors and CAFs, as well as in cancer cell
invasion. For example, in gemcitabine-treated human pancreatic
ductal adenocarcinoma CAFs, the expression levels of Snail and
microRNA (miR)-146a were revealed to be increased in exosomes, and
this enhanced cell proliferation and chemoresistance (71). In gastric carcinoma, exosomal
miR-522 derived from CAFs was shown to inhibit ferroptosis by
suppressing arachidonate lipoxygenase 15 and reducing lipid
reactive oxygen species accumulation, resulting in chemoresistance
and tumor progression (72)
(Fig. 2).
Cellular aging is a response to cellular stress
caused by molecular damage. Common triggers of senescence include
replicative exhaustion (replicative senescence), hyperactivation of
oncogenes (oncogene-induced senescence), and persistent damage to
DNA and chromatin structures (73). Cellular senescence is regarded as a
vital intrinsic tumor-suppressing mechanism. Various anticancer
therapies induce senescence in malignant cells by inducing
genotoxic stress, overactivating mitotic signals or inducing
oxidative stress; this blocks the growth of tumor cells and
promotes immune cell infiltration (74). However, some patients experience
relapse, metastasis and/or therapeutic resistance. The TME is a
complex biological system consisting of tumor cells and numerous
nontumor components. These nontumor components, such as the ECM,
fibroblasts and immune cells, can have profound effects on tumor
progression during aging (75,76).
This review focuses on the impact of aging of CAFs on tumor
progression.
Tumor cells can induce the aging of stromal cells
through cytokine secretion. It has been reported that the specific
expression of matrix metalloproteinase 1 in large-cell carcinoma
can induce fibroblast senescence and promote the progression of
lung cancer (76). In addition,
the gut microbiota can induce aging of hepatic stellate cells
through the enterohepatic circulation of metabolites, leading to
the development of liver cancer (77-79).
Cancer cells cease to proliferate after TIS; however, senescent
cells still exhibit metabolic activity and undergo changes in their
secretory proteomes. They secrete proinflammatory factors, growth
factors and proteases, collectively referred to as SASP (80). The accumulation of therapy-induced
senescent cells leads to chronic inflammation and
immunosuppression, which can have long-term adverse effects
(74). Since most anticancer
treatments are administered systemically, they become problematic
when nonmalignant cells in nontumor areas undergo senescence in
response to these therapies (81).
The present review aims to concentrate on recent studies that refer
to the induction of senescence in CAFs by antitumor therapies, such
as chemotherapy, radiotherapy (RT) and targeted therapies.
Chemotherapy and RT are conventional and widely
utilized treatments for cancer. High doses of chemotherapy or RT
can effectively induce the apoptosis of cancer cells; however, they
also pose a risk of damaging the surrounding tissues, causing
serious side effects in patients (74). Consequently, an alternative
therapeutic strategy is to induce senescence in malignant cells to
permanently halt their ability to proliferate without triggering
apoptosis. An extensively studied mechanism of cellular aging is
activation of the DNA damage response (DDR) (7). The DDR is closely associated with the
relapse and development of tumors, and it also provides therapeutic
opportunities for tumor treatment (82). When cells are exposed to various
endogenous and exogenous stressors (such as replication stress,
ultraviolet, drugs and ionization radiation), DDR-associated
proteases (such as ATM, ATR, CHK1 and PARP) are activated, which
eventually results in senescence (83). Camptothecin, doxorubicin and
etoposide, the most commonly used topoisomerase inhibitors in
chemotherapy for various types of cancer, can effectively block DNA
replication by causing misalignment of DNA strands after supercoil
unwinding (84). Similarly,
bioalkylating agents, another widely used type of chemotherapeutic
drug, trigger DNA damage-mediated aging reactions by causing DNA
strand crosslinking, abnormal base pairing or DNA strand breaks.
During cell division, these crosslinked DNA strands break,
triggering DDR-mediated senescence. Cisplatin is an alkylating
agent widely used in anticancer treatments (85). Microtubule inhibitors, such as
paclitaxel can arrest tumor cells in the mitotic phase by
disrupting the regular dynamics of microtubule spindles (86). These cytotoxic compounds, commonly
used as standard cancer therapies for various tumor types, have
dual effects. They induce a senescent phenotype in cancer cells and
exert an anticancer effect. However, they also induce senescence in
cellular components of the TME. CAFs play an essential role in
promoting tumorigenic signals inside the tumor stroma. Senescent
CAFs can enhance the differentiation and proliferation of
neighboring tumor cells via the secretion of SASP factors. In a
xenotransplant tumor model of breast cancer, senescent fibroblasts
have been reported to promote the growth of breast cancer in mice
through the secretion of matrix metalloproteinases (86). It has also been demonstrated that
CAFs are prone to p53-mediated senescence during chemotherapy,
leading to drug resistance in mouse lung cancer models (87). Similarly, a recent study revealed
that docetaxel and cisplatin treatment can strongly induce a
senescence phenotype in prostate- and ovary-associated fibroblasts
in vitro. These senescent fibroblasts exhibit enhanced
malignant behavior through alterations in metabolism and activation
of SASP (88).
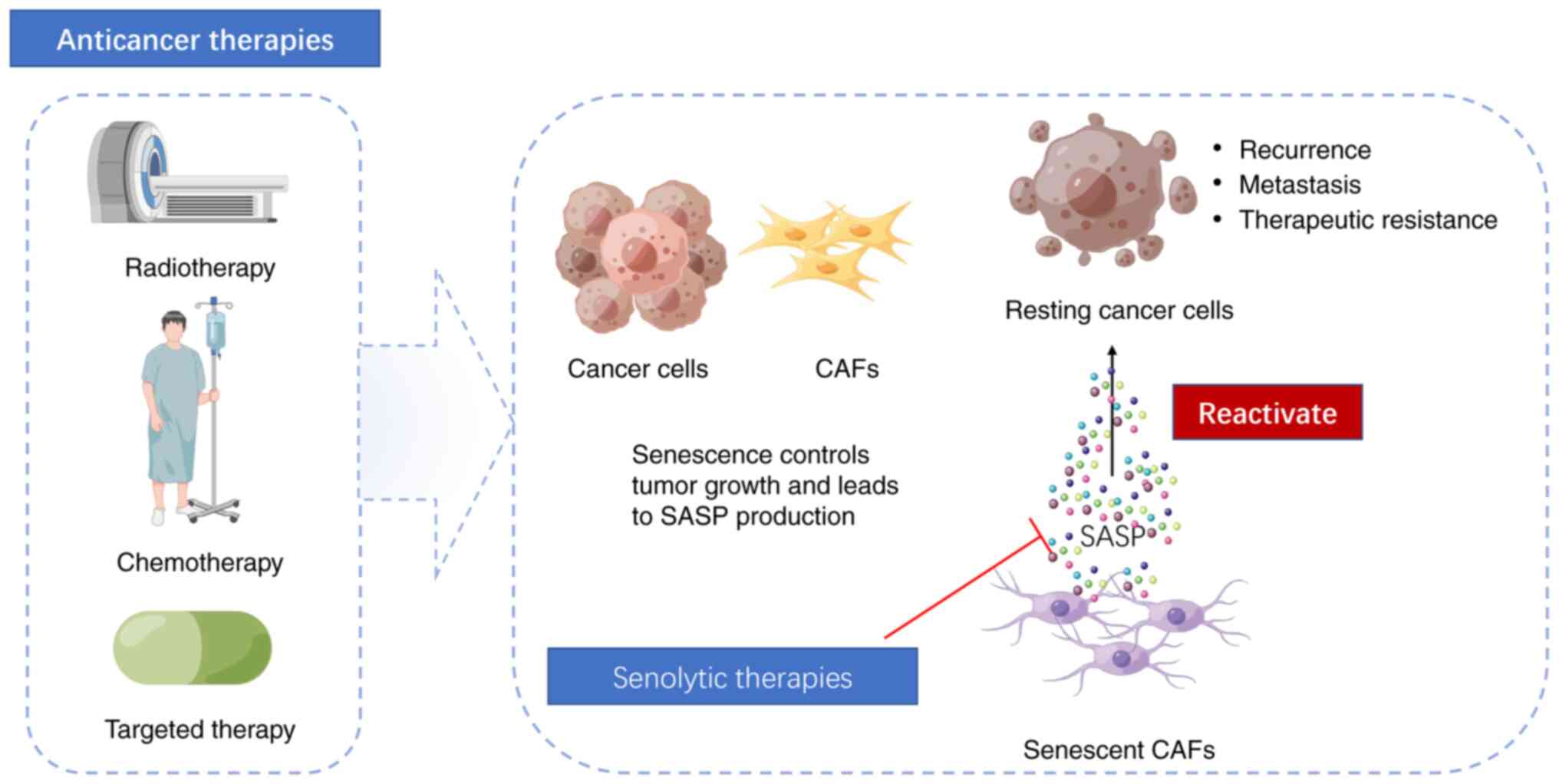
The clinical dose of cancer therapy, while
effectively inducing tumor cell apoptosis or senescence (first
punch), can also lead to senescence of other elements in the TME,
which can contribute to tumor relapse, metastasis and the
development of drug resistance to cancer treatment. Eliminating
these senescent cells is crucial for reducing the risk of tumor
progression (second punch) (5).
Senolytics, which are a type of drug that selectively eliminates
aging cells, have emerged as potential therapeutics for addressing
this issue. In the context of tumors, senolytic strategies
typically involve a combination of a senescence-inducing drug and
another drug targeting senescent cells to induce the synthesis of
lethal substances. By specifically targeting and eliminating these
senescent cells, senolytics hold promise in preventing tumor
progression and improving the efficacy of cancer treatment. A key
characteristic of aging cells is the alteration of chromatin
structure, which affects gene expression. The present review aims
to provide a brief introduction to the application of senolytic
drugs in targeting senescent fibroblasts or stromal cells in
tumors. However, there are detailed studies that focus on the
introductions and applications of these drugs in other diseases
(96,97).
Studies have revealed that the expression of
anti-apoptotic proteins often increases in senescent cells
(74,98). Senolytic drugs can target
upregulated anti-apoptotic pathways in senescent cells and
reactivate the apoptotic pathway to eliminate aging cells (99). Navitoclax, a selective BCL
inhibitor, is primarily active against BCL family members (such as
BCL-2, BCL-XL and BCL-W). When used in combination with anticancer
treatments, navitoclax can potentially achieve a dual effect. In a
cell line and mouse model of glioblastoma, extensive senescence has
been shown to occur in the brain following RT. Aging stromal cells
promote the aggressive phenotype of glioblastoma via SASP, which
enhances its ability to invade and migrate in vitro and
in vivo. However, the use of navitoclax (ABT-263)
selectively eliminates the senescent cells and significantly
reduces glioma cell growth (100). Quercetin, a natural flavonol
compound, and dasatinib, a tyrosine kinase inhibitor, are widely
used to target senescent cells by interfering with Src and PI3K
signaling. In a liver cancer mouse model, dasatinib has been
reported to effectively inhibit senescent stellate cells, thereby
inhibiting tumor progression (101). FOXO4-DRI, an interfering peptide
that can regulate the activity of p53, disrupts the interactions
between p53 and FOXO4. This selective disruption leads to p53
nuclear exclusion and apoptosis of senescent cells (102). Research has demonstrated that RT
can induce senescence-like characteristics of CAFs in non-small
cell lung cancer. FOXO4-DRI enhances the radiosensitivity of NSCLC
cells and alleviates RT-induced pulmonary fibrosis by targeting
aging CAFs, both in vitro and in vivo (90) (Fig.
3).
The goal of tumor therapy is to achieve a complete
cure by eliminating malignant tumors. Increasing attention has been
paid to the function of the TME in tumor therapies, as it can
significantly impact therapeutic outcomes. Systemic anticancer
therapy induces senescence in cancer cells and triggers the
development of aging components in the TME. Among these components,
CAFs are critical in tumor progression. The one-two punch strategy
aims to address the dual behavior of TIS, which can have both
favorable and unfavorable effects on tumor therapies. This approach
acknowledges both the positive outcomes and the possible challenges
associated with senescence induction. By targeting both the
beneficial and harmful effects of TIS, this strategy can provide
significant therapeutic benefits and may be an effective approach
for future studies on cancer treatment.
Not applicable.
Funding: This research was funded by the National Natural
Science Foundation of China (grant no. 81773945) and the Basic
Public Welfare Research Project of Zhejiang Province (grant no.
LY23H280011).
Not applicable.
QZ and YL were involved in study conceptualization.
HF and SS contributed to the generation of the figures and table.
RJ performed the literature investigation. QZ and YL wrote the
original draft. YJ reviewed and edited the manuscript. ZC
supervised the study. Data authentication is not applicable. All
authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
de Visser KE and Joyce JA: The evolving
tumor microenvironment: From cancer initiation to metastatic.
Cancer Cell. 41:374–403. 2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Roma-Rodrigues C, Mendes R, Baptista PV
and Fernandes AR: Targeting tumor microenvironment for cancer
therapy. Int J Mol Sci. 20(840)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Chen Y, Zhang X, Yang H, Liang T and Bai
X: The ‘Self-eating’ of cancer-associated fibroblast: A potential
target for cancer. Biomed Pharmacother. 163(114762)2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Saw PE, Chen J and Song E: Targeting CAFs
to overcome anticancer therapeutic resistance. Trends Cancer.
8:527–555. 2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Prasanna PG, Citrin DE, Hildesheim J,
Ahmed MM, Venkatachalam S, Riscuta G, Xi D, Zheng G, Deursen JV,
Goronzy J, et al: Therapy-Induced senescence: Opportunities to
improve anticancer therapy. J Natl Cancer Inst. 113:1285–1298.
2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ewald JA, Desotelle JA, Wilding G and
Jarrard DF: Therapy-induced senescence in cancer. J Natl Cancer
Inst. 102:1536–1546. 2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kumari R and Jat P: Mechanisms of cellular
senescence: Cell cycle arrest and senescence associated Secretory
Phenotype. Front Cell Dev Biol. 9(645593)2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hwang HJ, Lee YR, Kang D, Lee HC, Seo HR,
Ryu JK, Kim YN, Ko YG, Park HJ and Lee JS: Endothelial cells under
therapy-induced senescence secrete CXCL11, which increases
aggressiveness of breast cancer cells. Cancer Lett. 490:100–110.
2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chen X and Song E: Turning foes to
friends: Targeting cancer-associated fibroblasts. Nat Rev Drug
Discov. 18:99–115. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Öhlund D, Handly-Santana A, Biffi G,
Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA,
Lee EJ, et al: Distinct populations of inflammatory fibroblasts and
myofibroblasts in pancreatic cancer. J Exp Med. 214:579–596.
2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kalluri R: The biology and function of
fibroblasts in cancer. Nat Rev Cancer. 16:582–598. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kalluri R and Zeisberg M: Fibroblasts in
cancer. Nat Rev Cancer. 6:392–401. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Filliol A, Saito Y, Nair A, Dapito DH, Yu
LX, Ravichandra A, Bhattacharjee S, Affo S, Fujiwara N, Su H, et
al: Opposing roles of hepatic stellate cell subpopulations in
hepatocarcinogenesis. Nature. 610:356–365. 2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Quante M, Tu SP, Tomita H, Gonda T, Wang
SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, et al: Bone
marrow-derived myofibroblasts contribute to the mesenchymal stem
cell niche and promote tumor growth. Cancer Cell. 19:257–272.
2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Karnoub AE, Dash AB, Vo AP, Sullivan A,
Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R and Weinberg
RA: Mesenchymal stem cells within tumour stroma promote breast
cancer metastasis. Nature. 449:557–563. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rhim AD, Mirek ET, Aiello NM, Maitra A,
Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK,
Vonderheide RH, et al: EMT and dissemination precede pancreatic
tumor formation. Cell. 48:349–361. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Fischer KR, Durrans A, Lee S, Sheng J, Li
F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al:
Epithelial-to-mesenchymal transition is not required for lung
metastasis but contributes to chemoresistance. Nature. 527:472–476.
2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Park D, Sahai E and Rullan A: SnapShot:
Cancer-Associated fibroblasts. Cell. 181:486–486.e1.
2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Glabman RA, Choyke PL and Sato N:
Cancer-Associated fibroblasts: Tumorigenicity and targeting for
cancer therapy. Cancers (Basel). 14(3906)2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Fiori ME, Di Franco S, Villanova L, Bianca
P, Stassi G and De Maria R: Cancer-associated fibroblasts as
abettors of tumor progression at the crossroads of EMT and therapy
resistance. Mol Cancer. 18(70)2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhao Y, Shen M, Wu L, Yang H, Yao Y, Yang
Q, Du J, Liu L, Li Y and Bai Y: Stromal cells in the tumor
microenvironment: Accomplices of tumor progression? Cell Death Dis.
14(587)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Biffi G and Tuveson DA: Diversity and
biology of cancer-associated fibroblasts. Physiol Rev. 101:147–176.
2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Fiaschi T, Marini A, Giannoni E, Taddei
ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P and
Chiarugi P: Reciprocal metabolic reprogramming through lactate
shuttle coordinately influences tumor-stroma interplay. Cancer Res.
72:5130–5140. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Singh S, Singh AP and Mitra R:
Cancer-Associated Fibroblasts: Major co-conspirators in tumor
development. Cancers (Basel). 16(211)2024.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kuzet SE and Gaggioli C: Fibroblast
activation in cancer: When seed fertilizes soil. Cell Tissue Res.
365:607–619. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Caja L, Dituri F, Mancarella S,
Caballero-Diaz D, Moustakas A, Giannelli G and Fabregat I: TGF-β
and the tissue microenvironment: Relevance in fibrosis and cancer.
Int J Mol Sci. 19(1294)2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Derynck R, Turley SJ and Akhurst RJ: TGFβ
biology in cancer progression and immunotherapy. Nat Rev Clin
Oncol. 18:9–34. 2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Chandra Jena B, Sarkar S, Rout L and
Mandal M: The transformation of cancer-associated fibroblasts:
Current perspectives on the role of TGF-β in CAF mediated tumor
progression and therapeutic resistance. Cancer Lett. 520:222–232.
2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Neri S, Miyashita T, Hashimoto H, Suda Y,
Ishibashi M, Kii H, Watanabe H, Kuwata T, Tsuboi M, Goto K, et al:
Fibroblast-led cancer cell invasion is activated by
epithelial-mesenchymal transition through platelet-derived growth
factor BB secretion of lung adenocarcinoma. Cancer Lett. 395:20–30.
2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Bronzert DA, Pantazis P, Antoniades HN,
Kasid A, Davidson N, Dickson RB and Lippman ME: Synthesis and
secretion of platelet-derived growth factor by human breast cancer
cell lines. Proc Natl Acad Sci USA. 84:5763–5767. 1987.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Shao ZM, Nguyen M and Barsky SH: Human
breast carcinoma desmoplasia is PDGF initiated. Oncogene.
19:4337–4345. 2000.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Montori M, Scorzoni C, Argenziano ME,
Balducci D, De Blasio F, Martini F, Buono T, Benedetti A, Marzioni
M and Maroni L: Cancer-Associated fibroblasts in
cholangiocarcinoma: Current knowledge and possible implications for
therapy. J Clin Med. 11(6498)2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Liu C, Zhang Y, Lim S, Hosaka K, Yang Y,
Pavlova T, Alkasalias T, Hartman J, Jensen L, Xing X, et al: A
zebrafish model discovers a novel mechanism of stromal
fibroblast-mediated. Clin Cancer Res. 23:4769–4779. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Nissen LJ, Cao R, Hedlund EM, Wang Z, Zhao
X, Wetterskog D, Funa K, Bråkenhielm E and Cao Y: Angiogenic
factors FGF2 and PDGF-BB synergistically promote murine tumor
neovascularization and metastasis. J Clin Invest. 117:2766–2777.
2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Nurmik M, Ullmann P, Rodriguez F, Haan S
and Letellier E: In search of definitions: Cancer-associated
fibroblasts and their markers. Int J Cancer. 146:895–905.
2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Yamamoto Y, Kasashima H, Fukui Y, Tsujio
G, Yashiro M and Maeda K: The heterogeneity of cancer-associated
fibroblast subpopulations: Their origins, biomarkers, and roles in
the tumor microenvironment. Cancer Sci. 114:16–24. 2023.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Arnold JN, Magiera L, Kraman M and Fearon
DT: Tumoral immune suppression by macrophages expressing fibroblast
activation protein-α and heme oxygenase-1. Cancer Immunol Res.
2:121–126. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Tchou J, Zhang PJ, Bi Y, Satija C,
Marjumdar R, Stephen TL, Lo A, Chen H, Mies C, June CH, et al:
Fibroblast activation protein expression by stromal cells and
tumor-associated macrophages in human breast cancer. Hum Pathol.
44:2549–2557. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Jin J, Zhu SJ, Zhu ZM, Yang YJ and Ding G:
Relationship between proliferation of vascular smooth muscle cells
and PDGF-AA and PDGFR-alpha expression in SHRs. Sheng Li Xue Bao.
54:145–148. 2002.PubMed/NCBI(In Chinese).
|
|
40
|
Smyth LCD, Rustenhoven J, Scotter EL,
Schweder P, Faull RLM, Park TIH and Dragunow M: Markers for human
brain pericytes and smooth muscle cells. J Chem Neuroanat.
92:48–60. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Shamsi F, Piper M, Ho LL, Huang TL, Gupta
A, Streets A, Lynes MD and Tseng YH: Vascular smooth muscle-derived
Trpv1(+) progenitors are a source of cold-induced thermogenic
adipocytes. Nat Metab. 3:485–495. 2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Sá da Bandeira D, Casamitjana J and Crisan
M: Pericytes, integral components of adult hematopoietic stem cell
niches. Pharmacol Ther. 171:104–113. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Koliaraki V, Pallangyo CK, Greten FR and
Kollias G: Mesenchymal cells in colon cancer. Gastroenterology.
152:964–979. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Togarrati PP, Dinglasan N, Desai S, Ryan
WR and Muench MO: CD29 is highly expressed on epithelial,
myoepithelial, and mesenchymal stromal cells of human salivary
glands. Oral Dis. 24:561–572. 2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Delangre E, Oppliger E, Berkcan S,
Gjorgjieva M, Correia de Sousa M and Foti M: S100 proteins in fatty
liver disease and hepatocellular carcinoma. Int J Mol Sci.
23(11030)2022.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Martin M, Zhang J, Miao Y, He M, Kang J,
Huang HY, Chou CH, Huang TS, Hong HC, Su SH, et al: Role of
endothelial cells in pulmonary fibrosis via SREBP2 activation. JCI
Insight. 6(e125635)2021.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kamphuis W, Kooijman L, Orre M, Stassen O,
Pekny M and Hol EM: GFAP and vimentin deficiency alters gene
expression in astrocytes and microglia in wild-type mice and
changes the transcriptional response of reactive glia in mouse
model for Alzheimer's disease. Glia. 63:1036–1056. 2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Suzuki-Inoue K: Platelets and
cancer-associated thrombosis: Focusing on the platelet activation
receptor CLEC-2 and podoplanin. Blood. 134:1912–1918.
2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Krishnan H, Rayes J, Miyashita T, Ishii G,
Retzbach EP, Sheehan SA, Takemoto A, Chang YW, Yoneda K, Asai J, et
al: Podoplanin: An emerging cancer biomarker and therapeutic
target. Cancer Sci. 109:1292–1299. 2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Lowy CM and Oskarsson T: Tenascin C in
metastasis: A view from the invasive front. Cell Adh Migr.
9:112–124. 2015.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Yoshida T, Akatsuka T and Imanaka-Yoshida
K: Tenascin-C and integrins in cancer. Cell Adh Migr. 9:96–104.
2015.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yue H, Li W, Chen R, Wang J, Lu X and Li
J: Stromal POSTN induced by TGF-β1 facilitates the migration and
invasion of ovarian cancer. Gynecol Oncol. 160:530–538.
2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Liu C, Feng X, Wang B, Wang X, Wang C, Yu
M, Cao G and Wang H: Bone marrow mesenchymal stem cells promote
head and neck cancer progression through Periostin-mediated
phosphoinositide 3-kinase/Akt/mammalian target of rapamycin. Cancer
Sci. 109:688–698. 2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Soikkeli J, Podlasz P, Yin M, Nummela P,
Jahkola T, Virolainen S, Krogerus L, Heikkilä P, von Smitten K,
Saksela O and Hölttä E: Metastatic outgrowth encompasses COL-I,
FN1, and POSTN up-regulation and assembly to fibrillar networks
regulating cell adhesion, migration, and growth. Am J Pathol.
177:387–403. 2010.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Walcher L, Kistenmacher AK, Suo H, Kitte
R, Dluczek S, Strauß A, Blaudszun AR, Yevsa T, Fricke S and
Kossatz-Boehlert U: Cancer stem cells-origins and biomarkers:
Perspectives for targeted personalized therapies. Front Immunol.
11(1280)2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Bonnans C, Chou J and Werb Z: Remodelling
the extracellular matrix in development and disease. Nat Rev Mol
Cell Biol. 15:786–801. 2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Winkler J, Abisoye-Ogunniyan A, Metcalf KJ
and Werb Z: Concepts of extracellular matrix remodelling in tumour
progression and metastasis. Nat Commun. 11(5120)2020.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Asif PJ, Longobardi C, Hahne M and Medema
JP: The role of cancer-associated fibroblasts in cancer invasion
and metastasis. Cancers (Basel). 13(4720)2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Eiró N, Fernandez-Garcia B, Vázquez J, Del
Casar JM, González LO and Vizoso FJ: A phenotype from tumor stroma
based on the expression of metalloproteases and their inhibitors,
associated with prognosis in breast cancer. Oncoimmunology.
4(e992222)2015.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Neri S, Ishii G, Hashimoto H, Kuwata T,
Nagai K, Date H and Ochiai A: Podoplanin-expressing
cancer-associated fibroblasts lead and enhance the local invasion
of cancer cells in lung adenocarcinoma. Int J Cancer. 137:784–796.
2015.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Gaggioli C, Hooper S, Hidalgo-Carcedo C,
Grosse R, Marshall JF, Harrington K and Sahai E: Fibroblast-led
collective invasion of carcinoma cells with differing roles for
RhoGTPases in leading and following cells. Nat Cell Biol.
9:1392–1400. 2007.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Pastushenko I, Mauri F, Song Y, de Cock F,
Meeusen B, Swedlund B, Impens F, Van Haver D, Opitz M, Thery M, et
al: Fat1 deletion promotes hybrid EMT state, tumour stemness and
metastasis. Nature. 589:448–455. 2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesenchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Lüönd F, Sugiyama N, Bill R, Bornes L,
Hager C, Tang F, Santacroce N, Beisel C, Ivanek R, Bürglin T, et
al: Distinct contributions of partial and full EMT to breast cancer
malignancy. Dev Cell. 56:3203–3221.e11. 2021.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Fares J, Fares MY, Khachfe HH, Salhab HA
and Fares Y: Molecular principles of metastasis: A hallmark of
cancer revisited. Signal Transduct Target Ther.
5(28)2020.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Ren Y, Jia HH, Xu YQ, Zhou X, Zhao XH,
Wang YF, Song X, Zhu ZY, Sun T, Dou Y, et al: Paracrine and
epigenetic control of CAF-induced metastasis: The role of HOTAIR
stimulated by TGF-ß1 secretion. Mol Cancer. 17(5)2018.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Nicolas AM, Pesic M, Engel E, Ziegler PK,
Diefenhardt M, Kennel KB, Buettner F, Conche C, Petrocelli V,
Elwakeel E, et al: Inflammatory fibroblasts mediate resistance to
neoadjuvant therapy in rectal cancer. Cancer Cell. 40:168–184.
2022.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Jia C, Wang G, Wang T, Fu B, Zhang Y,
Huang L, Deng Y, Chen G, Wu X, Chen J, et al: Cancer-associated
Fibroblasts induce epithelial-mesenchymal transition via the
transglutaminase 2-dependent IL-6/IL6R/STAT3 axis in hepatocellular
carcinoma. Int J Biol Sci. 16:2542–2558. 2020.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Wang Y, Lan W, Xu M, Song J, Mao J, Li C,
Du X, Jiang Y, Li E, Zhang R and Wang Q: Cancer-associated
fibroblast-derived SDF-1 induces epithelial-mesenchymal transition
of lung adenocarcinoma via CXCR4/β-catenin/PPARδ signalling. Cell
Death Dis. 12(214)2021.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Dai J, Su Y, Zhong S, Cong L, Liu B, Yang
J, Tao Y, He Z, Chen C and Jiang Y: Exosomes: Key players in cancer
and potential therapeutic strategy. Signal Transduct Target Ther.
5(145)2020.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Richards KE, Zeleniak AE, Fishel ML, Wu J,
Littlepage LE and Hill R: Cancer-associated fibroblast exosomes
regulate survival and proliferation of pancreatic cancer cells.
Oncogene. 36:1770–1778. 2017.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Zhang H, Deng T, Liu R, Ning T, Yang H,
Liu D, Zhang Q, Lin D, Ge S, Bai M, et al: CAF secreted miR-522
suppresses ferroptosis and promotes acquired chemo-resistance in
gastric cancer. Mol Cancer. 19(43)2020.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Martínez-Zamudio RI, Robinson L, Roux PF
and Bischof O: SnapShot: Cellular senescence pathways. Cell.
170:816–816.e11. 2017.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Wang L, Lankhorst L and Bernards R:
Exploiting senescence for the treatment of cancer. Nat Rev Cancer.
22:340–355. 2022.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Bahcecioglu G, Yue X, Howe E, Guldner I,
Stack MS, Nakshatri H, Zhang S and Zorlutuna P: Aged breast
extracellular matrix drives mammary epithelial cells to an
invasive. Adv Sci (Weinh). 8(e2100128)2021.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Bancaro N, Calì B, Troiani M, Elia AR,
Arzola RA, Attanasio G, Lai P, Crespo M, Gurel B, Pereira R, et al:
Apolipoprotein E induces pathogenic senescent-like myeloid cells in
prostate. Cancer Cell. 41:602–619.e11. 2023.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Nguyen PT, Kanno K, Pham QT, Kikuchi Y,
Kakimoto M, Kobayashi T, Otani Y, Kishikawa N, Miyauchi M, Arihiro
K, et al: Senescent hepatic stellate cells caused by deoxycholic
acid modulates malignant. J Cancer Res Clin Oncol. 146:3255–3268.
2020.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Yoshimoto S, Loo TM, Atarashi K, Kanda H,
Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et
al: Obesity-induced gut microbial metabolite promotes liver cancer
through senescence secretome. Nature. 499:97–101. 2013.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Loo TM, Kamachi F, Watanabe Y, Yoshimoto
S, Kanda H, Arai Y, Nakajima-Takagi Y, Iwama A, Koga T, Sugimoto Y,
et al: Gut Microbiota Promotes Obesity-Associated Liver Cancer
through PGE(2)-Mediated. Cancer Discov. 7:522–538. 2017.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Chambers CR, Ritchie S, Pereira BA and
Timpson P: Overcoming the senescence-associated secretory phenotype
(SASP): A complex mechanism of resistance in the treatment of
cancer. Mol Oncol. 15:3242–3255. 2021.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Ewald JA, Desotelle JA, Wilding G and
Jarrard DF: Therapy-induced senescence in cancer. J Natl Cancer
Inst. 102:1536–1546. 2010.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Groelly FJ, Fawkes M, Dagg RA, Blackford
AN and Tarsounas M: Targeting DNA damage response pathways in
cancer. Nat Rev Cancer. 23:78–94. 2023.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Shiloh Y: ATM and related protein kinases:
Safeguarding genome integrity. Nat Rev Cancer. 3:155–168.
2003.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Buzun K, Bielawska A, Bielawski K and
Gornowicz A: DNA topoisomerases as molecular targets for anticancer
drugs. J Enzyme Inhib Med Chem. 35:1781–1799. 2020.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Aasland D, Götzinger L, Hauck L, Berte N,
Meyer J, Effenberger M, Schneider S, Reuber EE, Roos WP, Tomicic
MT, et al: Temozolomide induces senescence and repression of DNA
repair pathways in glioblastoma cells via activation of ATR-CHK1,
p21, and NF-κB. Cancer Res. 79:99–113. 2019.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Mikuła-Pietrasik J, Witucka A, Pakuła M,
Uruski P, Begier-Krasińska B, Niklas A, Tykarski A and Książek K:
Comprehensive review on how platinum- and taxane-based chemotherapy
of ovarian cancer affects biology of normal cells. Cell Mol Life
Sci. 76:681–697. 2019.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Burdelya LG, Komarova EA, Hill JE, Browder
T, Tararova ND, Mavrakis L, DiCorleto PE, Folkman J and Gudkov AV:
Inhibition of p53 response in tumor stroma improves efficacy of
anticancer treatment by increasing antiangiogenic effects of
chemotherapy and radiotherapy in mice. Cancer Res. 66:9356–9361.
2006.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Pardella E, Pranzini E, Nesi I, Parri M,
Spatafora P, Torre E, Muccilli A, Castiglione F, Fambrini M, Sorbi
F, et al: Therapy-Induced stromal senescence promoting
aggressiveness of prostate and ovarian cancer. Cells.
11(4026)2022.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Li M, You L, Xue J and Lu Y: Ionizing
radiation-induced cellular senescence in normal, non-transformed
cells and the involved DNA damage response: A mini review. Front
Pharmacol. 9(522)2018.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Meng J, Li Y, Wan C, Sun Y, Dai X, Huang
J, Hu Y, Gao Y, Wu B, Zhang Z, et al: Targeting senescence-like
fibroblasts radiosensitizes non-small cell lung cancer and reduces
radiation-induced pulmonary fibrosis. JCI Insight.
6(e146334)2021.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Wagner V and Gil J: Senescence as a
therapeutically relevant response to CDK4/6 inhibitors. Oncogene.
39:5165–5176. 2020.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Coppé JP, Rodier F, Patil CK, Freund A,
Desprez PY and Campisi J: Tumor suppressor and aging biomarker
p16(INK4a) induces cellular senescence without the associated
inflammatory secretory phenotype. J Biol Chem. 286:36396–36403.
2011.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Capparelli C, Chiavarina B,
Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, Andò S, Howell
A, Martinez-Outschoorn UE, Sotgia F and Lisanti MP: CDK inhibitors
(p16/p19/p21) induce senescence and autophagy in cancer-associated
fibroblasts, ‘fueling’ tumor growth via paracrine interactions,
without an increase in neo-angiogenesis. Cell Cycle. 11:3599–3610.
2012.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Guan X, LaPak KM, Hennessey RC, Yu CY,
Shakya R, Zhang J and Burd CE: Stromal senescence by prolonged
CDK4/6 inhibition potentiates tumor growth. Mol Cancer Res.
15:237–249. 2017.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Campisi J, Kapahi P, Lithgow GJ, Melov S,
Newman JC and Verdin E: From discoveries in ageing research to
therapeutics for healthy ageing. Nature. 571:183–192.
2019.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Chaib S, Tchkonia T and Kirkland JL:
Cellular senescence and senolytics: The path to the clinic. Nat
Med. 28:1556–1568. 2022.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Yosef R, Pilpel N, Tokarsky-Amiel R, Biran
A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, et
al: Directed elimination of senescent cells by inhibition of BCL-W
and BCL-XL. Nat Commun. 7(11190)2016.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Jochems F, Thijssen B, De Conti G, Jansen
R, Pogacar Z, Groot K, Wang L, Schepers A, Wang C, Jin H, et al:
The Cancer SENESCopedia: A delineation of cancer cell senescence.
Cell Rep. 36(109441)2021.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Fletcher-Sananikone E, Kanji S, Tomimatsu
N, Di Cristofaro LFM, Kollipara RK, Saha D, Floyd JR, Sung P,
Hromas R, Burns TC, et al: Elimination of radiation-induced
senescence in the brain tumor microenvironment attenuates
glioblastoma recurrence. Cancer Res. 81:5935–5947. 2021.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Li F, Huangyang P, Burrows M, Guo K,
Riscal R, Godfrey J, Lee KE, Lin N, Lee P, Blair IA, et al: FBP1
loss disrupts liver metabolism and promotes tumorigenesis through a
hepatic stellate cell senescence secretome. Nat Cell Biol.
22:728–739. 2020.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Baar MP, Brandt RMC, Putavet DA, Klein
JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van
Willigenburg H, Feijtel DA, et al: Targeted apoptosis of senescent
cells restores tissue homeostasis in response to chemotoxicity and
aging. Cell. 169:132–147.e16. 2017.PubMed/NCBI View Article : Google Scholar
|