Introduction
Acute lung injury (ALI) is characterized by dyspnea
and hypoxemia as clinical features, and pulmonary interstitial
edema, alveolar epithelial injury, and the acute excessive
inflammatory response process are triggered by various pathologies,
such as infection, trauma, and shock (1). ALI is accompanied by a high
morbidity and mortality rate, and no specific treatment exists
(2). As a result, how to target
the biomarkers of ALI to inhibit the inflammatory response and the
molecular and cellular events during inflammation, to activate the
host immune response or produce a balanced relationship between
immune and inflammatory responses have attracted scholars'
attention (3,4).
High mobility group box 1 protein (HMGB1), a late
inflammatory cytokine, is a highly conserved nuclear DNA-binding
protein that activates macrophages and dendritic cells (DCs) to
produce inflammatory cytokines, mediating systemic inflammatory
responses (5-8). Several studies have reported that
HMGB1 plays a substantial role in the pathogenesis of ALI and
sepsis (5,6,9).
The serum and tissue levels of HMGB1 are elevated during ALI and
sepsis, and HMGB1 inhibitors can alleviate the inflammatory
response, tissue injury, and organ dysfunction (9,10).
However, to the best of our knowledge no data has been reported
about the exact mechanism of action of HMGB1 in the pathogenesis of
ALI.
Macrophages, the main proinflammatory cells, can
differentiate into classically activated macrophages (M1) and
alternatively activated macrophages (M2) (11). Under physiological conditions,
there is a balance between M1 and M2 macrophages. M1 macrophages
produce interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α) and
IL-6, which stimulate and activate neutrophils, leading to the
release of proteases and oxidant-induced lung damage (12). M2 macrophages can accelerate
resolution and lung repair in the acute respiratory distress
syndrome (13). In ALI,
polarization of macrophages into the M1 phenotype by adapting to
the modified microenvironment results in the production of a great
number of inflammatory factors (14). Activated macrophages not only
secrete HMGB1 into the extracellular environment, but also express
receptors at their surface to which HMGB1 binds (8). Receptor for advanced glycation end
products (RAGE), Toll-like receptor 2 (TLR2) and TLR4 have been
reported as the main receptors of HMGB1 that transmit intracellular
signals, leading to activation of the nuclear factor-κB (NF-κB)
pathway (5,7,8).
Therefore, it is possible that HMGB1 activates macrophages via
TLR2, TLR4, and RAGE/NF-κB signaling pathways, and induces
polarization of macrophage M1 in ALI.
The absent in melanoma 2 (AIM2) inflammasome,
comprising AIM2, the adaptor apoptosis-associated speck-like
protein containing a CARD (ASC) and caspase-1, is a major
intracellular polyprotein complex of the innate immune system
expressed by macrophages, DCs, and other immune cells (15,16). In the development of infection and
ALI, AIM2 inflammasomes are activated, leading to the maturation of
caspase-1, pro-IL-18 and pro-IL-1β, in addition to their release in
the serum and tissue, inducing lung inflammation (15-17). In the process of lung
inflammation, the expression of AIM2 in macrophages would elevate
and the polarization of macrophage M1 polarization would develop. A
study also found that M1 polarized macrophages express elevated
AIM2 gene expression (18). As a
result, in the process of lung injury, macrophages could on one
hand activate AIM2 and release inflammatory mediators and lead to
inflammatory injury and on the other hand, could develop towards M1
polarization, resulting lung tissue injury. A study revealed that
inflammasomes induce the secretion of HMGB1 in the nucleus
(19). However, HMGB1 can
activate AIM2 inflammasomes to attend to the process of
proinflammatory response and innate immunity (20,21). Thus, it was inferred that HMGB1
participates in the process of lipopolysaccharide (LPS)-induced ALI
in mice by activating AIM2 inflammasomes in macrophages, as well as
inducing polarization of M1 macrophages via TLR2, TLR4 and
RAGE/NF-κB signaling pathways.
Materials and methods
Animals
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Wuhan University
(Wuhan, China), as well as being in compliance with the Animal
Welfare Act. A total of 150 male C57BL/6 mice, aged 6-8 weeks old
and weighing 20-22 g, were purchased from the Hubei Experimental
Animal Research Center of Hubei province (Wuhan, China) and bred in
the Animal Biosafety Level-III Laboratory of Wuhan University. In
the laboratory, the mice were kept on a 12-h light and dark cycle
at 25°C, with a humidity of 45-55% in a ventilated cage, and with
free access to food and water. The animals were assigned to 10
groups (control, LPS, anti-HMGB1, LPS+anti-HMGB1, rHMGB1,
LPS+rHMGB1, FPS-ZM1, LPS+FPS-ZM1, LPS+RS and LPS+LPS-RS; n=4-6 per
group) and all experiments were repeated more than three times.
LPS-induced model of ALI and experimental
treatment
Male mice were anesthetized by intraperitoneal
(i.p.) injection of 1% pentobarbital sodium solution (80 mg/kg,
Biyuntian Institute of Biotechnology) and randomly assigned to the
following groups: Control, LPS, positive control, and treatment
group (n=4-6 per each group). Mice in the LPS and treatment groups
then received an i.p., injection of LPS (15 mg/kg; Sigma-Aldrich,
Merck KGaA) diluted in 200 µl sterile PBS. The control and
positive control groups were administered only PBS by the same way
and volume. The positive control and treatment groups received an
intranasal (i.n.) injection of anti-HMGB1 (2.5 µg/g in 40
µl PBS, 2 h after LPS challenge; BioLegend, Inc.) (22) or rHMGB1 (0.5 µg/g in 40
µl PBS, 2 h after LPS challenge; R&D Systems, Inc.)
(23-25), an i.p., injection of LPS from
Rhodobacter sphaeroides (LPS-RS), a TLR2/4 antagonist (0.1
mg/mg in 200 µl endotoxin-free water, 1 h before LPS
challenge; InvivoGen) (25-27) or FPS-ZM1, a RAGE antagonist (1.5
mg/kg in 200 µl PBS, 1 h before LPS challenge; EMD
Millipore) (25,28). All mice were sacrificed (cervical
dislocation) 24 h after the last treatment and the blood, lung,
bronchoalveolar lavage fluid (BALF), and spleen of mice were
extracted for sample preparation.
Lung wet-to-dry (W/D) ratio
The left lung of the mice was collected and
immediately weighed to obtain the 'wet' weight, and then placed in
an incubator at 80°C for 48 h to obtain the 'dry' weight. The ratio
of W/D was quantified by dividing the lung wet weight by the lung
dry weight.
Myeloperoxidase (MPO) activity
detection
MPO activity in lung tissue was measured using an
MPO assay kit (A044; Nanjing Jiancheng Bioengineering Institute) in
accordance with the manufacturer's protocol. Parts of the right
lung tissues were homogenized and centrifuged (4°C; 1,006.2 × g; 30
min). The supernatant was incubated with a substrate buffer (1:10).
The enzymatic activity was determined at 450 nm using a
spectrophotometer.
Histological analysis of the lungs
The left lung of mice was removed and fixed in 4%
paraformaldehyde buffer (room temperature; 24 h), dehydrated,
embedded, and sectioned into 5-µm slices, which were stained
with hematoxylin and eosin (H&E) (room temperature; staining
with hematoxylin for 5 min, followed by HCl-alcohol differentiation
for 20 sec, flushing with running water, incubation with 1% ammonia
for 30 sec to return the blue color, further washing in water and
staining with eosin for 3 min) to evaluate lung inflammation. A
light microscope with magnification of ×200 was used to observe the
lung tissue. A total of five samples were chosen in each group and
five images were acquired for every sample. The pathological
changes of lung tissues were assessed by the lung injury scores
(29,30). Lung injury scores were categorized
as follows: Neutrophil infiltration (0-4), interstitial edema
(0-4), hemorrhage (0-4) and hyaline membrane formation, necrosis,
congestion (0-4). The severity of microscopic injury was judged
according to the following scoring system: 0, Normal; 1, Minimal
(<25%); 2, Mild (25-50%); 3, Moderate (50-70%); and 4, Severe
(>75%).
BALF collection
BALF was collected from mice sacrificed 24 h after
LPS administration to analyze the cellular components and cytokine
production in BALF. The lungs were lavaged three times in 0.5 ml
PBS and BALF was centrifuged at 503.1 × g for 7 min at 4°C. The
collected supernatants were stored at -80°C for the next step.
Total and differential inflammatory cells in BALF were counted
using Wright-Giemsa staining (3-5 drops of methanol fixing agent
was added to the cells at room temperature for 1 min; Wright-Giemsa
stain was then added at room temperature for 8 min, and the cells
were finally flushed with running water for 30-60 sec;
Wright-Giemsa Stain kit; WGK-1; ScyTek; Shenzhen Xinbosheng
Bioengineering Institute) for cytospin preparations.
Immunohistochemical analysis
For immunohistochem-istry of AIM2, left lung
sections (5-µm thickness) were deparaffinized with xylene,
rehydrated in a series of graded concentrations of ethanol and
submerged in an antigen repair solution (pH 6.0), containing EDTA.
All lung tissue sections were treated with 3%
H202 for 20 min to clear away endogenous
peroxidase, incubated at polyclonal anti-AIM2 antibody (1:100; cat.
no. ab93015; Abcam) at 4°C overnight and then, incubated in a
horseradish peroxidase (HRP)-linked anti-rabbit secondary antibody
(Goat anti-Rabbit IgG (H+L)-HRP; 1:50; cat. no. BS13278; Bioworld
Technology, St.) for 30 min. Diaminobenzidine (VECTASTAIN Elite ABC
HRP kit; PK-6200; Vector Laboratories) was used to stain the
sections (diaminobenzidine staining for 30 min, followed by
flushing with running water, staining with hematoxylin for 5 min,
HCl-alcohol differentiation for 20 sec, flushing with running
water, incubation with 1% ammonia for 30 sec to return the blue
color, dehydration in alcohol and sealing with neutral gum). The
process of staining and the following steps were all at room
temperature. The IPP 6.0 software (Media Cybernetics, Inc.) was
applied for image analysis.
ELISA
The expression levels of TNF-α, IL-1β, IL-18, IL-6,
IL-10 and MCP-1 in BALF and cell culture supernatant were measured
using ELISA kits (Mouse TNF-α ELISA kit; cat. no. EK2822/2-96T;
Mouse IL-1β ELISA kit; cat. no. EK201B2/2-96T; Mouse IL-18 ELISA
kit; cat. no. EK2182-96T; Mouse IL-6 ELISA kit; EK2062/2-96T; Mouse
IL-10 ELISA kit; cat. no. EK2012/2-96T; Mouse MCP-1 ELISA kit; cat.
no. EK2872/2-96T; MultiSciences), according to the manufacturer's
protocol. The concentrations of cytokines were expressed as
pg/ml.
RNA isolation, cDNA synthesis and
RT-qPCR
Total RNA was extracted from the lower right lung
tissues using the TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and 1 µg RNA was reverse transcribed into
cDNA with ReverTra Ace qPCR RT kit (Toyobo Life Science) (reaction
conditions: 37°C for 15 min; 98°C for 5 min; holding at 4°C).
Additionally, RT-qPCR was performed using SYBR Premix Ex Taq™ kit
(Takara Bio Inc.). All the primers were obtained from Sangon
Biotech Co., Ltd. The PCR primer sequences are listed in Table I. The amplification was optimized
by QuantStudio™ 6 Flex (Applied Biosystems) using the following PCR
procedure: Initial denaturation at 95°C for 30 sec, with 40 cycles
of denaturation at 95°C for 5 sec, as well as annealing at 60°C for
20-30 sec. The conditions of melt curve analysis were at 95°C for
15 sec, 59-61°C for 60 sec and at 95°C for 15 sec. Differences in
expression levels between the groups were calculated by the
2−ΔΔCq method (31)
using GAPDH as an internal reference gene.
 | Table IPrimer sequences and product
sizes. |
Table I
Primer sequences and product
sizes.
| Gene name | Primer sequences
(5′-3′) | Product size
(bp) |
|---|
| TLR2 | | 167 |
| Sense |
5′-CAGTCCCAAAGTCTAAAGTCG-3′ | |
| Antisense |
5′-TCTACGGGCAGTGGTGAAA-3′ | |
| TLR4 | | 151 |
| Sense |
5′-TGGGTCAAGGAACAGAAGCA-3′ | |
| Antisense |
5′-TCACACTGACCACTGACACA-3′ | |
| RAGE | | 187 |
| Sense |
5′-CGGGACTCTTTACACTGCG-3′ | |
| Antisense |
5′-CCTTCAGGCTCAACCAACA-3′ | |
| NF-κB | | 112 |
| Sense |
5′-GGACCTATGAGACCTTCAAGAG-3′ | |
| Antisense |
5′-ACAGAAGTTGAGTTTCGGGTAG-3′ | |
| AIM2 | | 382 |
| Sense |
5′-CACACTCGACGTGGCAGATAGGAC-3′ | |
| Antisense |
5′-CAGCACCGTGACAACAAGTGG-3′ | |
| ASC | | 121 |
| Sense |
5′-CACCAGCCAAGACAAGATGA-3′ | |
| Antisense |
5′-CTCCAGGTCCATCACCAAGT-3′ | |
| Caspase-1 | | 190 |
| Sense |
5′-AACAGAACAAAGAAGATGGCACA-3′ | |
| Antisense |
5′-CCAACCCTCGGAGAAAGAT-3′ | |
| GAPDH | | 150 |
| Sense |
5′-TGTGTCCGTCGTGGATCTGA-3′ | |
| Antisense |
5′-TTGCTGTTGAAGTCGCAGGAG-3′ | |
Western blot analysis
Nuclear and cytoplasmic proteins were extracted from
the upper right lung tissue using radio-immunoprecipitation assay
lysis buffer (Beyotime Institute of Biotechnology). The method of
protein determination was a BCA assay (cat. no. P0010; Beyotime
Institute of Biotechnology), and involved calculating the
absorbance value at optical density (OD)568 with a DG-3022A enzyme
labeling instrument (Nanjing East China Electronics Group Co.,
Ltd.). The linear regression equation was calculated according to
the standard protein concentration and its corresponding OD.
According to the OD value of the protein sample, the protein
concentration of the sample was calculated via the regression
equation. The proteins (40 µg/lane) were resolved on 10%
SDS-PAGE gels by electrophoresis and then transferred onto
polyvinylidene difluoride membranes (EMD Millipore) using a Mini
Trans-Blot system (Bio-Rad Laboratories Inc.). The membranes were
blocked (for 2 h at room temperature with agitation) with
Tris-buffered saline and 20% Tween-20 containing 5% non-fat dried
milk, and incubated overnight at 4°C with antibodies (1:1,000)
against TLR2 (anti-TLR 2 monoclonal; cat. no. 122765; Cell
Signaling Technology, Inc.), RAGE (anti-RAGE; cat. no. 6996S; Cell
Signaling Technology, Inc.), phosphorylated (p)-NF-κB p65
(anti-p-NF-κB p65; cat. no. 4025S; Cell Signaling Technology,
Inc.), ASC (ASC monoclonal antibody; cat. no. 13833S; Cell
Signaling Technology, Inc.), AIM2 (anti-AIM2 monoclonal; cat. no.
12948S), TLR4 (anti-TLR4; cat. no. ab83444; Abcam), NF-κB p65
(anti-NF-κB p65; cat. no. ab207297; Abcam) and caspase-1
(anti-caspase-1; cat. no. IMG-5028; Novus Biologicals Ltd.).
HRP-conjugated secondary anti-rabbit antibody (1:50,000; cat. no.
BM2006; BOSTER Biological Technology Co., Ltd.) was added at 37°C
for 2 h. Chemiluminescence images were revealed with Pierce ECL
Western Blotting Substrate (Thermo Fisher Scientific, Inc.) using
BandScan 5.0 gel image software (Glyko, Inc.; Novato) for
quantification of the bands.
Generation of bone marrow-derived
macrophages
Primary macrophages were prepared from bone marrow
progenitors. Bone marrow mononuclear cells were prepared from femur
bone marrow suspensions of male C57BL/6 mice (age, 5-8 weeks old)
and then cultured in RPMI-1640 medium supplemented with 10% fetal
bovine serum (FBS; HyClone; GE Healthcare Life Science), 0.1% 100X
penicillin-streptomycin solution (Beijing Solarbio Science &
Technology Co., Ltd.) at 37°C in a 5% CO2 incubator.
After 4 h, the non-adherent cells were collected. The adherent
cells were suspended (1-2×106 cells/ml) in a medium
containing 10 ng/ml macrophage-colony stimulating factor
(PeproTech, Inc.) and 10 ng/ml recombinant mouse IL-4 (PeproTech,
Inc.). The medium was replaced on 3rd and 5th days. On 7th day, and
the obtained primary macrophages were incubated with or without LPS
(100 ng/ml), anti-HMGB1 (10 µg/ml) (22,32), rHMGB1 (50 µg/ml) (25,32,33), LPS-RS (1,000 ng/ml) (25,27), or FPS-ZM1 (1 µM) (25,28) for 24 h. Macrophages and
supernatants were collected for subsequent RT-qPCR, western
blotting and ELISA assays.
Flow cytometry
Lung mononuclear cells (MNCs) were re-suspended in
FACS buffer (cat. no. 00-4222; eBioscience, Inc.) (2×106
cells/ml) and stained with a PE-cluster of differentiation (CD) 11c
antibody (cat. no. 561044; eBioscience; Thermo Fisher Scientific,
Inc.) and APC-Cy7-F4/80 antibody (cat. no. 123117; BioLegend, Inc.)
to characterize the alveolar macrophages (AMs) as CD11c and F4/80
double-positive cells. The antibodies fluorescein isothiocyanate
(FITC)-major histocompatibility complex (MHC) II (cat. no. 556643),
FITC-CD80 (cat. no. 553768), FITC-CD40 (cat. no. 561845),
FITC-CD206 (cat. no. 551135), FITC-IL-10 (cat. no. 554466;
eBioscience Inc.) and APC-CD86 (cat. no. 102513; BioLegend, Inc.)
were used to perform the second cell surface staining. All cells
were analyzed by flow cytometry (FACSAria III; BD Bioscences). The
results were analyzed using FlowJo (version 10.0.7r2; FlowJo
LLC)
Statistical analysis
Data are expressed as the mean ± standard deviation.
The differences were compared using the one-way analysis of
variance. The post hoc test used was the Bonferroni test. Data were
analyzed using the GraphPad Prism 5 software (GraphPad Software,
Inc.) and SPSS 22.0 software (IBM, Corps.). P<0.05 was
considered to indicate a statistically significant difference. The
experiments were performed more than three times.
Results
HMGB1 aggravates the inflammatory
response in LPS-induced ALI in a mouse model
To investigate the role of HMGB1 in LPS-induced ALI
in a mouse model, anti-HMGB1 and rHMGB1 were injected i.n., 2 h
after LPS, and all mice were sacrificed 24 h after the last
treatment. As shown in Fig. 1A and
B, no evident histological alteration of the left lung tissue
was observed in the control group and anti-HMGB1 alone group.
However, lung sections of the LPS group showed significant
pathological changes, including infiltration of inflammatory cells,
edema, hemorrhage and alveolar collapse (P<0.001). Slight
pathological changes were observed in the rHMGB1 alone group.
Treatment with anti-HMGB1 markedly ameliorated LPS-induced lung
injury and administration of rHMGB1 significantly aggravated the
pathological injury of lung tissue (P<0.001). The lung W/D ratio
in lung tissues was notably decreased in the LPS challenge group
compared with that of the control group. There were no significant
differences of W/D ratio in lung tissues between anti-HMGB1, rHMGB1
alone and control groups. Treatment with anti-HMGB1 significantly
decreased the W/D ratio in lung tissues and rHMGB1 significantly
increased that ratio (P<0.001; Fig. 1C and D). As neutrophils are the
main cells involved in inflammatory reactions in injured lungs, MPO
activity was measured to reflect neutrophil accumulation in lung
tissues. In the present study, LPS and single rHMGB1 administration
significantly increased MPO activity compared with the control
group (P<0.001; Fig. 1E and
F). Treatment with anti-HMGB1 significantly decreased MPO
activity in lung tissues of LPS-induced ALI mice, while rHMGB1 had
the opposite effect (P<0.001; Fig.
1E and F). The numbers of inflammatory cells to evaluate
pulmonary inflammation were also examined. After LPS challenge, the
numbers of total cells, macrophages, neutrophils and lymphocytes
significantly increased compared with those of the control group
(P<0.001). This increase in total cells, macrophages and
neutrophils was significantly attenuated by treatment with
anti-HMGB1 and aggravated by rHMGB1 (P<0.01; Fig. 1G and H). The levels of TNF-α,
IL-6, MCP-1, IL-1β and IL-18 production in BALF were detected by
ELISA. The results showed that their concentrations were
significantly increased in the LPS-challenged mice compared with in
the mice of the control group (P<0.05). Treatment with
anti-HMGB1 after LPS challenge significantly inhibited the
proinflammatory cytokine production (P<0.05) and treatment with
rHMGB1 increased their levels (Fig.
1I and J). These results suggest that HMGB1 may aggravate the
inflammatory response in LPS-induced ALI.
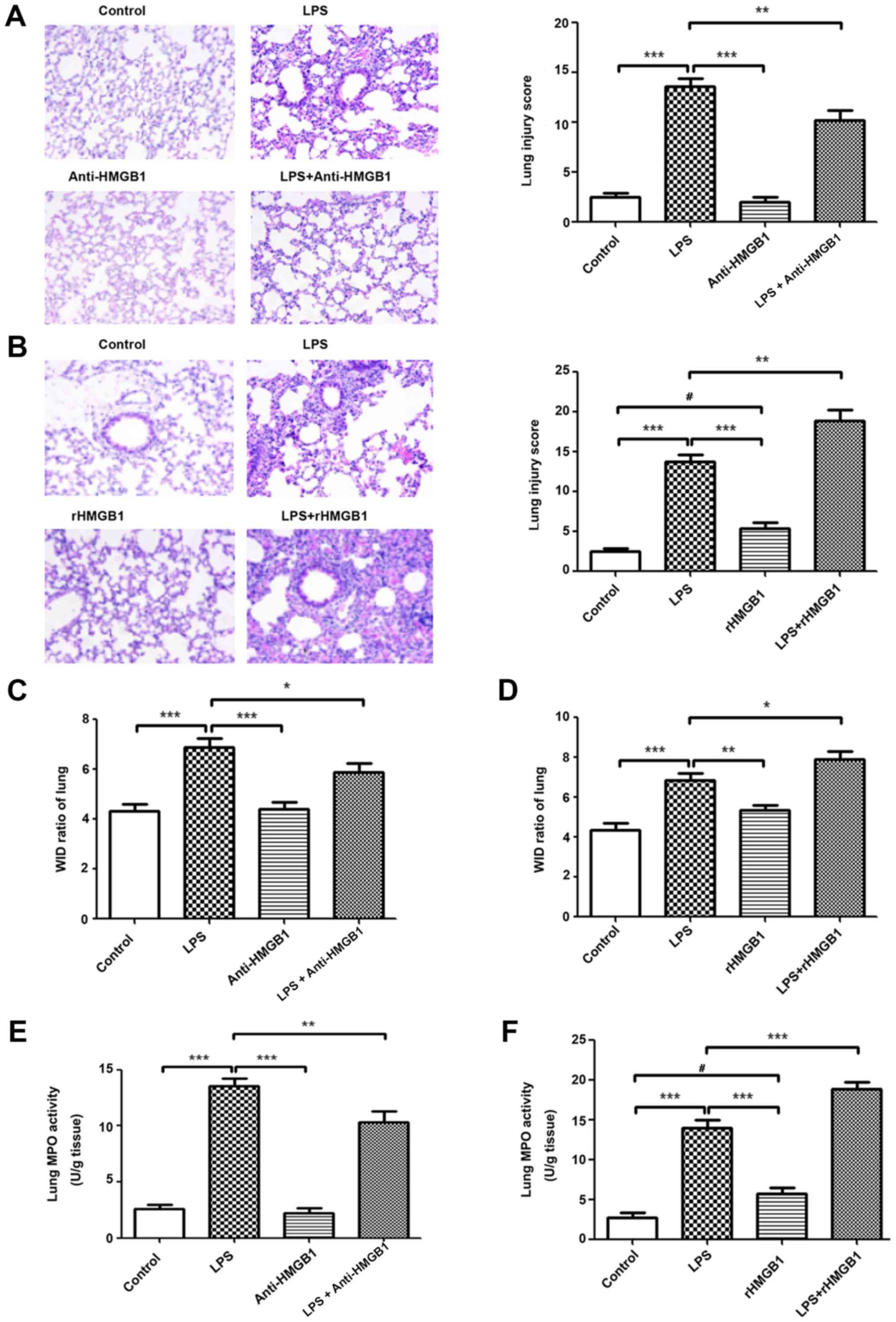 | Figure 1Extracellular HMGB1 affects the
inflammatory response in lung tissues in ALI. Histopathological
changes and injury scores in the left lung are shown for different
groups: (A) Control group, LPS group, Anti-HMGB1 group and LPS +
anti-HMGB1 group (H&E staining, ×200 magnification). (B)
Control group, LPS group, rHMGB1 group and LPS + rHMGB1 group
(H&E staining, ×200 magnification). Effects of (C) anti-HMGB1
and (D) rHMGB1 on the lung W/D ratio of LPS-induced ALI mice.
Effects of (E) anti-HMGB1 and (F) rHMGB1 on MPO activity in lung
tissues and infiltration of inflammatory cells in BALF of
LPS-induced ALI mice. All the experiments were repeated more than
three times (n=4-6 mice per each group). Data presented is from a
selected representative experiment. All data are expressed as the
mean ± standard deviation. *P<0.05,
**P<0.01 and ***P<0.001 vs. LPS group;
#P<0.05 vs. control group. Extracellular HMGB1
affects the inflammatory response in lung tissues in ALI. Effects
of (G) anti-HMGB1 and (H) rHMGB1 on MPO activity in lung tissues
and infiltration of inflammatory cells in BALF of LPS-induced ALI
mice. Effects of (I) anti-HMGB1 and (J) rHMGB1 on the ELISA results
of secretion of inflammatory cytokines (TNF-α, IL-6, MCP-1, IL-1β,
and IL-18) in BALF. All the experiments were repeated more than
three times (n=4-6 mice per each group). Data presented is from a
selected representative experiment. All data are expressed as the
mean ± standard deviation. *P<0.05,
**P<0.01 and ***P<0.001 vs. LPS group;
#P<0.05 vs. control group. LPS, lipopolysaccharide;
IL, interleukin; BALF, bronchoalveolar lavage fluid; MCP,
myeloper-oxidase; TNF, tumor necrosis factor; ALI, acute lung
injury; rHMGB1, recombinant High mobility group box 1; H&E,
hematoxylin and eosin; W/D, wet to dry. |
HMGB1 activates the AIM2 inflammasome
complex in macrophages
To further explore the mechanism of HMGB1 in ALI,
the present study attempted to study the relationship between HMGB1
and the AIM2 inflammasome. AIM2 was immunostained with anti-AIM2 to
further determine AIM2 expression in lung tissue sections. It was
observed that AIM2 was mainly expressed in inflammatory cells, such
as macrophages and neutrophils. There were more positively stained
cells in lung tissues of the LPS+rHMGB1 group compared with those
of the LPS group, whereas there were fewer positively stained cells
in the LPS+anti-HMGB1 group (Fig. 2A
and B). In the in vivo experiment, LPS significantly
upregulated the expression levels of AIM2, ASC and caspase-1,
except for pro-caspase-1, which is an inactive precursor of
caspase-1, as determined by western blot analysis (P<0.001).
This increase was aggravated by rHMGB1 administration; however,
anti-HMGB1 inhibited expression of LPS-induced the AIM2
inflammasome (Fig. 2C and E).
Similar results were obtained by RT-qPCR detection of AIM2, ASC and
caspase-1 in lung tissues (Fig. 2D
and F). To further study their relationships at the macrophage
level, bone marrow-derived macrophages (BMMs) primed with LPS and
treated with anti-HMGB1 or rHMGB1 were cultured. The expression
level of the inflammasome in BMMs was detected by western blotting
and RT-qPCR. As illustrated in Fig.
2G and H, the expression levels of AIM2, ASC and caspase-1
proteins significantly increased in the LPS group, and the
significant increase was greater in the LPS+rHMGB1 group
(P<0.05). In the LPS+anti-HMGB1 group, ASC showed a significant
decrease compared with the LPS group (Fig. 2H), although a significant decrease
in expression levels of AIM2, ASC and caspase-1 was observed in
Fig. 2G. The activated AIM2
inflammasome induces pro-IL-1β and pro-IL-18 cleavage into active
IL-1β and IL-18. That is to say, IL-1β and IL-18 in the culture
supernatant are downstream of the AIM2 inflammasome in BMMs. They
could indirectly reflect activation of the AIM2 inflammasome in
macrophages. As illustrated in Fig.
2I, the concentrations of IL-1β and IL-18 in culture
supernatants were significantly increased in LPS-primed groups
(P<0.01), with a maximum increase in the rHMGB1 group and
minimum elevation in the anti-HMGB1 group. These results suggest
that HMGB1 may activate the AIM2 inflammasome in macrophages,
accelerating infiltration of inflammatory cells and increasing the
level of its downstream inflammatory cytokines in LPS-induced
ALI.
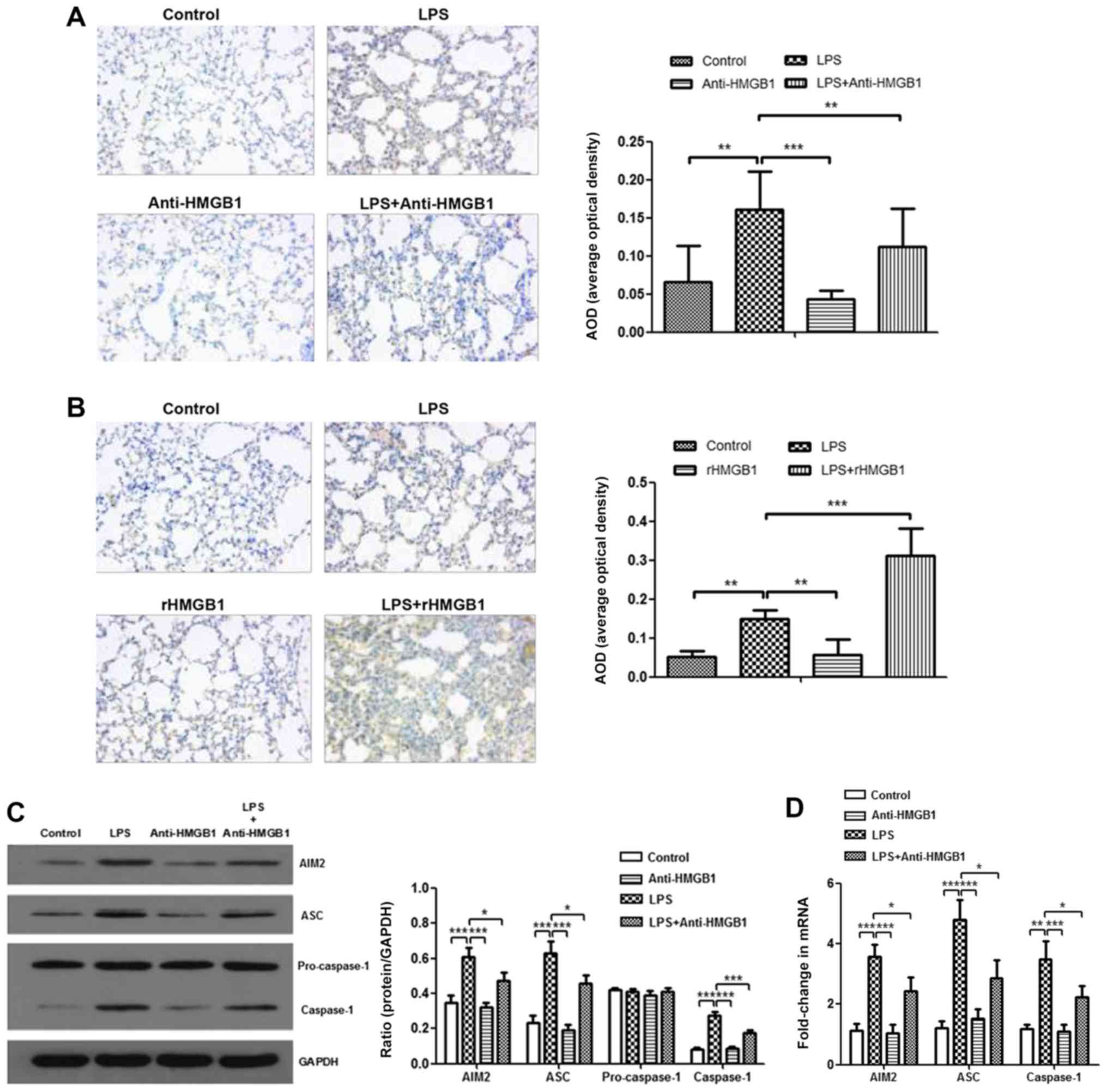 | Figure 2Expression level of AIM2 inflammasome
is upregulated by HMGB1. Effects of (A) anti-HMGB1 and (B) rHMGB1
on the expression level of AIM2 in mouse lung tissue was detected
by immunohistochemistry (magnification, ×200), and AOD was analyzed
in different groups. In the in vivo experiment, the
expression levels of AIM2 inflammasome and GAPDH were detected by
(C and E) western blotting with (C) anti-HMGB1 and (E) rHMGB1 and
RT-qPCR with (D) anti-HMGB1 and (F) rHMGB1. All experiments were
repeated more than three times (n=4-6 mice per each group). Data
presented is from a representative experiment. All data are
expressed as the mean ± standard deviation. *P<0.05,
**P<0.01 and ***P<0.001 vs. LPS group.
Expression level of AIM2 inflammasome is upregulated by HMGB1. In
an experiment with BMMs, the expression levels of the AIM2
inflammasome and GAPDH were also detected by (G) western blotting
and (H) RT-qPCR. (I) The expression levels of IL-1β and IL-18 in
culture supernatant of BMMs were measured by ELISA. All experiments
were repeated more than three times (n=4-6 mice per each group).
Data presented is from a representative experiment. All data are
expressed as the mean ± standard deviation. *P<0.05,
**P<0.01 and ***P<0.001 vs. LPS group.
LPS, lipopolysaccharide; IL, interleukin; rHMGB1, recombinant High
mobility group box 1; RT-q, reverse transcription-quantitative;
AIM2, absent in melanoma 2; AOD, average optical density; BMMs,
bone-marrow derived macrophages; ASC, apoptosis-associated
speck-like protein containing a CARD. |
HMGB1 induces polarization of M1
macrophages
AMs form the first line of defense in the
inflammatory response and phagocytosis of pathogens. A previous
study demonstrated that M1 AMs have been implicated in the
pathogenesis of ALI and M2 AMs were mainly described as having
anti-inflammatory or reparative functions (34). To explore whether HMGB1 had a
relationship with AMs in an LPS-induced ALI mouse model, the
present study attempted to investigate whether HMGB1 regulated
polarization of M1 or M2 macrophages and the function of AMs. Lung
interstitial macrophages (IMs) express the macrophage marker F4/80,
rather than the marker CD11c, whereas lung AMs are F4/80 and CD11c
double-positive MNCs (35,36).
Lung MNCs were subsequently stained with F4/80-APC-Cy7 and
CD11c-PE, and analyzed by flow cytometry. It was found that the
lung F4/80+ macrophages were divided into two subgroups
(IMs and AMs; Fig. 3A).
M1-related surface markers (MHC II, CD80, CD86 and CD40) and
M2-related markers (CD206 and IL-10) from the gating region of lung
AMs (F4/80+CD11c+) were then observed. The
percentage of MHC II, CD80, CD86 and CD40-expressing AMs was
significantly increased in ALI mice compared with in control mice.
It was also noted that their percentages further increased in the
ALI group that was administered rHMGB1 and decreased when treated
with anti-HMGB1. However, the percentage of CD206, IL-10-expressing
AMs showed an antipodal tendency (Fig. 3B and C). As described above, HMGB1
may mediate polarization of M1 macrophages in the ALI mouse model.
In vitro, it was also observed that HMGB1 regulated
polarization of M1 macrophages by detecting the M1-related
cytokines (TNF-α, IL-6 and MCP-1) and M2-related cytokine (IL-10)
in the culture supernatant of BMMs stimulated with anti-HMGB1 or
rHMGB1 by ELISA. The results showed that the expression levels of
TNF-α, IL-6 and MCP-1 increased, while the expression level of
IL-10 decreased in LPS-stimulated group compared with those in the
control group. Moreover, the changes were significantly enhanced by
rHMGB1 stimulation (P<0.05), however they were weakened by
anti-HMGB1 stimulation (Fig. 3D).
Therefore, HMGB1 can induce polarization of M1 macrophages in
vivo and in vitro.
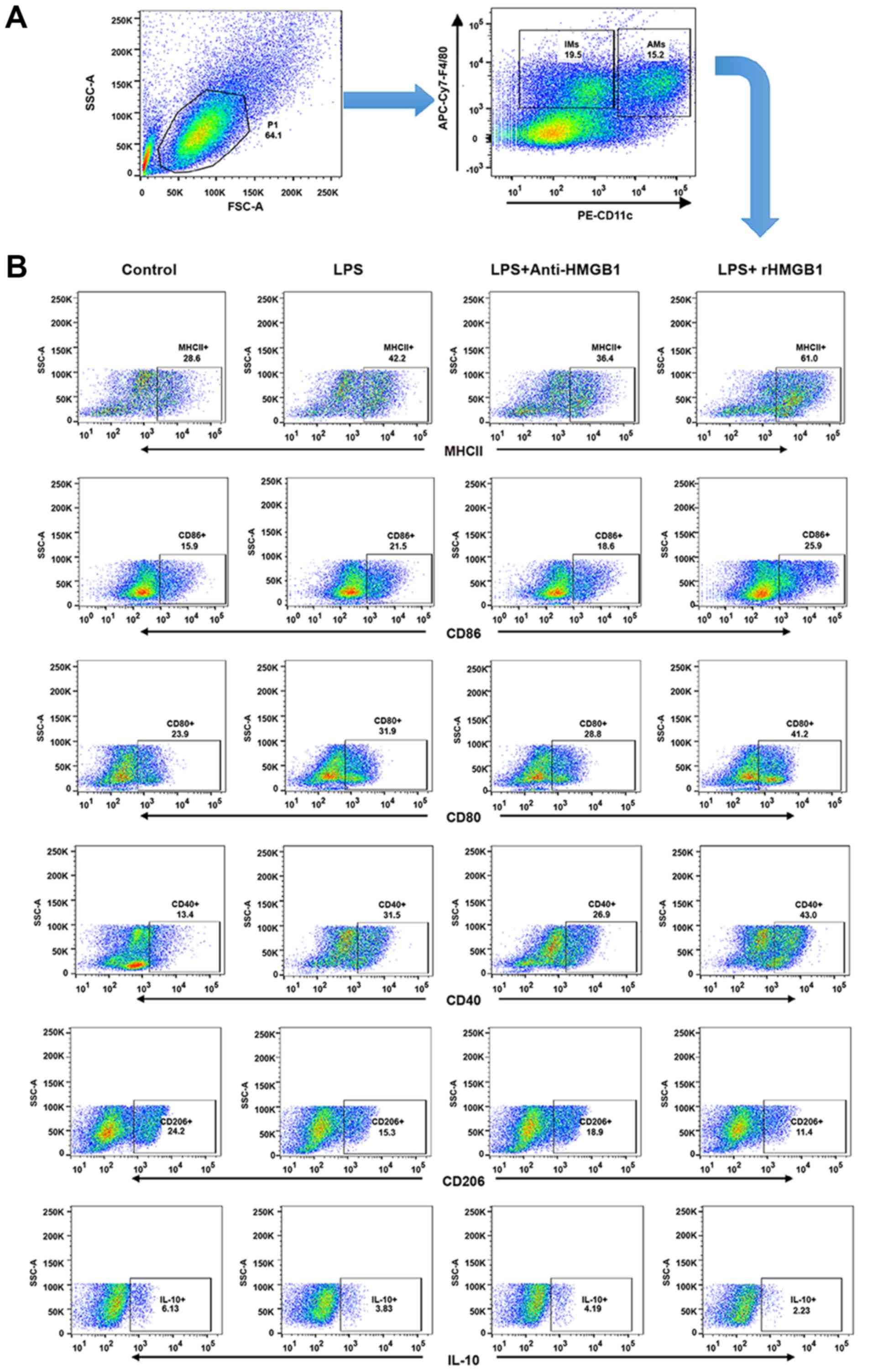 | Figure 3Upregulation surface markers and
cytokines of M1 macrophages by extracellular HMGB1 (A) Flow
cytometry was used to identify F4/80+ and
CD11c+ lung AMs isolated from lung MNCs of mice. IMs
were marked as F4/80+ and CD11c− MNCs. (B)
Detection of expression levels of MHC II+,
CD80+, CD86+, CD40+,
CD206+ and IL-10 on AM cell surface. All experiments
were repeated more than three times (n=4-6 mice per each group).
Data presented is from a representative experiment. All data are
expressed as the mean ± standard deviation. Upregulation surface
markers and cytokines of M1 macrophages by extracellular HMGB1. (C)
The percentages of lung AMs expressing MHC II, CD80, CD86, CD40,
CD206 and IL-10 were calculated. (D) AM activation is defined by
two distinct polarization states: M1 and M2. M1-related cytokines
(tumor necrosis factor-α, IL-6, and MCP-1) and M2-related cytokine
(IL-10) were detected in culture supernatant of BMMs by ELISA. All
experiments were repeated more than three times (n=4-6 mice per
each group). Data presented is from a representative experiment.
All data are expressed as the mean ± standard deviation.
*P<0.05, **P<0.01 and
***P<0.001 vs. LPS group. LPS, lipopolysaccharide;
IL, interleukin; MCP, myeloperoxidase; rHMGB1, recombinant High
mobility group box 1; CD, cluster of differentiation; MHC, major
histocompatibility complex; AMs, alveolar macrophages; MNCs,
mono-nuclear cells; BMMs, bone-marrow derived macrophages. |
HMGB1 acts on macrophages through TLR2,
TLR4 and RAGE/NF-κB signaling pathways
HMGB1 stimulates the inflammatory response through
several pattern-recognition receptors, including RAGE, TLR2 and
TLR4 (5,25,37,38). HMGB1 interacts with those
receptors by activating the NF-κB signaling pathway and downstream
inflammatory mediators (5,39,40).
However, whether HMGB1 acts on macrophages through TLR2, TLR4 and
RAGE/NF-κB signaling pathways in ALI needs to be further verified.
Treatment with anti-HMGB1 was found to inhibit upregulation of
expression levels of LPS-induced TLR2, TLR4, RAGE, NF-κB p65 and
p-NF-κB p65 in the animal experiments by western blot analysis
(Fig. 4A). Whether anti-HMGB1
affected mRNA expression of TLR2, TLR4, RAGE and NF-κB was also
examined. As displayed in Fig.
4B, the same inhibiting effect was observed. In addition,
administration of rHMGB1 enhanced expression levels of LPS-induced
TLR2, TLR4, RAGE, NF-κB p65 and p-NF-κB p65 proteins (Fig. 4C), as well as expression levels of
TLR2, TLR4, RAGE, and NF-κB mRNA (Fig. 4D). Western blotting and RT-qPCR
were also used to analyze LPS-primed BMMs stimulated with
anti-HMGB1 or rHMGB1. As shown in Fig. 4E, administration of LPS
significantly increased the expression levels of TLR2, TLR4, RAGE,
NF-κB p65 and p-NF-κB p65 compared with the control group
(P<0.01). However, stimulation of anti-HMGB1 significantly
decreased their expression levels induced by LPS (P<0.05) and
the increases were weakened upon rHMGB1 stimulation. Similar to the
present findings by western blot analysis, the expression levels of
TLR2, TLR4, RAGE and NF-κB mRNA were notably increased in BMMs
stimulated with LPS. However, these increases were markedly
inhibited by treatment with anti-HMGB1 and significantly promoted
by treatment with rHMGB1 (P<0.05; Fig. 4F). Thus, HMGB1 might act on
macrophages through TLR2, TLR4 and RAGE/NF-κB signaling
pathways.
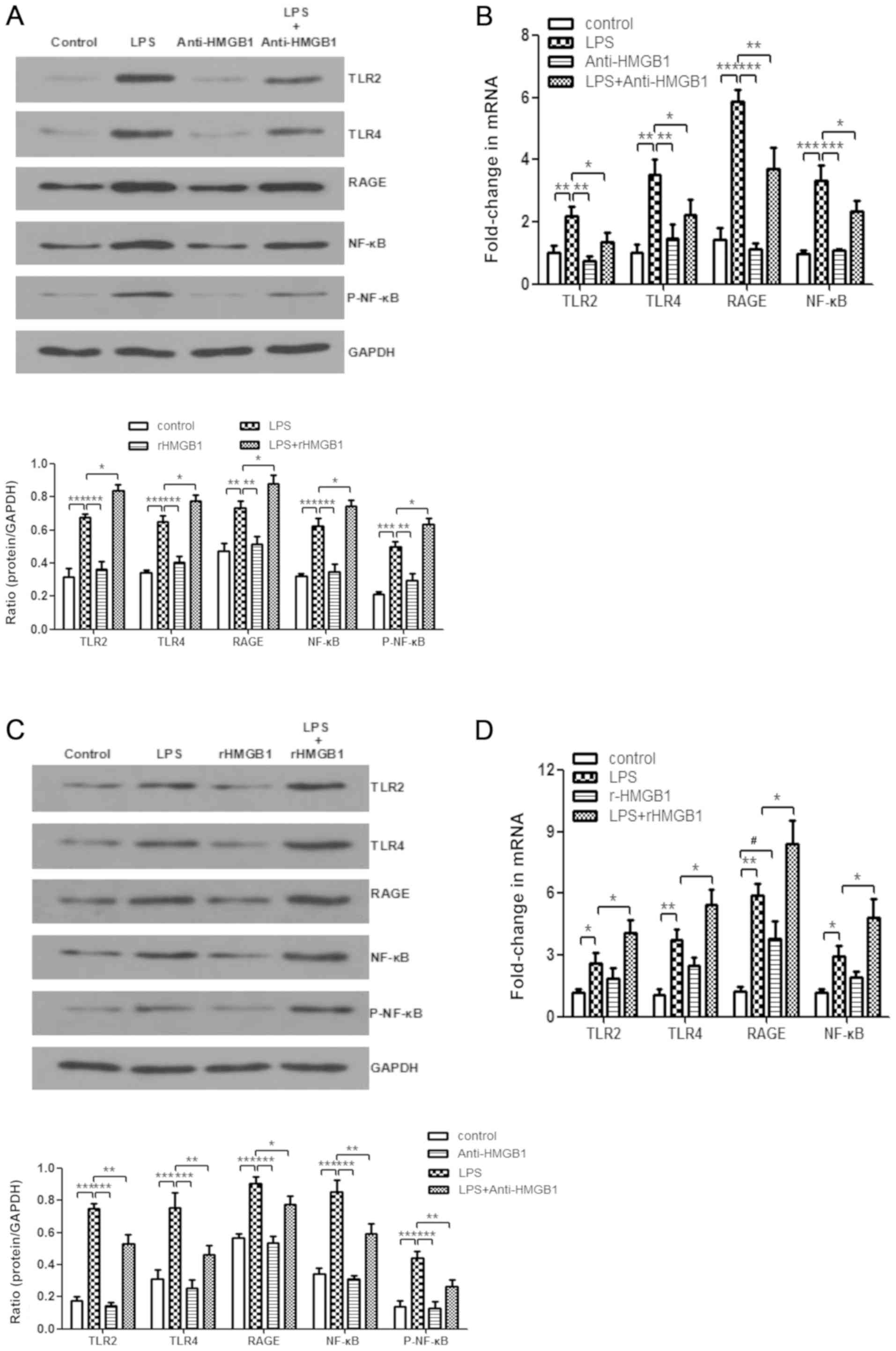 | Figure 4Enhanced expression levels of TLR2,
TLR4, RAGE and NF-κB in macrophages by extracellular HMGB1.
Expression levels of TLR2, TLR4, RAGE, NF-κB p65, p-NF-κB p65, and
GAPDH were detected by western blotting with (A) anti-HMGB1 and (C)
rHMGB1. The expression levels of TLR2, TLR4, RAGE, NF-κB, and GAPDH
were detected by RT-qPCR with (B) anti-HMGB1 and (D) rHMGB1 in the
right lungs of LPS-induced ALI mice and control mice. All
experiments were repeated more than three times (n=4-6 mice per
each group). Data presented is from a representative experiment.
All data are expressed as the mean ± standard deviation.
*P<0.05, **P<0.01 and
***P<0.001 vs. LPS group. #P<0.05 vs.
control group. Enhanced expression levels of TLR2, TLR4, RAGE and
NF-κB in macrophages by extracellular HMGB1. The expression levels
of TLR2, TLR4, RAGE, NF-κB, and GAPDH were detected by RT-qPCR with
anti-HMGB1 and rHMGB1 in the right lungs of LPS-induced ALI mice
and control mice, and in LPS-primed BMMs stimulated with (E)
anti-HMGB1 or (F) rHMGB1. All experiments were repeated more than
three times (n=4-6 mice per each group). Data presented is from a
representative experiment. All data are expressed as the mean ±
standard deviation. *P<0.05, **P<0.01
and ***P<0.001 vs. LPS group. #P<0.05
vs. control group. LPS, lipopolysaccharide; IL, interleukin; MCP,
myeloperoxidase; TNF, tumor necrosis factor; ALI, acute lung
injury; rHMGB1, recombinant High mobility group box 1; TLR,
toll-like receptor; RAGE, receptor for advanced glycation end
products; RT-q, reverse transcription-quantitative; p-NF,
phosphorylated-nuclear factor; BMMs, bone-marrow derived
macrophages. |
Inhibition of TLR2, TLR4 and RAGE reduces
the inflammatory response in LPS-induced ALI mouse model
The effects of TLR2/4 antagonist (LPS-RS) and RAGE
antagonist (FPS-ZM1) on histopathological changes in LPS-induced
ALI were measured by H&E staining. As depicted in Fig. 5A and B, no histological alteration
was observed in lung sections of LPS-RS, FPS-ZM1, and control
groups. Treatment with LPS-RS and FPS-ZM1 significantly ameliorated
LPS-induced lung injury (P<0.001). In accordance with these
results, treatment with LPS-RS or FPS-ZM1 also inhibited the
LPS-induced lung W/D ratio and MPO activity (Fig. 5C-E and G). In BALF, a reduced
number of inflammatory cells (total cells, macrophages and
neutrophils; Fig. 5F and H) and
attenuated levels of proinflammatory cytokines (TNF-α, IL-6, MCP-1,
IL-1β, and IL-18; Fig. 5I and J)
were noted in ALI mice that received LPS-RS or FPS-ZM1. These data
demonstrated that inhibition of TLR2, TLR4 and RAGE reduced the
inflammatory response in LPS-induced ALI mouse model.
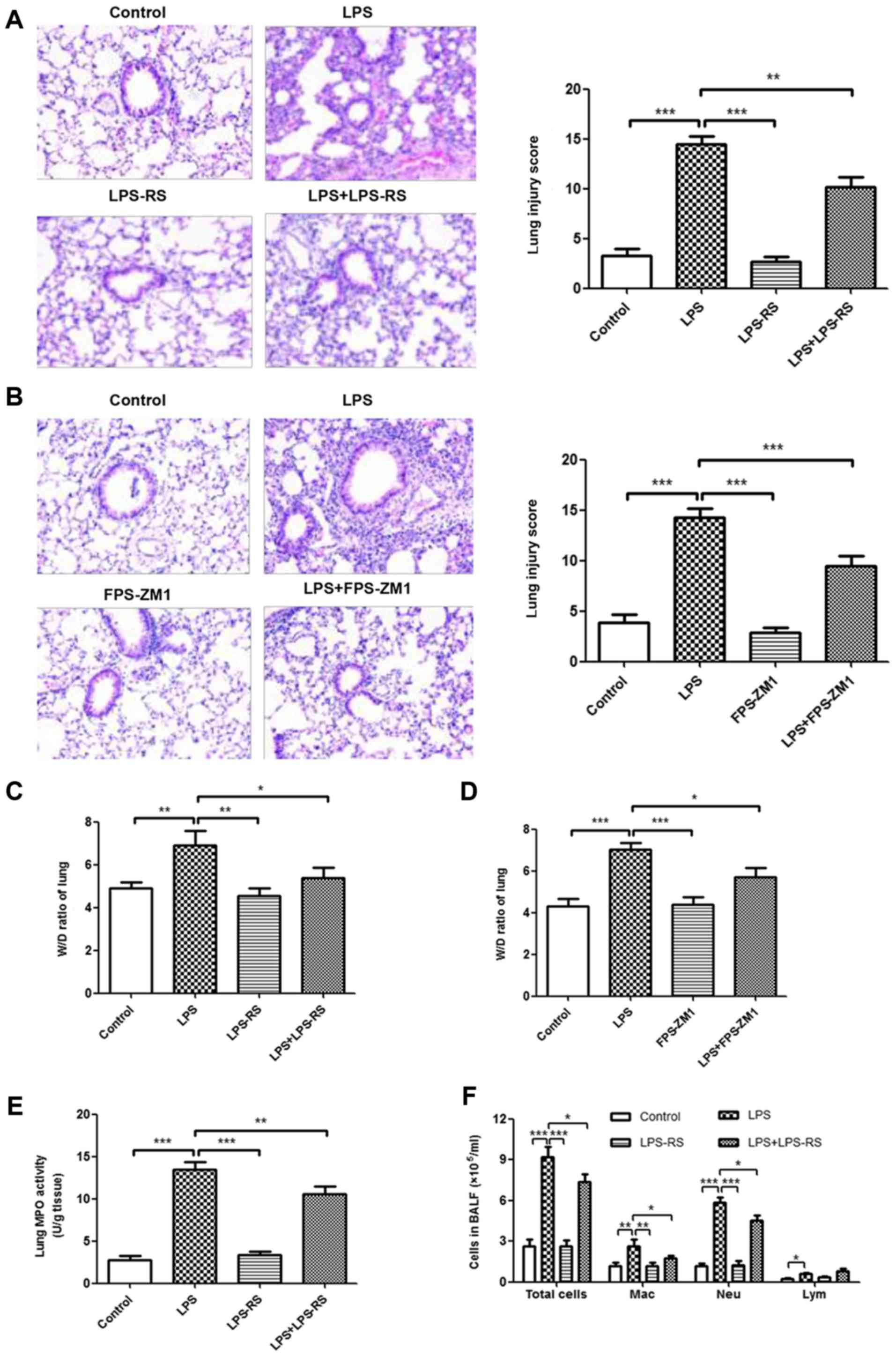 | Figure 5Reduced inflammatory response in lung
tissue in ALI by administration of TLR2/4 and RAGE antagonists.
Histopathological changes and injury scores in left lung are
illustrated for different groups: (A) Control group, LPS group,
LPS-RS group, and LPS + LPS-RS group (H&E staining, ×200). (B)
Control group, LPS group, FPS-ZM1 group and D: LPS+FPS-ZM1 group
(H&E staining, ×200). Effects of (C) LPS-RS and (D) FPS-ZM1 on
the lung W/D ratio of LPS-induced ALI mice. Effects of LPS-RS and
on (E) MPO activity in lung tissues and (F) infiltration of
inflammatory cells in BALF of LPS-induced ALI mice. All experiments
were repeated more than three times (n=4-6 mice per each group).
Data presented is from a representative experiment. All data are
expressed as the mean ± standard deviation. *P<0.05,
**P<0.01 and ***P<0.001 vs. LPS group.
Reduced inflammatory response in lung tissue in ALI by
administration of TLR2/4 and RAGE antagonists. Effects of FPS-ZM1
on (G) MPO activity in lung tissues and (H) infiltration of
inflammatory cells in BALF of LPS-induced ALI mice. ELISA results
of secretion of inflammatory cytokines (TNF-α, IL-6, MCP-1, IL-1β
and IL-18) treated with (I) LPS-RS and (J) FPS-ZM1 in BALF. All
experiments were repeated more than three times (n=4-6 mice per
each group). Data presented is from a representative experiment.
All data are expressed as the mean ± standard deviation.
*P<0.05, **P<0.01 and
***P<0.001 vs. LPS group. LPS, lipopolysaccharide;
IL, interleukin; MCP, myeloperoxidase; TNF, tumor necrosis factor;
ALI, acute lung injury; rHMGB1, recombinant High mobility group box
1; RT-q, reverse transcription-quantitative; NF, nuclear factor;
BMMs, bone-marrow derived macrophages; TLR, toll-like receptor;
RAGE, receptor for advanced glycation end products; BALF,
bronchoalveolar lavage fluid; H&E, hematoxylin and eosin; RS,
Rhodobacter sphaeroides; Mac, macrophage; Neu, neutrophil;
Lym, lymphocyte. |
Inhibition of TLR2, TLR4 and RAGE weakens
the activation of the AIM2 inflammasome
It was demonstrated that HMGB1 mediates the
activation of the AIM2 inflammasome in macrophages and that HMGB1
acts on macrophages through TLR2, TLR4, and RAGE/NF-κB signaling
pathways. Next, whether the AIM2 inflammasome was a downstream
mediator of TLR2, TLR4 and RAGE receptors was investigated.
LPS-induced increases of AIM2, ASC and caspase-1 protein levels, as
well as their mRNA expression levels, were significantly abolished
after treatment with LPS-RS or FPS-ZM1 (P<0.05; Fig. 6A-D). In vitro, compared
with those from the LPS-stimulated group, BMMs treated with LPS-RS
and FPS-ZM1 significantly decreased mRNA expression levels of AIM2,
ASC, and caspase-1 (P<0.05; Fig.
6E and F). These results suggest that inhibiting TLR2, TLR4 and
RAGE weakens the activation of AIM2 inflammasome.
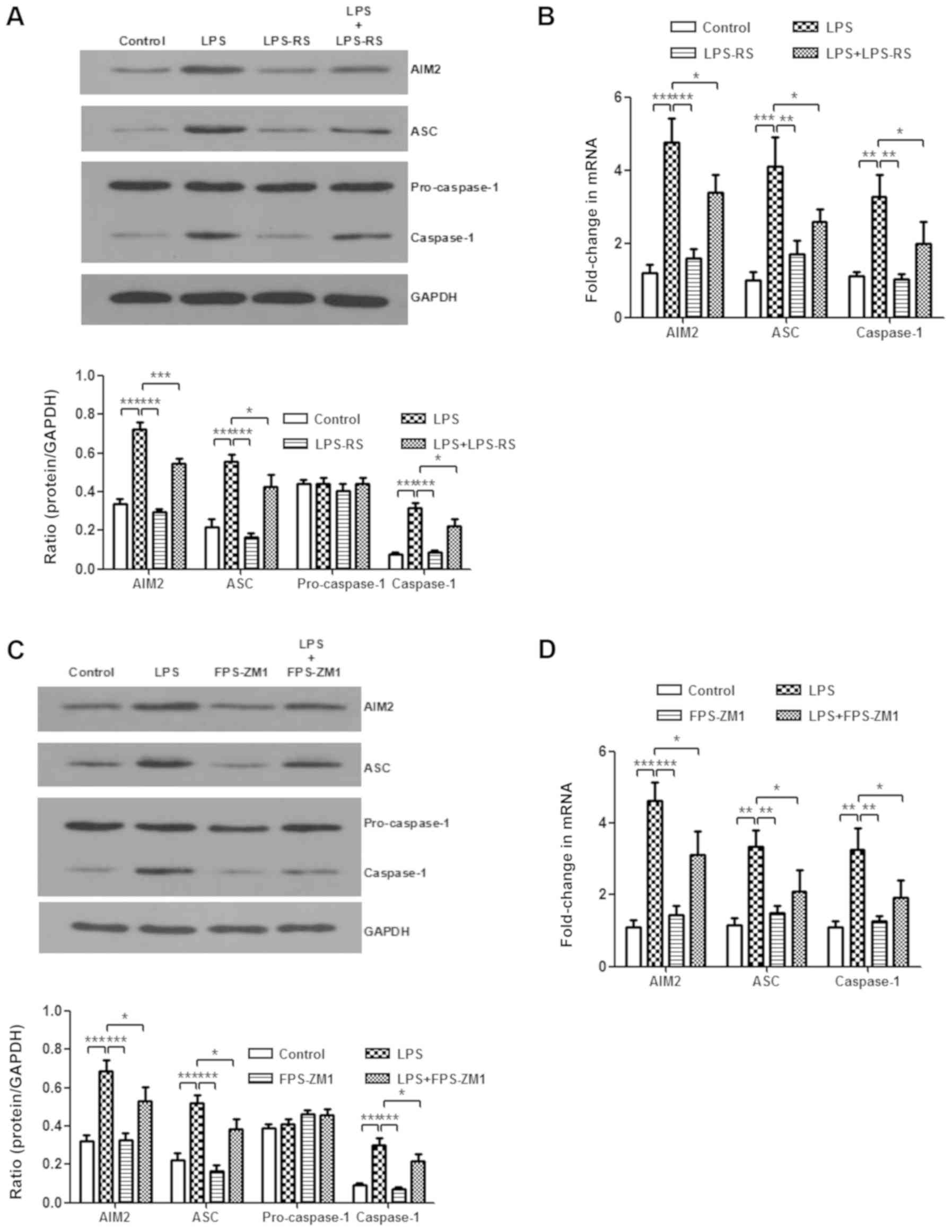 | Figure 6Expression level of the AIM2
inflammasome is downregulated by administration of Toll-like
receptor 2/4 or receptor for advanced glycation end products
antagonists. In the in vivo experiment, the expression
levels of AIM2 inflammasome and GAPDH were detected by western
blotting following treatment with (A) LPS-RS and (C) FPS-ZM1.
Additionally, RT-qPCR following treatment with (B) LPS-RS and (D)
FPS-ZM1 was performed. All experiments were repeated more than
three times (n=4-6 mice per each group). Data presented is from a
representative experiment. All data are expressed as the mean ±
standard deviation. *P<0.05, **P<0.01
and ***P<0.001 vs. LPS group. Expression level of the
AIM2 inflammasome is downregulated by administration of Toll-like
receptor 2/4 or receptor for advanced glycation end products
antagonists. In the in vitro experiment, the expression
levels of AIM2 inflammasome and GAPDH in BMMs were also detected by
(E) western blotting and (F) RT-qPCR. (G) The expression levels of
IL-1β and IL-18 in culture supernatant of BMMs were measured by
ELISA. All experiments were repeated more than three times (n=4-6
mice per each group). Data presented is from a representative
experiment. All data are expressed as the mean ± standard
deviation. *P<0.05, **P<0.01 and
***P<0.001 vs. LPS group. LPS, lipopolysaccharide;
IL, interleukin; rHMGB1, recombinant High mobility group box 1;
RT-q, reverse transcription-quantitative; AIM2, absent in melanoma
2; BMMs, bone-marrow derived macrophages; ASC, apoptosis-associated
speck-like protein containing a CARD; RS, Rhodobacter
sphaeroides. |
Discussion
In the present study, using in vivo and in
vitro experiments, whether extracellular HMGB1 could activate
the AIM2 inflammasome in macrophages, in addition to mediating the
polarization of M1 macrophages, to participate in mediating an
inflammatory response in ALI through TLR2, TLR4 and RAGE/NF-κB
signaling pathways was investigated. An inflammasome can be used as
upstream component to promote the secretion of HMGB1 in macrophages
and mediate inflammatory reactions. Previous studies demonstrated
that extracellular HMGB1 also activated the inflammasome through a
number of pathways to further promote the inflammatory response
(18-20). Additionally, HMGB1 and the
inflammasome have a synergistic influence on each other, causing
inflammasome-aggravated tissue damage. However, the relationship
between HMGB1 and AIM2 in LPS-induced ALI model has remained
elusive. On the other hand, HMGB1 was reported to induce
polarization of M1 macrophages in some studies (41-43). Macrophages could secrete and
increase the expression level of HMGB1 in the inflammatory
reaction. However, the relationship between HMGB1 and polarization
M1 macrophages in LPS-induced ALI model needs to be verified.
HMGB1 can activate NF-κB, mediating the inflammatory
response after binding to TLR2, TLR4 and RAGE on the surface of
inflammatory cells. This classical pathway has been widely
recognized and its application in ALI has been confirmed by
numerous studies (5-10). However, whether this pathway might
be appropriate for the regulation of HMGB1 on macrophages has been
confirmed for the first time in the present study to the best of
our knowledge. The present results showed that HMGB1 can activate
the AIM2 inflammasome in macrophages and promote the polarization
of M1 macrophages during the inflammatory reaction of LPS-ALI. The
current study confirms that both processes may act through the
above-mentioned pathways.
The authors' previous study demonstrated that
treatment with anti-HMGB1 had strong anti-inflammatory effects, as
shown by decreased infiltration of inflammatory cells, W/D ratio,
MPO activity in lung and cytokine production, macrophage and
neutrophil infiltration in BALF, as well as strong proinflamma-tory
influences induced by rHMGB1 administration. HMGB1, participating
in DNA replication and repair and transcriptional regulation of
gene expression, can be passively released by necrotic cells or
actively released by macrophages, DCs, and immune cells. HMGB1 is
also recognized as a cytokine, owing to its mediation of systemic
inflammatory responses through transducing cellular signals
(8). HMGB1 has been investigated
as a proinflammatory mediator in several acute and chronic immune
diseases (44-46). Previous studies demonstrated that
HMGB1 plays a pivotal role in monocyte/macrophage and neutrophil
activation in the early stage of ALI. HMGB1 not only is an
important late mediator of inflammation, but also positively
correlates with severity of disease (47). The increase of HMGB1 level in the
alveolar spaces might induce neutrophilic influx. HMGB1 is secreted
by macrophages and DCs into the alveoli, leading to a severe
systemic inflammatory response in ALI (48,49). Administration of rHMGB1 causes
pulmonary neutrophil accumulation, in addition to the release of
IL-1β, TNF-α and MIP-2, enhancing inflammatory responses (47,50). Inhibiting the expression level of
HMGB1 may represent an underlying approach for preventing or
minimizing ALI (51,52). All those above-mentioned studies
are in line with results of the present study.
Additionally, AIM2 is a member of the IFI20X/IFI16
(PYHIN) protein family (53). The
AIM2 inflammasome is expressed in macrophages and DCs, and its
components are AIM2, ASC, and caspase-1 (15,16). When the AIM2 inflammasomes are
activated, ASC, AIM2 and pro-caspase-1 interact with each other,
leading to their activation. The activated caspase-1 cleaves
pro-IL-1β and pro-IL-18 into active proinflammatory cytokines. The
activation of the AIM2 inflammasome is responsible for processing
and secretion of IL-1β and IL-18 (54,55). AIM2 is a major intracellular
polyprotein inflammatory pathway for the innate immune system in
infection through overproduction of IL-1β and IL-18 (56). Inhibition of the activation of the
AIM2 inflammasome is associated with improved ALI and innate immune
responses (16,56-58). To further investigate the
proinflamma-tory mechanism of HMGB1 in ALI, in the present
research, an attempt was made to study the relationship between
HMGB1 and AIM2 inflammasome in macrophages. Research conducted by
Liu et al (20) showed
that the HMGB1-DNA complex initially induced the activation of the
AIM2 inflammasome and also promoted swift release of
proinflammatory cytokines (IL-1β) via RAGE. The effects of HMGB1 on
activation of the LPS-induced AIM2 inflammasome in macrophages in
in vivo and in vitro experiments were observed. The
findings of the current study showed that HMGB1 enhanced the
activation of the LPS-induced AIM2 inflammasome in macrophages and
promoted the secretion of downstream inflammatory cyto-kines (IL-1β
and IL-18) induced by the AIM2 inflammasome. Therefore, these
results suggested that HMGB1 participates in the pathogenesis of
ALI through activation of the AIM2 inflammasome.
Macrophages are essential for innate immunity and
inflammatory responses, playing a substantial role in lung alveoli
and the alveolar space. Inflammation is related to macrophage
phenotype and function, and this process has been described during
the inflammatory stage of ALI (59). During lung inflammation,
macrophages exhibit two main differentiation states, M1 and M2. M1
macrophages produce the proinflammatory cytokines (TNF-α, IL-6,
IL-1β, IL-12 and MCP-1), and inducible nitric oxide synthase in
response to interferon-γ (IFN-γ). M2 macrophages produce
anti-inflammatory cytokines (IL-10 and IL-1RA) in response to IL-4
and IL-13 (34,60,61). M1-polarized macrophages express
specific surface markers, including MHC II, CD80, CD86, CD40 and
CCR2, whereas M2-polarized macrophages express CD206, dectin-1,
transferrin receptor, and CD200R (34). Increasing evidence suggests that
polarization of M1 macrophages induces tissue injury, which is
involved in the pathogenesis of ALI. In contrast, polarization of
M2 macrophages is characterized by reducing the proinflammatory
response (34,62,63). The present study observed that the
percentage of MHC II, CD80, CD86 and CD40-expressing AMs increased,
while the percentage of CD206, IL-10-expressing AMs decreased with
administration of rHMGB1. Moreover, opposite changes were found in
the anti-HMGB1 group. All the results reflected the HMGB1 induction
of polarization of M1 macrophages in LPS-induced ALI. It was also
observed that stimulation of rHMGB1 increased the expression levels
of M1-related cytokines (TNF-α, IL-6 and MCP-1), while decreased
the expression levels of M2-related cytokine (IL-10) in culture
supernatant of BMMs; however, stimulation of anti-HMGB1 induced the
opposite effect, which indirectly proved the above-mentioned
conclusion. Taken together, HMGB1 could induce polarization of M1
macrophages in vivo and in vitro.
HMGB1 has been reported to interact with at least
three receptors, RAGE, TLR2 and TLR4, to transmit cellular signals.
The RAGE pathway leads to the activation of NF-κB, as well as
promoting secretion of cytokines (TNF-α, IL-6 and IFN-γ).
Activation of TLR2 and TLR4 results in activation of NF-κB through
MyD88 (8). TLR2, TLR4 and RAGE
were expressed at high levels in the lungs by a variety of cells,
including macrophages, DCs and activated endothelial and vascular
smooth muscle cells (8,64,65). To explore the signaling pathway
between HMGB1 and the AIM2 inflammasome in macrophages, it was
evaluated whether TLR2, TLR4, and RAGE/NF-κB signaling pathways
participated in mediating these two inflammatory mediator effects
(5,25,39,40). The results of the present study
showed that extracellular HMGB1 could enhance the expression levels
of components of TLR2, TLR4 and RAGE/NF-κB signaling pathways in
macrophages. Consistent with the present results, several studies
showed that HMGB1 stimulated the inflammatory response through
RAGE, TLR2 and TLR4, and was associated with the upregulation of
RAGE, TLR2, TLR4, and NF-κB expression (23,25,38). In the current research, it was
found that HMGB1 upregulated the expression levels of components of
TLR2 TLR4 and RAGE/NF-κB signaling pathways. To further assess
whether HMGB1 could activate the AIM2 inflammasome in macrophages
through TLR2, TLR4 and RAGE/NF-κB signaling pathways, TLR2/4
(LPS-RS) and RAGE antagonists (FPS-ZM1) were used to intervene on
expression of AIM2 inflammasome in vivo and in vitro.
The present results showed that LPS-RS and FPS-ZM1 reduced
LPS-induced inflammatory response in ALI mouse model, weakened
activation and expression of LPS-induced AIM2 inflammasome in
macrophages, and inhibited AIM2 inflammasome-induced secretion of
downstream inflammatory cytokines (IL-1β and IL-18). Therefore, it
can be concluded that extracellular HMGB1 activated and upregulated
TLR2, TLR4, and RAGE/NF-κB signaling pathways to further activate
the AIM2 inflammasome in macrophages, participating in the
pathogenesis of ALI.
The current study illustrates the importance of the
mechanism of HMGB1 regulation on macrophages. In the treatment of
ALI induced by LPS, to downregulate polarization of M1 macrophages
or to promote polarization of M2 macrophages, or to inhibit the
expression of AIM2 inflammasome, or to apply anti-HMGB1 to decrease
HMGB1, or to apply TLR inhibitor, or RAGE inhibitor to downregulate
inflammatory response were used as control targets to successfully
treat ALI. As a result, to test the levels of inflammatory factors
and decrease their high level through these strategies in ALI
patients are the therapeutic goals of this study. Some
anti-inflammatory drugs are widely applied in clinical practice
though some inhibitors are still being applied in experimental
studies and are a certain distance away from clinical
application.
However, further studies are required to thoroughly
study the complex signaling pathways between extracellular HMGB1
and AIM2 inflammasomes in macrophages in ALI, in addition to via
TLR2, TLR4, and RAGE/NF-κB.
In conclusion, HMGB1 participated in the
pathological mechanism of LPS-induced ALI by activating the AIM2
inflammasomes in macrophages, as well as inducing polarization of
M1 macrophages through TLR2, TLR4 and RAGE/NF-κB signaling
pathways.
Abbreviations:
|
HMGB1
|
high mobility group box 1
|
|
AIM2
|
absent in melanoma 2
|
|
ALI
|
acute lung injury
|
|
NF-κB
|
nuclear factor-κB
|
|
W/D
|
wet-to-dry
|
|
MPO
|
myeloperoxidase
|
|
BALF
|
bronchoalveolar lavage fluid
|
|
DCs
|
dendritic cells
|
|
H&E
|
hematoxylin and eosin
|
Acknowledgments
Not applicable.
Funding
This study was financially supported by the National
Natural Science Foundation of China (grant no. 81571946). The
authors would like to thank staff in the Medical Science Research
Center of Zhongnan Hospital for their assistance (Wuhan,
China).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW and RL carried out the studies, participated in
data collection, and drafted the manuscript. BH and XR performed
the statistical analysis and participated in study design. JL and
ZP participated in acquisition, analysis, or interpretation of
data, and drafted the manuscript as well. All the authors studied
and approved the submitted version of manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Wuhan University
(Wuhan, China), as well as complying with the Animal Welfare
Act.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of
interest.
References
|
1
|
Matthay MA, Ware LB and Zimmerman GA: The
acute respiratory distress syndrome. J Clin Invest. 122:2731–2740.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bellani G, Laffey JG, Pham T, Fan E,
Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley
DF, et al LUNG SAFE Investigators; ESICM Trials Group:
Epidemiology, Patterns of Care, and Mortality for Patients With
Acute Respiratory Distress Syndrome in Intensive Care Units in 50
Countries. JAMA. 315:788–800. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Levy BD and Serhan CN: Resolution of acute
inflammation in the lung. Annu Rev Physiol. 76:467–492. 2014.
View Article : Google Scholar
|
|
4
|
Standiford TJ and Ward PA: Therapeutic
targeting of acute lung injury and acute respiratory distress
syndrome. Transl Res. 167:183–191. 2016. View Article : Google Scholar
|
|
5
|
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao
L, Huang J, Yu Y, Fan XG, Yan Z, et al: HMGB1 in health and
disease. Mol Aspects Med. 40:1–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bae JS: Role of high mobility group box 1
in inflammatory disease: Focus on sepsis. Arch Pharm Res.
35:1511–1523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lotze MT and Tracey KJ: High-mobility
group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal.
Nat Rev Immunol. 5:331–342. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Klune JR, Dhupar R, Cardinal J, Billiar TR
and Tsung A: HMGB1: Endogenous danger signaling. Mol Med.
14:476–484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ueno H, Matsuda T, Hashimoto S, Amaya F,
Kitamura Y, Tanaka M, Kobayashi A, Maruyama I, Yamada S, Hasegawa
N, et al: Contributions of high mobility group box protein in
experimental and clinical acute lung injury. Am J Respir Crit Care
Med. 170:1310–1316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sundén-Cullberg J, Norrby-Teglund A,
Rouhiainen A, Rauvala H, Herman G, Tracey KJ, Lee ML, Andersson J,
Tokics L and Treutiger CJ: Persistent elevation of high mobility
group box-1 protein (HMGB1) in patients with severe sepsis and
septic shock. Crit Care Med. 33:564–573. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee JW, Park JW, Shin NR, Park SY, Kwon
OK, Park HA, Lim Y, Ryu HW, Yuk HJ, Kim JH, et al: Picrasma
quassiodes (D. Don) Benn. attenuates lipopolysaccharide
(LPS)-induced acute lung injury. Int J Mol Med. 38:834–844. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duan JX, Zhou Y, Zhou AY, Guan XX, Liu T,
Yang HH, Xie H and Chen P: Calcitonin gene-related peptide exerts
anti-inflammatory property through regulating murine macrophages
polarization in vitro. Mol Immunol. 91:105–113. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu YC, Zou XB, Chai YF and Yao YM:
Macrophage polarization in inflammatory diseases. Int J Biol Sci.
10:520–529. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
de Zoete MR, Palm NW, Zhu S and Flavell
RA: Inflammasomes. Spring Harb Perspect Biol. 6:a0162872014.
View Article : Google Scholar
|
|
16
|
Zhang H, Luo J, Alcorn JF, Chen K, Fan S,
Pilewski J, Liu A, Chen W, Kolls JK and Wang J: AIM2 Inflammasome
Is Critical for Influenza-Induced Lung Injury and Mortality. J
Immunol. 198:4383–4393. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kang R, Chen R, Xie M, Cao L, Lotze MT,
Tang D and Zeh HJ III: The Receptor for Advanced Glycation End
Products Activates the AIM2 Inflammasome in Acute Pancreatitis. J
Immunol. 196:4331–4337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ngoungoure FP and Owona BA: Withaferin A
modulates AIM2 inflammasome and caspase-1 expression in THP-1
polarized macrophages. Exp Cell Res. 383:1115642019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vande Walle L, Kanneganti TD and Lamkanfi
M: HMGB1 release by inflammasomes. Virulence. 2:162–165. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu L, Yang M, Kang R, Dai Y, Yu Y, Gao F,
Wang H, Sun X, Li X, Li J, et al: HMGB1-DNA complex-induced
autophagy limits AIM2 inflammasome activation through RAGE. Biochem
Biophys Res Commun. 450:851–856. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun Q, Loughran P, Shapiro R, Shrivastava
IH, Antoine DJ, Li T, Yan Z, Fan J, Billiar TR and Scott MJ:
Redox-dependent regulation of hepatocyte absent in melanoma 2
inflammasome activation in sterile liver injury in mice.
Hepatology. 65:253–268. 2017. View Article : Google Scholar
|
|
22
|
Zhou H, Wang Y, Wang W, Jia J, Li Y, Wang
Q, Wu Y and Tang J: Generation of monoclonal antibodies against
highly conserved antigens. PLoS One. 4:e60872009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han L, Zhang M, Wang M, Jia J, Zhao M, Fan
Y and Li X: High Mobility Group Box-1 Promotes Inflammation-Induced
Lymphangiogenesis via Toll-Like Receptor 4-Dependent Signalling
Pathway. PLoS One. 11:e01541872016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang C, Liu XX, Huang KB, Yin SB, Wei JJ,
Hu YF, Gu Y and Zheng GQ: Preconditioning with recombinant
high-mobility group box 1 induces ischemic tolerance in a rat model
of focal cerebral ischemia-reperfusion. J Neurochem. 137:576–588.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen Y, Huang XJ, Yu N, Xie Y, Zhang K,
Wen F, Liu H and Di Q: HMGB1 Contributes to the Expression of
P-Glycoprotein in Mouse Epileptic Brain through Toll-Like Receptor
4 and Receptor for Advanced Glycation End Products. PLoS One.
10:e01409182015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abdelmageed ME, El-Awady MS, Abdelrahim M
and Suddek GM: LPS-RS attenuation of lipopolysaccharide-induced
acute lung injury involves NF-κB inhibition. Can J Physiol
Pharmacol. 94:140–146. 2016. View Article : Google Scholar
|
|
27
|
Krikun G, Trezza J, Shaw J, Rahman M,
Guller S, Abrahams VM and Lockwood CJ: Lipopolysaccharide appears
to activate human endometrial endothelial cells through
TLR-4-dependent and TLR-4-independent mechanisms. Am J Reprod
Immunol. 68:233–237. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deane R, Singh I, Sagare AP, Bell RD, Ross
NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ, et al: A
multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain
disorder in a mouse model of Alzheimer disease. J Clin Invest.
122:1377–1392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhong WT, Wu YC, Xie XX, Zhou X, Wei MM,
Soromou LW, Ci XX and Wang DC: Phillyrin attenuates LPS-induced
pulmonary inflammation via suppression of MAPK and NF-κB activation
in acute lung injury mice. Fitoterapia. 90:132–139. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xiao X, Yang M, Sun D and Sun S: Curcumin
protects against sepsis-induced acute lung injury in rats. J Surg
Res. 176:e31–e39. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
32
|
Zhang F, Huang G, Hu B, Fang LP, Cao EH,
Xin XF, Song Y and Shi Y: Anti-HMGB1 neutralizing antibody
ameliorates neutrophilic airway inflammation by suppressing
dendritic cell-mediated Th17 polarization. Mediators Inflamm.
2014:2579302014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang F, Huang G, Hu B, Qian GS and Song
Y: Recombinant HMGB1 A box protein inhibits Th17 responses in mice
with neutrophilic asthma by suppressing dendritic cell-mediated
Th17 polarization. Int Immunopharmacol. 24:110–118. 2015.
View Article : Google Scholar
|
|
34
|
Aggarwal NR, King LS and D'Alessio FR:
Diverse macrophage populations mediate acute lung inflammation and
resolution. Am J Physiol Lung Cell Mol Physiol. 306:L709–L725.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nie H, Wang A, He Q, Yang Q, Liu L, Zhang
G, Huang Y, Ding X, Yu H and Hu S: Phenotypic switch in lung
interstitial macrophage polarization in an ovalbumin-induced mouse
model of asthma. Exp Ther Med. 14:1284–1292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mathie SA, Dixon KL, Walker SA, Tyrrell V,
Mondhe M, O'Donnell VB, Gregory LG and Lloyd CM: Alveolar
macrophages are sentinels of murine pulmonary homeostasis following
inhaled antigen challenge. Allergy. 70:80–89. 2015. View Article : Google Scholar :
|
|
37
|
Pilzweger C and Holdenrieder S:
Circulating HMGB1 and RAGE as Clinical Biomarkers in Malignant and
Autoimmune Diseases. Diagnostics (Basel). 5:219–253. 2015.
View Article : Google Scholar
|
|
38
|
Fang F and Jiang D: IL-1β/HMGB1 signalling
promotes the inflammatory cytokines release via TLR signalling in
human intervertebral disc cells. Biosci Rep. 36:pii: e00379. 2016.
View Article : Google Scholar
|
|
39
|
Watanabe K, Karuppagounder V, Arumugam S,
Thandavarayan RA, Pitchaimani V, Sreedhar R, Afrin R, Harima M,
Suzuki H, Suzuki K, et al: Pruni cortex ameliorates skin
inflammation possibly through HMGB1-NFκB pathway in house dust mite
induced atopic dermatitis NC/Nga transgenic mice. J Clin Biochem
Nutr. 56:186–194. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kang N, Hai Y, Yang J, Liang F and Gao CJ:
Hyperbaric oxygen intervention reduces secondary spinal cord injury
in rats via regulation of HMGB1/TLR4/NF-κB signaling pathway. Int J
Clin Exp Pathol. 8:1141–1153. 2015.
|
|
41
|
Tian S, Zhang L, Tang J, Guo X, Dong K and
Chen SY: HMGB1 exacerbates renal tubulointerstitial fibrosis
through facilitating M1 macrophage phenotype at the early stage of
obstructive injury. Am J Physiol Renal Physiol. 308:F69–F75. 2015.
View Article : Google Scholar :
|
|
42
|
Son M, Porat A, He M, Suurmond J,
Santiago-Schwarz F, Andersson U, Coleman TR, Volpe BT, Tracey KJ,
Al-Abed Y, et al: C1q and HMGB1 reciprocally regulate human
macrophage polarization. Blood. 128:2218–2228. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Schaper F, de Leeuw K, Horst G, Bootsma H,
Limburg PC, Heeringa P, Bijl M and Westra J: High mobility group
box 1 skews macrophage polarization and negatively influences
phagocytosis of apoptotic cells. Rheumatology (Oxford).
55:2260–2270. 2016. View Article : Google Scholar
|
|
44
|
Cavone L, Cuppari C, Manti S, Grasso L,
Arrigo T, Calamai L, Salpietro C and Chiarugi A: Increase in the
Level of Proinflammatory Cytokine HMGB1 in Nasal Fluids of Patients
With Rhinitis and its Sequestration by Glycyrrhizin Induces
Eosinophil Cell Death. Clin Exp Otorhinolaryngol. 8:123–128. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cuppari C, Manti S, Chirico V, Caruso R,
Salpietro V, Giacchi V, Laganà F, Arrigo T, Salpietro C and
Leonardi S: Sputum high mobility group box-1 in asthmatic children:
A noninvasive sensitive biomarker reflecting disease status. Ann
Allergy Asthma Immunol. 115:103–107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gangemi S, Casciaro M, Trapani G,
Quartuccio S, Navarra M, Pioggia G and Imbalzano E: Association
between HMGB1 and COPD: A Systematic Review. Mediators Inflamm.
2015:1649132015. View Article : Google Scholar
|
|
47
|
Kim JY, Park JS, Strassheim D, Douglas I,
Diaz del Valle F, Asehnoune K, Mitra S, Kwak SH, Yamada S, Maruyama
I, et al: HMGB1 contributes to the development of acute lung injury
after hemorrhage. Am J Physiol Lung Cell Mol Physiol.
288:L958–L965. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nogueira-Machado JA and de Oliveira Volpe
CM: HMGB-1 as a target for inflammation controlling. Recent Pat
Endocr Metab Immune Drug Discov. 6:201–209. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lamkanfi M, Sarkar A, Vande Walle L,
Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD and Dixit
VM: Inflammasome-dependent release of the alarmin HMGB1 in
endotoxemia. J Immunol. 185:4385–4392. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Deng Y, Yang Z, Gao Y, Xu H, Zheng B,
Jiang M, Xu J, He Z and Wang X: Toll-like receptor 4 mediates acute
lung injury induced by high mobility group box-1. PLoS One.
8:e643752013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hidaka S, Iwasaka H, Hagiwara S and
Noguchi T: Gabexate mesilate inhibits the expression of HMGB1 in
lipopolysac-charide-induced acute lung injury. J Surg Res.
165:142–150. 2011. View Article : Google Scholar
|
|
52
|
Entezari M, Javdan M, Antoine DJ, Morrow
DM, Sitapara RA, Patel V, Wang M, Sharma L, Gorasiya S, Zur M, et
al: Inhibition of extracellular HMGB1 attenuates hyperoxia-induced
inflammatory acute lung injury. Redox Biol. 2:314–322. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ludlow LE, Johnstone RW and Clarke CJ: The
HIN-200 family: More than interferon-inducible genes? Exp Cell Res.
308:1–17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rathinam VA, Jiang Z, Waggoner SN, Sharma
S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, et
al: The AIM2 inflammasome is essential for host defense against
cytosolic bacteria and DNA viruses. Nat Immunol. 11:395–402. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hu S, Peng L, Kwak YT, Tekippe EM, Pasare
C, Malter JS, Hooper LV and Zaki MH: The DNA Sensor AIM2 Maintains
Intestinal Homeostasis via Regulation of Epithelial Antimicrobial
Host Defense. Cell Rep. 13:1922–1936. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fernandes-Alnemri T, Yu JW, Juliana C,
Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott
E, et al: The AIM2 inflammasome is critical for innate immunity to
Francisella tularensis. Nat Immunol. 11:385–393. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Man SM, Karki R and Kanneganti TD: AIM2
inflammasome in infection, cancer, and autoimmunity: Role in DNA
sensing, inflammation, and innate immunity. Eur J Immunol.
46:269–280. 2016. View Article : Google Scholar
|
|
58
|
Karki R, Man SM, Malireddi RKS, Gurung P,
Vogel P, Lamkanfi M and Kanneganti TD: Concerted activation of the
AIM2 and NLRP3 inflammasomes orchestrates host protection against
Aspergillus infection. Cell Host Microbe. 17:357–368. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Herold S, Mayer K and Lohmeyer J: Acute
lung injury: How macrophages orchestrate resolution of inflammation
and tissue repair. Front Immunol. 2:652011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lawrence T and Natoli G: Transcriptional
regulation of macrophage polarization: Enabling diversity with
identity. Nat Rev Immunol. 11:750–761. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Gordon S and Martinez FO: Alternative
activation of macrophages: Mechanism and functions. Immunity.
32:593–604. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Davis MJ, Tsang TM, Qiu Y, Dayrit JK,
Freij JB, Huffnagle GB and Olszewski MA: Macrophage M1/M2
polarization dynamically adapts to changes in cytokine
microenvironments in Cryptococcus neoformans infection. MBio.
4:e00264–e13. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lu G, Zhang R, Geng S, Peng L, Jayaraman
P, Chen C, Xu F, Yang J, Li Q, Zheng H, et al: Myeloid cell-derived
inducible nitric oxide synthase suppresses M1 macrophage
polarization. Nat Commun. 6:66762015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wolf L, Herr C, Niederstraßer J,
Beisswenger C and Bals R: Receptor for advanced glycation
endproducts (RAGE) maintains pulmonary structure and regulates the
response to cigarette smoke. PLoS One. 12:e01800922017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Fallah MP, Chelvarajan RL, Garvy BA and
Bondada S: Role of phosphoinositide 3-kinase-Akt signaling pathway
in the age-related cytokine dysregulation in splenic macrophages
stimulated via TLR-2 or TLR-4 receptors. Mech Ageing Dev.
132:274–286. 2011. View Article : Google Scholar : PubMed/NCBI
|














