Introduction
Atherosclerosis (AS) involves the coronary and
peripheral arteries and can have consequences such as ischemia of
the lower limbs and myocardium, or chronic heart failure (1). Atherothrombosis represents the
terminal manifestation of this pathology. Plaque rupture or erosion
and the intimal thickening of blood vessels cause occlusion, which
may cause patients to experience claudication, myocardial
infarction, stroke and other symptoms (2). Lower extremity atherosclerotic
occlusive disease (ASO) is the most common type of peripheral
vascular obliterans. ASO has an incidence rate of 12-14% in the
global population, and ~200 million individuals worldwide are known
to have ASO, which is life-threatening in certain cases (3). The first-line treatments for
occlusion are percutaneous transluminal angioplasty and stent
placement (4,5); however, in-stent restenosis or
intimal hyperplasia after interventional therapy remain a
challenge. Therefore, understanding the pathogenesis of occlusion
is the focus of current research.
Previous studies have revealed that vascular smooth
muscle cell (VSMC) phenotype switching is a pathological process in
various vascular diseases, including AS, postoperative restenosis
and aneurysms (6-8).
Phenotypic regulation is a prerequisite for VSMC proliferation and
migration from the capsular media to the arterial wall intima. The
major manifestations of this process include the transformation of
cells from a contractile (differentiated) to a synthetic
(proliferative) phenotype (9).
Contractile VSMCs are spindle-shaped and function as arterial
elastic constrictors. Smooth muscle cell (SMC)-specific
differentiation proteins, such as α-smooth muscle actin, smooth
muscle 22α and calmodulin, are the major components that contribute
to the contractility of the cells (10). By contrast, the synthetic phenotype
is characterized by the reduced expression of SMC-specific markers,
increased osteopontin (OPN) expression and transformation to a
rhomboid cell morphology, accompanied by increased proliferation
and migration capabilities, and the secretion of extracellular
matrix components (11).
Additionally, under the action of risk factors, such as high
pressure, lipid deposition, inflammation and infection, vascular
endothelial structure and function are impaired, which leads to the
synthesis and release of vasoactive substances and cytokines
(12), particularly
platelet-derived growth factor (PDGF). PDGF type BB (PDGF-BB) is
one of the first factors discovered to promote the phenotypic
transformation of VSMCs (13).
PDGF-BB acts on VSMC membrane receptors and activates various
intracellular signal transduction pathways, including the
Ras/Raf/MEK/ERK, PI3K/Akt, NF-κB and JAK2/STAT3 pathways, thereby
producing a dedifferentiation effect (10). In addition, basic fibroblast growth
factor and epidermal growth factor are also able to induce VSMC
phenotypic transformation (14).
Exploring the mechanisms responsible for the abnormal
proliferation, migration and phenotypic transformation of VSMCs may
provide an effective strategy for the treatment of arterial
occlusion.
Canopy FGF signaling regulator 2 (CNPY2) is a
cytoplasmic secreted protein that is widely expressed in various
tissues and organs, and is a member of the canopy family, which
also includes CNPY1, 3 and 4. Recently, it has been demonstrated
that CNPY2 is an angiogenic growth factor (15). CNPY2 has also been found to be
abnormally highly expressed in ApoE-/- mice and oxidized
low-density lipoprotein-stimulated mouse aortic endothelial cells.
In the former, it was found to aggravate the atherosclerotic
process via the induction of vascular endothelial cell damage
through the activation of protein kinase R-like endoplasmic
reticulum kinase signaling (16).
In addition, the hypoxia response element upstream of the CNPY2
promoter can bind to hypoxia-inducible factor-1α and thereby
promote the migration and proliferation of SMCs and tissue
angiogenesis when induced by hypoxia (17). Considering that arterial occlusion
can lead to chronic hypoxia in the limbs or organs, it is
reasonable to hypothesize that CNPY2 is involved in the
proliferation, migration and phenotypic transformation of
VSMCs.
When platelets come into contact with the injured
vessel wall, PDGF is released which drives intimal layer
proliferation and leads to occlusion (18). The present study aimed to examine
the potential changes in the expression of CNPY2 in response to
PDGF, and to determine the effects of CNPY2 on the proliferation,
migration and phenotypic transformation of VSMCs, as well as the
underlying mechanisms.
Materials and methods
Cells, cell culture and
treatments
Human VSMCs (T/G HA-VSMC cell line; passage 2) were
purchased from Shanghai Jinyuan Biotechnology Co., Ltd. (cat. no.
JY524) and cultured in Dulbecco's modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc.) containing 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 100 U/ml
penicillin and 0.1 mg/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) at 37˚C with 5% CO2. VSMCs were
passaged at 1:2, and passages 3-5 were used for the experiments. A
concentration of 20 ng/ml PDGF-BB (MilliporeSigma) was used to
stimulate the VSMCs at 37˚C for 24 h. In addition, in certain
experiments the cells were pre-treated with the Akt activator SC79
(MilliporeSigma) at 10 µM for 2 h (19).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the cells using VeZol
reagent (cat. no. R411; Vazyme Biotech Co., Ltd.). The HiScript II
1st Strand cDNA synthesis kit (cat. no. R211; Vazyme Biotech Co.,
Ltd.) was used to reverse transcribe the isolated RNA into cDNA
according to the manufacturer's protocols. qPCR was then performed
using synthetic primers and AceQ® qPCR SYBR Green Master
Mix (cat. no. Q111-02; Vazyme Biotech Co., Ltd.) with a Stepone™
Software v2.3 Detection System (Thermo Fisher Scientific, Inc.).
The conditions were 95˚C for 30 sec, and 40 cycles of 95˚C for 5
sec, 55˚C for 10 sec and 72˚C for 15 sec. The mRNA levels of CNPY2
were normalized to those of GAPDH and calculated using the
2-ΔΔCq method (20).
The primer sequences (5'-3') were as follows: CNPY2, forward,
TAGTGGGCCGGAATGGAGAA and reverse, AAACTTGAGGGTGCCGCTAA; GAPDH,
forward, GACTCATGACCACAGTCCATGC and reverse,
AGAGGCAGGGATGATGTTCTG.
Western blot analysis
Proteins were extracted from the cells
(5x106) using RIPA lysis buffer (Beyotime Institute of
Biotechnology). The protein concentration of the extract was
determined using a BCA kit (Beyotime Institute of Biotechnology). A
total of 30 µl protein/lane was then separated using 10% SDS-PAGE
and transferred to PVDF membranes (SEQ00010; MilliporeSigma). After
blocking the membranes with bovine serum albumin (BSA; 5%, v/v;
Elabscience Co., Ltd.) at room temperature for 2 h, the membranes
were incubated overnight at 4˚C with primary antibodies against
CNPY2 (PA5-100135; 1:1,000; Invitrogen; Thermo Fisher Scientific,
Inc.), OPN (22952-1-AP; 1:2,000; Proteintech), smooth muscle actin
(SMA; 14395-1-AP; 1:3,000; Proteintech), F-actin (MA1-80729; 1:500;
Invitrogen; Thermo Fisher Scientific, Inc.), phosphorylated (p-)Akt
(66444-1-Ig; 1:20,000; Proteintech), Akt (60203-2-Ig; 1:20,000;
Proteintech), p-mTOR (67778-1-Ig; 1:5,000; Proteintech), mTOR
(28273-1-AP; 1:5,000; Proteintech), p-GSK-3β (67558-1-Ig; 1:5,000;
Proteintech), GSK-3β (22104-1-AP; 1:4,000; Proteintech) and GAPDH
(PA1-16777; 1:5,000; Invitrogen; Thermo Fisher Scientific, Inc.).
The membranes were washed with TBST (0.05% Tween 20), and then
incubated for 120 min at room temperature with secondary antibodies
(cat. nos. 31460 and 31430; 1:100,000; Invitrogen; Thermo Fisher
Scientific, Inc.). Enhanced chemiluminescence Western Blotting
Substrate (32109; Pierce; Thermo Fisher Scientific, Inc.) was used
to visualize the bands, and the density of the protein bands was
semi-quantified using ImageJ software version 1.8.0 (National
Institutes of Health).
Cell transfection
To inhibit CNPY2, the two human CNPY2-targeting
pRNAT-U6.1/Neo short hairpin RNAs (shRNAs; 1 µg; sh-CNPY2#1 and
sh-CNPY2#2; Guangzhou Ribobio Co., Ltd.) were respectively
transfected into cells using Lipofectamine 2000®
(Invitrogen; Thermo Fisher Scientific, Inc.). Cells transfected
with the same plasmid carrying non-targeting shRNAs served as the
negative control (sh-NC). Target sequences were as follows:
sh-CNPY2#1, CGAACAGATCTTTGTGACCAT; sh-CNPY2#2,
CTTTGCAGTAAGCGAACAGAT; and sh-NC, TTCTCCGAACGTGTCACGT. Complexes of
Lipofectamine and shRNA in serum-free DMEM were added to VSMCs at
~70% confluence. Knockdown efficacy was determined 48 h after
transfection at 37˚C (21).
Cell Counting Kit-8 (CCK-8) assay
The cells were plated in 96-well plates
(1x103/well) and then subjected to the aforementioned
treatments. Then, 100 µl CCK-8 solution (Dojindo Laboratories,
Inc.) was added to the cells for 2 h, after which the optical
density was measured at 450 nm using a microplate reader (Thermo
Fisher Scientific, Inc.).
5-Ethynyl-2'-deoxyuridine (EdU)
staining
The cells were plated in six-well plates
(1x105/well) and subjected to treatment. The cells were
then incubated at 37˚C in 20 µM EdU solution from an EdU Cell
Proliferation Image Kit (cat. no. KTA2030; Abbkine Scientific Co.,
Ltd.) for 2 h, and fixed in 4% formaldehyde for 30 min at room
temperature (22). The cells were
permeated by 0.3% Triton X-100 for 10 min and subsequently exposed
to reaction solution (Abbkine Scientific Co., Ltd.) for 30 min,
followed by DAPI staining (cat. no. E607303; Sangon Biotech Co.,
Ltd.) for 30 min at room temperature. Images were captured and
analyzed using an inverted fluorescence microscope (Axio Observer
Z1; Zeiss AG).
Flow cytometry analysis
The cells were plated in six-well plates
(1x105/well) and were then subjected to treatment. The
Cell Cycle Assay Kit (cat. no. E-CK-A351; Elabscience
Biotechnology, Inc.) was used for analysis of the cell cycle. Cells
were harvested after digestion with 0.25% trypsin (Vazyme Biotech
Co., Ltd.), and the supernatant was discarded. The cells were
washed with ice-cold PBS and collected after centrifugation at 850
x g for 5 min at 4˚C. Precooled 70% ethanol was used to immobilize
the cells at 4˚C for 2 h and then the cells were washed with PBS.
Subsequently, the cells were incubated with RNase reagent for 30
min at 37˚C and propidium iodide for 30 min at 4˚C in the dark
(23). The cell cycle status was
then assessed using flow cytometry (BD Bioscience; excitation
wavelength 488 nm) and analyzed using FlowJo version 10
software.
Transwell migration assay
The treated VSMCs (1x104/well) in
serum-free DMEM were plated in the upper chambers of 24-well
Transwell plates (pore size, 8.0 µm; Corning, Inc.) and the lower
chambers were supplemented with 10% FBS in DMEM. Following 24 h of
incubation at 37˚C, the cells on the lower surface of the Transwell
membranes were fixed with 4% formaldehyde for 20 min and stained
with 0.1% crystal violet at 25˚C for 15 min (24). The migratory cells were observed
under a light microscope (BX51W; Olympus Corporation).
Wound healing assay
A cell layer of the treated VSMCs were scratched
using a pipette tip to form a straight scratch wound. The cell
layer was incubated with serum-free DMEM for 24 h. Images of the
wound were captured using a light microscope (BX51W; Olympus
Corporation) at 0 and 24 h. Cell migration was analyzed using
ImageJ software version 1.8.0 (National Institutes of Health).
Immunofluorescence (IF) analysis
The treated cells were fixed with 4%
paraformaldehyde at room temperature for 10 min and blocked with 1%
BSA (MilliporeSigma) at room temperature for 30 min, followed by
overnight incubation with F-actin primary antibody (MA1-80729;
1:20; Invitrogen; Thermo Fisher Scientific, Inc.) at 4˚C, and then
incubation with goat anti-mouse Alexa Fluor™ 488
secondary antibody (A-11001; 1:2,000; Invitrogen; Thermo Fisher
Scientific, Inc.) at room temperature for 1 h. Cell nuclei were
then stained with DAPI for 5 min at room temperature. Six random
high-power fields were selected, and images were obtained using a
fluorescence microscope (Axio Observer Z1).
Statistical analysis
Data are presented as the mean ± SD (n≥3).
Statistically significant differences between experimental
parameters among the groups were analyzed using one-way ANOVA
followed by Tukey's test or by unpaired Student's t-test using
GraphPad Prism Software Version 6.0 (Dotmatics). P<0.05 was
considered to indicate a statistically significant difference.
Results
Knockdown of CNPY2 inhibits the
PDGF-BB-induced proliferation and cell cycle of VSMCs
After stimulation of the VSMCs with PDGF-BB, the
expression of CNPY2 was detected using RT-qPCR and western blot
analysis. The results revealed that compared with the untreated
control group, the expression of CNPY2 in the PDGF-BB-stimulated
cells was significantly increased (Fig. 1A). A CNPY2 interference plasmid was
then constructed, and the transfection efficacy was detected using
RT-qPCR and western blot analysis (Fig. 1B). Since the interference
efficiency achieved with sh-CNPY2#2 was stronger than that of
sh-CNPY2#1, sh-CNPY2#2 was selected for use in the follow-up
experiments and is henceforth referred to as sh-CNPY2. CCK-8 and
EdU assays were used to detect cell viability and proliferation,
respectively. The results revealed that the viability and
proliferative ability of cells in the PDGF-BB group were increased
compared with those in the control group. In addition, cell
viability and proliferation in the PDGF-BB + sh-CNPY2 group were
decreased compared with those in the PDGF-BB + sh-NC group
(Fig. 1C and D). Flow cytometric analysis revealed that
the number of cells in the G0/G1 phase
decreased, and that in the S phase increased following stimulation
with PDGF-BB, indicating that the cells were proliferating. In the
PDGF-BB + sh-CNPY2 group compared with the PDGF-BB + sh-NC group,
the number of cells in the G0/G1 phase
increased, while that of cells in the S phase decreased, indicating
that sh-CNPY2 blocked cell proliferation (Fig. 1E and F).
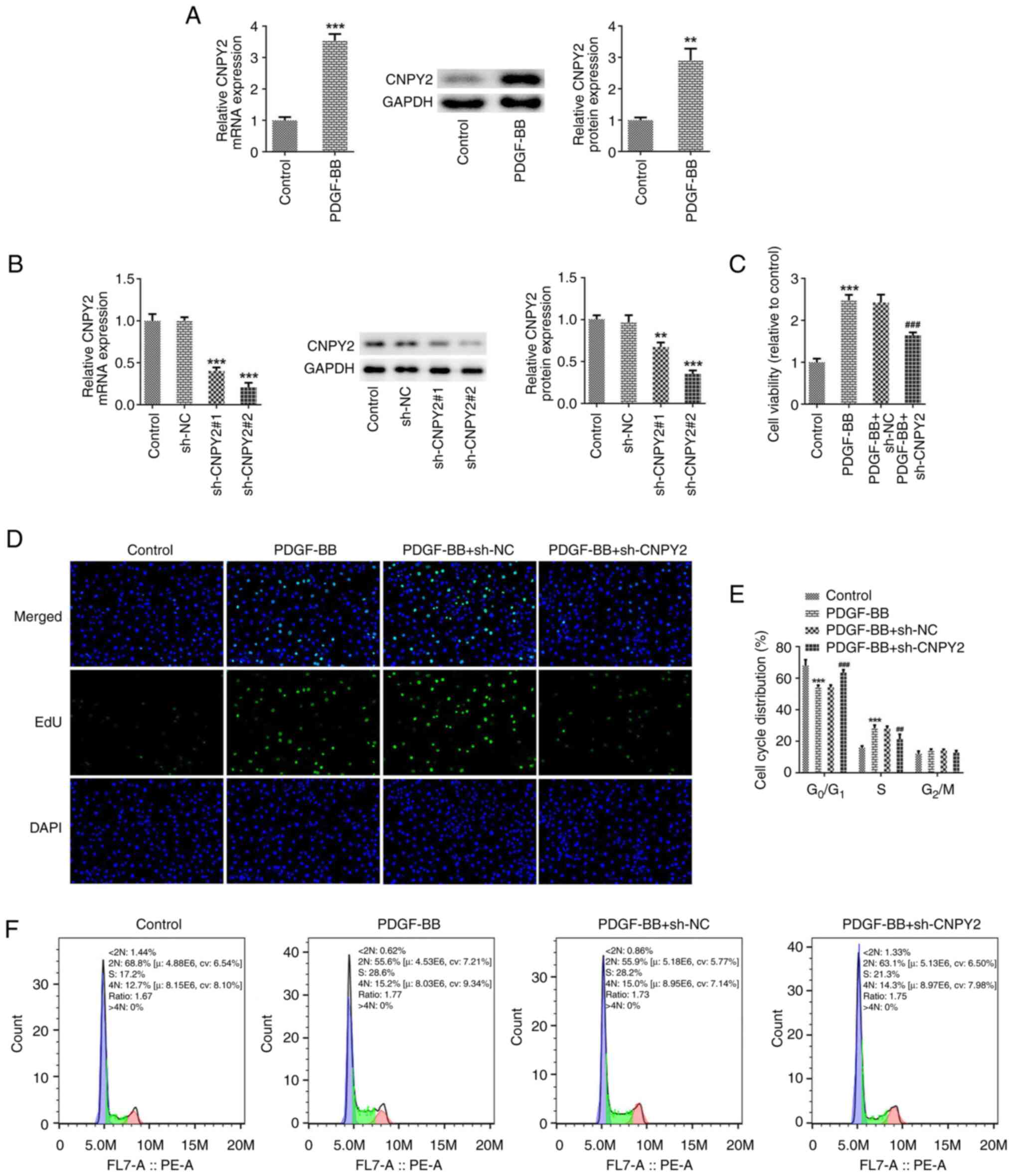 | Figure 1Knockdown of CNPY2 attenuates the
PDGF-BB-induced proliferation and cell cycling of VSMCs. (A)
Following the stimulation of VSMCs with PDGF-BB, the expression of
CNPY2 was detected using RT-qPCR and western blot analysis (n=5).
**P<0.01 and ***P<0.001 vs. control.
(B) CNPY2 interference plasmids were constructed, and the
transfection efficacy was detected using RT-qPCR and western blot
analysis (n=5). **P<0.01 and ***P<0.001
vs. sh-NC. (C) Cell Counting Kit-8 assay was used to detect cell
viability (n=5). (D) EdU staining was used to detect cell
proliferation and representative images are shown (magnification,
x200; n=3). (E) Quantification of cell cycle distribution using
flow cytometry, and (F) representative flow cytometry plots for the
cell cycle analysis (n=5). ***P<0.001 vs. control;
##P<0.01 and ###P<0.001 vs. PDGF-BB +
sh-NC. CNPY2, canopy FGF signaling regulator 2; PDGF-BB,
platelet-derived growth factor type BB; VSMCs, vascular smooth
muscle cells; RT-qPCR, reverse transcription-quantitative PCR; EdU,
5-ethynyl-2'-deoxyuridine; sh, short hairpin; NC, negative
control. |
Knockdown of CNPY2 inhibits the
PDGF-BB-induced migration and phenotypic transformation of
VSMCs
The results of wound healing and Transwell assays
revealed that the migratory ability of the VSMCs significantly
increased following stimulation with PDGF-BB, and the knockdown of
CNPY2 expression significantly attenuated the PDGF-BB-induced
migration of the cells (Fig. 2A
and B). When observed under a
microscope, it was observed that following stimulation with
PDGF-BB, the morphology of the VSMCs flattened and the cell bodies
increased in size, exhibiting a low degree of differentiation.
However, compared with those in the PDGF-BB + sh-NC group, the cell
bodies of the PDGF-BB + sh-CNPY2 group were normalized and
exhibited a high degree of differentiation (Fig. 2C). The IF staining of cytoskeletal
F-actin also showed that the cell morphology was flattened
following stimulation with PDGF-BB, and revealed that the content
of myofilaments and structural proteins decreased. However, in
comparison with the contents of myofilaments and structural
proteins in the PDGF-BB + sh-NC group, those in the PDGF-BB +
sh-CNPY2 group were increased (Fig.
2D). Western blot analysis demonstrated that in the PDGF-BB
group compared with the control group, the expression of OPN was
significantly increased, while that of SMA and F-actin was
significantly decreased. In addition, in the PDGF-BB + sh-CNPY2
group, OPN expression was significantly decreased while SMA and
F-actin expression was significantly increased compared with that
in the PDGF-BB + sh-NC group (Fig.
2E).
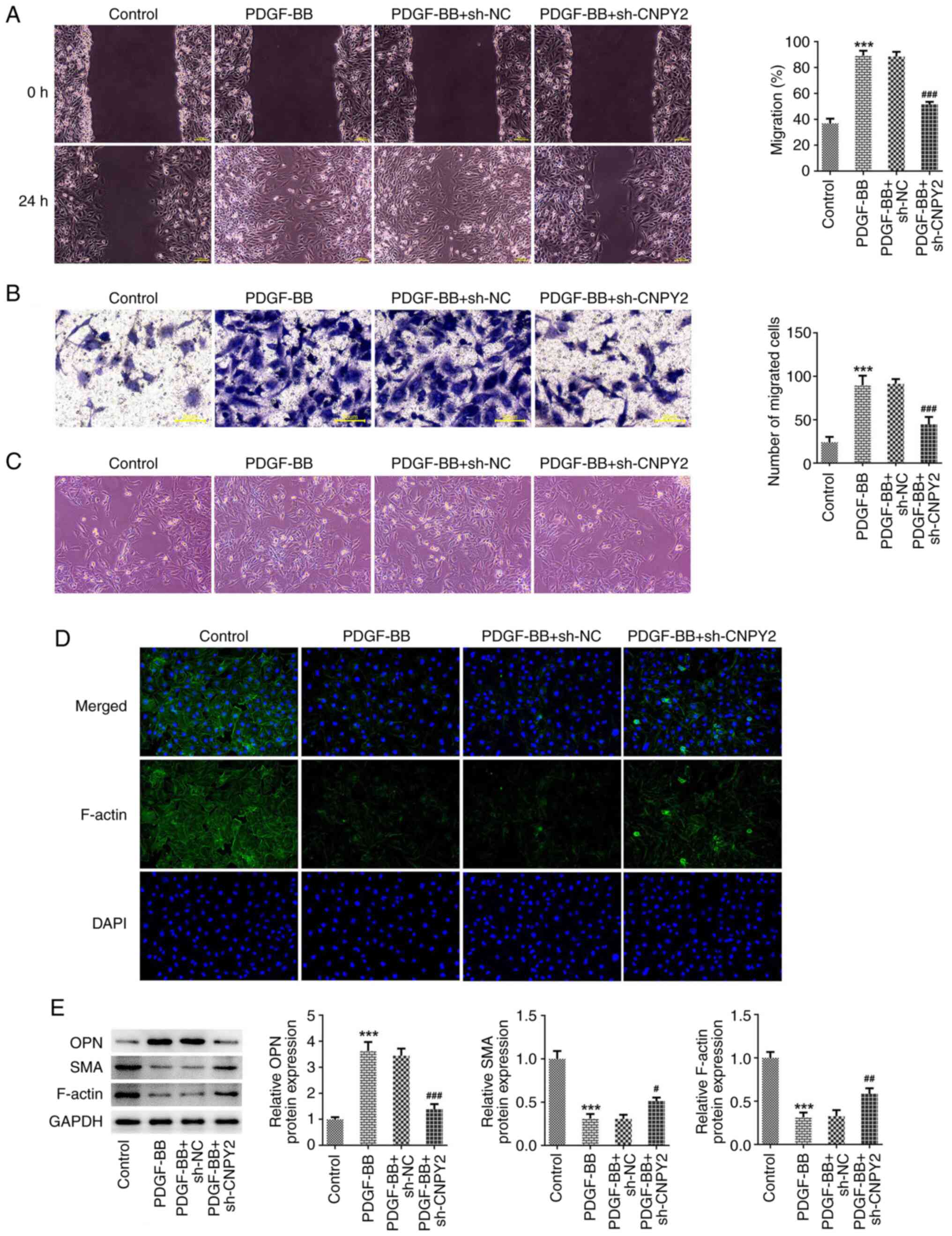 | Figure 2Knockdown of CNPY2 inhibits the
PDGF-BB-induced migration and phenotypic transformation of VSMCs.
(A) Wound healing (scale bar, 100 µm) and (B) Transwell assays
(scale bar, 50 µm) were used to detect the migration ability of the
VSMCs (n=3). (C) Images of cell morphology were captured
(magnification, x100; n=3). (D) Immunofluorescence staining was
used to detect cytoskeletal F-actin (magnification, x200; n=3). (E)
Western blot analysis of phenotypic transformation-associated
proteins (n=5). ***P<0.001 vs. control;
#P<0.05, ##P<0.01 and
###P<0.001 vs. PDGF-BB + sh-NC. CNPY2, canopy FGF
signaling regulator 2; PDGF-BB, platelet-derived growth factor type
BB; VSMCs, vascular smooth muscle cells; sh, short hairpin; NC,
negative control; OPN, osteopontin; SMA, smooth muscle actin. |
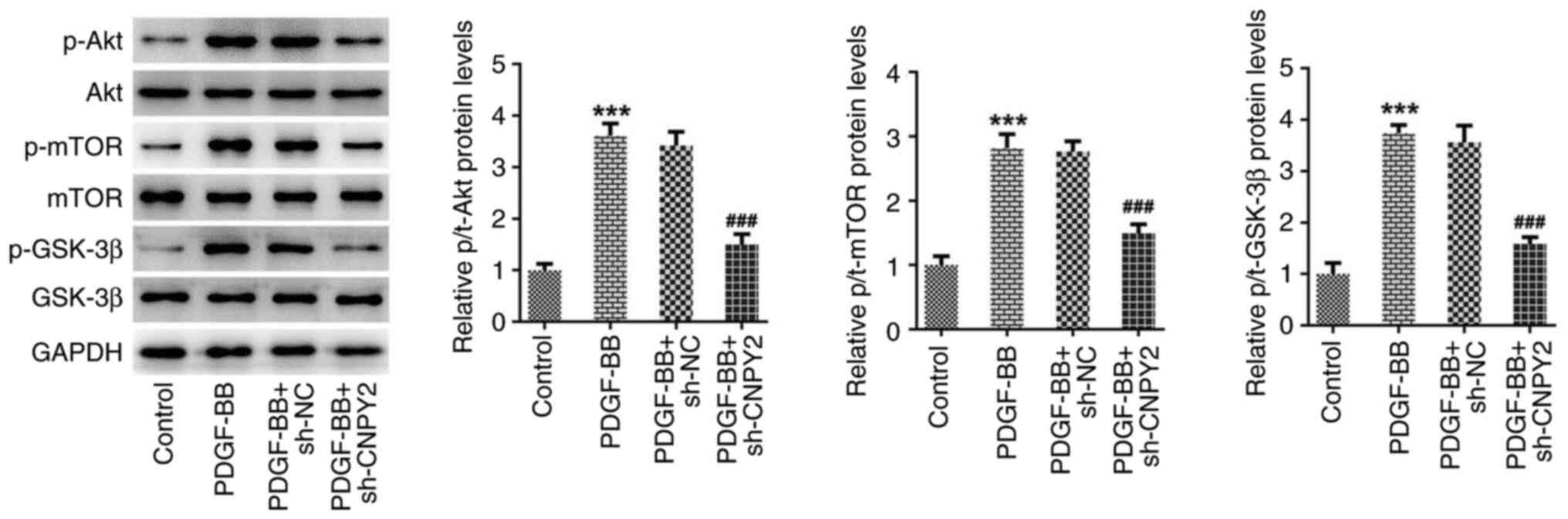
CNPY2 activates the Akt/mTOR/GSK-3β
signaling pathway
Western blot analysis of the proteins in the
Akt/mTOR/GSK-3β signaling pathway revealed that the ratios of
p-Akt, p-mTOR and p-GSK-3β protein levels to those of the
respective total proteins in the cells was significantly increased
following stimulation with PDGF-BB. However, the knockdown of CNPY2
expression significantly inhibited the PDGF-BB-induced upregulation
of p-Akt, p-mTOR and p-GSK-3β protein levels (Fig. 3).
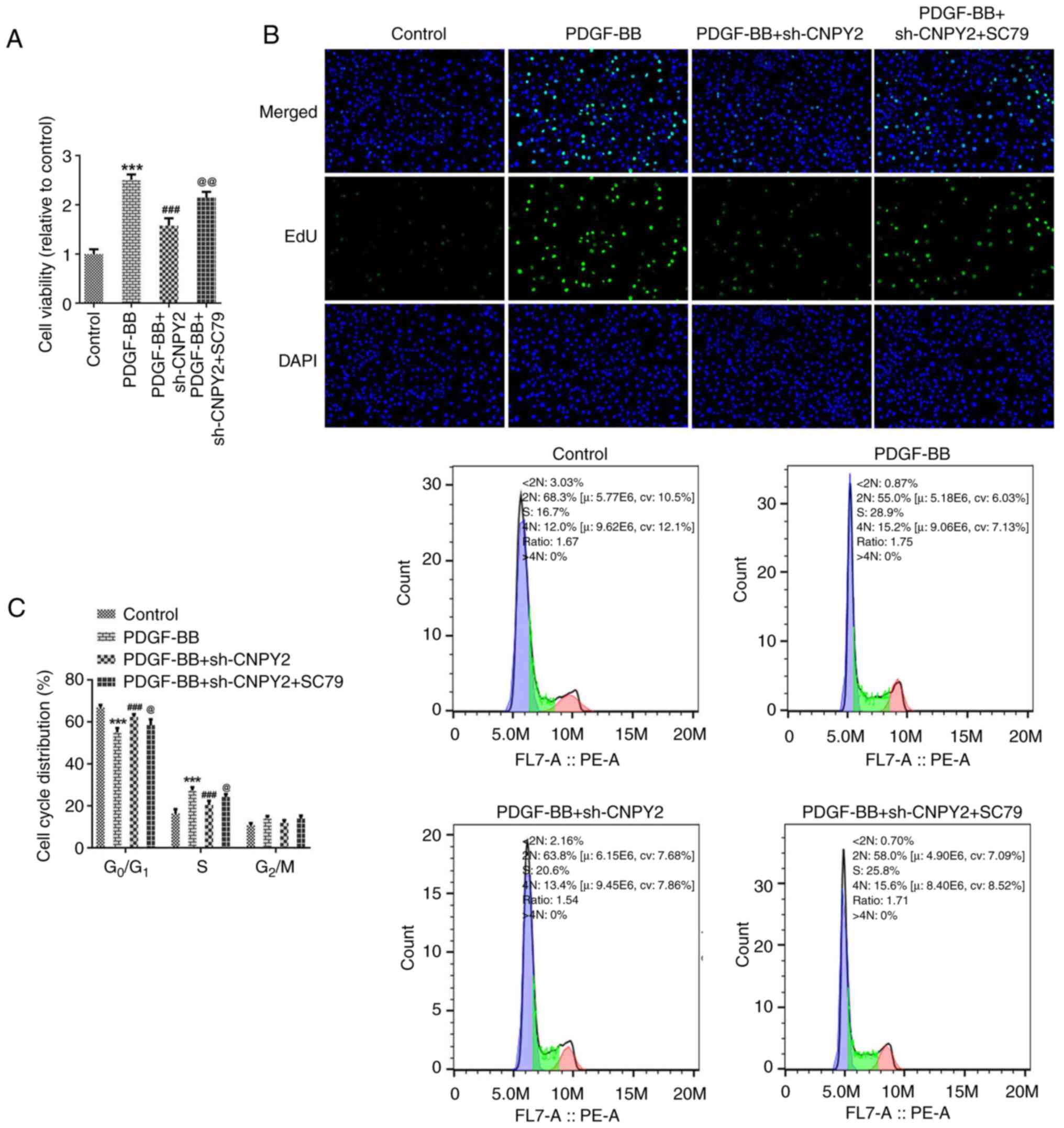
CNPY2 regulates the PDGF-BB-induced
abnormal proliferation, migration and phenotypic transformation of
VSMCs via the activation of Akt/mTOR/GSK-3β signaling
To further explore the regulatory mechanisms of
CNPY2, the cells were pretreated with the Akt activator SC79. The
results of CCK-8 and EdU staining revealed that the cell viability
and proliferative capacity of the PDGF-BB + sh-CNPY2 + SC79 group
were significantly increased compared with those of the PDGF-BB +
sh-CNPY2 group (Fig. 4A and
B). Also, flow cytometric analysis
demonstrated that the cell cycle and cell proliferation were
accelerated in the PDGF-BB + sh-CNPY2 + SC79 group compared with
the PDGF-BB + sh-CNPY2 group (Fig.
4C). The results of the wound healing and Transwell assays
indicated that the migratory abilities of the VSMCs were
significantly increased in the PDGF-BB + sh-CNPY2 + SC79 group
compared with the PDGF-BB + sh-CNPY2 group (Fig. 5A and B). In addition, IF staining demonstrated
that the content of myofilaments and structural proteins in the
PDGF-BB + sh-CNPY2 + SC79 group was reduced compared with that in
the PDGF-BB + sh-CNPY2 group (Fig.
5C). Furthermore, western blotting indicated that OPN
expression in the PDGF-BB + sh-CNPY2 + SC79 group was significantly
increased compared with that in the PDGF-BB + sh-CNPY2 group, while
SMA and F-actin expression was significantly decreased (Fig. 5D).
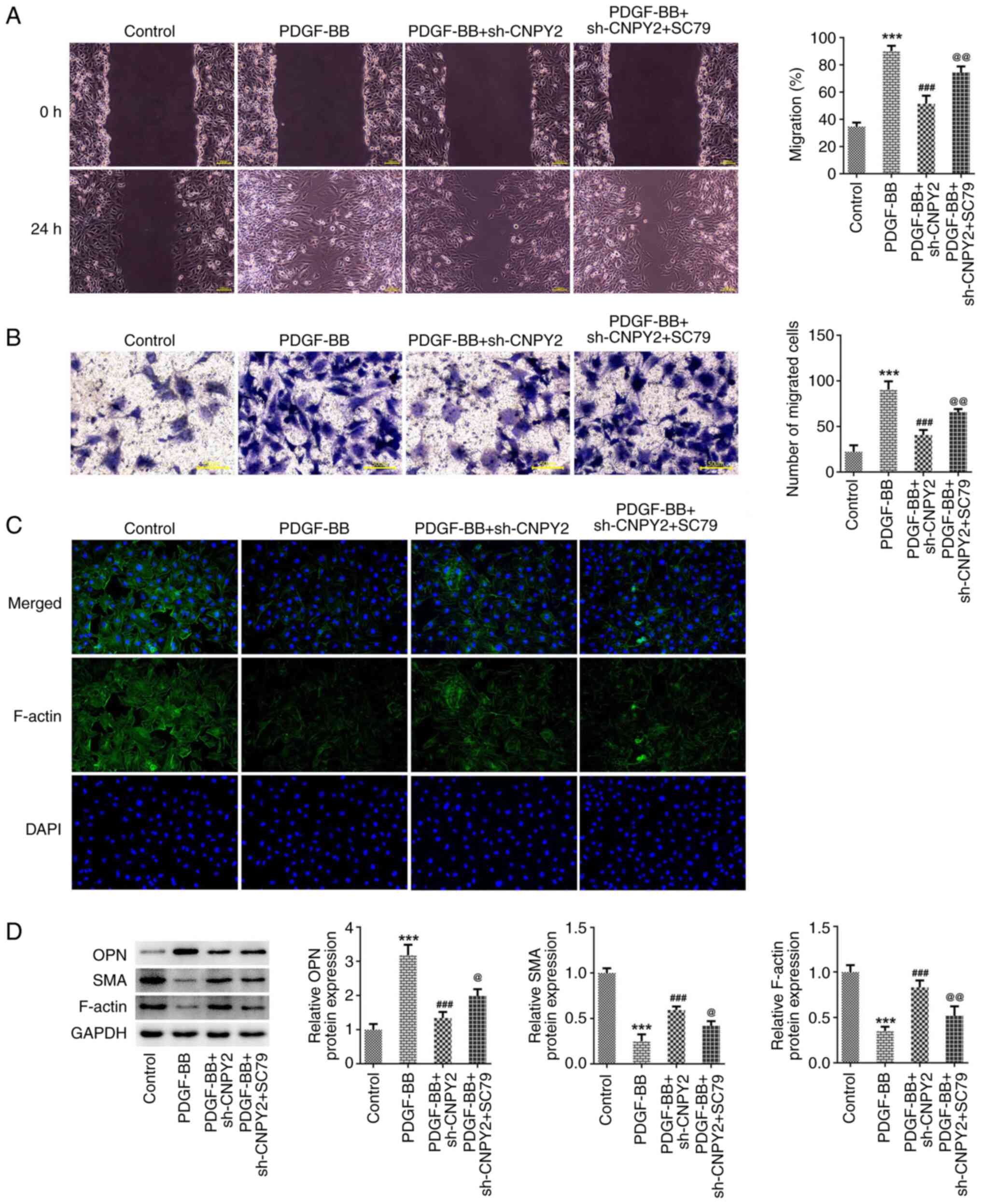 | Figure 5CNPY2 knockdown-induced reductions in
the PDGF-BB-induced phenotypic transformation of VSMCs are
attenuated by activation of the Akt signaling pathway. (A) Wound
healing (scale bar, 100 µm) and (B) Transwell assays (scale bar, 50
µm) were performed to detect the migration ability of the VSMCs.
(C) Immunofluorescence staining was used to detect cytoskeletal
F-actin (magnification, x200; n=3). (D) Western blot analysis was
used to detect the expression of phenotypic
transformation-associated proteins (n=5). ***P<0.001
vs. control; ###P<0.001 vs. PDGF-BB;
@P<0.01 and @@P<0.001 vs. PDGF-BB +
sh-CNPY2. CNPY2, canopy FGF signaling regulator 2; PDGF-BB,
platelet-derived growth factor type BB; VSMCs, vascular smooth
muscle cells; sh, short hairpin; OPN, osteopontin; SMA, smooth
muscle actin; SC79, Akt pathway activator. |
Discussion
In the present study, PDGF-BB was used to stimulate
VSMCs in order to explore the regulatory mechanisms of their
proliferation, migration and phenotypic transformation. Following
its release in response to vascular injury, PDGF-BB serves as a
critical stimulating factor and is a major regulator of VSMC growth
and proliferation (25). In the
present study, following the stimulation of VSMCs by PDGF-BB, it
was found that cell viability, proliferation and migration were
significantly increased, and phenotypic transformation occurred.
Additionally, upregulated CNPY2 expression was observed in
PDGF-BB-stimulated VSMCs. Although a previous study has
investigated the potential function of CNPY2 in AS, it only found
that CNPY2 aggravated oxidized low-density lipoprotein-induced
cellular inflammation and apoptosis (16), and did not reveal its pivotal role
in VSMC phenotypic switching. Given that VSMC phenotypic switching
plays a decisive role in lipoprotein retention, plaque formation
and postoperative restenosis (26), elucidation of the role of CNPY2 in
VSMC phenotypic switching is important as it highlights the
significance of this factor in the pathology of diseases associated
with AS. A previous study demonstrated that the proliferation of
white blood cells and endothelial cells in CNPY2 knockout mice was
significantly less than that in non-knockout mice, resulting in
impaired angiogenesis in the mice (27). Furthermore, the study found that
endogenous CNPY2 expression was significantly reduced in the
cardiac tissue of patients with end-stage heart failure compared
with that of control patients. These studies indicate that CNPY2
mediates the maintenance of normal myocardial function and has an
active role in repair after cardiac injury. Therefore, in ASO, it
is necessary to maintain its level of expression; CNPY2 expression
that is too low or too high is not conducive to a healthy vascular
environment.
In the present study, after knocking down CNPY2,
proteins associated with the downstream Akt/mTOR/GSK-3β signaling
pathway were evaluated and this signaling pathway was found to be
activated. The addition of the Akt signaling pathway activator SC79
significantly attenuated the inhibitory effects of CNPY2 knockdown
on PDGF-BB-induced VSMC proliferation, migration and phenotypic
transformation. Based on these results, the present study reveals
for the first time, to the best of our knowledge, that
Akt/mTOR/GSK-3β signaling mediates the regulation of VSMC
phenotypic switching by CNPY2. However, previous studies have
revealed the regulation of Akt signaling by CNPY2 in other disease
models. For example, one study showed that hypoxia-induced CNPY2
promotes the glycolysis of cervical cancer cells via activation of
the Akt pathway (28). In another,
CNPY2 was shown to enhance epithelial-mesenchymal transformation in
non-small cell lung cancer via activation of the Akt/GSK-3β pathway
(29). The involvement of Akt
signaling in ASO has also been demonstrated, as microRNA-21 was
found to be upregulated in the arteries of ASO injury rat models,
and was shown to regulate the function of VSMCs through the Akt and
ERK1/2 pathways (30). Previous
studies have also revealed that Akt signaling contributes to the
triggering of platelet activation and thrombosis (31,32).
The investigation of strategies to block Akt signaling may provide
a solution to arterial occlusion. In addition, the discovery of the
potential regulatory mechanism of CNPY2 in VSMCs may lead to CNPY2
becoming a disease marker or a target for new therapies.
In conclusion, the present study demonstrates that
CNPY2 regulates the abnormal proliferation, migration and
phenotypic transformation of PDGF-BB-stimulated VSMCs by activating
the Akt/mTOR/GSK-3β signaling pathway. However, this regulatory
mechanism of CNPY2 is based on in vitro experiments only,
and in vivo experiments and the analysis of clinical tissues
are required to fully uncover its features in the future.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YY and HS conceived and designed the study. HS, XW,
LM, XL and WJ performed the experiments and wrote the manuscript.
YY and HS processed the experimental data. YY and HS confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hutchings G, Kruszyna Ł, Nawrocki MJ,
Strauss E, Bryl R, Spaczyńska J, Perek B, Jemielity M, Mozdziak P,
Kempisty B, et al: Molecular mechanisms associated with
ROS-dependent angiogenesis in lower extremity artery disease.
Antioxidants (Basel. 10(735)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Grover SP and Mackman N: Tissue factor in
atherosclerosis and atherothrombosis. Atherosclerosis. 307:80–86.
2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhang R, Lai ZC and Liu CW:
Femoral-popliteal arteriosclerosis obliterans:Review of
evidence-based studies on drug-eluting endovascular treatment.
Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 41:256–260. 2019.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
4
|
Schicho A, Bäumler W, Verloh N, Beyer LP,
Schierling W, Uller W, Gößmann H, Stroszczynski C and Dollinger M:
Percutaneous aspiration thrombectomy for arterial thromboembolic
occlusion following percutaneous transluminal angioplasty:
Technical success rates and clinical outcomes. Rofo. 194:291–295.
2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Machado M, Borges de Almeida G, Sequeira
M, Pedro F, Fior A, Carvalho R, Fragata I, Reis J and Nunes AP:
Percutaneous transluminal angioplasty and stenting in acute stroke
caused by basilar artery steno-occlusive disease: The experience of
a single stroke centre. Interv Neuroradiol. 28:547–555.
2022.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Cao G, Xuan X, Hu J, Zhang R, Jin H and
Dong H: How vascular smooth muscle cell phenotype switching
contributes to vascular disease. Cell Commun Signal.
20(180)2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Petsophonsakul P, Furmanik M, Forsythe R,
Dweck M, Schurink GW, Natour E, Reutelingsperger C, Jacobs M, Mees
B and Schurgers L: Role of vascular smooth muscle cell phenotypic
switching and calcification in aortic aneurysm formation.
Arterioscler Thromb Vasc Biol. 39:1351–1368. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhang L, Tao Y, Yang R, Hu Q, Jia J, Yu M,
He B, Shen Z, Qin H, Yu Z and Chen P: Euonymine inhibits in-stent
restenosis through enhancing contractile phenotype of vascular
smooth muscle cells via modulating the PTEN/AKT/mTOR signaling
pathway. Phytomedicine. 107(154450)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Green ID, Liu R and Wong JJL: The
expanding role of alternative splicing in vascular smooth muscle
cell plasticity. Int J Mol Sci. 22(10213)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zhang F, Guo X, Xia Y and Mao L: An update
on the phenotypic switching of vascular smooth muscle cells in the
pathogenesis of atherosclerosis. Cell Mol Life Sci.
79(6)2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang G, Luo Y, Gao X, Liang Y, Yang F, Wu
J, Fang D and Luo M: MicroRNA regulation of phenotypic
transformations in vascular smooth muscle: Relevance to vascular
remodeling. Cell Mol Life Sci. 80(144)2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Medrano-Bosch M, Simón-Codina B, Jiménez
W, Edelman ER and Melgar-Lesmes P: Monocyte-endothelial cell
interactions in vascular and tissue remodeling. Front Immunol.
14(1196033)2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Holycross BJ, Blank RS, Thompson MM, Peach
MJ and Owens GK: Platelet-derived growth factor-BB-induced
suppression of smooth muscle cell differentiation. Circ Res.
71:1525–1532. 1992.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Chen PY, Qin L, Li G, Tellides G and
Simons M: Fibroblast growth factor (FGF) signaling regulates
transforming growth factor beta (TGFβ)-dependent smooth muscle cell
phenotype modulation. Sci Rep. 6(33407)2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Chen KQ, Zhang YQ, Wang ZB and Wang SZ:
Progress in research on CNPY2 in diseases. Mini Rev Med Chem.
24:391–402. 2024.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Huang H, Tang N, Li Y, Huo Q, Chen Q and
Meng Q: The role of CNPY2 in endothelial injury and inflammation
during the progress of atherosclerosis. J Mol Histol. 54:195–205.
2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Guo J, Zhang Y, Mihic A, Li SH, Sun Z,
Shao Z, Wu J, Weisel RD and Li RK: A secreted protein (Canopy 2,
CNPY2) enhances angiogenesis and promotes smooth muscle cell
migration and proliferation. Cardiovasc Res. 105:383–393.
2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Weyand CM and Goronzy JJ: Pathogenic
mechanisms in giant cell arteritis. Cleve Clin J Med. 69 (Suppl
2):SII28–SII32. 2002.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Xu TY, Qing SL, Zhao JX, Song J, Miao ZW,
Li JX, Yang FY, Zhao HY, Zheng SL, Li ZY, et al: Metrnl deficiency
retards skin wound healing in mice by inhibiting AKT/eNOS signaling
and angiogenesis. Acta Pharmacol Sin. 44:1790–1800. 2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhao MJ, Jiang HR, Sun JW, Wang ZA, Hu B,
Zhu CR, Yin XH, Chen MM, Ma XC, Zhao WD and Luan ZG: Roles of
RAGE/ROCK1 pathway in HMGB1-induced early changes in barrier
permeability of human pulmonary microvascular endothelial cell.
Front Immunol. 12(697071)2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhou J, Jiang YY, Chen H, Wu YC and Zhang
L: Tanshinone I attenuates the malignant biological properties of
ovarian cancer by inducing apoptosis and autophagy via the
inactivation of PI3K/AKT/mTOR pathway. Cell Prolif.
53(e12739)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Gao Q, Chen K, Gao L, Zheng Y and Yang YG:
Thrombospondin-1 signaling through CD47 inhibits cell cycle
progression and induces senescence in endothelial cells. Cell Death
Dis. 7(e2368)2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhang Q, Lu S, Li T, Yu L, Zhang Y, Zeng
H, Qian X, Bi J and Lin Y: ACE2 inhibits breast cancer angiogenesis
via suppressing the VEGFa/VEGFR2/ERK pathway. J Exp Clin Cancer
Res. 38(173)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Salabei JK, Cummins TD, Singh M, Jones SP,
Bhatnagar A and Hill BG: PDGF-mediated autophagy regulates vascular
smooth muscle cell phenotype and resistance to oxidative stress.
Biochem J. 451:375–388. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Liu YX, Yuan PZ, Wu JH and Hu B: Lipid
accumulation and novel insight into vascular smooth muscle cells in
atherosclerosis. J Mol Med (Berl). 99:1511–1526. 2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yin W, Guo J, Zhang C, Alibhai FJ, Li SH,
Billia P, Wu J, Yau TM, Weisel RD and Li RK: Knockout of canopy 2
activates p16INK4a pathway to impair cardiac repair. J
Mol Cell Cardiol. 132:36–48. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Tian T, Dong Y, Zhu Y, Chen Y, Li X, Kuang
Q, Liu X, Li P, Li J and Zhou L: Hypoxia-induced CNPY2 upregulation
promotes glycolysis in cervical cancer through activation of AKT
pathway. Biochem Biophys Res Commun. 551:63–70. 2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Dou Y, Lei JQ, Guo SL, Zhao D, Yue HM and
Yu Q: The CNPY2 enhances epithelial-mesenchymal transition via
activating the AKT/GSK3β pathway in non-small cell lung cancer.
Cell Biol Int. 42:959–964. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Huang S, Xu T, Huang X, Li S, Qin W, Chen
W and Zhang Z: miR-21 regulates vascular smooth muscle cell
function in arteriosclerosis obliterans of lower extremities
through AKT and ERK1/2 pathways. Arch Med Sci. 15:1490–1497.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Dong J, Lin J, Wang B, He S, Wu C,
Kushwaha KK, Mohabeer N, Su Y, Fang H, Huang K and Li D:
Inflammatory cytokine TSLP stimulates platelet secretion and
potentiates platelet aggregation via a TSLPR-dependent PI3K/Akt
signaling pathway. Cell Physiol Biochem. 35:160–174.
2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Tang Z, Shi H, Chen C, Teng J, Dai J,
Ouyang X, Liu H, Hu Q, Cheng X, Ye J, et al: Activation of platelet
mTORC2/Akt pathway by anti-β2GP1 antibody promotes thrombosis in
antiphospholipid syndrome. Arterioscler Thromb Vasc Biol.
43:1818–1832. 2023.PubMed/NCBI View Article : Google Scholar
|