Introduction
Hypoxic/ischemic (HI) brain damage (HIBD) is a
serious complication of parturition caused by the deficiency of
blood and oxygen supply in the neonatal brain (1). Moreover, HIBD is a major cause of
acute neonatal brain injury, leading to high mortality and serious
neurological deficits, such as behavioral, social and cognitive
deficits (2). It has been
reported that mortality occurs in ~20% of neonates with HIBD during
the postnatal period, and an additional 25% of neonates with HIBD
develop irreversible and lifelong mental and physical disabilities
(3). Therapeutic hypothermia is
the current method for the treatment of HI encephalopathy. However,
mortality and significant neurologic disability after hypothermia
treatment still occur in ~50% of patients (4,5).
Thus, the underlying mechanism of HIBD requires further
investigation in order to facilitate the development of novel
therapeutic methods.
Long non-coding RNAs (lncRNAs), which are non-coding
RNA transcripts, have gained increased attention due to their roles
in several physiological and pathological processes (6). For example, lncRNAs play important
roles in RNA processing, stabilization, metabolism and translation
(7,8). Previous studies have reported that
cerebral lncRNA expression profiles are extensively altered during
brain damage. For instance, ischemia leads to temporal changes in
the cerebral transcriptome that affects the neurologic outcome
after brain damage (9,10). Moreover, lncRNAs such as Fos
Downstream Transcript, Metastasis Associated Lung Adenocarcinoma
Transcript 1 and growth arrest-specific 5 have been shown to be
involved in ischemic stroke (11-13). Thus, it was speculated that
lncRNAs are potential regulators of neonatal brain HI injury.
Brain-derived neurotrophic factor (BDNF) is a
canonical nerve growth factor highly expressed in the brain, and
the binding of BDNF protein to its receptor contributes to neuronal
survival (14). It has also been
revealed that BDNF plays an important role in stress responses and
brain disorders (15).
lncRNA-BDNF-antisense (AS) is the antisense RNA transcript of BDNF
(16). BDNF-AS is widely
expressed in various human tissues, and may have crosstalk with
BDNF expression in the regulation of neural cell function (17). Previous studies have revealed that
inhibition of BDNF-AS exerts neuroprotective effects on ischemic
damage in retinal ganglion neurons (18,19). However, direct evidence of the
role of BDNF-AS in HI neonatal brain damage is still lacking.
Based on the potential crosstalk between BDNF-AS and
BDNF, the present study hypothesized that BDNF-AS serves an
important role in HI-induced brain injury. Therefore, the role of
BDNF-AS in neonatal HIBD was investigated, as well as the
underlying mechanism of BDNF-AS-mediated brain injury.
Materials and methods
HI neonatal brain injury model
A total of 30 pregnant C57BL/6J (wild-type) mice
(age, 14-16 weeks; weight, 30–40 g) were purchased from Nanjing
Qinglongshan Experimental Animal Center. The day of birth was
considered day 0. After birth, the pups were housed with their dam
under a 12:12-h light-dark cycle at a temperature of 23±3°C and
relative humidity of 50±5%, with food and water available ad
libitum throughout the study. The male 7-day-old postnatal (P7)
pups (weight, 10-15 g) were anesthetized with isoflurane (3% in a
mixture of medical air and oxygen 70:30 ratio). A 5.0 silk surgical
suture was used to ligate the right common carotid artery. The
artery was cut between two ligation sites. After the surgery, the
pups were recovered for 1 h and then placed in the hypoxic
incubator containing 8% oxygen and 92% nitrogen at 37°C for 2 days,
5 days, 7 days or 8 weeks. Sham animals were selected as the
control group, and underwent anesthesia and the common carotid
artery was exposed without ligation and hypoxia. The normal brains
from P7 mice without any treatment were used as the normal group.
In total, six animals were used in each experimental group. All
procedures were approved by the Animal Care and Use Committee of
Nanjing University.
The male pups received a stereotaxic injection of
BDNF-AS short hairpin (sh)RNA (5′-
CCGGCCGGCATTGGAACTCCCAGTGTTCAAGACGCACTGGGAGTTCCAATGCCTTTTTTG-3′) or
scrambled shRNA
(5′-AATGCCAGTTCCGGTTTTTTGGCCGGCCTGGGAACTGGCATTTGTTCAAGACCCCAGCAC-3′)
of 2 µl concentrated BDNF-AS shRNA viral stocks
(3×109 particles/µl) or scrambled shRNA viral
stocks (3×109 particles/µl) into the hippocampus
after HI treatment. The injection was performed using a Hamilton
syringe and conducted unilaterally at the following coordinates
calculated from Bregma and the skull surface: Anterior, -2.4;
lateral, +1.5 (right side); ventral, -2.0 (20). Then, 48 h after HI, measurement of
the brain infarct size and immunostaining of Glial fibrillary
acidic protein (GFAP), CD11b/c or neuronal nuclei (NeuN) were
performed in the cornu ammonis 1 hippocampal region. Subsequently,
8 weeks after HI, the neurobehavioral function of the brain was
evaluated by water maze or rotarod tests.
Isolation of primary hippocampal neuron
and stress induction
Animal experiments were approved by the Animal Care
and Use Committee of Nanjing University, and were conducted
according to the suggested method of euthanasia of fetuses by the
University of California, Los Angeles (https://rsawa.research.ucla.edu/arc/euthanasia-fetuses/).
The pregnant mice at ~18 days post-fertilization were euthanized by
cervical dislocation. The use of anesthesia to euthanize the
pregnant female is not recommended, as anesthesia is known to cause
brain cell death (21,22). The fetuses were euthanized by
decapitation. Sterile scissors were used to open the cranium of pup
from back of the neck to the nose. The entire brain was removed
with the forceps. Then, a small section of meninges surrounding the
hippocampus was grasped with the sterile forceps and pulled gently
away. The hippocampi were treated with 0.25% trypsin
(Sigma-Aldrich; Merck KGaA) supplemented with 100 ng/ml DNase
(Roche Diagnostics) at 37°C for 30 min and dissociated using
repeated passage through a series of fire-polished constricted
Pasteur pipettes. The digestion was terminated with DMEM (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 5% FBS (Gibco;
Thermo Fisher Scientific, Inc.). The hippocampal neurons were
distributed in 8-well poly-l-lysine-treated chamber slide and
cultured in the neurobasal medium (DMEM; 4.5 g/liter glucose; 100
U/ml penicillin; 100 µg/ml streptomycin; 2 mM glutamine) at
5×104 cell/well. Cells were incubated at 37°C with 5%
CO2 and 95% relative humidity. Half of the medium was
replaced once a week. Hippocampal cultures were used after a
culturing period of 10-14 days. For hypoxic induction, cells were
cultured in a NAPCO 7001 incubator (Precision Scientific Company)
with 1% oxygen, 6% CO2 and balance nitrogen at 37°C for
24 and 48 h. For oxidative stress induction, cells were exposed to
H2O2 (150 µM) to mimic oxidative
stress for 24 and 48 h.
Detection of brain infarct size
The male pups were anesthetized after neurological
evaluation by intraperitoneal injection of ketamine (80 mg/kg) and
xylazine (4 mg/kg) and euthanized by cervical dislocation. The
death of mice was verified using the following criteria, including
lack of pulse, breathing, corneal reflex, failure of a response to
firm toe pinch, graying of mucus membranes and an inability to
auscultate respiratory or heart sounds. The brain was quickly
removed and placed in ice-cold sterile saline for 5 min. The
infarct size of brain was detected with 2,3,5-triphenyltetrazolium
chloride mono-hydrate (TTC; Sigma-Aldrich) staining (23). The slices of pup brains were cut
into 2-mm thick sections and were immersed in 2% TTC solution for 5
min at 37°C. The slices were then fixed by 10% formaldehyde at 4°C
overnight. The caudal and the rostral surfaces of each slice were
imaged using a fluorescence microscope (magnification, ×400). The
percentage of infarct size for each slice was traced and analyzed
using the Image-Pro plus version 7.0 software (Media Cybernetics,
Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR) assay
In HI neonatal brain injury model, the male pups
were anaesthetized with 3% isoflurane, decapitated and the
striatum, hippocampi and cortex were excised and stored in liquid
nitrogen for RNA extraction. Total RNAs were extracted from the
primary hippocampal neurons or infarcted brain tissues using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Ultraviolet
analysis and formaldehyde gel electrophoresis were performed to
detect RNA quality. RT was performed using the miScript II RT kit
(Invitrogen; Thermo Fisher Scientific, Inc.).
The temperature protocols for RT were as follows:
37°C for 60 min, 95°C for 5 min and maintenance at 4°C. Following
this step, qPCR assays were performed using the Brilliant SYBR
Green II RT-qPCR kit (Agilent Technologies, Inc.) according to the
manufacturer's instructions. qPCR assays were performed in a final
volume of 25 µl and each PCR reaction mixture consisted of
the specific primers and SYBR Green Supermix. The thermocycling
protocols were as follows: Initial denaturation at 95°C for 2 min,
followed by 40 cycles at 95°C for 10 sec, 55°C for 30 sec and 72°C
for 30 sec, followed by the final extension at 72°C for 60 sec. All
qPCR assays were performed in triplicate and normalized to GAPDH.
All reactions were carried out on the Applied Biosystems 7500 RT
PCR system (Thermo Fisher Scientific, Inc.). Relative gene
expression was analyzed using the 2−ΔΔCq method
(24). The primers were designed
as follows: BDNF-AS forward, 5′-CCGTGAGAAGATCTCATTGGG-3′ and
reverse, 5′-GGGTCACAAGTCACGTAGCA-3′; and GAPDH forward,
5′-GTCAAGGCTGAGAACGGGAA-3′ and reverse,
5′-AAATGAGCCCCAGCCTTCTC-3′.
Cell Counting Kit (CCK)-8 assay
CCK-8 assay kit was performed to detect cell
viability (Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. Primary hippocampal cells were seeded
onto 96-well plates at the density of 5×104 cells/well.
Then, ~10 µl CCK-8 solution was added to each well of the
plate after hypoxic stress induction (1% oxygen, 6% CO2
and balance nitrogen) with or without BDNF-AS expression
intervention and incubated for 3 h at 37°C. Cell viability was
detected by a microplate reader (Molecular Devices, LLC) at 450
nm.
Calcein - AM/propidium iodide (PI)
staining
Calcein-AM/PI double staining was performed to
detect the apoptosis of hippocampal cells. Hippocampal cells were
fixed in 4% paraformaldehyde (Beyotime Institute of Biotechnology)
for 10 min at 37°C, stained using Calcein-AM (10 µmol/l;
Biosharp Life Sciences) for 10 min at 37°C and then stained with PI
(10 µmol/l; BD Pharmingen; BD Biosciences) for 10 min at
37°C. The 490 nm excitation filter was used to observe the alive
cells. The 545 nm excitation filter was used to observe the dead or
dying cells. Calcein-AM/PI double staining was observed in ten
selected visual fields under an IX71 inverted light microscope
(Olympus Corporation) at magnification, ×500.
Hoechst staining
Primary hippocampal cells were fixed in 4%
paraformaldehyde (Beyotime Institute of Biotechnology) for 15 min
at room temperature. After permeabilizing with Triton-X 100, cells
were stained with Hoechst 33342 solution (10 µg/ml; Biosharp
Life Sciences) for an additional 10 min at 37°C. Hoechst staining
was observed in ten selected visual fields under an IX71 inverted
fluorescent microscope (Olympus Corporation) at magnification,
×500.
Immunofluorescence staining
The male pups were anesthe-tized after neurological
evaluation by intraperitoneal injection of ketamine (80 mg/kg) and
xylazine (4 mg/kg) and euthanized by cervical dislocation. The
brains were quickly removed and the slices of brain were cut into
2-mm thick sections. The brain slices were fixed in 4%
paraformaldehyde (Beyotime Institute of Biotechnology) for 10 min
at room temperature. Then, brain slices were blocked with 5% BSA
(Beyotime Institute of Biotechnology) for 1 h at room temperature,
incubated antibodies against CD11b/c (Abcam; 1:100; cat. no.
ab226482), GFAP (Abcam; 1:100; cat. no. ab7260) and NeuN (Abcam;
1:100; cat. no. ab128886) overnight at 4°C, followed by the Alexa
Fluor 594-conjugated secondary antibody (Invitrogen; Thermo Fisher
Scientific, Inc.; 1:200; cat. no. R37117) for 1 h at room
temperature. Fluorescent mounting medium was used to mount the
slices. All slices were observed using a fluorescent microscope.
CD11b/c and GFAP staining was observed in ten selected visual
fields under an IX71 inverted fluorescent microscope (Olympus
Corporation) at magnification, ×200. NeuN staining was observed in
ten selected visual fields at magnification, ×500.
Western blotting
Primary hippocampal cells were lysed in RIPA lysis
buffer (Beyotime Institute of Biotechnology) and the concentration
of protein was determined using a bicinchoninic acid assay
(Beyotime Institute of Biotechnology). A total of 30 µg
total proteins were separated by 10-12% SDS-PAGE and transferred to
PVDF membranes (Beyotime Institute of Biotechnology). The membranes
were blocked with 5% BSA (Sigma-Aldrich; Merck KGaA) for 1 h at
room temperature and then incubated with the primary antibody
overnight at 4°C using antibodies against BDNF (1:1,000; cat. no.
ab108319; Abcam), tropomyosin receptor kinase B (TrkB; 1:1,000;
cat. no. ab33655; Abcam), phosphorylated (p)-TrkB (phospho S479;
1:2,000; cat. no. ab228507; Abcam), Akt (1:2,000; cat. no. ab18785;
Abcam), p-Akt (phosphor T308; 1:2,000; cat. no. ab38449; Abcam) or
GAPDH (1:2,000; cat. no. ab181602; Abcam). Each PVDF membrane was
washed three times in Tris buffered saline-0.1% Tween-20 for 10 min
per wash and then incubated with the horseradish
peroxidase-conjugated secondary antibody (1:1,000; cat. no. A0208;
Beyotime Institute of Biotechnology) at room temperature for 1 h.
The membranes were washed three times in Tris buffered saline-0.1%
Tween-20 for 10 min per wash and then visualized using enhanced
chemiluminescence methods (Nanjing KeyGen Biotech Co., Ltd.).
Quantity One software version 4.6.5 (Bio-Rad Laboratories, Inc.)
was used to analyze protein bands, which were expressed as a
relative level against the internal reference.
Lentiviral overexpression of BDNF-AS
The lentiviral vector expressing BDNF-AS cDNA (cat.
no. LQP0010926) was purchased from Guangzhou RiboBio Co., Ltd. The
controlled lentiviral vector expressing a non-specific cDNA (cat.
no. LQP0010926-C) was also purchased from Guangzhou RiboBio Co.,
Ltd. The primary hippocampal cells were transduced with the
lentiviral vector containing either BDNF-AS (1×1013
U/ml) or non-specific cDNA (1×1013 U/ml), in the
presence of 8 µg/ml polybrene (EMD Millipore) for 48 h.
RT-qPCR assays were conducted to verify the transduction
efficiency.
Estimation of antioxidants and lipid
peroxidation products
Tissue lipid peroxide level was determined by
thiobarbituric acid reactive substances (TBARS) using a reagent kit
(BioAssay Systems; cat. no. DTBA-100) according to the
manufacturer's instruction. The activities of superoxide dismutase
(SOD; cat. no. A001-3-2) and glutathione peroxidase (GPx; cat. no.
A005-1-2) were measured using kits obtained from the Institute of
Biological Engineering of Nanjing Jianchen, according to the
manufacturer's protocol. The content of protein in the homogenate
was determined using a bicinchoninic acid assay (Beyotime Institute
of Biotechnology).
Neurobehavioral assay
Rotarod test and water maze test was performed to
detect the neurobehavioral function (25). To measure motor coordination, the
mice were assessed on the rotarod apparatus. The rotarod consisted
of the horizontal cylinder divided into four lanes. In total, three
consecutive block trials were administered, in which the rotarod
rotated at a constant speed of 5 RPM (rotations per min) for two
trials for 300 sec, followed by two trials of acceleration by 3 RPM
every 5 sec and finally two trials of acceleration by 5 RPM every 3
sec. The time that each mouse was able to remain on the rotating
rod before falling was measured. All of the tests were repeated
three times in a blinded fashion.
The Morris water maze test was performed for the
assessment of spatial learning and memory. A circular pool (1.3 m
in diameter; 50 cm in depth) was filled with room-temperature
water. In addition, 1 cm below the surface of the water, a
trans-parent platform was placed in the southeastern quadrant. The
water maze tests consisted of cued learning, spatial learning and
probe trial. In the cued learning trial, the mice were released
from different starting points and allowed to swim for 60 sec to
search for the platform. The escape latency time was defined by the
time spent to find the hidden platform by the mice. In the spatial
learning trial, the mice were allowed to find and climb onto the
escape platform, and performed ten trials per day in five blocks of
two consecutive trials (120 sec trial; 120 sec inter-trial interval
during which time the mouse remained on the escape platform). The
cumulative distance from the platform and escape latency to find
the platform in each test was recorded. In the probe trial, the
mice were released from a new start position in the pool without
the platform. The amount of time spent in the southeastern quadrant
was recorded. All behavioral tests were recorded by video tracking
software (TopScan; version 2.0; CleverSys, Inc.).
Statistics analysis
Data are presented as the mean ± SEM, and analyzed
using SPSS statistics software (version 21.0; SPSS, Inc.). Each
experiment was repeated three times. Comparisons between two groups
were determined using Student's t-test (unpaired; 2-tailed).
Comparisons between multiple groups were determined using one-way
or two-way ANOVA followed by Tukey's test. As for two-way ANOVA,
the parameters BDNF-AS expression and training location, were used
for the interaction analysis of neurobehavioral function. P<0.05
was considered to indicate a statistically significant
difference.
Results
Detection of BDNF-AS and BDNF expression
levels after HI stress in vivo and in vitro
The normal brains were collected from P7 mice
without any treatment. The P7 mice were anaesthetized with 3%
isoflurane, decapitated and the striatum, hippocampi and cortex
were excised and stored in liquid nitrogen for RNA extraction.
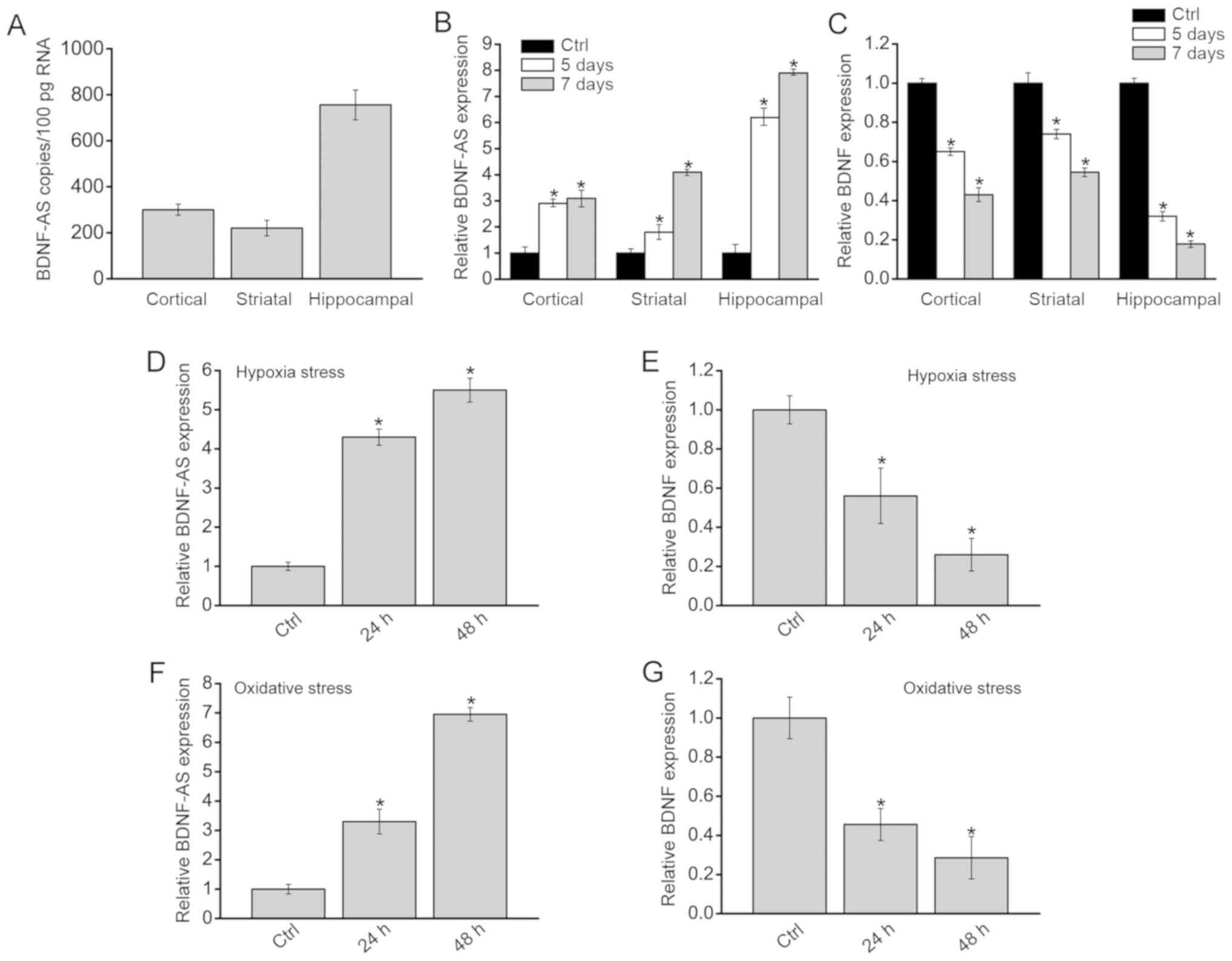
Absolute quantification of BDNF-AS expression demonstrated that
BDNF-AS expression was higher in hippocampi compared with the
cortex or striatum in the normal brain (Fig. 1A). P7 mice received ligation of
the unilateral common carotid artery, and were exposed to 8%
O2 for ~30 min. Then, 5 and 7 days after HI stress, the
striatum, hippocampi and cortex were excised. RT-qPCR assays
demonstrated that BDNF-AS and BDNF expression levels were compared
in the striatum, hippocampi and cortex between HI brains and normal
brains. The results indicated that BDNF-AS expression was
significantly increased in HI brains compared with Sham group
(Control; Fig. 1B), while BDNF
expression was reduced in these brains (Fig. 1C). The highest significant
difference was detected in the hippocampal cells of HI brains
compared with the control hippocampal neurons.
Primarily isolated hippocampal neurons were also
exposed to hypoxia stress or oxidative stress. BDNF-AS expression
was significantly upregulated after hypoxia stress or oxidative
stress (Fig. 1D and E), while
BDNF demonstrated an opposite expression pattern (Fig. 1F and G). Collectively, these
results suggested that HI stress led to increased BDNF-AS
expression in vivo and in vitro.
BDNF-AS silencing protects hippocampal
cells against HI stress in vitro
To determine the role of BDNF-AS in hippocampal
cells in vitro, BDNF-AS shRNA transfection was used to
silence BDNF-AS expression in the primary hippocampal cells
(Fig. 2A). CCK-8 results
suggested that hypoxia-induced decrease in cell viability was
partially rescued by BDNF-AS silencing (Fig. 2B).
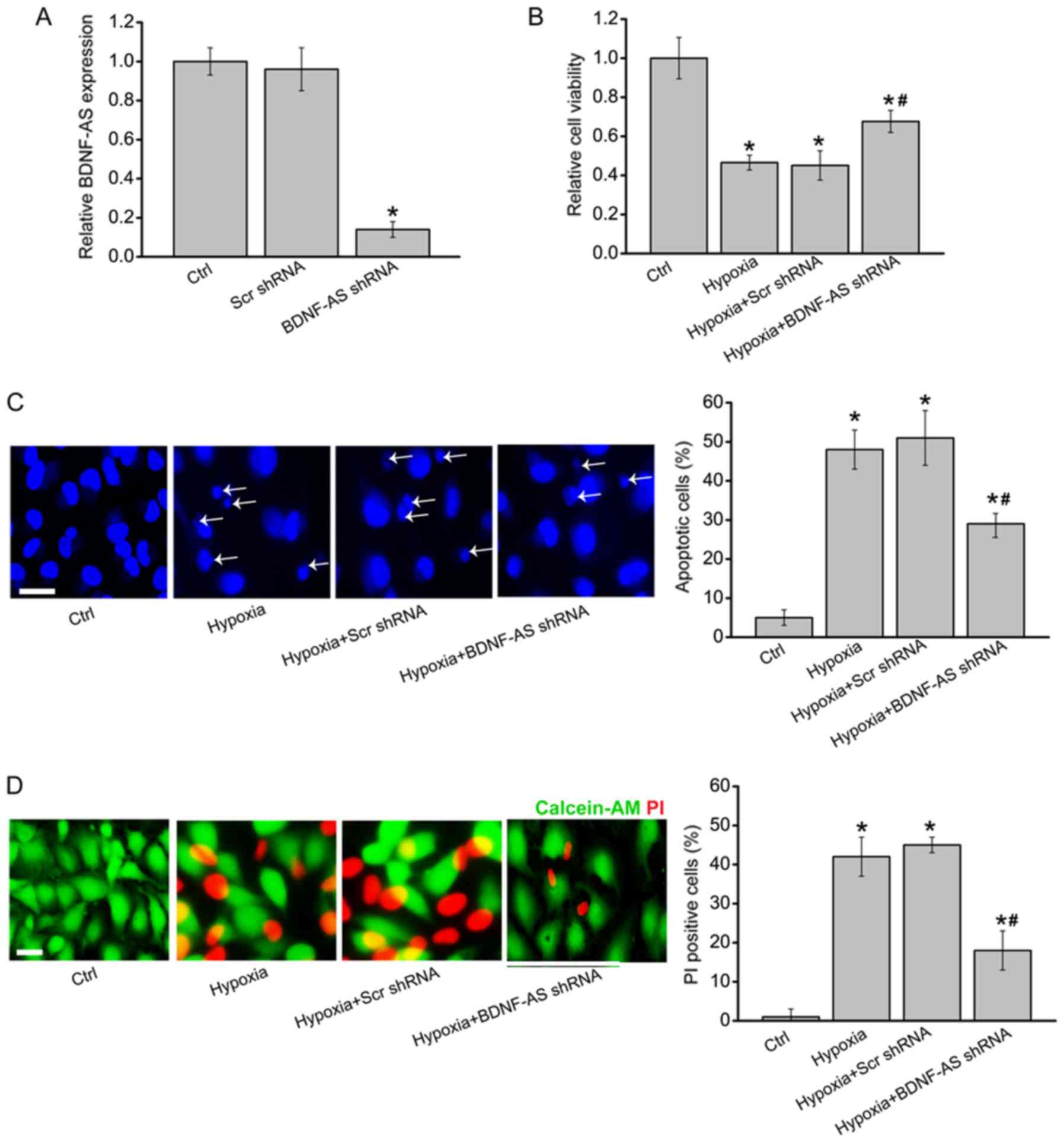 | Figure 2BDNF-AS silencing protects
hippocampal cells against hypoxic stress in vitro. (A)
Primary hippocampal cells were transfected with Scr shRNA, BDNF-AS
shRNA or left untreated (Ctrl) for 48 h. Reverse
transcription-quantitative PCR was performed to determine BDNF-AS
expression (n=4). Primary hippocampal cells were transfected with
Scr shRNA, BDNF-AS shRNA or left untreated, and then exposed to
hypoxia chamber (1% O2) for 48 h. The untreated group
was used as the Ctrl group. (B) Cell viability was determined by
Cell Counting Kit-8 assays (n=4). (C) Apoptotic cells were detected
by Hoechst staining. Scale bar, 50 µm. Magnification, ×500.
Apoptotic nuclei are indicated by the white arrows (n=4). (D) Dead
or dying cells were determined by Calcein-AM/PI staining. Green,
live cells; Red, dead or dying cell. Scale bar, 50 µm.
Magnification, ×500 (n=4). Error bars represent the standard error
of the mean. *P<0.05 vs. Ctrl group;
#P<0.05 vs. hypoxia + Scr shRNA group. Scr,
scrambled; shRNA, short hairpin RNA; PI, propidium iodide; BDNF,
brain-derived neurotrophic factor; AS, antisense; Ctrl,
control. |
Hoechst 33342 staining and Calcein-AM/PI staining
demonstrated that compared with hypoxia group, BDNF-AS silencing
significantly decreased the number of apoptotic nuclei (condensed
or fragmented) and PI-positive cells (dying or dead cells), thus
suggesting that hypoxia-induced cell apoptosis was reduced by
BDNF-AS silencing (Fig. 2C and
D). Therefore, these results indicated that BDNF-AS silencing
protected primary hippocampal cells against HI stress in
vitro.
BDNF-AS silencing attenuates HI-induced
brain injury in vivo
The present study further investigated whether
BDNF-AS silencing protected against HI-induced brain injury in
vivo. BDNF-AS shRNA or scrambled shRNA (used as the negative
control) were administered before HI treatment. Compared with the
scrambled shRNA group, BDNF-AS shRNA injection significantly
decreased BDNF-AS expression (Fig.
3A). Furthermore, BDNF-AS silencing significantly attenuated
HI-induced brain injury as demonstrated by a decreased brain
infarct size (Fig. 3B).
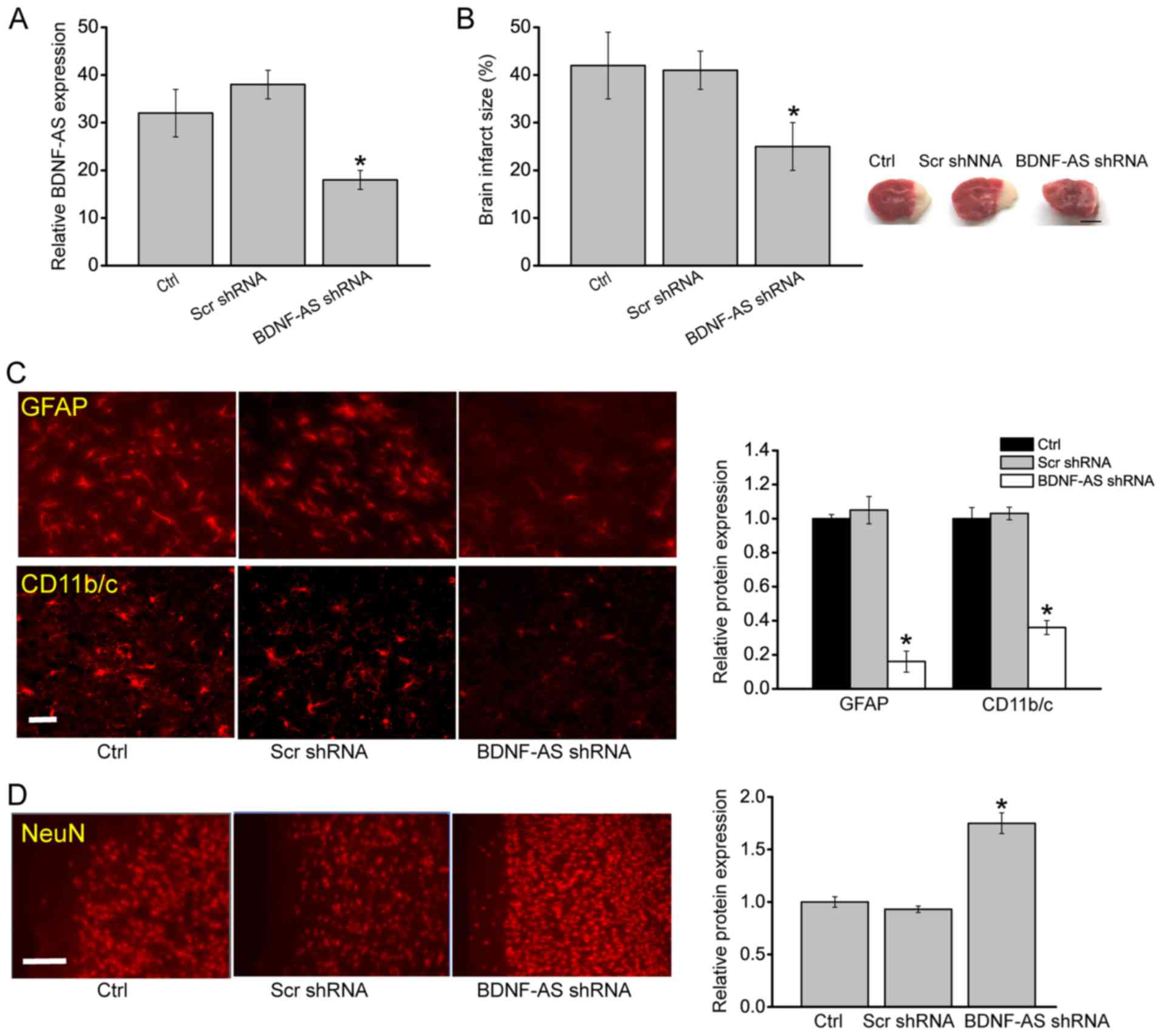 | Figure 3BDNF-AS silencing attenuates
HI-induced brain injury in vivo. (A) Reverse
transcription-quantitative PCR was performed to detect BDNF-AS
expression in the hippocampus in Sham, Scr-shRNA-injected and
BDNF-AS shRNA-injected C57Bl/6 pups (n=6). Sham animals were used
as the Ctrl group, which underwent anesthesia and the common
carotid artery was exposed without ligation and hypoxia. (B) P7
mice (C57Bl/6) received BDNF-AS shRNA or Scr shRNA prior to HI
treatment, and brain infarct size was determined 48 h after HI
(n=6). Scale bar, 2 mm. Immunostaining and quantification of (C)
GFAP and CD 11b/c, or (D) NeuN in the cornu ammonis 1 hippocampal
regions of P7 mice (C57Bl/6) injected with BDNF-AS shRNA and Scr
shRNA. Fig. 3C: Scale bar, 20 μm;
magnification, ×200. Fig. 3D:
Scale bar, 50 μm; magnification, ×500. (n=6). Error bars represent
the standard error of the mean. *P<0.05 vs. Ctrl group. HI,
hypoxic/ischemic; Scr, scrambled; shRNA, short hairpin RNA; BDNF,
brain-derived neurotrophic factor; AS, antisense; Ctrl, control;
P7, postnatal day 7; GFAP, Glial fibrillary acidic protein; NeuN,
neuronal nuclei. |
The degree of astrocytosis was examined by GFAP
staining, which is an astrocytic marker, and the degree of
microgliosis by CD11b/c staining, which is a microglial marker
(14). BDNF-AS silencing led to
decreased GFAP and CD11b/c staining signaling in the hippocampus
(Fig. 3C). NeuN staining was also
performed to detect the neurons in the hippocampus. It was found
that BDNF-AS silencing led to increased NeuN staining signaling in
the hippocampus compared with control group (Fig. 3D). The neonatal brain is usually
vulnerable to oxidative stress (3). The results indicated that BDNF-AS
silencing led to decreased biological activities of SOD and GPx,
and reduced level of TBARS in the damaged hemispheres after HI
injury (Table I).
 | Table IEffects of systemic BDNF-AS silencing
on the activities of SOD and GPx, and TBARS levels. |
Table I
Effects of systemic BDNF-AS silencing
on the activities of SOD and GPx, and TBARS levels.
| Experimental
group | SOD, U/mg
protein | GPx, U/mg
protein | TBARS, U/mg
protein |
|---|
| Scr shRNA | 4.73±0.52 | 0.063±0.023 | 1.98±0.37 |
| BDNF-AS shRNA | 2.08±0.28 | 0.036±0.029 | 1.12±0.54 |
BDNF-AS silencing ameliorates brain
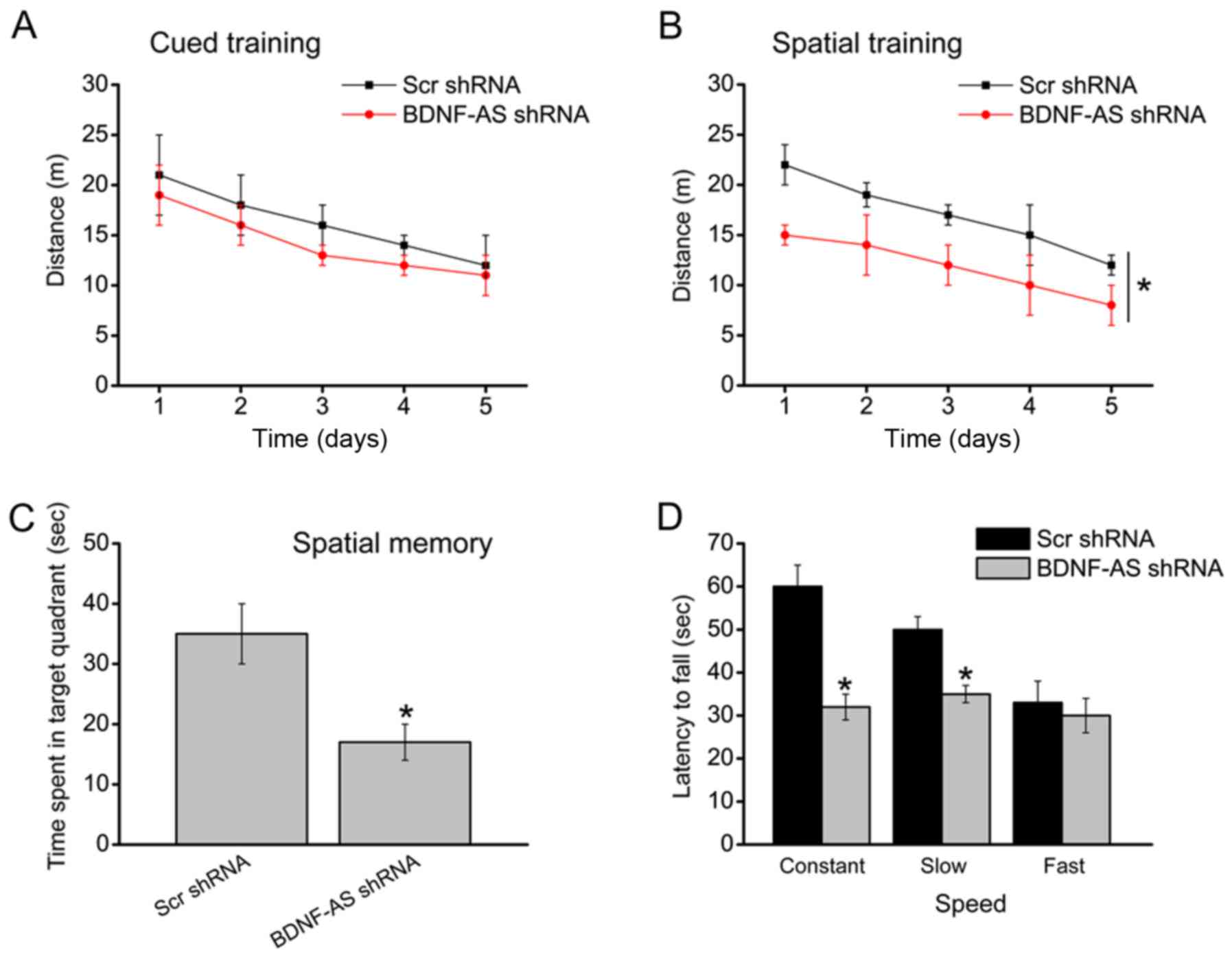
neurological function in vivo
Subsequently, whether BDNF-AS silencing was able to
ameliorate brain neurological function in vivo was
investigated. A total of 8 weeks after HIBD, the water maze
experimental results demonstrated that BDNF-AS silencing did not
affect the swimming distance to reach the platform in the cued
learning task (Fig. 4A). However,
BDNF-AS silencing improved the spatial learning ability as
indicated by shorter swimming distance to reach the platform
(Fig. 4B). A probe experiment was
then conducted to determine the effects of BDNF-AS silencing on
spatial memory. The results suggested that the animal injected with
BDNF-AS shRNA did not show a preference for the target quadrant
(Fig. 4C). Moreover, BDNF-AS
silencing improved the motor function during the constant and slow
acceleration trials, but did not improved the motor function during
the fast acceleration trials in the rotarod task (Fig. 4D).
BDNF-AS regulates hippocampal cell
function by regulating BDNF-mediated signaling
As BDNF-AS is the antisense RNA of BDNF (16), the present study determined
whether intervention of BDNF-AS expression affected the expression
of BNDF. RT-qPCR results suggested that BDNF-AS overexpression
significantly decreased the expression of BDNF mRNA, suggesting
that BDNF-AS affected BDNF expression at the transcriptional level
(Fig. 5A). It was also determined
whether the intervention of BDNF-AS expression affected the
activation of BDNF-mediated signaling. BDNF-AS overexpression led
to decreased expression levels of BDNF, p-Akt and p-TrkB (Fig. 5B), indicating that BDNF-AS
affected hippocampal cell function via BDNF-mediated signaling.
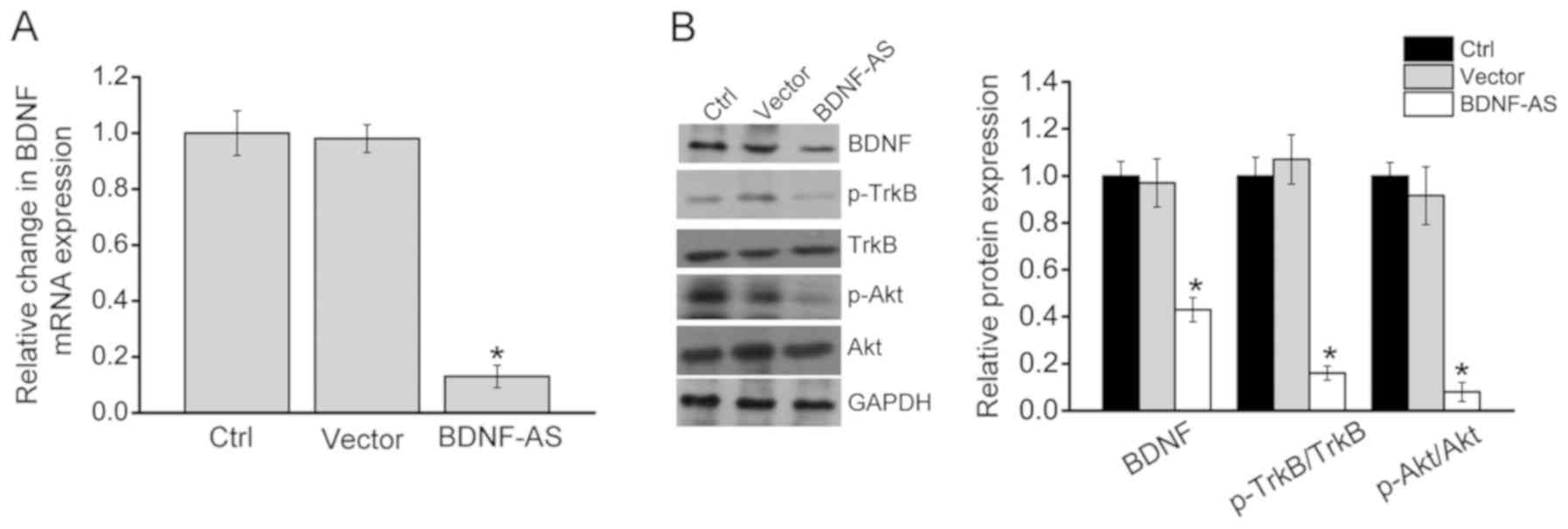 | Figure 5BDNF-AS regulates hippocampal cell
function by regulating BDNF-mediated signaling. Hippocampal cells
were transfected with pcDNA 3.0 (vector), pcDNA3.0- BDNF-AS or left
untreated (Ctrl). (A) Reverse transcription-quantitative PCR was
conducted to detect BDNF mRNA expression (n=4). (B) Western
blotting was performed to detect the expression levels of BDNF,
p-Akt, Akt, TrkB and p-TrkB (n=4). Error bars represent the
standard error of the mean. *P<0.05 vs. Ctrl group. Scr,
scrambled; shRNA, short hairpin RNA; BDNF, brain-derived
neurotrophic factor; AS, antisense; Ctrl, control; p-,
phosphorylated; TrkB, tropomyosin receptor kinase B; OE,
overexpression. |
Discussion
Previous studies have reported that lncRNAs are
aberrantly dysregulated and play important roles in brain injury
(26,27). The present study determined the
role of BDNF-AS in HI-induced hippocampal cell injury in
vivo and in vitro. It was found that BDNF-AS was
significantly upregulated HI-induced neonatal brain injury and
hippocampal cell injury. Furthermore, BDNF-AS silencing protected
against HI-induced brain injury in vivo and primary
hippocampal cell injury in vitro. Mechanistically, BDNF-AS
affected neonatal brain injury via the inverse regulation of BDNF
expression.
BDNF-AS is the antisense RNA of BDNF, which is
expressed in various human tissues, and may have reciprocal neural
functions to BDNF, such as supporting the survival of existing
neurons, as well as encouraging growth and differentiation of new
neurons and synapses (16).
BDNF-AS protects local anesthetic-induced neurotoxicity in dorsal
root ganglion neurons (18).
Moreover, knockdown of BDNF-AS suppresses neuronal cell apoptosis
in the acute spinal cord injury (28). Thus, these studies suggest that
BDNF-AS regulates the apoptosis of neural cells. However, to the
best of our knowledge, the current understanding of underlying role
of BDNF-AS in neonatal brain injury is limited. The present results
indicated that BDNF-AS knockdown protected hippocampal cells
against HI-induced brain injury. The environment, together with the
gene regulatory network, directs hippocampal cells to rest,
proliferation, differentiate or undergo apoptosis (29). For instance, hypoxic stress and
oxidative stress are the two major pathological drivers during
neonatal brain injury (3). In the
present study, primary hippocampal cells were exposed to 1% oxygen
or H2O2 (150 µM) to mimic hypoxic
stress or oxidative stress, and it was found that hypoxic and
oxidative stress led to increased expression of BDNF-AS. Thus,
BDNF-AS may direct hippocampal cells to adapt hypoxic or ischemic
conditions by regulating oxidative stress or hypoxic
stress-responsive genes.
BDNF is important for neuronal proliferation,
maturation, differentiation and maintenance (14). Furthermore, BDNF synchronizes
neuronal and glial maturation and enhances neuronal cell survival
(30). BDNF upregulation is
speculated to have beneficial effects in a number of neurological
disor-ders, such as Alzheimer's disease, Huntington's disease and
Parkinson's disease (31). Since
BDNF-AS is transcribed oppositely by the BDNF gene, the present
study investigated whether BDNF-AS regulates BDNF expression in
hippocampal cells. BDNF-AS overexpression decreased the expression
of BDNF mRNA. Moreover, BDNF-AS knockdown affected the activation
of the BDNF-mediated signaling pathway as indicated by decreased
expression levels of BDNF, p-Akt and p-TrkB. Therefore, the present
results suggested that BDNF-AS knock-down induced the activation of
the BDNF/TrkB/PI3K/Akt signaling pathway after HI-induced
neurotoxicity (32).
The neonatal brain is usually characterized by a low
concentration of anti-oxidants and high level of oxygen
consumption, which is why the neonatal brain is vulnerable to
oxidative stress injury (33). As
a result, it is beneficial to decrease oxidative damage and
increase the anti-oxidant defense during neonatal brain injury. The
present results indicated that BDNF-AS knockdown affected the
activities of anti-oxidant enzymes, SOD and GPx, and the level of
TBARS, which is a lipid peroxidation index (34). In addition, newborns and premature
babies experience free radical oxidative injury (35). Thus, it was demonstrated that
BDNF-AS silencing plays a neuroprotective role in HI brain injury
at least partially via the modulation of anti-oxidant enzyme
activity.
In conclusion, the present study identified a
potential role of BDNF-AS in the pathogenesis of HI-induced
neonatal brain injury and its underlying molecular mechanism. It
was demonstrated that BDNF-AS knockdown could improve brain
function by reducing the infarct size and improving the
neurological function, suggesting that BDNF-AS may be a promising
target for the treatment of HIBD.
Funding
The present study was supported by the Project of
Clinical Advanced Techniques, Primary Research & Development
Plan of Jiangsu Province (grant no. BE2017719), the Pediatric
Medical Innovation Team of Jiangsu Province (grant no.
CXTDA2017022), the Project of National Youth Found (grant no.
81601355) and the Project of Postdoctoral Fund of Jiangsu Province
(grant no. 1701162C).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
RBZ and ZKX were the major contributors to the
experimental design. RBZ established the animal models. LXQ, MFW
and LHZ performed the western blot analysis and cell culture. RBZ
and ZKX were involved in writing the manuscript, the analysis and
interpretation of data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experimental procedures adhered to the
principles stated in the Guide for the Care and Use of Laboratory
Animals (updated 2011; National Institutes of Health) and were
approved by the Animal Care and the Use Committee of Nanjing
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
The authors would like to thank Dr Li-Hua Zhu
(Jiangsu Health Vocational College) for the helpful discussion for
paper revision.
References
|
1
|
Zhu C, Kang W, Xu F, Cheng X, Zhang Z, Jia
L, Ji L, Guo X, Xiong H, Simbruner G, et al: Erythropoietin
improved neurologic outcomes in newborns with hypoxic-ischemic
encephalopathy. Pediatrics. 124:e218–e226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rocha-Ferreira E and Hristova M:
Antimicrobial peptides and complement in neonatal hypoxia-ischemia
induced brain damage. Front Immunol. 6:562015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Johnston MV, Trescher WH, Ishida A and
Nakajima W: Neurobiology of hypoxic-ischemic injury in the
developing brain. Pediat Res. 49:735–741. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Blackmon LR and Stark AR: American Academy
of Pediatrics Committee on Fetus and Newborn: Hypothermia: A
neuroprotective therapy for neonatal hypoxic-ischemic
encephalopathy. Pediatrics. 117:942–948. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tagin MA, Woolcott CG, Vincer MJ, Whyte RK
and Stinson DA: Hypothermia for neonatal hypoxic ischemic
encephalopathy: An updated systematic review and meta-analysis.
Arch Pediat Adol Med. 166:558–566. 2012. View Article : Google Scholar
|
|
6
|
Quinn JJ and Chang HY: Unique features of
long non-coding RNA biogenesis and function. Nat Rev Genet.
17:47–62. 2016. View Article : Google Scholar
|
|
7
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fatica A and Bozzoni I: Long non-coding
RNAs: New players in cell differentiation and development. Nat Rev
Genet. 15:7–21. 2014. View
Article : Google Scholar
|
|
9
|
Zhang J, Yuan L, Zhang X, Hamblin MH, Zhu
T, Meng F, Li Y, Chen YE and Yin KJ: Altered long non-coding RNA
transcriptomic profiles in brain microvascular endothelium after
cerebral ischemia. Exp Neurol. 277:162–170. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao F, Qu Y, Liu J, Liu H, Zhang L, Feng
Y, Wang H, Gan J, Lu R and Mu D: Microarray profiling and
co-expression network analysis of LncRNAs and mRNAs in neonatal
rats following hypoxic-ischemic brain damage. Sci Rep. 5:138502015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mehta SL, Kim T and Vemuganti R: Long
noncoding RNA FosDT promotes ischemic brain injury by interacting
with REST-associated chromatin-modifying proteins. J Neurosci.
35:16443–16449. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao RB, Zhu LH, Shu JP, Qiao LX and Xia
ZK: GAS5 silencing protects against hypoxia/ischemia-induced
neonatal brain injury. Biochem Biophys Res Commun. 497:285–291.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang X, Tang X, Liu K, Hamblin MH and Yin
KJ: Long noncoding RNA Malat1 regulates cerebrovascular pathologies
in ischemic stroke. J Neurosci. 37:1797–1806. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rossi C, Angelucci A, Costantin L, Braschi
C, Mazzantini M, Babbini F, Fabbri ME, Tessarollo L, Maffei L,
Berardi N and Caleo M: Brain-derived neurotrophic factor (BDNF) is
required for the enhancement of hippocampal neurogenesis following
environmental enrichment. Eur J Neurosci. 24:1850–1856. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Angelucci F, Mathe AA and Aloe L:
Neurotrophic factors and CNS disorders: Findings in rodent models
of depression and schizophrenia. Prog Brain Res. 146:151–165. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Y, Xu F, Xiao H and Han F: Long
noncoding RNA BDNF-AS inversely regulated BDNF and modulated
high-glucose induced apoptosis in human retinal pigment epithelial
cells. J Cell Biochem. 119:817–823. 2018. View Article : Google Scholar
|
|
17
|
Zheng X, Lin C, Li Y, Ye J, Zhou J and Guo
P: Long noncoding RNA BDNF-AS regulates ketamine-induced
neurotoxicity in neural stem cell derived neurons. Biomed
Pharmacother. 82:722–728. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, Yan L, Cao Y, Kong G and Lin C:
Long noncoding RNA BDNF-AS protects local anesthetic induced
neurotoxicity in dorsal root ganglion neurons. Biomed Pharmacother.
80:207–212. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo CC, Jiao CH and Gao ZM: Silencing of
lncRNA BDNF-AS attenuates Aβ25-35-induced neurotoxicity in PC12
cells by suppressing cell apoptosis and oxidative stress. Neurol
Res. 40:795–804. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cetin A, Komai S, Eliava M, Seeburg PH and
Osten P: Stereotaxic gene delivery in the rodent brain. Nat Protoc.
1:3166–3173. 2006. View Article : Google Scholar
|
|
21
|
Klaunberg BA, O'malley J, Clark T and
Davis JA: Euthanasia of mouse fetuses and neonates. Contemp Top Lab
Anim Sci. 43:29–34. 2004.PubMed/NCBI
|
|
22
|
Artwohl J, Brown P, Corning B and Stein S:
Report of the ACLAM task force on rodent euthanasia. J Am Assoc Lab
Anim Sci. 45:98–105. 2006.PubMed/NCBI
|
|
23
|
Kramer M, Dang J, Baertling F, Denecke B,
Clarner T, Kirsch C, Beyer C and Kipp M: TTC staining of damaged
brain areas after MCA occlusion in the rat does not constrict
quantitative gene and protein analyses. J Neurosci Methods.
187:84–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Merritt JR and Rhodes JS: Mouse genetic
differences in voluntary wheel running, adult hippocampal
neurogenesis and learning on the multi-strain-adapted plus water
maze. Behav Brain Res. 280:62–71. 2015. View Article : Google Scholar
|
|
26
|
Yin KJ, Hamblin M and Chen YE: Non-coding
RNAs in cerebral endothelial pathophysiology: Emerging roles in
stroke. Neurochem Int. 77:9–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xin JW and Jiang YG: Long noncoding RNA
MALAT1 inhibits apoptosis induced by oxygen-glucose deprivation and
reoxygenation in human brain microvascular endothelial cells. Exp
Ther Med. 13:1225–1234. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang H, Li D, Zhang Y, Li J, Ma S, Zhang
J, Xiong Y, Wang W, Li N and Xia L: Knockdown of lncRNA BDNF-AS
suppresses neuronal cell apoptosis via downregulating miR-130b-5p
target gene PRDM5 in acute spinal cord injury. RNA Biol.
15:1071–1080. 2018.PubMed/NCBI
|
|
29
|
Hagg T: Molecular regulation of adult CNS
neurogenesis: An integrated view. Trends Neurosci. 28:589–595.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Waterhouse EG, An JJ, Orefice LL, Baydyuk
M, Liao GY, Zheng K, Lu B and Xu B: BDNF promotes differentiation
and maturation of adult-born neurons through GABAergic
transmission. J Neurosci. 32:14318–14330. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lu B, Nagappan G and Lu Y: BDNF and
synaptic plasticity, cognitive function, and dysfunction. Handb Exp
Pharmacol. 220:223–250. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yoshii A and Constantine-Paton M:
Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity,
and disease. Dev Neurobiol. 70:304–322. 2010.PubMed/NCBI
|
|
33
|
Vannucci SJ and Hagberg H:
Hypoxia-ischemia in the immature brain. J Exp Biol. 207:3149–3154.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kumral A, Gonenc S, Acikgoz O, Sonmez A,
Genc K, Yilmaz O, Gokmen N, Duman N and Ozkan H: Erythropoietin
increases glutathione peroxidase enzyme activity and decreases
lipid peroxidation levels in hypoxic-ischemic brain injury in
neonatal rats. Biol Neonate. 87:15–18. 2005. View Article : Google Scholar
|
|
35
|
Vexler ZS and Ferriero DM: Molecular and
biochemical mechanisms of perinatal brain injury. Semin Neonatol.
6:99–108. 2001. View Article : Google Scholar : PubMed/NCBI
|