Contents
Introduction
Tumor-associated macrophages
TAMs and hypoxia
TAMs and angiogenesis
TAMs and the tumor matrix
TAMs and toll-like receptors
Clinical applications
Conclusion
Introduction
Tumors of epithelial cell origin grow in a complex
and dynamic stroma consisting of various cell types, matrix
proteins and soluble factors. This microenvironment provides all
the necessary stimuli for tumor viability, growth, and
invasiveness. Essential components of this microenvironment are
inflammatory cells and the pro-inflammatory cytokines they produce.
The role of inflammation in cancer progression was first described
by Virchow in 1867 (1). Recent
epidemiologic evidence suggests that inflammation resulting from
pathogen stimulation could be a promoter of tumorigenesis by
driving the immune response through different inflammatory cells
that are recruited to and reside in the tumor microenvironment
(2). Processes that enhance the
aggressiveness of tumor behavior include angiogenesis, hypoxia,
interaction between cell membrane receptors and tumor
microenvironment cytokines and basement membrane degradation.
The process of metastasis and invasiveness of tumor
cells is complex and determines clinical outcomes of cancer
patients. Many studies have identified a link between this process
and some of the cellular elements in the tumor microenvironment.
Cells in the tumor microenvironment secrete a wide array of
products that modulate the behavior of tumor cells in a
tumor-enhancing and tumor-suppressing fashion. Of these cells,
macrophages have gained a considerable interest in recent years.
Macrophages are the most abundant inflammatory cells in the solid
tumor microenvironment, and in this review, we examined how
macrophages enhance the invasiveness of breast cancer.
Tumor-associated macrophages
Tumor-associated macrophages (TAMs) have been
described in many solid tumors, including breast cancer. They make
up a big proportion of the cellular elements in different tumors.
In breast cancer, macrophages can constitute as much as 50% of the
cell mass (3). Multiple clinical
studies have shown a correlation between breast cancer prognosis
and the number of macrophages in the tumor mass. In a recent report
by Bingle et al, a meta-analysis showed that an increased
macrophage density was associated with poor prognosis in more than
80% of breast cancer cases (4).
Origin of TAMs
Macrophages are derived from the mononuclear
phagocyte system (5). Progenitor
cells in the bone marrow can give rise to two types of cells:
neutrophils and macrophages. The macrophage pathway is committed to
produce monoblasts through serial divisions of promonoblasts from
the colony forming unit, granulocyte-macrophage (CFU-GM).
Peripheral blood monocytes, derived from bone marrow progenitor
cells that give rise to monoblasts and promonocytes, eventually
leave the blood stream and enter the tissue where they
differentiate into macrophages (6). In the tissues, they have different
roles based on the tissue in which they reside. Leek and Harris
(5) described macrophages as the
‘Swiss army knife’ of the immune system. However, when they are
activated appropriately, macrophages have specific and distinct
functions. Many signals influencing macrophages have been
identified and include hypoxia, lipids, and cytokines (e.g., CCL2,
CCL3 and CSF-1), TNF-α, INF-γ and MIF (7). Macrophage migration into tissues is
not haphazard; rather, this process is specifically orchestrated by
various chemoattractants. Among the chemoattractants, CCL2
(formally referred to as monocyte chemoattractant prorein-1, MCP-1)
is suggested to be important in tumor progression. Fujimoto et
al showed recently that stromal CCL2 is associated with
recruitment of macrophages to the breast cancer stroma and plays a
role in relapse-free survival (8).
Macrophage migration inhibitory factor (MIF) is another
chemoattractant involved in tumor progression. MIF, an upstream
regulator of host immunity, constitutes an important link between
chronic inflammation and cancer. By interacting with CXCR2 and
CXCR4, MIF recruits leukocytes and activates cellular inflammatory
responses (9). Its levels were
found to be elevated in different cancers, particularly prostate
and breast cancer. Of particular interest, intratumoral MIF levels
were inversely related to nodal status, and this determined an
interaction between MIF and intratumoral interleukins (10). A series of experiments with breast
cancer cell lines and macrophage cell lines showed that MIF was a
major gene product that was upregulated when breast cancer cells
were cocultured with macrophages. MIF secreted by tumor cells
increased the macrophage metalloproteinase production rates and
facilitated tumor cell invasion (11).
Macrophage polarization
After entering peripheral tissues, the pattern of
macrophage activation depends on the surrounding microenvironment,
which accounts for the heterogeneity among macrophage populations
and their plasticity. Typically, macrophages are divided into two
main groups, the M1 and M2 macrophages.
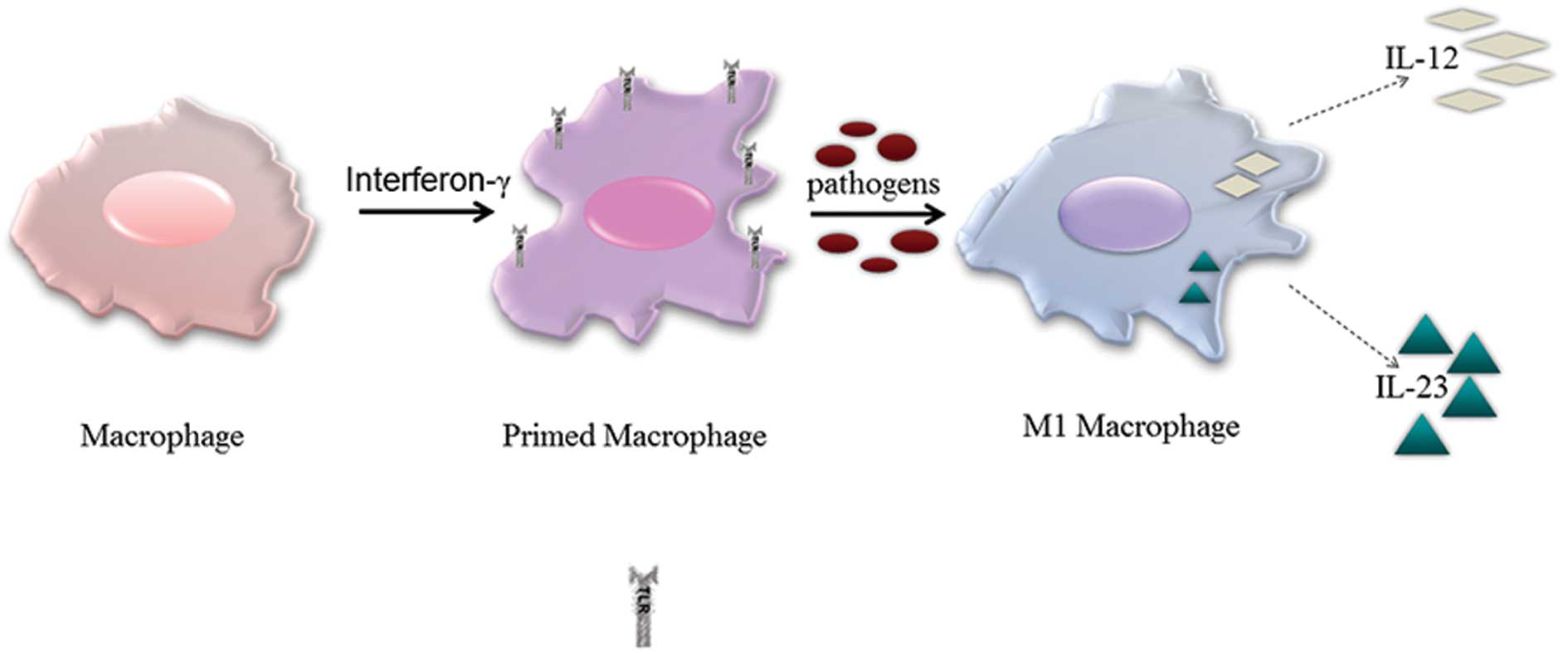
M1 macrophages (also known as ‘classically’
activated macrophages) are an integral cellular component of the
immune system. These cells play an important role in protection
against intracellular pathogens and cancer cells by virtue of the
type I immune response they activate. They are ‘primed’ by the
cytokine INF-γ for activation either by tumor necrosis factor-α
(TNF-α) or (and more importantly) by activation of toll-like
receptors (TLRs) via exposure to microbes or microbial products
such as bacterial LPS (12). Once
activated, M1 macrophages start secreting pro-inflammatory
cytokines such as interferons and interleukins. M1 macrophages
particularly secrete high levels of IL-12 and in humans they also
secrete high levels of IL-23 (13). M1 macrophages also function as
antigen-presenting cells; a function that is regarded as a
potentially antitumor function (14). These cells have an amplified
capacity to kill intracellular pathogens by generating toxic oxygen
species and by activating the inducible NO synthase (iNOS) gene to
produce nitric oxide (NO). Therefore, M1 macrophages can defend the
host from tumor cells and infections (14–16)
(Fig. 1).
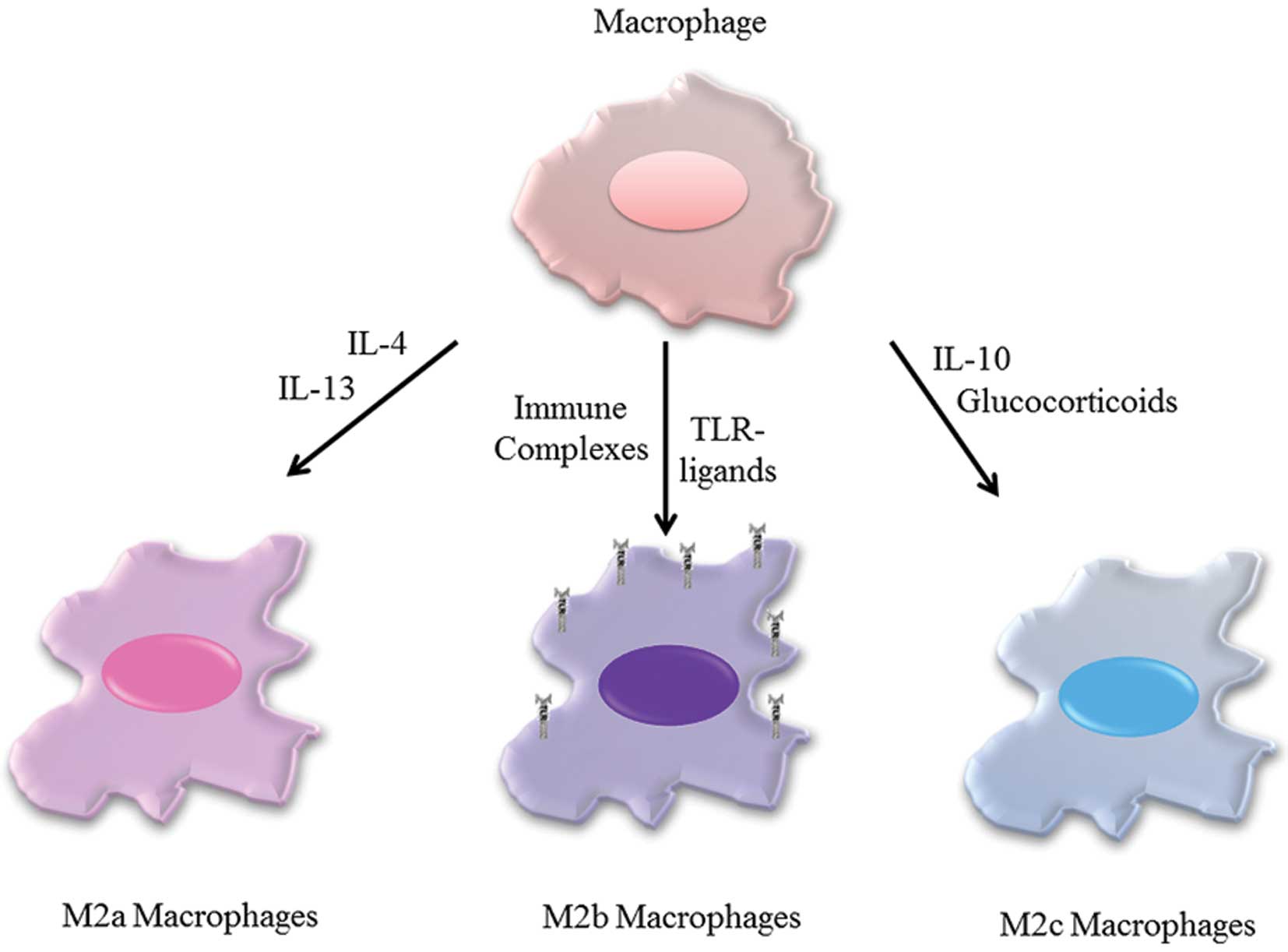
The M2 group of macrophages is referred to as
tumor-associated macrophages as they generally promote tumor growth
and metastasis (17). They are
also referred to as ‘alternatively activated’ macrophages (M2).
Unlike M1 macrophages that are activated by INF-γ, M2 macrophages
are activated by other cytokines or by immune complexes. This
activation induces a T-helper 2 type of response and as such, they
are appropriately called M2 macrophages (reflecting this Th2-type
response). However, it has recently been suggested that this M1/M2
dichotomous assignment of macrophages might be too simple and does
not convey the different functions and activation patterns of M2
macrophages. An alternative classification was proposed by
Mantovani et al whereby M2 macrophages are divided into
three sub-populations depending on the particular stimulus that
activates them (17). M2a
(alternative) macrophages are induced by exposure to IL-4 and IL-13
while M2b macrophages are induced by toll-like receptor ligands
(e.g., LPS) and immune complexes. Both the M2a and M2b groups
promote the Th2 immune response, by secreting Th2 type cytokines
and by recruiting Th2 cells (17).
The M2c macrophage subgroup represents macrophages that are
activated by IL-10 and glucocorticoid hormones. In general, an M2c
response is responsible for suppressing inflammation which is an
immunologically a Th-1 type of response (Fig. 2).
Macrophages seem to be directed by the tumor
microenvironment and in turn depending on the surrounding milieu
will acquire a trophic property as opposed to the original immune
role they had (18). It is clear
that different stimuli polarize macrophages into different
phenotypes. In vitro models have shown that macrophages can
change their polarization pattern according to the different
stimuli (19). This suggests their
dynamic interaction with the tumor microenvironment changing it
constantly favoring tumor cell invasion and subsequent
metastasis.
TAMs and hypoxia
Hypoxia and macrophage recruitment
Lower oxygen concentrations in tumors have been
verified and reproduced in many studies. In solid tumors the
chaotic disorganized array of vascularization with blind ended
vessels and leaky endothelial lining leads to a low oxygen tension
(hypoxic condition) in such tumors (20). Breast cancer, like other solid
malignancies, is hypoxic (21),
making the oxygen supply insufficient for the rapidly growing
tumor. In addition to the areas of low oxygen supply, necrotic
areas in a tumor bed develop an acute drop in oxygen with ensuing
cell death. This hypoxic environment within the tumor is one factor
that influences macrophage function as debris in this area attracts
macrophages (22). The research
done by Turner et al showed that once attracted, macrophages
are entrapped in necrotic tumor areas (23). One explanation for this phenomenon
is that upregulation of mitogen-activated protein kinase
phosphatase (MKP-1) dephosphorylates chemoattractant receptors for
VEGF and CCL2 (VEGFR and CCR2, respectively), thus aborting their
chemotactic response in TAMs (24,25).
This accumulation of TAMs in hypoxic areas has been associated with
aggressive tumor behavior in breast cancer (26).
Hypoxia upregulates VEGF expression in
TAMs
Hypoxia is known to regulate gene expression in both
cancer and inflammatory cells within the tumor microenvironment.
Hypoxia-induced gene expression in macrophages is in part due to
the upregulation of the transcription factors HIF-1α and HIF-2α
(27). HIF-1α is a marker for
hypoxia and in breast cancer it has been linked to aggressive
malignant behavior (28). Indeed,
HIF-1α-dependent genes are associated with an increased patient
mortality in some cancers (29).
So far VEGF remains the most notable gene upregulated by HIF-1α and
HIF-2α secondary to hypoxic stress in tumor microenvironment. Lewis
et al showed that TAMs express VEGF almost exclusively in
perinecrotic and poorly vascularized areas in breast cancer
(30). In breast cancer, the
overexpression of HIF-2α in TAMs was related to increased tumor
vascularity from the upregulation of VEGF expression by the effect
of HIF-2α (31).
Hypoxia promotes immune evasion by
TAMs
Hypoxia-induced HIF-1α also promotes tumor
progression by promoting immune evasion. Under hypoxic conditions
TAMs secrete more immunosuppressive cytokines such as IL-10, which
inhibits immune effector T-cells (32). In fact, using a murine model of
breast cancer Doedens et al recently reported that tumor
growth is decreased with deletion of HIF1α in macrophages, despite
normal levels of VEGF and tumor vascularization. In this model,
TAMs suppressed the antitumor activity of tumor-infiltrating T
cells (33). The loss of HIF-1α in
myeloid cells reversed the hypoxia-induced suppression of T-cell
activation resulting in tumor progression. Overall, this study
demonstrated that T cells of the adaptive immune response are
suppressed by the myeloid derived cells of innate immune system in
a HIF-1α-dependent and hypoxia-induced fashion.
Hypoxic TAMs promote metastatic behavior
of tumor cells
Promoting tumor metastatic behavior is another
mechanism whereby macrophages in hypoxic breast cancers increase
tumor aggressiveness. Macrophage migratory inhibitory factor (MIF)
is one of the gene products increased by HIF-1α upregulation in
hypoxic conditions. It was found that MIF is released by hypoxic
macrophages (34). Recently, Oda
et al showed that the effect of MIF on HIF-1α is
p53-dependent process and that MIF stabilizes HIF-1α protein under
hypoxic conditions (35). MIF's
stimulatory effect on the release of matrix metalloproteinases
[e.g., MIF-induced increased expression of MMP-9 (36)], which degrade the basement
membranes in tumors, opens the gates for tumor cells to escape and
initiate a nidus of metastatic foci.
Together the above studies reveal that hypoxic
conditions drive accumulation of TAMs in necrotic and poorly
vascularized areas of breast cancer. Once in the tumor, the
microenvironment influences macrophages to enhance the aggressive
behavior of tumors. These hypoxia-mobilized pro-tumoral activities
of TAMs may serve as potential targets for breast cancer therapy.
By interfering at various levels prior to metastatic and invasive
tumor activity the progression induced by macrophages may be
reversed. For example, since radiation therapy is known to induce
hypoxia in tumor beds, blocking VEGF or CCL2 could potentially stop
the recruitment of macrophages to these hypoxic tumor areas and
slow or stop tumor progression.
TAMs and angiogenesis
TAMs secrete VEGF, promoting tumor
angiogenesis
Focal TAM infiltration was shown to have an effect
on prognosis in breast cancer. It is well known now that TAMs can
affect tumors in different ways, and one of the major pathways is
by stimulating angiogenesis (26).
TAMs in breast carcinomas express numerous tumor-promoting factors,
the most prominent of which is the proangiogenic vascular
endothelial growth factor (VEGF) (30). In a series of breast cancer cases,
Leek et al found that there was a positive correlation
between high mean vessel density and increased macrophage index in
the tumor, as well as an inverse relationship between macrophage
counts and relapse-free and overall survival (26). Subsequently, they showed that there
is an association between vascular endothelial growth factor (VEGF)
and macrophage infiltration (37)
increasing the evidence for a proangiogenic role for TAMs in breast
cancer. Therefore, it became reasonable to think that the effect on
prognosis which macrophages have is in part related to their
proangiogenic effect. Indeed, breast cancer spheroids containing
macrophages induce overexpression of VEGF and an increase in the
number of vessels when injected in dorsal skin folds of nude mice
(38). Further evidence comes from
a study that showed the level of post-surgery circulating serum
VEGF levels were directly dependent on macrophage infiltration of
the primary tumor as well as the presence of somatic mutation of
p53 gene (39). However,
this relationship needs to be further investigated to validate its
effect on the metastatic activity of macrophage rich tumors.
Using an elegant transgenic mouse model, Lin et
al demonstrated that inhibition of macrophage maturation and
infiltration into tumors delayed the formation of a high-density
vessel network. In this model, genetically deleting colony
stimulating factor-1 (CSF-1), a key chemoattractant of macrophages,
makes PyMT mice susceptible to mammary cancer. However, restoring
CSF-1 showed that tumor-associated macrophages play a key role in
fostering tumor angiogenesis which is an essential step in the
tumor progression to malignancy (40). They later elaborated that
genetically restoring VEGF to the tumor microenvironment in these
macrophage-depleted animals restores tumor progression through
macrophage-produced VEGF stimulating tumor angiogenesis (41).
TAMs selectively have an angiogenic gene
signature
Gene expression profiling has been utilized to
understand the different macrophage populations in breast cancer.
In an experimental mouse model, Ojalvo et al were able to
use a flow cytometry technique to isolate a specific TAM population
from late stage mammary tumors that was previously shown to be
associated with an invasive mammary carcinoma. For comparison, they
isolated a similar macrophage population from the spleens of mice
that did not have mammary tumors. Gene expression signatures for
460 genes from both populations were studied. They found that the
transcript abundance was differentially regulated between the two
populations. Macrophage functions such as suppression of immune
activation, matrix remodeling and secretion of tumor angiogenesis
mediators were higher in the TAM population from the mammary tumor
(42). This indicates that a TAM
gene expression signature in mouse tumors could be used to assess
expression of TAMs in human breast cancer. These data suggest TAMs
may regulate tumor angiogenesis and therefore support further
investigation into the role of other transcriptional mediators of
TAM function within the tumor microenvironment (42). In addition, gene expression
profiling may be a promising methodology to explain the different
populations of TAMs and their potential functions. It also may be
helpful in differentiating tumors by their aggressive
potential.
From the highlighted studies, it is clear that
macrophages contribute to tumor growth, invasion, and metastasis
through an angiogenic pathway. Clinically, macrophages could be a
potential target of anticancer therapy, where possibly a treatment
would limit metastatic spread and thus increase overall survival.
In the angiogenesis pathway, clinical trials have already been
studying the targeting of proangiogenic factors such as VEGF.
Therefore, an indirect targeted therapy against macrophages or
their products is already underway, yet they are not selectively
aimed at macrophages which could provide excellent opportunities
for more targeted therapies (43).
Moreover, detailed gene expression profiling of tumor-associated
macrophages will eventually be used to determine what pathway(s) to
target for each particular tumor.
TAMs and the tumor matrix
Tumor cells and inflammatory cells of the tumor
microenvironment are surrounded by extracellular matrix (ECM). The
growth of tumor cells locally and their ability to invade and
metastasize depends partially on how they can get around this
matrix as well as by how they can modulate it to help their
evasion.
TAMs and matrix degrading enzymes
TAMs produce several enzymes which can degrade the
extracellular matrix (ECM). Such enzymes include several
metalloproteinases (e.g., MMP-2 and MMP-9) as well as
urokinase-type plasminogen activator (uPA) that degrade the ECM
(44). Dissolution of the ECM
leads to cleavages through which tumor cells can evade and
metastasize. Clinically, uPA has been under investigation in breast
cancer. In one study, it was shown that those patients whose uPA
activity was elevated in breast cancer tissue had a significantly
shorter disease-free interval compared to those with low levels of
activity (45). Recently uPA and
plasminogen activator inhibitor type 1 (PAI-1) have received
greater interest in breast cancer research and treatment and are
possible novel tumor biological markers. In fact, uPA is possibly a
potent independent prognostic factor in breast cancer (46). Currently, the NNBC-3 trial is
investigating treatment of node-negative early stage breast cancer
classified as high or low risk based on uPA and its inhibitor PAI-1
(47). Therefore, this draws
attention to the importance of the ECM and macrophages in breast
cancer progression and even targeted therapy.
Macrophages and fibrillary collagen in
ECM
Fibrillary collagen is an important substrate of the
extracellular matrix. Using multi-photon microscopy Wyckoff et
al revealed these structures and found that surrounding the
tumor there is a dense ring of fibrillary collagen (48). Moreover, tumor cells were seen
moving alone the collagen fibers ten times faster than they do
through the stroma. Tumor cells move directly towards vessels along
the collagen fibers. Ingman et al, found that depleting
macrophages during the process of mammary tumor development in a
mouse model, reduced the extent of collagen I synthesis and
restoration of macrophages resulted in correcting this defect
(49). Further studies are needed
to understand how exactly macrophage relate to this collagen
fibrillogenesis and tumor invasiveness.
TAMs and toll-like receptors
Toll-like receptors (TLRs) are a family of
transmembrane receptors that recognize conserved molecular patterns
of microbial origins. TLRs play an important role in tissue repair
and their role in cancer progression is being more appreciated
(50). In breast cancer, there is
an increased evidence for an overexpression of certain TLRs in the
more aggressive breast cancer subtypes (51). It is well known that TLRs are
expressed on mononuclear inflammatory cells (52). The relationship of TLRs and
macrophages in breast cancer is in its infancy. The data are still
limited but based on some other tumor models, there seems to be a
role for macrophages and TLRs in breast cancer. In a Lewis lung
carcinoma cell line model, Kim et al showed that cancer
cells can activate macrophages leading to production of IL-6 and
TNF-α via TLR2 and TLR6 activation (53). In a mouse model of
inflammation-induced liver cancer by diethylnitrosamine (DEN),
debris from hepatocytes activated macrophages via the MyD88
pathway. Interestingly, gender difference favoring the development
of this tumor in female mice was identified. Naugler et al
(54) showed that the genetic
deletion of IL-6 decreased the incidence of liver cancer and the
presence of estradiol (E2) conferred protection from developing
liver cancer even in IL-6-deficient mice after IL-6 was exogenously
restored (IL-6-deficient mice with exogenously restored IL-6
developed liver cancer in male mice and in ovariectomized female
mice). Although this model is based on a hepatic carcinoma in
vivo model, this interesting finding, of TLR activation pathway
seemed to be protective in the presence of estrogen receptor
(particularly ERα) which raises the question of whether there is a
protective role for estradiol in women with breast cancer that
would be lost in chemical/physiological menopause, that is mediated
through TLRs and tumor-associated macrophages. If this is proven
then targeting this pathway may confer protection from disease
progression. Indeed, ERα may be related to the downregulation of
certain TLRs in breast cancer (55).
On the other hand, some studies showed a positive
role for TLR signaling in stimulating a regulatory immune response
in tumor microenvironment. The high mobility group box 1 protein
(HMGB1) is released from apoptotic cells and activates TLR 4 on
dendritic cells, stimulating them to function as cross-presenters
of antigens (MHC I), thus stimulating T-cell responses (56,57).
Mutation of TLR4 gene in humans was found to have an
increased frequency of metastasis in breast cancer patients
(56). The counterpart mutation in
TLR4 in mouse models showed a decreased response to both
chemotherapy and radiation therapy for mammary cancer (56). In a cell line model, hypoxic stress
was found to increase the TLR4 expression in macrophages in a
HIF1α-regulated manner (58).
These results suggest that TLR4 activity in immune cells of the
tumor microenvironment and the innate immunity are involved in the
inflammatory process that is hypoxia-driven. However, further
studies are required to evaluate the role of different TLRs and
macrophage activation in breast cancer.
Clinical applications
Research studying the malignant potential of
transformed epithelial cells, such as mammary cells, showed that
these cells acquire a malignant potential; however, this alone is
not sufficient to render them able to grow and sustain tumor growth
unless in a permissive environment (59). The breast cancer microenvironment
is rich with inflammatory cells of which TAMs play a significant
role. Augmented by tumor hypoxia, TAMs may potentially affect the
clinical course of the tumor by potentiating an invasion permissive
environment. Additionally, TAMs secrete proangiogenic factors such
as VEGF stimulating the growth of tumor vasculature to sustain the
growing malignant mass (18). They
also secrete extracellular degrading proteases (e.g., uAP) allowing
for tumor cells to escape and metastasize. Moreover, TAMs suppress
the proinflammatory response that is initiated against tumor cells
allowing them to grow and divide.
Clinically, the role of TAMs is gaining importance.
For example, in a study by Allavena et al, trabectedin
(Yondelis®) which is a novel antitumor agent of marine
origin was found to affect differentiated macrophages as well as
freshly isolated TAMs at effective therapeutic doses (60). The selective cytotoxic effect of
Yondelis on TAMs significantly reduced their production of IL-6 and
CCL2 (a major monocyte chemoattractant recruiting them to tumor
tissue). This compound can be seen as the prototype of a new class
of breast cancer therapies since it targets inflammation-driven
tumors with high TAM component.
As we discussed, tumor-associated macrophages play
an important role in enhancing the vasculature of tumors, by
secreting the vascular endothelial growth factor (VEGF). Anti-VEGF
antibodies such as bevacizumab, can stop this process of
vasculogenesis that a tumor mass needs in order to sustain its
viability. Additionally, therapies that target VEGF receptor 2 may
decrease macrophage infiltration in the tumor microenvironment
(61). This can be a potential
therapeutic target, as not only would it work by its
anti-angiogenic activity, but also by decreasing the macrophage
recruitment which is the source of VEGF secretion and further
angiogenesis. The addition of a VEGF inhibitor to chemotherapy has
been shown to increase progression-free survival for metastatic
breast cancer, but not overall survival (62,63).
Clearly, there appears to be a subset of patients who benefit from
the addition of anti-angiogenesis therapy to chemotherapy, and
macrophage density in such tumors may be the guide to the subset of
patients who benefit most from this therapy.
The bisphosphonate zoledronic acid
(Zometa®) is of particular interest in the treatment of
women with breast cancer. Not only does it prevent demineralization
of bones from hormone manipulation therapy and decreases skeletal
related events in metastatic breast cancer, but it also increases
disease-free survival in young women when given early in the
treatment have been reported (64). Many studies on this
amino-bisphosphonate showed that its anticancer activity is related
to inhibiting angiogenesis and decreasing the recruitment and
differentiation of monocytes into TAMs (65). In one laboratory model, the
amino-bisphosphonate zoledronic acid was found to suppress MMP-9
expression by infiltrating macrophages and inhibit its ECM
degrading activity (66). These
data suggest zoledronic acid could be one of the new medications
targeting TAMs by an ‘unconventional MMP-9 inhibition’.
Further work in immunotherapy aimed at activating
macrophages toward the M1 lineage and therefore, depleting or
reducing other macrophage populations (M2) would be another way of
targeting macrophages in the clinical setting. Activation of TLR-9
by ligands such as CpG-oligodeoxynucleotides has been used in an
early clinical trial of lymphoma and non-small cell lung cancer. In
an in vitro model, the addition of TLR-9 CpG oligonucleotide
and an IL-10 receptor antibody induced a shift in the residence and
recruited tumor-infiltrating macrophages from the M2 into the M1
type in mouse mammary tumors (67). Therefore, although the therapeutic
targeting of TAMs is still in early stages, it is nevertheless
promising.
Conclusion
The tumor microenvironment is rich in inflammatory
cells and their products. Current research done indicates that
among these cells, TAMs play a key role in breast cancer
progression and invasiveness. Recruited by tumor cells, TAMs are
preferentially molded to serve the need of the cancer cells in a
pro-tumoral function instead of an anticancer role and as such,
potentiate their malignant behavior. More research needs to be done
in the field to understand the intricate relationship between
macrophages and breast cancer in order to translate the findings
into meaningful clinical trials of therapeutics with TAMs as a
target.
Acknowledgements
This work was supported by the
University of Chicago Breast Cancer SPORE NCI P50 CA125183. Partial
support from Avon Foundation. E.O. was supported by NIH/NCI T32
CA09566. PI: O. Olopade. ‘Basic Research Training Grant in Medical
Oncology’. Project period: 7/1/11-6/30/13.
References
|
1.
|
Balkwill F and Mantovani A: Inflammation
and cancer: back to Virchow? Lancet. 357:539–545. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Lewis CE, Leek R, Harris A and McGee JO:
Cytokine regulation of angiogenesis in breast cancer: the role of
tumor-associated macrophages. J Leukoc Biol. 57:747–751.
1995.PubMed/NCBI
|
|
4.
|
Bingle L, Brown NJ and Lewis CE: The role
of tumour-associated macrophages in tumour progression:
implications for new anticancer therapies. J Pathol. 196:254–265.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Leek RD and Harris AL: Tumor-associated
macrophages in breast cancer. J Mammary Gland Biol Neoplasia.
7:177–189. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Gordon S and Taylor PR: Monocyte and
macrophage heterogeneity. Nat Rev Immunol. 5:953–964. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Winston BW, Krein PM, Mowat C and Huang Y:
Cytokine-induced macrophage differentiation: a tale of 2 genes.
Clin Invest Med. 22:236–255. 1999.PubMed/NCBI
|
|
8.
|
Fujimoto H, Sangai T, Ishii G, et al:
Stromal MCP-1 in mammary tumors induces tumor-associated macrophage
infiltration and contributes to tumor progression. Int J Cancer.
125:1276–1284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Bernhagen J, Krohn R, Lue H, et al: MIF is
a noncognate ligand of CXC chemokine receptors in inflammatory and
atherogenic cell recruitment. Nat Med. 13:587–596. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Bando H, Matsumoto G, Bando M, et al:
Expression of macrophage migration inhibitory factor in human
breast cancer: association with nodal spread. Jpn J Cancer Res.
93:389–396. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Hagemann T, Wilson J, Kulbe H, et al:
Macrophages induce invasiveness of epithelial cancer cells via
NF-kappa B and JNK. J Immunol. 175:1197–1205. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Mosser DM: The many faces of macrophage
activation. J Leukoc Biol. 73:209–212. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Verreck FA, de Boer T, Langenberg DM, et
al: Human IL-23-producing type 1 macrophages promote but
IL-10-producing type 2 macrophages subvert immunity to
(myco)bacteria. Proc Natl Acad Sci USA. 101:4560–4565. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Solinas G, Germano G, Mantovani A and
Allavena P: Tumor-associated macrophages (TAM) as major players of
the cancer-related inflammation. J Leukoc Biol. 86:1065–1073. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Lamagna C, Aurrand-Lions M and Imhof BA:
Dual role of macrophages in tumor growth and angiogenesis. J Leukoc
Biol. 80:705–713. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Benoit M, Desnues B and Mege JL:
Macrophage polarization in bacterial infections. J Immunol.
181:3733–3739. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Mantovani A, Sica A, Sozzani S, Allavena
P, Vecchi A and Locati M: The chemokine system in diverse forms of
macrophage activation and polarization. Trends Immunol. 25:677–686.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Pollard JW: Tumour-educated macrophages
promote tumour progression and metastasis. Nat Rev Cancer. 4:71–78.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Brown JM and Giaccia AJ: The unique
physiology of solid tumors: opportunities (and problems) for cancer
therapy. Cancer Res. 58:1408–1416. 1998.PubMed/NCBI
|
|
21.
|
Vaupel P, Kelleher DK and Hockel M: Oxygen
status of malignant tumors: pathogenesis of hypoxia and
significance for tumor therapy. Semin Oncol. 28:29–35. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Murdoch C, Giannoudis A and Lewis CE:
Mechanisms regulating the recruitment of macrophages into hypoxic
areas of tumors and other ischemic tissues. Blood. 104:2224–2234.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Turner L, Scotton C, Negus R and Balkwill
F: Hypoxia inhibits macrophage migration. Eur J Immunol.
29:2280–2287. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Grimshaw MJ and Balkwill FR: Inhibition of
monocyte and macrophage chemotaxis by hypoxia and inflammation - a
potential mechanism. Eur J Immunol. 31:480–489. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Wain JH, Kirby JA and Ali S: Leucocyte
chemotaxis: examination of mitogen-activated protein kinase and
phosphoinositide 3-kinase activation by monocyte chemoattractant
proteins-1, -2, -3 and -4. Clin Exp Immunol. 127:436–444. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Leek RD, Lewis CE, Whitehouse R, Greenall
M, Clarke J and Harris AL: Association of macrophage infiltration
with angiogenesis and prognosis in invasive breast carcinoma.
Cancer Res. 56:4625–4629. 1996.PubMed/NCBI
|
|
27.
|
Burke B, Tang N, Corke KP, et al:
Expression of HIF-1alpha by human macrophages: implications for the
use of macrophages in hypoxia-regulated cancer gene therapy. J
Pathol. 196:204–212. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Kimbro KS and Simons JW: Hypoxia-inducible
factor-1 in human breast and prostate cancer. Endocr Relat Cancer.
13:739–749. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Giaccia A, Siim BG and Johnson RS: HIF-1
as a target for drug development. Nat Rev Drug Discov. 2:803–811.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Lewis JS, Landers RJ, Underwood JC, Harris
AL and Lewis CE: Expression of vascular endothelial growth factor
by macrophages is upregulated in poorly vascularized areas of
breast carcinomas. J Pathol. 192:150–158. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Leek RD, Talks KL, Pezzella F, et al:
Relation of hypoxia-inducible factor-2 alpha (HIF-2 alpha)
expression in tumor-infiltrative macrophages to tumor angiogenesis
and the oxidative thymidine phosphorylase pathway in Human breast
cancer. Cancer Res. 62:1326–1329. 2002.
|
|
32.
|
Murata Y, Ohteki T, Koyasu S and Hamuro J:
IFN-gamma and pro-inflammatory cytokine production by
antigen-presenting cells is dictated by intracellular thiol redox
status regulated by oxygen tension. Eur J Immunol. 32:2866–2873.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Doedens AL, Stockmann C, Rubinstein MP, et
al: Macrophage expression of hypoxia-inducible factor-1 alpha
suppresses T-cell function and promotes tumor progression. Cancer
Res. 70:7465–7475. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Schmeisser A, Marquetant R, Illmer T, et
al: The expression of macrophage migration inhibitory factor 1alpha
(MIF 1alpha) in human atherosclerotic plaques is induced by
different proatherogenic stimuli and associated with plaque
instability. Atherosclerosis. 178:83–94. 2005. View Article : Google Scholar
|
|
35.
|
Oda S, Oda T, Nishi K, et al: Macrophage
migration inhibitory factor activates hypoxia-inducible factor in a
p53-dependent manner. PLoS One. 3:e22152008. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Yu X, Lin SG, Huang XR, et al: Macrophage
migration inhibitory factor induces MMP-9 expression in macrophages
via the MEK-ERK MAP kinase pathway. J Interferon Cytokine Res.
27:103–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Leek RD, Hunt NC, Landers RJ, Lewis CE,
Royds JA and Harris AL: Macrophage infiltration is associated with
VEGF and EGFR expression in breast cancer. J Pathol. 190:430–436.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Bingle L, Lewis CE, Corke KP, Reed MW and
Brown NJ: Macrophages promote angiogenesis in human breast tumour
spheroids in vivo. Br J Cancer. 94:101–107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Vicioso L, Gonzalez FJ, Alvarez M, et al:
Elevated serum levels of vascular endothelial growth factor are
associated with tumor-associated macrophages in primary breast
cancer. Am J Clin Pathol. 125:111–118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Lin EY, Li JF, Gnatovskiy L, et al:
Macrophages regulate the angiogenic switch in a mouse model of
breast cancer. Cancer Res. 66:11238–11246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Lin EY, Li JF, Bricard G, et al: Vascular
endothelial growth factor restores delayed tumor progression in
tumors depleted of macrophages. Mol Oncol. 1:288–302. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Ojalvo LS, King W, Cox D and Pollard JW:
High-density gene expression analysis of tumor-associated
macrophages from mouse mammary tumors. Am J Pathol. 174:1048–1064.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Tang X, Mo C, Wang Y, Wei D and Xiao H:
Anti-tumour strategies aiming to target tumour-associated
macrophages. Immunology. 138:93–104. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Nagakawa Y, Aoki T, Kasuya K, Tsuchida A
and Koyanagi Y: Histologic features of venous invasion, expression
of vascular endothelial growth factor and matrix
metalloproteinase-2 and matrix metalloproteinase-9, and the
relation with liver metastasis in pancreatic cancer. Pancreas.
24:169–178. 2002. View Article : Google Scholar
|
|
45.
|
Duffy MJ, O'Grady P, Devaney D, O'Siorain
L, Fennelly JJ and Lijnen HJ: Urokinase-plasminogen activator, a
marker for aggressive breast carcinomas. Preliminary report.
Cancer. 62:531–533. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Ulisse S, Baldini E, Sorrenti S and
D'Armiento M: The urokinase plasminogen activator system: a target
for anti-cancer therapy. Curr Cancer Drug Targets. 9:32–71. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Kantelhardt EJ, Vetter M, Schmidt M, et
al: Prospective evaluation of prognostic factors uPA/PAI-1 in
node-negative breast cancer: phase III NNBC3-Europe trial (AGO,
GBG, EORTCPBG) comparing 6xFEC versus 3xFEC/3xDocetaxel. BMC
Cancer. 11:1402011. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Wyckoff JB, Wang Y, Lin EY, et al: Direct
visualization of macrophage-assisted tumor cell intravasation in
mammary tumors. Cancer Res. 67:2649–2656. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Ingman WV, Wyckoff J, Gouon-Evans V,
Condeelis J and Pollard JW: Macrophages promote collagen
fibrillogenesis around terminal end buds of the developing mammary
gland. Dev Dyn. 235:3222–3229. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Rakoff-Nahoum S and Medzhitov R: Toll-like
receptors and cancer. Nat Rev Cancer. 9:57–63. 2009. View Article : Google Scholar
|
|
51.
|
Gonzalez-Reyes S, Marin L, Gonzalez L, et
al: Study of TLR3, TLR4 and TLR9 in breast carcinomas and their
association with metastasis. BMC Cancer. 10:6652010. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Siednienko J and Miggin SM: Expression
analysis of the Toll-like receptors in human peripheral blood
mononuclear cells. Methods Mol Biol. 517:3–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Kim S, Takahashi H, Lin WW, et al:
Carcinoma-produced factors activate myeloid cells through TLR2 to
stimulate metastasis. Nature. 457:102–106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Naugler WE, Sakurai T, Kim S, et al:
Gender disparity in liver cancer due to sex differences in
MyD88-dependent IL-6 production. Science. 317:121–124. 2007.
View Article : Google Scholar
|
|
55.
|
Sandholm J, Kauppila JH, Pressey C, et al:
Estrogen receptor-alpha and sex steroid hormones regulate Toll-like
receptor-9 expression and invasive function in human breast cancer
cells. Breast Cancer Res Treat. 132:411–419. 2012. View Article : Google Scholar
|
|
56.
|
Apetoh L, Ghiringhelli F, Tesniere A, et
al: Toll-like receptor 4-dependent contribution of the immune
system to anticancer chemotherapy and radiotherapy. Nat Med.
13:1050–1059. 2007. View
Article : Google Scholar
|
|
57.
|
Apetoh L, Tesniere A, Ghiringhelli F,
Kroemer G and Zitvogel L: Molecular interactions between dying
tumor cells and the innate immune system determine the efficacy of
conventional anti-cancer therapies. Cancer Res. 68:4026–4030. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Kim SY, Choi YJ, Joung SM, Lee BH, Jung YS
and Lee JY: Hypoxic stress upregulates the expression of Toll-like
receptor 4 in macrophages via hypoxia-inducible factor. Immunology.
129:516–524. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Tlsty TD and Coussens LM: Tumor stroma and
regulation of cancer development. Annu Rev Pathol. 1:119–150. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Allavena P, Signorelli M, Chieppa M, et
al: Anti-inflammatory properties of the novel antitumor agent
yondelis (trabectedin): inhibition of macrophage differentiation
and cytokine production. Cancer Res. 65:2964–2971. 2005. View Article : Google Scholar
|
|
61.
|
Dineen SP, Lynn KD, Holloway SE, et al:
Vascular endothelial growth factor receptor 2 mediates macrophage
infiltration into orthotopic pancreatic tumors in mice. Cancer Res.
68:4340–4346. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Miller K, Wang M, Gralow J, et al:
Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic
breast cancer. N Engl J Med. 357:2666–2676. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Miles DW, Chan A, Dirix LY, et al: Phase
III study of bevacizumab plus docetaxel compared with placebo plus
docetaxel for the first-line treatment of human epidermal growth
factor receptor 2-negative metastatic breast cancer. J Clin Oncol.
28:3239–3247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Gnant M, Mlineritsch B, Schippinger W, et
al: Endocrine therapy plus zoledronic acid in premenopausal breast
cancer. N Engl J Med. 360:679–691. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Green JR and Guenther A: The backbone of
progress - preclinical studies and innovations with zoledronic
acid. Crit Rev Oncol Hematol. 77(Suppl 1): S3–S12. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Giraudo E, Inoue M and Hanahan D: An
amino-bisphosphonate targets MMP-9-expressing macrophages and
angiogenesis to impair cervical carcinogenesis. J Clin Invest.
114:623–633. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
67.
|
Guiducci C, Vicari AP, Sangaletti S,
Trinchieri G and Colombo MP: Redirecting in vivo elicited tumor
infiltrating macrophages and dendritic cells towards tumor
rejection. Cancer Res. 65:3437–3446. 2005.PubMed/NCBI
|