1. Introduction
Generalized pustular psoriasis (GPP) is a rare and
severe auto-inflammatory skin disease with life-threatening
potential that is characterized by recurrent and sudden episodic
generalized erythematous eruptions with neutrophil-filled pustules1
(1,2). GPP is accompanied by high fever,
leukocytosis and elevated serum levels of C-reactive protein in the
acute phase, and can be triggered by infections, pregnancy or drugs
(1,2). GPP is an extremely rare form of
psoriasis with an estimated prevalence of 7.46 patients per million
in Japan (3) and 1.76 patients
per million in France (4), and
represents about 1% of all clinical types of psoriasis (5-8).
Histologically, GPP is characterized by Kogoj's spongiform pustule
and Munro's microabscesses with a large number of infiltrating
neutrophils (9,10). GPP is clinically heterogeneous in
presentation and progression, and currently lacks consistent
classification. Concerning clinical presentation, GPP is considered
one of the distinct subtypes of pustular psoriasis (PP), which can
present as a recurrent systemic illness (GPP) or chronic localized
form affecting palms and/or soles (palmoplantar pustulosis, PPP),
or digits/nail beds (acrodermatitis continua of Hallopeau, ACH)
(1,11,12). Since GPP often presents in
individuals with an existing history of psoriasis vulgaris (PV), it
can be divided into two subtypes, namely GPP alone and GPP with PV.
Patients affected by GPP alone generally carry genetic variations
of IL36RN and show more severe clinical symptoms, early
acute onset of the disease, repeated and persistent attacks, and
systemic inflammation (13,14). According to age of onset, GPP can
be classified into pediatric-onset GPP (≤18 years) and adult-onset
GPP, with pediatric-onset GPP manifesting mostly as GPP alone and
occurring with recurrent and sudden systemic inflammation (15-17). GPP, especially the
pediatric-onset GPP form, is considered to be an independent
subtype of psoriasis which differs from PV and requires a distinct
diagnosis.
Although the first GPP case was reported in 1910,
its etiology and detailed pathogenesis have been only recently
described in the literature. In 2011, the identification of
loss-of-function mutations in IL36RN gene emphasized the key
role of this pathway in the pathogenesis of GPP (18). Since then, an increasing number
of genetic variants in CARD14, AP1S3, and MPO
pathogenic genes have been found to be associated with GPP in
affected individuals (19-21). Subsequent to the identification
of disease-causing genes, the pathogenesis of GPP has progressively
been characterized and new specific biological agents have been
developed.
In the present review, the aim was to assess current
knowledge on the genetic basis and molecular details of the
cutaneous pathomechanisms and specific treatments available on GPP
and relative clinical courses.
2. Mutation update on disease-causing gene
associated with GPP
In recent years, a number of allelic variations and
mutations in IL36RN, CARD14, AP1S3 genes, as well as
in the latest identified pathogenic MPO gene have been found
to be associated with GPP (18-21). Among those genes, IL36RN
mutations are the most frequent genetic abnormality (22,23), CARD14 mutations are
primarily present in GPP with PV and rarely in GPP alone(24,25). The pathogenic variants of
AP1S3 were mainly found in individuals of European origin
and rarely in East Asians (20,26).
Disease-causing gene IL36RN
Pathogenic mechanism underlying IL36RN
mutations
Mutations in IL36RN gene are likely to be the
main molecular genetic basis defect in patients affected by GPP
(22). Interleukin-36 (IL-36)
refers to three related IL-1 family cytokines, IL-36α, IL-36β, and
IL-36γ, which can activate the downstream pro-inflammatory nuclear
factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK)
pathways by binding to IL-36 receptor (IL-36R). Subsequently,
IL-36s induce the release of inflammatory mediators and chemotaxis
that promote activation of neutrophils, macrophages, dendritic
cells, and T cells, ultimately causing the amplification of
inflammatory responses (27).
IL-36 receptor antagonist (IL-36Ra) encoded by IL36RN gene
is specifically expressed by epidermal keratinocytes (28) and can compete with IL-36 via
binding to IL-36R, thereby blocking the inflammatory responses
caused by IL-36 itself (29).
The loss of function mutations in IL36RN gene results in the
inability of IL-36Ra to antagonize and limit the pro-inflammatory
effects of IL-36 (18,30), thereby leading to increased
expression of pro-inflammatory cytokine regulated by transcription
factor NF-κB and MAPK, such as IL-8, CXCL1-3, IL-1, and even IL-36
itself, thus forming a vicious cycle of enhancing inflammation.
IL-8 and CXCL1-3 are strong neutrophil chemokines and the
upregulation of their expression contributes to the neutrophils
infiltrating in skin pustules and systemic inflammation of GPP
patients (31).
Identification of the IL36RN gene
mutations in GPP patients
In 2011, Marrakchi et al (18) first reported that 9 familial
Tunisian GPP patients carried the c.80T>C (p.Leu27Pro)
homozygous missense mutation in IL36RN, which determines
increased keratinocyte expression of the inflammatory cytokines in
GPP patients, such as IL-8, IL-36α, IL-36β, and IL-36γ. Therefore,
IL36RN was identified as a causative gene for GPP patients
and the disease caused by IL-36Ra decrease was defined as
deficiency of interleukin thirty-six-receptor antagonist (DITRA)
(18,28,32). Notably, patients with DITRA
primarily involved the skin and presented with high-grade fever and
general malaise during an attack, in contrast to deficiency of
interleukin-1-receptor antagonist (DIRA), an autoinflammatory
disease related to activation of the IL-1 pathway, even if they
suffered from similar skin manifestations (18,32,33,34). Then, Onoufriadis et al
(30) revealed the c.338C>T
(p.Ser113Leu) homozygous missense substitution and the c.338C>T
(p.Ser113Leu) and c.142C>T (p.Arg48Trp) compound heterozygote
missense mutations in IL36RN gene in sporadic GPP cases in
the UK. Subsequently, a set of functional relevant variants in
IL36RN gene, such as c.28C>T (p.Arg10X, c.104A>G
(p.Lys35Arg), c.140A>G (p.Asn47Ser), c.227C>T (p.Pro76Leu),
c.304C>T (p.Arg102Trp), c.305G>A (p.Arg102Gln), c.368C>G
(p.Thr123Arg), c.368C>T (p.Thr123Met), and c.115+6T>C
(p.Arg10ArgfsX1), were identified in GPP patients of Eastern Asia
(15,35-37). According to sequencing and
functional analysis of GPP patients from different populations, a
total of 25 possible pathogenic variants in the IL36RN gene
have been reported thus far (Fig.
1A and Table I). The
majority of these genetic variants are missense substitutions, or
to a lesser extent, nonsense mutations. The latter include
c.28C>T (p.Arg10X), c.41C>A (p.Ser14X), c.280G>T (p.
Glu94X) and c.338C>A (p.Ser113X) mutations that generate
termination codons after the base substitutions. In addition, the
c.115+6T>C mutation in a splicing site of IL36RN causes
the skipping of exon3 at mRNA level, leading to a frameshift and
premature protein termination (p.Arg10ArgfsX1) (37). Furthermore, small fragment
deletions (c.420_426del and c.295-300del) have been also identified
in GPP patients. The c.420_426del mutation in exon 5 results in a
frameshift starting from the amino acid 140, as well as in
premature stop codon formation at position 170 (38). On the other hand, the
c.295-300del variant leads to thr99 and phe100 amino acid deletion
(23). Although the c.338C>T
substitution is the most frequent variant in Europeans (39) c.115+6T>C is the most common in
Asian populations (37,40-42). In vitro functional assays
have shown that IL36RN gene pathogenic mutations lead to a
decrease in the expression or activity of IL36Ra and increase of
IL-36-dependent pro-inflammatory factors activated by NF-κB pathway
(i.e., IL-1β, IL-8, IL-36). For instance, the c.80T>C
(p.Leu27Pro), c.28C>T (p.Arg10X), c.280G>T (p.Glu94X),
c.368C>G (p.Thr123Arg), c.368C>T (p.Thr123Met) and
c.227C>T (p.Pro76Leu) homozygous missense mutations result in
functional impairment of IL-36Ra protein expression and capacity to
suppress downstream inflammatory responses (38). However, the function of some
variants remains to be elucidated. Interestingly, homozygous or
heterozygous variants in IL36RN gene, such as c.115+6T>C,
have also been identified in healthy cohorts (15). Findings of those studies indicate
that the onset of GPP depends on a combination of multiple genetic
factors, rather than a single inherited gene.
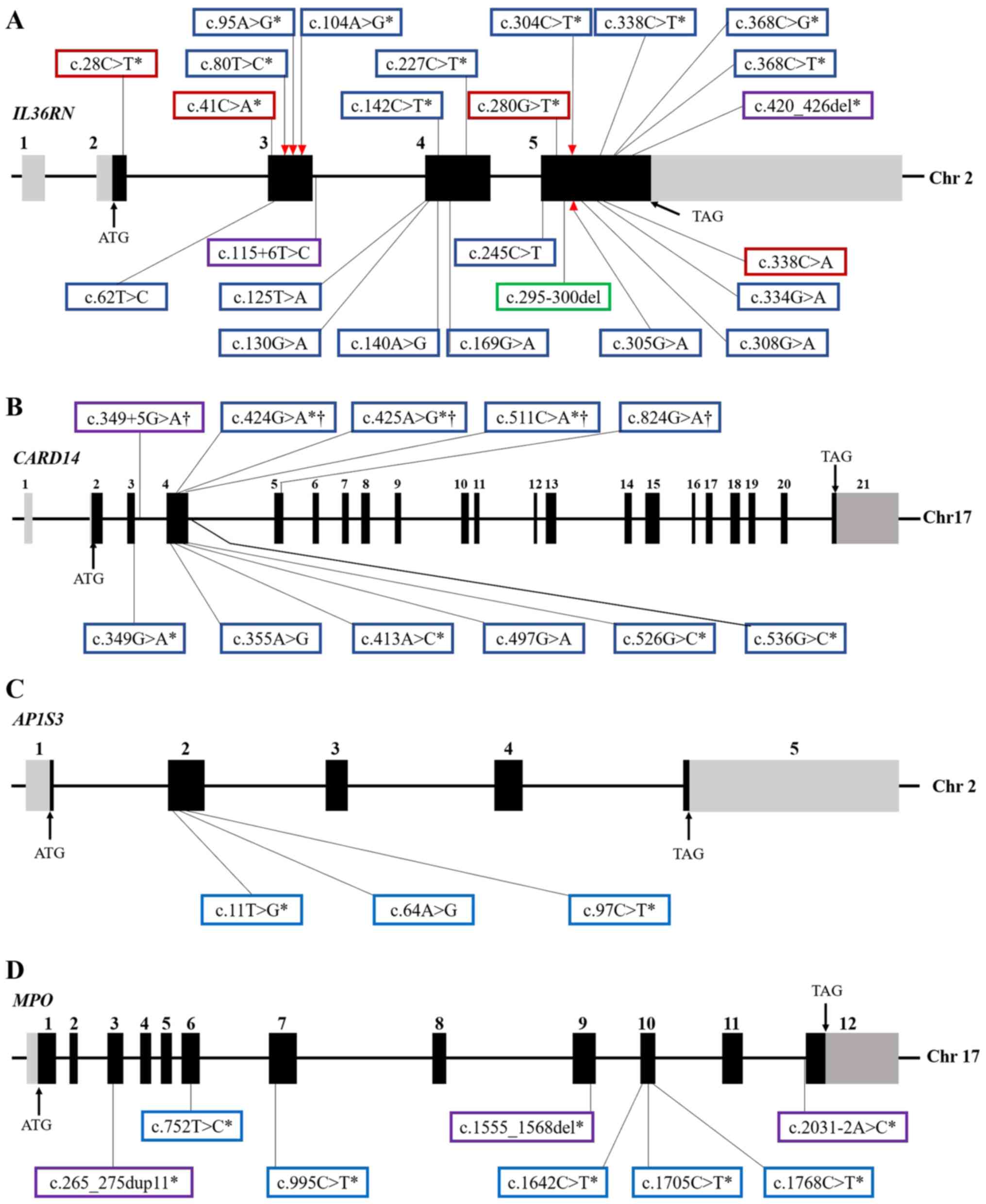 | Figure 1Genomic structure of GPP-related
genes and location of the identified variants. Exons and relative
non-coding introns of (A) IL36RN, (B) CARD14, (C)
AP1S3 and (D) MPO genes were shown by solid black and
gray boxes, respectively. Blue, red, purple and green boxes
represent, respectively, missense, nonsense and frameshift
mutations, as well as small fragment deletions. Asterisks indicate
the mutations that have been validated by functional assays. The
red triangle represents the affected IL-36R binding site after
nucleotide substitution, and daggers indicate that the
CARD14 mutations only characterized in PV patients. |
 | Table IMutations of IL36RN gene and
related characteristics in GPP patients. |
Table I
Mutations of IL36RN gene and
related characteristics in GPP patients.
| Nucleotide
variations | Amino acid
variations | Variant type | Status of the
mutations | Origin | Protein expression
in vitro | Inflammation
inhibition in vitro | (Refs.) |
|---|
| c.28C>T | p.Arg10X | Nonsense | Hom/CHet |
Japanese/Palestinian | None | Impaired | (13,35-38, 43,44) |
| c.41C>A | p.Ser14X | Nonsense | Hom | Algerian | None | Impaired | (38) |
| c.62T>C | p.Leu21Pro | Missense | Hom | Pakistani | Not reported | Not reported | (45) |
| c.80T>C | p.Leu27Pro | Missense | Hom | Tunisian | None | Impaired | (18,38,46) |
| c.95A>G | p.His32Arg | Missense | Hom | Iraqi | Reduced | Impaired | (23,38,47) |
| c.104A>G | p.Lys35Arg | Missense | Het/CHet | British | Unchanged | Unchanged | (22,38,39) |
| c.125T>A | p.Ile42Asn | Missense | Hom | Japanese | Not reported | Not reported | (48) |
| c.130G>A | p.Val44Met | Missense | CHet | Chinese/German | Not reported | Not reported | (22,23,42) |
| c.140A>G | p.Asn47Ser | Missense | Hom/CHet/Het | Chinese | Not reported | Not reported | (15,16,42, 49) |
| c.142C>T | p.Arg48Trp | Missense | Het/CHet | British/German | Reduced | Reduced | (22,23,30, 38,47) |
| c.169G>A | p.Val57Ile | Missense | Het | Chinese | Not reported | Not reported | (16) |
| c.227C>T | p.Pro76Leu | Missense | Hom/CHet/Het |
Chinese/Turkish/German/Bosnian/Syrian/Malay | None | Impaired | (15,16,22, 23,38,42, 47,49) |
| c.245C>T | p.Pro82Leu | Missense | Het | Chinese | Not reported | Not reported | (16) |
| c.280G>T | p.Glu94X | Nonsense | CHet | German | None | Impaired | (23,39,47) |
| c.304C>T | p.Arg102Trp | Missense | Hom/CHet/Het |
British/Turkish/East Asian | Unchanged | Unchanged | (22,38,39, 42) |
| c.305G>A | p.Arg102Gln | Missense | Het | Chinese | Not reported | Not reported | (15) |
| c.308G>A | p.Arg103Gln | Missense | Het | German | Not reported | Not reported | (23) |
| c.334G>A | p.Glu112Lys | Missense | CHet | Chinese | Not reported | Not reported | (49) |
| c.338C>T | p.Ser113Leu | Missense | Hom/CHet/Het |
British/German/Iraqi/Swiss/Russian | Reduced | Reduced | (17,22,23, 30,38,39, 47,50) |
| c.338C>A | p.Ser113X | Nonsense | CHet | Russian | Not reported | Not reported | (23) |
| c.368C>G | p.Thr123Arg | Missense | CHet | Japanese | None | Impaired | (37,38) |
| c.368C>T | p.Thr123Met | Missense | CHet |
Japanese/Chinese | None | Impaired | (16,36,38) |
| c.115+6T>C | p.Arg10ArgfsX1 | Frameshift | Hom/CHet/Het |
Japanese/Chinese/Malay/Korean/German | Not reported | Not reported | (13,15-17, 22,37,39, 42,49, 51-53) |
| c.295-300del |
p.Thr99_Phe100del | Small fragment | CHet deletion | German | Not reported | Not reported | (23) |
| c.420_426del |
p.Gly141MetfsX29 | Frameshift | Hom |
Spanish/Algerian | None | Impaired | (17,38) |
Genotype-phenotype correlation
Since some GPP cases are accompanied by PV, Sugiura
et al (13) first
screened the IL36RN gene in two subgroups of GPP patients in
the Japanese population (GPP alone and GPP with PV, respectively),
showing that all the GPP patients without PV (n=11) harbored
homozygous or compound heterozygous mutations in IL36RN gene
(13,48), whereas only 2 out of 20 cases of
GPP with PV carried compound heterozygous mutations. Since the
frequency of IL36RN mutations in patients of GPP alone was
much higher than that observed in patients with both GPP and PV
forms, Sugiura et al (13) suggested that GPP alone represents
a distinct subtype of GPP and is etiologically distinguishable from
GPP occurring with PV. In 2014, the genetic heterogeneity in
different subtypes of GPP also was validated in a study analyzing
IL36RN mutations in GPP Chinese patients (16). Consistently, the meta-analysis of
233 GPP patients by Hussain et al in 2015 (17) revealed that carriage of
IL36RN mutations manifested early onset of the disease
(17±2.4 years vs. 33±1.5 years; P=5.9×10−3), higher risk
of systemic inflammation (83 vs. 56%; P=1.5×10−3), and
lower prevalence of PV (36.1 vs. 68.7%, P=5×10−4). Of
note, findings of that study also demonstrated that the number of
mutant alleles of IL36RN gene also correlated with a younger
age of onset. In 2017, further genotype-phenotype correlation
analysis of 66 Chinese children with GPP alone also validated that
IL36RN-positive cases manifested a more severe clinical
phenotype, characterized by early onset, severe inflammation in
skin lesions, and high recurrence rate following treatment with
low-dose acitretin (42). In
2019, a survey including a cohort of 251 cases of GPP patients from
multiple countries also showed IL36RN gene mutations
associated with the age of onset, prevalence of PV, and recurrence
rate of GPP (22). Taken
together, the aforementioned studies demonstrated that
IL36RN gene mutations are, not only related to the
pathogenesis of GPP, but also to the clinical phenotype associated
to GPP (Table II).
| Table IIStudies of correlation between
IL36RN mutations and clinical phenotype. |
Table II
Studies of correlation between
IL36RN mutations and clinical phenotype.
| Studies, year | Origin | No. of patients
enrolled | Correlation between
IL36RN mutations and clinical presentations
| (Refs.) |
|---|
| Low prevalence of
PV | Early age of
onset | Severe
inflammation | High recurrence
rate |
|---|
| Sugiura et
al, 2013 | Japanese | 31 | Y | N/A | N/A | N/A | (13) |
| Li et al,
2014 | Chinese | 62 | Y | N/A | N/A | N/A | (16) |
| Hussain et
al, 2015 | European, Asian,
African | 233 | Y | Y | Y | N/A | (17) |
| Wang et al,
2017 | Chinese | 66 | N/A | Y | Y | Y | (42) |
| Twelves et
al, 2019 | European, East
Asian, Malay | 251 | Y | Y | N/A | Y | (22) |
Disease-causing gene CARD14
CARD14 gene, also known as CARMA2
gene, encodes caspase recruitment domain family member 14 (CARD14)
which mediates the activation of TRAF2-dependent NF-κB signaling in
keratinocytes (54,55). CARD14 expression is mostly
restricted to the basal layer of epidermis in healthy skin, whereas
it is upregulated in the granular layers in GPP-affected skin
(19).
In 2012, Jordan et al (19) identified the c.349G>A
(p.Gly117Ser) and c.349+5G>A heterozygosity in CARD14
gene in European ancestry with psoriasis, and the c.413A>C
(p.Glu138Ala) variant in a sporadic pediatric case with GPP. The
gain-of-function mutations of c.413A>C and c.349+5G>A caused
enhanced NF-κB activation in keratinocytes and upregulation of a
subset of psoriasis-associated genes, in particular chemokine (C-C
motif) ligand 20 (CCL20), and IL8 genes. Then, the
group of Jordan continued to expand the number of cohorts (56), further screening more than 6,000
psoriasis patients and 4,000 controls in multiple regions. Those
studies identified 15 novel rare missense mutations, among which
the c.425A>G (p.Glu142Gly) and c.424G>A (p.Glu142Lys)
mutations resulting in, respectively, 4- and 5-fold activation of
NF-κB, as compared with wild-type allele. On the other hand,
c.511C>A (p.His171Asn) and c.536G>A (p.Arg179His) variants
significantly activated the NF-κB pathway after the stimulation of
tumor necrosis factor-α (TNF-α). The expression of 13 inflammatory
genes (e.g., CCL20, IL8, IL6, colony
stimulating factor 2, CSF2) was also described to be
upregulated in keratinocytes of patients with CARD14 gene
variants (56).
The aforementioned studies have shown that the
gain-of-function mutations of CARD14 gene are associated
with psoriasis, but the relationship between CARD14 gene and
GPP remains to be adequately elucidated. In 2014, Sugiura et
al (24) found that 4 out of
19 cases of GPP with PV carried the c.526G>C (p.Arg179His)
heterozygous missense mutations in CARD14 gene in a Japanese
cohort, and the frequency of allelic mutations was significantly
higher than that of controls (3/100) and of patients with PV
(4/100). Thus, Sugiura et al suggested that c.526G>C
mutation is an important risk factor for GPP with PV, and is
distinct from the PV form. However, no pathogenic variants in the
CARD14 gene were identified in 11 patients affected by GPP
without PV, which supports that GPP alone is a heterogeneous
disease and genetically different from GPP with PV. Subsequently,
Qin et al (57)
identified two novel heterozygous mutations, the c.355A>G
(p.Met119Val) and c.497G>A (p.Arg166His), in 62 Chinese patients
suffering from GPP with PV, with the frequency of allelic mutations
being significantly higher than that of control (0/365), but
similar to that detected in patients with PV (2/174). In 2015, a
significant association between pathogenic c.526G>C mutation and
GPP in Asian populations was revealed by the analysis of 105
individuals affected by GPP (58). Subsequently, the group of Mössner
(23) and Twelves (22) identified CARD14 variants
in patients, even though they were rarely found in patients with
GPP. Mössner et al (23)
also identified 3 heterozygous missense mutations in CARD14
gene, the c.206G>A (p.Arg69Gln), c.349G>A, and c.536G>A,
in 51 GPP cases, and Twelves et al (22) reported that only 3 out of 251 GPP
patients harbored the c.526G>C heterozygous mutation. Taken
together, 10 possible pathogenic variants of CARD14 gene
have been identified (Fig. 1B,
Table III), even though they
are not common in GPP patients. Among them, the c.526G>C
missense mutation, found in the Asian population, is the most
common. Mutations in CARD14 gene are mainly presented in GPP
patients concomitantly affected by PV and rarely showing GPP alone
(20,26). CARD14 gene mutations
specific for PV and GPP patients have not been characterized yet.
Therefore, the correlation between CARD14 gene mutations and
the onset of GPP remains to be further elucidated.
| Table IIIMutations of CARD14 gene and
related characteristics in GPP patients. |
Table III
Mutations of CARD14 gene and
related characteristics in GPP patients.
| Nucleotide
variations | Amino acid
variations | Variants type | Status of the
mutations | Origin | Effect on NF-κB
activation (vs. wild-type) | (Refs.) |
|---|
| c.349G>A | p.Gly117Ser | Missense | Het |
European/German | 3.71 | (19,23,56) |
| c.355A>G | p.Met119Val | Missense | Het | Chinese | Not reported | (57) |
| c.413A>C | p.Glu138Ala | Missense | Het | Haitian | 8.95 | (19,56) |
| c.497G>A | p.Arg166His | Missense | Het | Chinese | Not reported | (57) |
| c.526G>C | p.Asp176His | Missense | Het |
Japanese/Chinese | 2.78 | (22,24,25, 56,58) |
| c.536G>A | p.Arg179His | Missense | Het | German | 1.38 (2.19 with
TNF-α stimulation) | (23,56) |
| c.424G>A† | p.Glu142Lys | Missense | Het | Not reported | 4.03 | (56) |
| c.425A>G† | p.Glu142Gly | Missense | Het | Not reported | 5 | (56) |
| c.511C>A† | p.His171Asn | Missense | Het | Not reported | 0.68 (5.95 with
TNF-α stimulation) | (56) |
| c.824G>A† | p.Arg275His | Missense | Het | Not reported | Not reported | (56) |
|
c.349+5G>A† | Alter splice of
intron | Frameshift | Het | Taiwanese | Not reported | (19) |
Disease-causing gene AP1S3
AP1S3 gene, encoding the core subunit σ1C of
adaptor protein complex 1 (AP-1), is responsible for the
stabilization of AP-1 heterotetramers involved in vesicular
trafficking between the trans-Golgi network and endosomes. Findings
have shown that loss-of-function mutations of AP1S3 gene are
relevant in GPP. In 2014, Setta-Kaffetzi et al (20) identified heterozygosity for the
c.11T>G (p.Phe4Cys) and c.97C>T (p.Arg33Trp) missense
mutations in AP1S3 gene in 15 European patients with various
forms of pustular psoriasis (i.e., PPP, ACH, and GPP) and not
harboring IL36RN and CARD14 gene mutations (Fig. 1C). In parallel, these pathogenic
variants were not detected in 70 cases from Africa and Asia. In
vitro functional assays demonstrated that the substitution of
c.11T>G causes a significant reduction in protein expression,
and silencing of AP1S3 in human keratinocytes and HEK293
cells abolishes endosomal translocation of toll-like receptor-3
(TLR3) and TLR3-dependent expression of interferon-ß1 (IFNB1)
following induction with polyinosinic-polycytidylic acid
[poly(I:C)], an agonist of TLR3 involved in responses to viral
infections. Thus, Setta-Kaffetzi et al (20) proposed that defects in vesicular
trafficking may be an important pathological basis for
auto-inflammatory in pustular psoriasis. In 2016, Mahil et
al (26) further
demonstrated that knockout of AP1S3 gene disrupts autophagy
in keratinocytes, thereby resulting in abnormal accumulation of
p62, which mediates NF-κB activation and upregulation of IL-1,
IL-36α and other cytokines. Subsequently, the c.11T>G and
c.97C>T heterozygous mutations in AP1S3 gene were
detected in two European patients with GPP, and the novel
c.64A>G (p.Thr22Ala) homozygous variant was identified in a
daughter of a consanguineous marriage (23). All these subjects carried
additional homozygous or compound-heterozygous IL36RN
mutations, as shown in the study of Mössner et al (23). Similarly, Twelves et al
(22) found that 8 out of 251
GPP cases carry c.11T>G (4 cases) or c.97C>T (4 cases)
heterozygous mutations in the AP1S3 gene, with two carriers
of c.11T>G mutation in AP1S3 gene also harboring the
known pathogenic IL36RN variants. Of note, AP1S3
pathogenic variants are mainly found in Europeans and rarely in
East Asians, and variant frequency of AP1S3 in GPP patients
of European descent is 10.8% (4/37).
Disease-causing gene MPO
MPO gene encodes myeloperoxidase, a lysosomal
hemoprotein located in the azurophilic granules of neutrophils. The
correlation between MPO deficiency and the onset of GPP has been
characterized only recently (21,59). Although previously described in a
single case with pustular psoriasis (60,61), MPO deficiency was not recognized
as a genetic risk factor of GPP until 2020 (4), when genetic variants in MPO
gene were screened in GPP and in conditions phenotypically related
to GPP, such as acral pustular psoriasis (APP) and acute
generalized exanthematous pustulosis (AGEP). Vergnano et al
(21) first identified the
c.2031-2A>C homozygous mutation due to A-C transition in the 3′
end of intron 11 in MPO gene in patients with GPP or APP,
and resulting in activation of a cryptic 3′ splice site located 109
bp upstream of canonical 3′ splice site, thereby causing a 119-bp
fragment insertion and a shift of the reading frame leading to
premature protein truncation. In addition, the c.2031-2A>C and
c.1705C>T (p. Arg569Trp) compound heterozygous mutations and
c.1555_1568del homozygous variant have been found in patients with
AGEP. Of note, all three variants have been repeatedly observed in
individuals affected by myeloperoxidase deficiency (MPOD) in which
MPO gene variants cause impairment of MPO protein function
(62-64). Phenome-wide association studies
(pheWAS), which provide a way to identify important relationships
between genetic variants and a wide array of phenotypes, and in
vitro functional analysis demonstrated that mutations in
MPO gene cause an increase of neutrophil accumulation and
activity, as well as a reduction in the number of apoptotic
neutrophils induced by phorbol myristate acetate (PMA), thus
suggesting a role of MPO mutations in GPP pathogenesis. Haskamp
et al (59) further
confirmed the important role of MPO gene defects in the
pathogenesis of GPP. In fact, they showed that 15 out of 74
patients with GPP carried 8 variants in MPO gene, including
the following: The c.265_275dup11 (p.Ser94Alafs*24),
c.2031-2A>C, c.1768C>T (p.Arg590Cys) homozygous variants, the
c.995C>T (p.Ala332Val) and c.2031-2A>C (p.Phe678*)
compound heterozygous variants and the c.752T>C (p. Met251Thr),
c.995C>T (p.Ala332Val), c.2031-2A>C, c.1705C>T,
c.1642C>T (p.Arg548Trp), and c.1555_1568del
(p.Met519Profs*21) heterozygous variants (Fig. 1D). All these variants were
validated as loss-of-function mutations, and, among them, 5
missense mutations (c.1768C>T, c.1705C>T, c.1642C>T,
c.995C>T and c.752T>C) reduced MPO activity in HEK cells at
different extent (Fig. 1D).
While the c.265_275dup11 homozygous mutation determined a lack of
MPO expression in neutrophils, the c.2031-2A>C substitution in a
splicing site as well as the c.1555_1568del deletion resulted in a
premature termination codon and truncated MPO protein. Functional
experiments further demonstrated that all four affected individuals
showed MPO activity inversely correlating with the activity of NE,
CTSG and PR3, three serine proteases that cleave IL-36 precursors
into pro-inflammatory forms. These data strongly suggest that MPO
deficiency may be involved in the pathogenesis of GPP through
regulating the activity of neutrophil and monocytic proteases, and
in turn activating pro-inflammatory IL-36 signals. In addition, MPO
deficiency caused the reduction of neutrophil extracellular traps
(NET) formation in PMA-induced pathway and impaired phagocytosis of
neutrophils by monocytes, thereby tolerating the persistence of
unfavorable neutrophils and blocking resolution of skin
inflammation. Notably, dosage of mutant alleles of MPO gene
in individuals affected by GPP also correlated with the age of
onset, which is similar to the genotype-phenotype correlation of
IL36RN gene and further validates the genetic correlation of
GPP. Thus, the novel findings that MPO gene is a pathogenic
gene for GPP provide new insights for the elucidation of GPP
pathogenesis, even if the in-depth pathogenic mechanism and new
pathogenic variants of MPO gene remain to be identified.
3. IL-1/IL-36-chemokine-neutrophil axis is a
potent driver of disease pathology in GPP
Among the mutations identified in the
disease-causing genes IL36RN, CARD14, AP1S3 and the
newly identified MPO in GPP patients, those present in
IL36RN play a pathogenic dominant role. In addition, the
IL-1/IL-36-chemokine-neutrophil axis is considered a core
pathogenic molecular pathway.
In the present study, we found that all four
disease-causing genes share some common pathogenic molecular
pathways. IL36RN, CARD14 and AP1S3 gene mutations can
activate pro-inflammatory signaling pathways via NF-κB, and further
result in an increased expression of CXCL1-3, IL-1, IL-8, and even
IL-36 pro-inflammatory cytokines (18-21). In addition, MPO gene
deficiency also promotes the activation of IL-36 signals by
regulating the activity of NE, CTSG and PR3 serine proteases
(Fig. 2) (59).
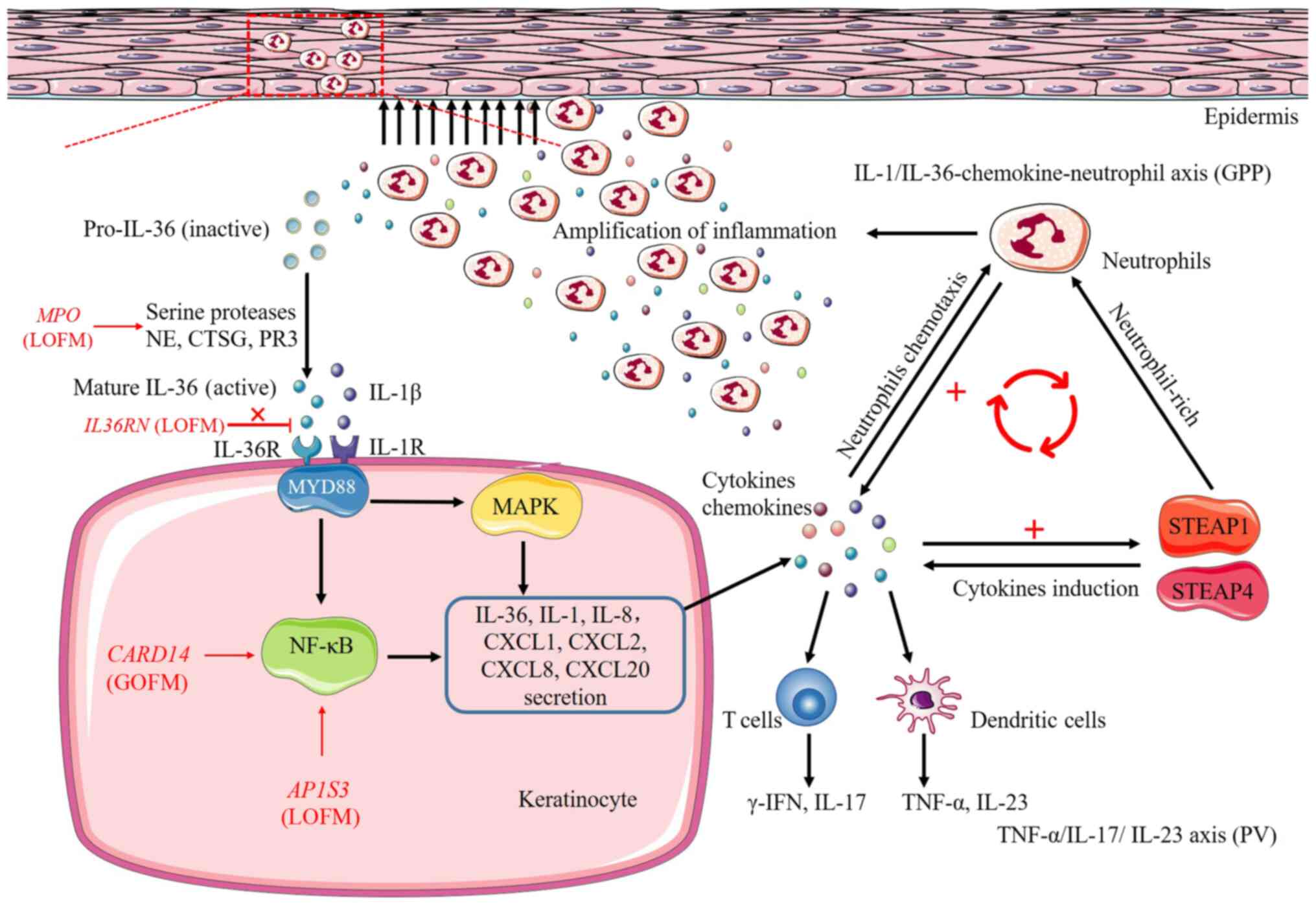 | Figure 2Pathways and processes of
inflammatory responses induced by IL36RN, CARD14,
AP1S3 and MPO genes. Loss-of-function mutations in
both IL36RN and MPO genes cause upregulation of IL-36
signaling, the former result in the inability of IL-36Ra to
antagonize and limit the pro-inflammatory effects of IL-36, the
latter upregulate the activity of NE, CTSG and PR3, three serine
proteases that cleave IL-36 precursors into pro-inflammatory forms.
Upregulated IL-36 signaling further activates the downstream
pro-inflammatory NF-κB and MAPK pathways by binding to IL-36
receptor, further leading to secretion of chemokines/cytokines,
IL-36, IL-1, IL-8, CXCL1, CXCL2, CXCL8, CXCL20, from the
keratinocyte and resulting in the activation of neutrophils, T
cells and dendritic cells. Secretion of cytokines also promotes
neutrophil-rich, cytokines induction, thereby amplifying the
pro-inflammatory responses in the skin by two inflammatory-related
proteins STEAP1 and STEAP4, ultimately forming a vicious cycle of
enhancing inflammation. In addition, CARD14 gain-of-function
mutations and AP1S3 loss-of-function mutations hyperactivate
NF-κB pathway and are involved in the processes of inflammatory
responses. Red or black arrows, secretion or activation; ┴,
inhibition; MyD88, myeloid differentiation primary response 88;
NF-κB, nuclear factor-κB; MAPK, mitogen-activated protein kinase;
LOFM, loss-of-function mutations; GOFM, gain-of-function mutations;
STEAP1, six-transmembrane epithelial antigens of prostate 1;
STEAP4, six-transmembrane epithelial antigens of prostate 4. |
Recently, the new disease concept of
autoinflammatory keratinization disease (AiKD) has been designated
to comprise inflammatory keratinization disorders with genetic
autoinflammatory pathomechanisms (65). GPP associated with IL36RN
and CARD14 mutations are included and early-onset GPP is
considered a typical one (66,67). Thus, initial genetic causative
factors related to the hyperactivation of innate immunity or
autoinflammation play dominant roles in the pathogenesis of GPP
(65-68). Unanimously, transcriptomic
analysis revealed that GPP patients share with patients affected by
plaque-type psoriasis the expression of common molecules and
pathways related to neutrophil chemotaxis; however, the
pathomechanisms operating in GPP patients are more related to
innate immunity inflammation (29,69) and those present in plaque
psoriasis are more dependent on adaptive immunity responses
(29). Thus, it is believed that
the IL-1/IL-36 inflammatory axis is central to the disease
pathology in GPP, whereas the TNF-α/IL-17/IL-23 axis appears to
plays a more important role in plaque psoriasis (31,70,71). A gene expression study found that
IL-17, TNF-α, IL-1, IL-36 and interferons (IFNs) were overexpressed
both in GPP and plaque psoriasis lesions, whereas GPP lesions
exhibited a higher mRNA level of IL-1 and IL-36 and lower of IL-17
and IFN-γ, as compared with plaque psoriasis lesions (29). Consequently, a high expression of
CXCL1, CXCL2, CXCL8 and IL-8 neutrophil chemoattractants is
observed in GPP lesions. Liang et al (69) further demonstrated that GPP, PPP
and AGEP pustular skin disorders have a common molecular basis
responsible for neutrophil chemotaxis. Of note, overexpression of
two inflammatory-related proteins, namely six-transmembrane
epithelial antigens of prostate1 and 4 (STEAP1 and STEAP4), was
revealed in the three pustular skin disorders. Those molecules
promoted neutrophil-rich, pro-inflammatory responses in the skin by
favoring induction of IL-1/IL-36 cytokines and CXCL1 and IL-8
neutrophil chemokines in the skin microenvironment. By contrast,
STEAP1 and STEAP4 are not upregulated in plaque psoriasis,
consistent with a weak induction of neutrophil-activating cytokines
in PV. This confirms that neutrophil recruitment is preferentially
active in pustular psoriasis, which is distinct from plaque-type
psoriasis mostly characterized by IL-17/IL-23 immunity responses
(Boehner et al, Mudigonda et al, Coimbra et
al, Grine et al, Fanoni et al) (14,72-75). Thus, IL-1/IL-36 inflammatory axis
can be considered a pivotal pathogenic pathway typically activated
in GPP. Its targeting by novel biological drugs potentially
represent an effective therapeutic strategy for GPP treatment.
4. Novel biologics treatment for GPP based
on pathoimmunology
At present, no standard guidelines for the treatment
of GPP have been established, and no specific therapeutic agents
for GPP have been approved in the United States or Europe. GPP
management currently refers to guidelines for psoriasis vulgaris.
However, new biologics targeting cytokines, including
TNF-α/IL-17/IL-23 and IL-1/IL-36 axis inhibitors, that are related
to pathological immunology bring bright prospects for the treatment
of GPP. The most relevant biological treatments have been
summarized in Table IV. While
TNF-α/IL-17/IL-23 axis is preferentially blocked in plaque
psoriasis, IL-1/IL-36-chemokine-neutrophil axis appears to be a
more promising therapeutic target in GPP.
| Table IVSummary of biologics treatment for
GPP. |
Table IV
Summary of biologics treatment for
GPP.
| Type | Drug | Properties | Therapeutic
target | IL36RN
mutations of patients enrolled | (Refs.) |
|---|
| TNF-α
inhibitors | Etanercept | Recombinant
DNA-derived TNF receptor-IgG fusion protein | TNF-α | c.80T>C | (80-84) |
| Infliximab | Chimeric monoclonal
antibody | TNF-α | c.115+6T>C | (51,82,83, 85,86) |
| Adalimumab | Fully human
monoclonal antibody | TNF-α | N/A | (82,83,87, 88) |
| IL-17
inhibitors | Ixekizumab | Monoclonal
antibody | IL-17A | N/A | (89-91) |
| Secukinumab | Monoclonal
antibody | IL-17A | c.115+6T>C | (90,92-94) |
| Brodalumab | Monoclonal
antibody | IL-17R | N/A | (95) |
| IL-23
inhibitors | Ustekinumab | Monoclonal
antibody | IL-12/23 p40 | c.227C>T | (96,97) |
| IL-1R
antagonist | Anakinra | Human recombinant
IL-1RA protein | IL-1R | c.142C>T,
C.338C>T | (76,98) |
| IL-1β
antagonists | Gevokizumab | Monoclonal
antibody | IL-1β | N/A | (77) |
| Canakinumab | Monoclonal
antibody | IL-1β | N/A | (78) |
| IL-36R
antagonist | BI655130 | Monoclonal
antibody | IL-36R | c.80T>C,
c.115+6T>C | (79) |
IL-1 targeting with biologics has been previously
performed in GPP patients using the IL-1α receptor antagonist
(IL-1-RA) anakinra and the IL-1β monoclonal antibodies gevokizumab
and canakinumab. Hüffmeier et al (76) reported successful treatment with
anakinra, produced by genetic recombination technology, in a
patient with GPP carrying the mutation of IL36RN gene.
Gevokizumab is an effective monoclonal antibody blocking the
pro-inflammatory cytokine IL-1β and its signal transduction in
inflammatory cells. Mansouri et al (77) reported the 79 and 65% reduction
in Psoriasis Activity and Severity Index (PASI) score at weeks 4
and 12 after treatment with gevokizumab in two patients with
severe, recalcitrant GPP. Skendros et al (78) reported a case of abrupt and
severe form of GPP with hypereosinophilia and cholestatic
hepatitis, completely cleared after treatment with canakinumab,
leading to anakinra discontinuation for persistent hypersensitivity
skin reactions. The new monoclonal antibody BI655130 targeting
IL-36 receptor can effectively block the IL-36 signaling pathway
and alleviate inflammatory response in GPP patients. A study on the
treatment of GPP with BI655130 showed that all 7 GPP patients
carrying homozygous IL36RN mutation (n=3), or heterozygous
mutation in CARD14 (n=1) or wild-type alleles (n=4)
significantly responded to BI655130 after 4-week therapy (79). The finding suggested that IL-36R
inhibition with a single dose of BI655130 can effectively alleviate
the severity of GPP regardless of the presence of the
disease-causing gene mutation and has great potential for future
clinical treatment of GPP. No serious adverse reactions and
recurrences related to therapy were reported in the abovementioned
studies. However, since clinical studies are very limited and
current data mainly derive from case reports or small single-arm
studies, further clinical investigations on larger populations are
required in order to determine the clinical efficacy, duration of
effect, and adverse events associated with the drug.
5. Conclusion and perspectives
The advances in our understanding of the genetic
variation underlying GPP has provided an outstanding framework for
basic research on the pathogenesis and treatment of GPP. These
advances have suggested several new theories while simultaneously
generating significant challenges. Evidence on the correlation
between genotype and clinical phenotype of GPP characterized by
various studies suggested that GPP is a heterogeneous disease with
distinct clinical manifestations and genetic characteristics, and
requires a separate diagnosis and treatment. Previous studies
reported that some GPP patients carry two or three disease-causing
gene variants or multiple mutations in one disease-causing gene
(17,22,23,59), and some healthy subjects who
carry the homozygous mutation of c.115+6T>C in IL36RN
gene, which theoretically leads to the complete loss of IL36RN
function, did not develop GPP until adulthood (15). Thus, it is suggested that the
genetic basis for the onset of GPP is an oligogenic rather than a
purely monogenic inheritance. The pathogenic variants in all four
genes found in patients with GPP can work together and promote skin
inflammation by increasing the production of pro-inflammatory
cytokines in keratinocytes, which ultimately shift the balance
towards substantial inflammation. Studies also found that large
number of GPP patients did not carry any known genetic variations
in IL36RN, CARD14, AP1S3 and MPO genes
(23), which suggests that some
novel variants located in introns or regulatory regions and other
genetic factors outside these four genes are expected to contribute
to the pathogenesis of the GPP. Therefore, further screening and
validating more pathogenic variants or novel pathogenic genes may
provide key insights into disease pathogenesis, as well as the
corresponding treatment and prevention strategies of GPP. We found
that the function of numerous possible pathogenic variants reported
remains to be validated. Therefore, functional research models
in vitro and in vivo are required to be established
for further elucidating the pathological mechanism. Although great
progress in therapy of GPP with biologics has been made, current
treatment studies are limited owing to a lack of data from controls
and the number of patient cohorts due to GPP rarity. Thus, it is
necessary to expand the patient cohorts from different countries
and ethnicities to provide more reliable data on long-term
maintenance of safety, efficacy and the impact of
withdrawal/re-treatment with new biologics.
Availability of data and materials
Not applicable
Authors' contributions
JZ and JL conceived and designed the review. JZ and
QL conducted formal literature search and analysis. QL, YC and XW
contributed to the raw data reviewing. JZ was involved in the
original draft preparation, JL was involved in the writing and
review of the manuscript. All the authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to express their gratitude
to EditSprings (https://www.editsprings.com/) for the expert
linguistic services provided.
Funding
This review is supported by the Sichuan Science and Technology
Program (grant nos. 2019YFS0332, 2019YFS0038, and 2020YFQ0045).
References
|
1
|
Navarini AA, Burden AD, Capon F, Mrowietz
U, Puig L, Köks S, Kingo K, Smith C and Barker JN; ERASPEN Network:
European consensus statement on phenotypes of pustular psoriasis. J
Eur Acad Dermatol Venereol. 31:1792–1799. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baker H and Ryan TJ: Generalized pustular
psoriasis. A clinical and epidemiological study of 104 cases. Br J
Dermatol. 80:771–793. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ohkawara A, Yasuda H, Kobayashi H, Inaba
Y, Ogawa H, Hashimoto I and Imamura S: Generalized pustular
psoriasis in Japan: Two distinct groups formed by differences in
symptoms and genetic background. Acta Derm Venereol. 76:68–71.
1996.PubMed/NCBI
|
|
4
|
Augey F, Renaudier P and Nicolas JF:
Generalized pustular psoriasis (Zumbusch): A French epidemiological
survey. Eur J Dermatol. 16:669–673. 2006.
|
|
5
|
Ito T, Takahashi H, Kawada A, Iizuka H,
Nakagawa H, Japanese Society For and Psoriasis Research:
Epidemiological survey from 2009 to 2012 of psoriatic patients in
Japanese society for psoriasis research. J Dermatol. 45:293–301.
2018. View Article : Google Scholar
|
|
6
|
Takahashi H, Nakamura K, Kaneko F and
Nakagawa H: Analysis of psoriasis patients registered with the
Japanese society for psoriasis research from 2002-2008. J Dermatol.
38:1125–1129. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen L, Huang X, Xiao Y, Su J, Shen M and
Chen X: Prevalence and risk factors of atopic dermatitis,
psoriasis, acne, and urticaria in China. Zhong Nan Da Xue Xue Bao
Yi Xue Ban. 45:449–455. 2020.In English, Chinese. PubMed/NCBI
|
|
8
|
Talaee R, Hajheydari Z, Moghaddam AY,
Moraveji SA and Ravandi BF: Prevalence of oral mucosal lesions and
their association with severity of psoriasis among psoriatic
patients referred to dermatology clinic: A cross-sectional study in
Kashan/Iran. Open Access Maced J Med Sci. 5:978–982. 2017.
View Article : Google Scholar
|
|
9
|
Griffiths CE and Barker JN: Pathogenesis
and clinical features of psoriasis. Lancet. 370:263–271. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Steffen C: William John Munro and Munro's
abscess, and Franz Kogoj and Kogoj's spongiform pustule. Am J
Dermatopathol. 24:364–368. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Crowley JJ, Pariser DM and Yamauchi PS: A
brief guide to pustular psoriasis for primary care providers.
Postgrad Med. 1–15. 2020.Online Ahead of Print.
|
|
12
|
Benjegerdes KE, Hyde K, Kivelevitch D and
Mansouri B: Pustular psoriasis: Pathophysiology and current
treatment perspectives. Psoriasis (Auckl). 6:131–144. 2016.
|
|
13
|
Sugiura K, Takemoto A, Yamaguchi M,
Takahashi H, Shoda Y, Mitsuma T, Tsuda K, Nishida E, Togawa Y,
Nakajima K, et al: The majority of generalized pustular psoriasis
without psoriasis vulgaris is caused by deficiency of
interleukin-36 receptor antagonist. J Invest Dermatol.
133:2514–2521. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Boehner A, Navarini AA and Eyerich K:
Generalized pustular psoriasis-a model disease for specific
targeted immunotherapy, systematic review. Exp Dermatol.
27:1067–1077. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li M, Han J, Lu Z, Li H, Zhu K, Cheng R,
Jiao Q, Zhang C, Zhu C, Zhuang Y, et al: Prevalent and rare
mutations in IL-36RN gene in Chinese patients with generalized
pustular psoriasis and psoriasis vulgaris. J Invest Dermatol.
133:2637–2639. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li X, Chen M, Fu X, Zhang Q, Wang Z, Yu G,
Yu Y, Qin P, Wu W, Pan F, et al: Mutation analysis of the IL36RN
gene in Chinese patients with generalized pustular psoriasis
with/without psoriasis vulgaris. J Dermatol Sci. 76:132–138. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hussain S, Berki DM, Choon SE, Burden AD,
Allen MH, Arostegui JI, Chaves A, Duckworth M, Irvine AD,
Mockenhaupt M, et al: IL36RN mutations define a severe
autoinflammatory phenotype of generalized pustular psoriasis. J
Allergy Clin Immunol. 135:1067–1070.e9. 2015. View Article : Google Scholar
|
|
18
|
Marrakchi S, Guigue P, Renshaw BR, Puel A,
Pei XY, Fraitag S, Zribi J, Bal E, Cluzeau C, Chrabieh M, et al:
Interleukin-36-receptor antagonist deficiency and generalized
pustular psoriasis. N Engl J Med. 365:620–628. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jordan CT, Cao L, Roberson ED, Pierson KC,
Yang CF, Joyce CE, Ryan C, Duan S, Helms CA, Liu Y, et al: PSORS2
is due to mutations in CARD14. Am J Hum Genet. 90:784–795. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Setta-Kaffetzi N, Simpson MA, Navarini AA,
Patel VM, Lu HC, Allen MH, Duckworth M, Bachelez H, Burden AD,
Choon SE, et al: AP1S3 mutations are associated with pustular
psoriasis and impaired Toll-like receptor 3 trafficking. Am J Hum
Genet. 94:790–797. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vergnano M, Mockenhaupt M, Benzian-Olsson
N, Paulmann M, Grys K, Mahil SK, Chaloner C, Barbosa IA, August S,
Burden AD, et al: Loss-of-function myeloperoxidase mutations are
associated with increased neutrophil counts and pustular skin
disease. Am J Hum Genet. 107:539–543. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Twelves S, Mostafa A, Dand N, Burri E,
Farkas K, Wilson R, Cooper HL, Irvine AD, Oon HH, Kingo K, et al:
Clinical and genetic differences between pustular psoriasis
subtypes. J Allergy Clin Immunol. 143:1021–1026. 2019. View Article : Google Scholar :
|
|
23
|
Mössner R, Wilsmann-Theis D, Oji V,
Gkogkolou P, Löhr S, Schulz P, Körber A, Prinz JC, Renner R,
Schäkel K, et al: The genetic basis for most patients with pustular
skin disease remains elusive. Br J Dermatol. 178:740–748. 2018.
View Article : Google Scholar
|
|
24
|
Sugiura K, Muto M and Akiyama M: CARD14
c.526G>C (p.Asp176His) is a significant risk factor for
generalized pustular psoriasis with psoriasis vulgaris in the
Japanese cohort. J Invest Dermatol. 134:1755–1757. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li L, You J, Fu X, Wang Z, Sun Y, Liu H
and Zhang F: Variants of CARD14 are predisposing factors for
generalized pustular psoriasis (GPP) with psoriasis vulgaris but
not for GPP alone in a Chinese population. Br J Dermatol.
180:425–426. 2019. View Article : Google Scholar
|
|
26
|
Mahil SK, Twelves S, Farkas K,
Setta-Kaffetzi N, Burden AD, Gach JE, Irvine AD, Képíró L,
Mockenhaupt M, Oon HH, et al: AP1S3 mutations cause skisn
autoinflammation by disrupting keratinocyte autophagy and
up-regulating IL-36 production. J Invest Dermatol. 136:2251–2259.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Towne JE, Garka KE, Renshaw BR, Virca GD
and Sims JE: Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal
through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to
NF-kappaB and MAPKs. J Biol Chem. 279:13677–13688. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Buhl AL and Wenzel J: Interleukin-36 in
infectious and inflammatory skin diseases. Front Immunol.
10:11622019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Johnston A, Xing X, Wolterink L, Barnes
DH, Yin Z, Reingold L, Kahlenberg JM, Harms PW and Gudjonsson JE:
IL-1 and IL-36 are dominant cytokines in generalized pustular
psoriasis. J Allergy Clin Immunol. 140:109–120. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Onoufriadis A, Simpson MA, Pink AE, Di
Meglio P, Smith CH, Pullabhatla V, Knight J, Spain SL, Nestle FO,
Burden AD, et al: Mutations in IL36RN/IL1F5 are associated with the
severe episodic inflammatory skin disease known as generalized
pustular psoriasis. Am J Hum Genet. 89:432–437. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Furue K, Yamamura K, Tsuji G, Mitoma C,
Uchi H, Nakahara T, Kido-Nakahara M, Kadono T and Furue M:
Highlighting interleukin-36 signalling in plaque psoriasis and
pustular psoriasis. Acta Derm Venereol. 98:5–13. 2018. View Article : Google Scholar
|
|
32
|
Cowen EW and Goldbach-Mansky R: DIRA,
DITRA, and new insights into pathways of skin inflammation: What's
in a name? Arch Dermatol. 148:381–384. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aksentijevich I, Masters SL, Ferguson PJ,
Dancey P, Frenkel J, van Royen-Kerkhoff A, Laxer R, Tedgård U,
Cowen EW, Pham TH, et al: An autoinflammatory disease with
deficiency of the interleukin-1-receptor antagonist. N Engl J Med.
360:2426–2437. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Reddy S, Jia S, Geoffrey R, Lorier R,
Suchi M, Broeckel U, Hessner MJ and Verbsky J: An autoinflammatory
disease due to homozygous deletion of the IL1RN locus. N Engl J
Med. 360:2438–2444. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sugiura K, Takeichi T, Kono M, Ogawa Y,
Shimoyama Y, Muro Y and Akiyama M: A novel IL36RN/IL1F5 homozygous
nonsense mutation, p.Arg10X, in a Japanese patient with adult-onset
generalized pustular psoriasis. Br J Dermatol. 167:699–701. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kanazawa N, Nakamura T, Mikita N and
Furukawa F: Novel IL36RN mutation in a Japanese case of early onset
generalized pustular psoriasis. J Dermatol. 40:749–751. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Farooq M, Nakai H, Fujimoto A, Fujikawa H,
Matsuyama A, Kariya N, Aizawa A, Fujiwara H, Ito M and Shimomura Y:
Mutation analysis of the IL36RN gene in 14 Japanese patients with
generalized pustular psoriasis. Hum Mutat. 34:176–183. 2013.
View Article : Google Scholar
|
|
38
|
Tauber M, Bal E, Pei XY, Madrange M,
Khelil A, Sahel H, Zenati A, Makrelouf M, Boubridaa K, Chiali A, et
al: IL36RN mutations affect protein expression and function: A
basis for genotype-phenotype correlation in pustular diseases. J
Invest Dermatol. 136:1811–1819. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Setta-Kaffetzi N, Navarini AA, Patel VM,
Pullabhatla V, Pink AE, Choon SE, Allen MA, Burden AD, Griffiths
CE, Seyger MM, et al: Rare pathogenic variants in IL36RN underlie a
spectrum of psoriasis-associated pustular phenotypes. J Invest
Dermatol. 133:1366–1369. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sugiura K, Nakasuka A, Kono H, Kono M and
Akiyama M: Impetigo herpetiformis with IL36RN mutations in a
Chinese patient: A founder haplotype of c.115+6T>C in East Asia.
J Dermatol Sci. 79:319–320. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shiratori T, Fukai K, Yasumizu M, Taguchi
R, Tsuruta D, Abe Y, Hozumi Y and Suzuki T: IL36RN gene analysis of
two Japanese patients with generalized pustular psoriasis. Int J
Dermatol. 54:e60–e62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Y, Cheng R, Lu Z, Guo Y, Yan M, Liang
J, Huang P, Li M and Yao Z: Clinical profiles of pediatric patients
with GPP alone and with different IL36RN genotypes. J Dermatol Sci.
85:235–240. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Renert-Yuval Y, Horev L, Babay S, Tams S,
Ramot Y, Zlotogorski A and Molho-Pessach V: IL36RN mutation causing
generalized pustular psoriasis in a Palestinian patient. Int J
Dermatol. 53:866–868. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ueda Y, Komine M, Kamiya K, Tsuda H,
Maekawa T, Murata S and Ohtsuki M: Generalized pustular psoriasis
in a 92-year-old man with a homozygous nonsense mutation in IL36RN.
J Dermatol. 45:326–328. 2018. View Article : Google Scholar
|
|
45
|
Ellingford JM, Black GC, Clayton TH, Judge
M, Griffiths CE and Warren RB: A novel mutation in IL36RN underpins
childhood pustular dermatosis. J Eur Acad Dermatol Venereol.
30:302–305. 2016. View Article : Google Scholar :
|
|
46
|
Rossi-Semerano L, Piram M, Chiaverini C,
De Ricaud D, Smahi A and Koné-Paut I: First clinical description of
an infant with interleukin-36-receptor antagonist deficiency
successfully treated with anakinra. Pediatrics. 132:e1043–e1047.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Körber A, Mössner R, Renner R, Sticht H,
Wilsmann-Theis D, Schulz P, Sticherling M, Traupe H and Hüffmeier
U: Mutations in IL36RN in patients with generalized pustular
psoriasis. J Invest Dermatol. 133:2634–2637. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Takeichi T, Togawa Y, Okuno Y, Taniguchi
R, Kono M, Matsue H, Sugiura K and Akiyama M: A newly revealed
IL36RN mutation in sibling cases complements our IL36RN mutation
statistics for generalized pustular psoriasis. J Dermatol Sci.
85:58–60. 2017. View Article : Google Scholar
|
|
49
|
Wang TS, Chiu HY, Hong JB, Chan CC, Lin SJ
and Tsai TF: Correlation of IL36RN mutation with different clinical
features of pustular psoriasis in Chinese patients. Arch Dermatol
Res. 308:55–63. 2016. View Article : Google Scholar
|
|
50
|
Abbas O, Itani S, Ghosn S, Kibbi AG,
Fidawi G, Farooq M, Shimomura Y and Kurban M: Acrodermatitis
continua of Hallopeau is a clinical phenotype of DITRA: Evidence
that it is a variant of pustular psoriasis. Dermatology. 226:28–31.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sugiura K, Endo K, Akasaka T and Akiyama
M: Successful treatment with infliximab of sibling cases with
generalized pustular psoriasis caused by deficiency of
interleukin-36 receptor antagonist. J Eur Acad Dermatol Venereol.
29:2054–2056. 2015. View Article : Google Scholar
|
|
52
|
Song HS, Yun SJ, Park S and Lee ES: Gene
mutation analysis in a Korean patient with early-onset and
recalcitrant generalized pustular psoriasis. Ann Dermatol.
26:424–425. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liang J, Zhang H, Guo Y, Yang K, Ni C, Yu
H, Kong X, Li M, Lu Z and Yao Z: Coinheritance of generalized
pustular psoriasis and familial Behçet-like autoinflammatory
syndrome with variants in IL36RN and TNFAIP3 in the heterozygous
state. J Dermatol. 46:907–910. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Scudiero I, Zotti T, Ferravante A,
Vessichelli M, Vito P and Stilo R: Alternative splicing of
CARMA2/CARD14 transcripts generates protein variants with
differential effect on NF-κB activation and endoplasmic reticulum
stress-induced cell death. J Cell Physiol. 226:3121–3131. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bertin J, Wang L, Guo Y, Jacobson MD,
Poyet JL, Srinivasula SM, Merriam S, DiStefano PS and Alnemri ES:
CARD11 and CARD14 are novel caspase recruitment domain
(CARD)/membrane-associated guanylate kinase (MAGUK) family members
that interact with BCL10 and activate NF-kappa B. J Biol Chem.
276:11877–11882. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jordan CT, Cao L, Roberson ED, Duan S,
Helms CA, Nair RP, Duffin KC, Stuart PE, Goldgar D, Hayashi G, et
al: Rare and common variants in CARD14, encoding an epidermal
regulator of NF-kappaB, in psoriasis. Am J Hum Genet. 90:796–808.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Qin P, Zhang Q, Chen M, Fu X, Wang C, Wang
Z, Yu G, Yu Y, Li X, Sun Y, et al: Variant analysis of CARD14 in a
Chinese Han population with psoriasis vulgaris and generalized
pustular psoriasis. J Invest Dermatol. 134:2994–2996. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Berki DM, Liu L, Choon SE, David Burden A,
Griffiths CEM, Navarini AA, Tan ES, Irvine AD, Ranki A, Ogo T, et
al: Activating CARD14 mutations are associated with generalized
pustular psoriasis but rarely account for familial recurrence in
psoriasis vulgaris. J Invest Dermatol. 135:2964–2970. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Haskamp S, Bruns H, Hahn M, Hoffmann M,
Gregor A, Löhr S, Hahn J, Schauer C, Ringer M, Flamann C, et al:
Myeloperoxidase modulates inflammation in generalized pustular
psoriasis and additional rare pustular skin diseases. Am J Hum
Genet. 107:527–538. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
De Argila D, Dominguez JD,
Lopez-Estebaranz JL and Iglesias L: Pustular psoriasis in a patient
with myeloperoxidase deficiency. Dermatology. 193:2701996.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Stendahl O, Coble BI, Dahlgren C, Hed J
and Molin L: Myeloperoxidase modulates the phagocytic activity of
polymorphonuclear neutrophil leukocytes. Studies with cells from a
myeloperoxidase-deficient patient. J Clin Invest. 73:366–373. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kizaki M, Miller CW, Selsted ME and
Koeffler HP: Myeloperoxidase (MPO) gene mutation in hereditary MPO
deficiency. Blood. 83:1935–1940. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Marchetti C, Patriarca P, Solero GP,
Baralle FE and Romano M: Genetic studies on myeloperoxidase
deficiency in Italy. Jpn J Infect Dis. 57:S10–S12. 2004.PubMed/NCBI
|
|
64
|
Marchetti C, Patriarca P, Solero GP,
Baralle FE and Romano M: Genetic characterization of
myeloperoxidase deficiency in Italy. Hum Mutat. 23:496–505. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Akiyama M, Takeichi T, McGrath JA and
Sugiura K: Autoinflammatory keratinization diseases. J Allergy Clin
Immunol. 140:1545–1547. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Akiyama M: Early-onset generalized
pustular psoriasis is representative of autoinflammatory
keratinization diseases. J Allergy Clin Immunol. 143:809–810. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kanazawa N: Designation of
autoinflammatory skin manifestations with specific genetic
backgrounds. Front Immunol. 11:4752020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Takeichi T and Akiyama M: Familial or
sporadic porokeratosis as an autoinflammatory keratinization
disease. J Dermatol. 46. pp. e125–e126. 2019, View Article : Google Scholar
|
|
69
|
Liang Y, Xing X, Beamer MA, Swindell WR,
Sarkar MK, Roberts LW, Voorhees JJ, Kahlenberg JM, Harms PW,
Johnston A and Gudjonsson JE: Six-transmembrane epithelial antigens
of the prostate comprise a novel inflammatory nexus in patients
with pustular skin disorders. J Allergy Clin Immunol.
139:1217–1227. 2017. View Article : Google Scholar
|
|
70
|
Ogawa E, Sato Y, Minagawa A and Okuyama R:
Pathogenesis of psoriasis and development of treatment. J Dermatol.
45:264–272. 2018. View Article : Google Scholar
|
|
71
|
Nestle FO, Conrad C, Tun-Kyi A, Homey B,
Gombert M, Boyman O, Burg G, Liu YJ and Gilliet M: Plasmacytoid
predendritic cells initiate psoriasis through interferon-alpha
production. J Exp Med. 202:135–143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Mudigonda P, Mudigonda T, Feneran AN,
Alamdari HS, Sandoval L and Feldman SR: Interleukin-23 and
interleukin-17: Importance in pathogenesis and therapy of
psoriasis. Dermatol Online J. 18:12012.PubMed/NCBI
|
|
73
|
Coimbra S, Oliveira H, Reis F, Belo L,
Rocha S, Quintanilha A, Figueiredo A, Teixeira F, Castro E,
Rocha-Pereira P and Santos-Silva A: Interleukin (IL)-22, IL-17,
IL-23, IL-8, vascular endothelial growth factor and tumour necrosis
factor-α levels in patients with psoriasis before, during and after
psoralen-ultraviolet A and narrowband ultraviolet B therapy. Br J
Dermatol. 163:1282–1290. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Grine L, Dejager L, Libert C and
Vandenbroucke RE: An inflammatory triangle in psoriasis: TNF, type
I IFNs and IL-17. Cytokine Growth Factor Rev. 26:25–33. 2015.
View Article : Google Scholar
|
|
75
|
Fanoni D, Venegoni L, Vergani B, Tavecchio
S, Cattaneo A, Leone BE, Berti E and Marzano AV: Evidence for a
role of auto-inflammation in early-phase psoriasis. Clin Exp
Immunol. 198:283–291. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hüffmeier U, Wätzold M, Mohr J, Schön MP
and Mössner R: Successful therapy with anakinra in a patient with
generalized pustular psoriasis carrying IL36RN mutations. Br J
Dermatol. 170:202–204. 2014. View Article : Google Scholar
|
|
77
|
Mansouri B, Richards L and Menter A:
Treatment of two patients with generalized pustular psoriasis with
the interleukin-1β inhibitor gevokizumab. Br J Dermatol.
173:239–241. 2015. View Article : Google Scholar
|
|
78
|
Skendros P, Papagoras C, Lefaki I,
Giatromanolaki A, Kotsianidis I, Speletas M, Bocly V, Theodorou I,
Dalla V and Ritis K: Successful response in a case of severe
pustular psoriasis after interleukin-1β inhibition. Br J Dermatol.
176:212–215. 2017. View Article : Google Scholar
|
|
79
|
Bachelez H, Choon SE, Marrakchi S, Burden
AD, Tsai TF, Morita A, Turki H, Hall DB, Shear M, Baum P, et al:
Inhibition of the interleukin-36 pathway for the treatment of
generalized pustular psoriasis. N Engl J Med. 380:981–983. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Fialová J, Vojáčková N, Vaňousová D and
Hercogová J: Juvenile generalized pustular psoriasis treated with
etanercept. Dermatol Ther. 27:105–108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Esposito M, Mazzotta A, Casciello C and
Chimenti S: Etanercept at different dosages in the treatment of
generalized pustular psoriasis: A case series. Dermatology.
216:355–360. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Viguier M, Aubin F, Delaporte E, Pagès C,
Paul C, Beylot-Barry M, Goujon C, Rybojad M and Bachelez H; Groupe
de Recherche sur le Psoriasis de la Société Française de
Dermatologie: Efficacy and safety of tumor necrosis factor
inhibitors in acute generalized pustular psoriasis. Arch Dermatol.
148:1423–1425. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Saikaly SK and Mattes M: Biologics and
pediatric generalized pustular psoriasis: An emerging therapeutic
trend. Cureus. 8:e6522016.PubMed/NCBI
|
|
84
|
Cuperus E, Koevoets R, van der Smagt JJ,
Toonstra J, de Graaf M, Frenkel J and Pasmans SGMA: Juvenile
interleukin-36 receptor antagonist deficiency (DITRA) with
c.80T>C (p.Leu27Pro) mutation successfully treated with
etanercept and acitretin. JAAD Case Rep. 4:192–195. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Adachi A, Komine M, Hirano T, Tsuda H,
Karakawa M, Murata S and Ohtsuki M: Case of generalized pustular
psoriasis exacerbated during pregnancy, successfully treated with
infliximab. J Dermatol. 43:1439–1440. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Pan J, Qiu L, Xiao T and Chen HD: Juvenile
generalized pustular psoriasis with IL36RN mutation treated with
short-term infliximab. Dermatol Ther. 29:164–167. 2016. View Article : Google Scholar
|
|
87
|
Morita A, Yamazaki F, Matsuyama T,
Takahashi K, Arai S, Asahina A, Imafuku S, Nakagawa H, Hasegawa Y,
Williams D, et al: Adalimumab treatment in Japanese patients with
generalized pustular psoriasis: Results of an open-label phase 3
study. J Dermatol. 45:1371–1380. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Matsumoto A, Komine M, Karakawa M,
Kishimoto M and Ohtsuki M: Adalimumab administration after
infliximab therapy is a successful treatment strategy for
generalized pustular psoriasis. J Dermatol. 44:202–204. 2017.
View Article : Google Scholar
|
|
89
|
Egawa G, Honda T and Kabashima K:
Long-term efficacy of ixekizumab in erythrodermic and generalized
pustular psoriasis patients. J Eur Acad Dermatol Venereol.
33:2592019. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Wilsmann-Theis D, Schnell LM,
Ralser-Isselstein V, Bieber T, Schön MP, Hüffmeier U and Mössner R:
Successful treatment with interleukin-17A antagonists of
generalized pustular psoriasis in patients without IL36RN
mutations. J Dermatol. 45:850–854. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Okubo Y, Mabuchi T, Iwatsuki K, Elmaraghy
H, Torisu-Itakura H, Morisaki Y and Nakajo K: Long-term efficacy
and safety of ixekizumab in Japanese patients with erythrodermic or
generalized pustular psoriasis: Subgroup analyses of an open-label,
phase 3 study (UNCOVER-J). J Eur Acad Dermatol Venereol.
33:325–332. 2019. View Article : Google Scholar
|
|
92
|
Ho PH and Tsai TF: Successful treatment of
refractory juvenile generalized pustular psoriasis with secukinumab
monotherapy: A case report and review of published work. J
Dermatol. 45:1353–1356. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Imafuku S, Honma M, Okubo Y, Komine M,
Ohtsuki M, Morita A, Seko N, Kawashima N, Ito S, Shima T and
Nakagawa H: Efficacy and safety of secukinumab in patients with
generalized pustular psoriasis: A 52-week analysis from phase III
open-label multi-center Japanese study. J Dermatol. 43:1011–1017.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Gabeff R, Safar R, Leducq S, Maruani A,
Sarrabay G, Touitou I and Samimi M: Successful therapy with
secukinumab in a patient with generalized pustular psoriasis
carrying homozygous IL36RN p.His32Arg mutation. Int J Dermatol.
58:e16–e17. 2019. View Article : Google Scholar
|
|
95
|
Yamasaki K, Nakagawa H, Kubo Y and Ootaki
K; Japanese Brodalumab Study Group: Efficacy and safety of
brodalumab in patients with generalized pustular psoriasis and
psoriatic erythroderma: Results from a 52-week, open-label study.
Br J Dermatol. 176:741–751. 2017. View Article : Google Scholar
|
|
96
|
Storan ER, O'Gorman SM and Markham T:
Generalized pustular psoriasis treated with ustekinumab. Clin Exp
Dermatol. 41:689–690. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Arakawa A, Ruzicka T and Prinz JC:
Therapeutic efficacy of interleukin 12/interleukin 23 blockade in
generalized pustular psoriasis regardless of IL36RN mutation
status. JAMA Dermatol. 152:825–828. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Viguier M, Guigue P, Pages C, Smahi A and
Bachelez H: Successful treatment of generalized pustular psoriasis
with the interleukin-1-receptor antagonist Anakinra: Lack of
correlation with IL1RN mutations. Ann Intern Med. 153:66–67. 2010.
View Article : Google Scholar : PubMed/NCBI
|














