Introduction
Lung cancer remains the leading cause of
cancer-related mortality in the world, with an overall 5-year
survival rate of 15%. Approximately 85% of lung cancer cases are
non-small cell lung cancer (NSCLC) (1). Tumor recurrence and metastasis is the
major cause of lung cancer treatment failure and death.
Epithelial-mesenchymal transition (EMT) plays an important role in
tumor progression and is a key feature of metastasis in many cancer
types, including lung cancer (2,3). EMT
characteristics include perturbations of several signaling pathways
including the loss of E-cadherin expression, which is a major step
in metastasis of NSCLC (4).
In the past few years, microRNAs (miRNAs) have
emerged as promising molecular factors with potential for clinical
applications in cancer diagnosis and therapy (5–8).
MicroRNAs are small endogenous non-coding RNAs that range 19–24
nucleotides in length. MicroRNAs regulate the expression of
numerous genes either via translational silencing or by inducing
mRNA degradation of the targeted genes (5). Moreover, it has been estimated that
one miRNA can modulate as many as 200 genes, and over 30% of human
coding genes are under miRNA regulation (9,10).
Increasing evidence indicates that in human cancers, miRNAs can act
either as oncogenes or as tumor suppressor genes (11,12).
To date, more than 1,400 miRNA sequences have been identified in
human cells (13).
The microRNA-200 (miR-200) family, represented by
miR-200a, -200b, -200c, -141 and -429, is a marker and powerful
regulator of the EMT process. Its functions include maintaining the
epithelial phenotype of tissues through suppression of the
EMT-inducing transcription factors zinc finger E-box binding
homeobox 1 and 2 (ZEB1 and ZEB2) (14,15).
It has been shown that suppression of ZEB1 in undifferentiated
mesenchymal-like cells leads to restoration of epithelial phenotype
with increased expression of epithelial phenotype marker,
E-cadherin (16), which mediates
cell-cell adhesion (14). In many
cancer types, E-cadherin is somatically inactivated via mutation,
truncation or epigenetic silencing, a loss that enables the cells
to acquire a highly invasive phenotype with the characteristics of
EMT (17). Recent studies have
shown that restoring miR-200c expression decreases cell migration
but does not result in E-cadherin re-expression in some cells, thus
suggesting that miR-200c targets other genes involved in tumor
progression and metastasis (18).
The goal of this study is to identify potential
targets of miR-200 family essential in NSCLC metastasis and
clinical outcome. Our previous studies identified prognostic
biomarkers associated with metastasis in early stage NSCLC tumors
not treated with chemotherapy (19–21).
Specifically, a 35-gene signature was identified (19) and validated (20) as predictive of tumor metastasis in
434 NSCLC patients, including lung adenocarcinoma, squamous cell
lung cancer and large cell lung cancer. This signature could
identify more aggressive tumors from stage IA NSCLC (20). In another genome-wide DNA
microarray analysis of data from the Director’s Challenge study
(22), 12- and 15-gene prognostic
signatures were identified and validated using multi-center NSCLC
patient cohorts (n=442) (21). All
the identified prognostic biomarkers were confirmed with
quantitative RT-PCR analysis and some were validated with western
blot assays of independent snap-frozen human NSCLC tumors (20). Based on these results, we sought to
identify key miRNAs that regulate multiple NSCLC prognostic marker
genes, to reveal molecular regulatory events in metastasis with
implications on clinical outcome. The following experimental
analyses were carried out in this study. First, bioinformatics
methods were used to predict miRNA regulators of the identified
signature genes in NSCLC. Then, miRNAs that could potentially
regulate multiple prognostic biomarkers were identified. Based on
the bioinformatic prediction results, the miR-200 family was
selected to further determine a regulation of the predicted target
genes and to determine putative molecular networks in EMT and tumor
metastasis.
Materials and methods
Patient samples and gene expression
profiling
A total of 130 lung squamous cell carcinoma (SCC)
samples were analyzed in this study. The patient characteristics
were described in a previous publication (23). Genome-wide mRNA expression profiles
of the tumor samples were quantified with the Affymetrix U133A Gene
Chip (23). Microarray data were
extracted and calculated using the Affymetrix MAS 5 software after
global scaling of the average intensity to 600. The mRNA microarray
data were available from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) with accession
no. GSE4573. Out of 130 SCC tumors, 57 samples were screened for
miRNA expression profiles with TaqMan miRNA assays (Applied
Biosystems Inc., Foster City, CA) (24). The miRNA expression data was
available from GEO with accession no. GSE16025. The mRNA and miRNA
profiles of these matched 57 tumor samples were used for further
analysis.
MicroRNA target prediction
The mRNA expression levels of the lung cancer
prognostic markers identified in our previous studies (19–21)
were retrieved from the genome-wide expression profiles of the 57
SCC tumor samples. Pearson’s correlation coefficient between each
mRNA and all available miRNAs was computed. Significant mRNA-miRNA
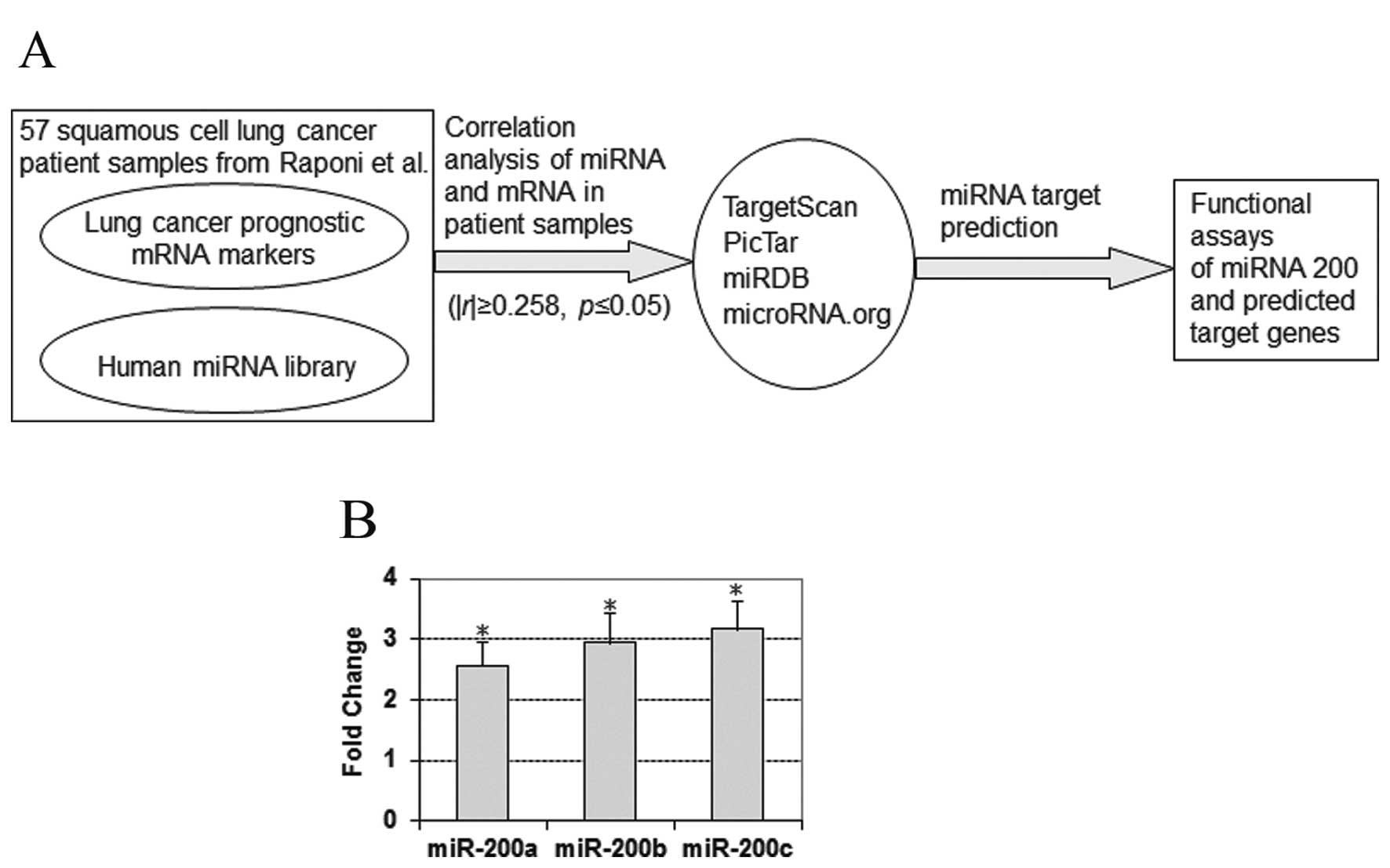
pairs (|r|≥0.258; p≤0.05) were selected for target prediction. Four
Bioinformatics tools were used in the study for miRNA target
prediction, including TargetScan (http://www.targetscan.org/) (10), PicTar (http://www.pictar.mdc-berlin.de/) (25), miRDB (http://www.mirdb.org/miRDB/) and microRNA.org (http://www.microrna.org/microrna/home.do) (26). These computational methods use
sequence complementary base-pairing between miRNA and mRNA in
target prediction. Files and databases containing miRNA and
predicted target genes were downloaded from the online websites of
these toolsets. These files were then analyzed with in-house
software script written in R to identify miRNA-mRNA gene pairs
showing significant expression correlation in SCC tumor samples. In
addition, TarBase (http://www.diana.cslab.ece.ntua.gr/tarbase/) (27) was used to retrieve experimentally
validated miRNA targets.
The miRNA targets predicted by TargetScan are based
on the presence of conserved 8mer, 7mer and 6mer sites that match
the seed region of each miRNA (10). TargetScan also predicts
non-conserved sites, additional types of seed matches that are
preferentially conserved in different species, and sites with
mismatches in the seed region that are compensated by conserved 3′
pairing (28). PicTar predicts
miRNA targets by searching 3′ UTR alignments with predicted sites
(25). The miRDB uses an
algorithm, MirTarget2, based on Support Vector Machine, to predict
miRNA targets. The microRNA.org uses miRanda algorithm to
predict miRNA targets based on sequence and contextual features of
the predicted miRNA-mRNA duplex (26).
MicroRNA and mRNA binding sites
The binding sites between each miRNA-mRNA pair was
retrieved from the Homo sapiens microRNA.org database
with the ‘Target mRNA’ module. The searching parameters were set
with mirSVR score ≤0 and PhastCons score ≥0.
Cell lines and cell culture
Small airway epithelial cells (SAEC) and normal
human bronchial/tracheal epithelial cells (NHBE) were obtained from
Lonza Walkersville Inc. (Walkersville, MD). Human non-small cell
lung cancer cell line H1299 and human immortalized lung epithelial
cell line BEAS-2B were purchased from the American Type Culture
Collection (ATCC, Manassas, VA). SAEC cells were cultured according
to the supplier’s recommendations in SABM medium supplemented with
52 μg/ml bovine pituitary extract, 0.5 ng/ml human
recombinant epidermal growth factor (EGF), 0.5 μg/ml
epinephrine, 1 μg/ml hydrocortisone, 10 μg/ml
transferrin, 5 μg/ml insulin, 6.5 ng/ml triiodothyronine, 50
μg/ml gentamicin/amphotericin B (GA-1000) and 50
μg/ml fatty acid-free bovine serum albumin. NHBE cells were
cultured according to the supplier’s recommendations in BEBM media
supplemented with bovine pituitary extract (52 μg/ml),
hydrocortisone (0.5 μg/ml), human epidermal growth factor
(hEGF, 0.5 ng/ml), epinephrine (0.5 μg/ml), insulin (5
μg/ml), triiodothyronine (6.5 ng/ml), transferrin (10
μg/ml), gentamicin (50 μg/ml), amphotericin B (50
ng/ml), bovine serum albumin (1.5 μg/ml). H1299 and BEAS-2B
cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with penicillin, streptomycin, L-glutamine and 10%
fetal bovine serum. All cells were cultured at 37°C in humidified
incubator with 95% air and 5% CO2.
Virus transduction
Human miRIDIAN shMIMIC lentiviral miRNA particles
(hsa-miR-200a: UAACACUGUCUGGUA ACGAUGU, hsa-miR-200b:
UAAUACUGCCUGGUAAUG AUGA, hsa-miR-200c: UAAUACUGCCGGGUAAUGA UGGA,
and control scrambled microRNA were purchased from Open Biosystems
(Huntsville, AL) and used for infection of target cells in the
presence of 4 μg/ml of polybrene.
Western blot analysis
Cells were lysed in 1X SDS lysis buffer (50 mM
Tris-HCl, pH 6.8, 2% SDS, 10% glycerol). Total protein was
quantified by the BCA method. β-mercaptoethanol was added to
lysates to a final concentration 100 mM. Equal amounts of total
protein were separated by 4–12% SDS-PAGE and transferred to a PVDF
membrane. Membranes were blocked 1 h with 5% non-fat milk in 1X PBS
containing 0.05% Tween-20. Membranes were then incubated for 1 h at
room temperature with primary antibodies. After incubation with the
primary antibody, membranes were washed thrice in 1X PBS with 0.05%
Tween-20 for 5 min each and blocked for 7 min in blocking solution.
Membranes were incubated for 1 h at room temperature with
horseradish peroxidase (HRP) conjugated donkey anti-mouse IgG or
donkey anti-rabbit IgG in 1X PBS with 0.05% Tween-20. Membranes
were then washed five times for 5 min in PBS-Tween-20 and finally
developed with HyGLO Western Blotting Substrate (Denville
Scientific) according to the instructions of the manufacturer.
Protein band intensity was determined using FluorChem® Q
software (AlphaInnotech, Santa Clara, CA). Relative protein level
was determined after normalization to tubulin and relative to
negative control (miR-scr) samples. The following antibodies were
used: ATRX (Santa Cruz Biotechnology, catalog no. SC-55584),
DLC1 (BD Biosciences, catalog no. 612020), HFE (Santa
Cruz Biotechnology, catalog no. SC-130375), ZEB1 (Sigma,
catalog no. HPA027524), HNRNPA3 (Santa Cruz Biotechnology,
catalog no. SC-133665), E-cadherin (BD Biosciences, catalog
no. 610181), GAPDH (Millipore, catalog no. MAB374) and
tubulin (Sigma, catalog no. T9026).
RNA isolation
Total RNA was extracted using the
mirVana® kit (Ambion Inc., Austin, TX) according to the
manufacturer’s protocol. To ensure a good RNA quality, the quality
and integrity of the total RNA was evaluated using 28S/18S ratio
and a visual image of the 28S and 18S bands were evaluated on the
2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA
isolated using this method yielded a very good quality, with a RIN
number ≥9. Concentration of the total RNA was assessed using the
NanoDrop-1000 Spectrophotometer (NanoDrop Technologies,
Germany).
Quantitative real-time RT-PCR
Complementary DNA (cDNA) was generated using total
RNA according to the TaqMan® MicroRNA Reverse
Transcription protocol (Applied Biosystems Inc.). Quantitative
RT-PCR for microRNA was performed using TaqMan MicroRNA assays
(Applied Biosystems Inc.). Human U47 small nuclear RNA was used as
an endogenous control. The expression levels of miRNAs were
quantified using ABI 7500 quantitative real-time instrument and SDS
software (Applied Biosystems Inc.). The abundance of miRNA is
expressed as Ct (threshold fluorescence) which gives the number of
cycles required to reach threshold fluorescence. Real-time PCR for
target genes was determined using total RNA and cDNA was generated
using a High-Capacity cDNA Reverse Transcription kit and TaqMan
gene expression assays (Applied Biosystems Inc.). E-cadherin (CDH1)
mRNA was measured using SYBR-Green Master mix and CDH1 specific
primers according to manufacturer’s protocol (Applied Biosystems
Inc.). All qRT-PCR reactions were performed on 7500 instrument
(Applied Biosystems Inc.). In the qRT-PCR analysis of E-cadherin,
the dissociation curve showed the absence of a secondary peak,
indicating no presence of primer dimer. Specificity of the PCR
product obtained from SYBR-Green reactions was verified by
sequencing. The expression level of each gene was determined by
following formulas: fold change = 2−ΔΔCt, where ΔCt
(cycle threshold) = Cttarget gene - Ctendogenous
control gene, and ΔΔCt = ΔCttreated sample -
ΔCtcontrol sample. The expression level of the analyzed
genes is reported as fold change relative to negative miR-scrambled
(-src) infected samples. The human UBC gene was used as an
endogenous control gene.
In this study, a predicted gene was considered a
confirmed target if the mRNA level was significantly downregulated
or the protein level was downregulated at least 15% relative to
negative control samples. Not all of the predicted targets were
analyzed at the protein level due to the lack of specificity of
commercially available antibodies.
Functional pathway analysis
Ingenuity pathway analysis (IPA) software (Ingenuity
Systems, Redwood City, CA) was used to derive curated molecular
interactions reported in the scientific literature. These
interactions included both physical and functional interactions, as
well as interactions representing pathway relevance. In this study,
in order to delineate molecular networks of genes interacting with
the miR-200 family and novel molecular targets, a core analysis was
employed to identify the most relevant canonical pathways,
biological functions and physiological processes from the
interactions reported in the IPA database. We then selected
pathways that were statistically significant with a p<0.05 in
adjusted Benjamini-Hochberg tests.
Statistical analysis
The statistical significance of the difference
between groups was determined by un-paired t-tests at p≤0.05. The
qRT-PCR expression data are presented as mean ± SEM.
Results
Prediction of miR-200 novel molecular
targets
To screen for potential miRNA regulators of our
previously identified lung cancer prognostic gene signatures
(19–21), the correlation between the mRNA
expression of these biomarkers and all available human miRNA
expression profiles in 57 squamous cell lung cancer tumors was
computed. For all miRNA-mRNA gene pairs showing significant
correlation (|r|>0.258, p≤0.05, Pearson’s correlation analysis)
in the lung SCC tumors, 4 bioinformatics toolsets (TargetScan,
PicTar, miRDB and microRNA.org) were used to determine
whether or not a given gene is a predicted target of the
corresponding miRNA (Fig. 1A). A
total of 233 miRNA-mRNA gene pairs were predicted as a target pair
by at least one bioinformatic method. The correlation analysis and
the prediction results may be viewed on our website (http://www.wvucancer.org/guoLab/Publications). Due to
alternative splicing, each gene may have multiple probes in DNA
microarray data. This will lead to discrepant gene expression among
different probes for the same gene and discordant correlation
between mRNA and miRNA. The mRNA-miRNA pairs with a negative
correlation for at least one probe set were selected for further
analysis.
Several miRNAs, including the miR-200 family, were
predicted to target multiple prognostic biomarkers. We focused on
the miR-200 family because of its reported role in tumor
metastasis. The miRNA-200 family is represented by miR-200a,
miR-200b, miR-200c, miR-141 and miR-429, based on their genomic
location and primary sequence. Based on sequence similarity, the
miR-200 family is divided into two subclasses: one class includes
miR-200b, -200c and -429, and the other class includes miR-200a and
miR-141 (29). The members in each
subclass share the same seed sequence. We analyzed the expression
levels of the miR-200s in squamous cell lung cancer patient primary
tumors and normal lung tissues in the cohort from Raponi et
al(24). The results show that
miR-200a had a 2.56-fold overexpression (p<1.35e-7; unpaired
t-tests) in the tumors, miR-200b exhibited a 2.94-fold
overexpression (p<1.32e-6; unpaired t-tests) and miR-200c showed
a 3.16-fold overexpression (p<0.001; unpaired t-tests) in the
patient tumors (Fig. 1B). However,
during metastasis, previous studies showed that the miR-200 family
expression is lost in mesenchymal subtypes of epithelial cancers
and negatively correlates with cancer cell invasion (15,16).
The predicted targets for hsa-miR-200a include
deleted in liver cancer 1 gene (DLC1), E2F transcription
factor 4 (E2F4), and AHNAK nucleoprotein (desmoyokin)
(AHNAK); for hsa-miR-200b: DLC1, ubiquitin-like
modifier activating enzyme 6 (UBA6), ubiquitin-conjugating
enzyme E2I (UBE2I) and heterogeneous nuclear
ribonucleoprotein A3 (HNRNPA3); for hsa-miR-200c: alpha
thalassemia/mental retardation syndrome X-linked gene
(ATRX), hereditary hemochromatosis (HFE), DLC1
and thrombospondin 1 (THBS) (Tables I and II). To confirm the regulation of
miR-200a, -200b, -200c on the predicted targets, expression of
these prognostic biomarkers genes at the mRNA and protein levels
were examined following re-expression of miR-200a, -200b, -200c in
H1299 and BEAS-2B cells.
 | Table I.The predicted target genes analyzed
in this study. |
Table I.
The predicted target genes analyzed
in this study.
| Gene symbol | Gene name | Assay ID | Function | Pathway | Remarks |
|---|
| AHNAK | AHNAK nucleoprotein
(desmoyokin) | Hs00225285_m1 | Protein-protein
binding | Signal
transduction | NSCLC prognostic
biomarker (19,20) |
| ATRX | Alpha
thalassemia/mental retardation syndrome X-linked | Hs00230877_m1 | Transcriptional
regulator, chromatin remodeling | Transcription | NSCLC prognostic
biomarker (19,20) |
| DLC1 | Deleted liver
cancer 1 | Hs00183436_m1 | Regulation of small
GTP-binding proteins | Signal
transduction | Tumor suppressor
gene (60) and NSCLC prognostic
marker (21) |
| E2F4 | E2F transcription
factor-4 | Hs00608098_m1 | Transcriptional
factor, cell cycle, apoptosis | Transcription | NSCLC prognostic
maker (19,20) |
| HFE |
Hemochromatosis | Hs00373474_m1 | Regulation of body
iron metabolism | Iron
metabolism | NSCLC prognostic
biomarker (21) |
| HNRNPA3 | Heterogeneous
nuclear ribonucleo protein-A3 | Hs00864845_s1 | Cytoplasmic RNA
binding and trafficking, protein binding | Signal
transduction | NSCLC prognostic
biomarker (19,20) |
| THBS1 | Thrombospondin
1 | Hs00962914_m1 | Extracellular
adhesive glycoprotein | Protein
interaction | NSCLC prognostic
biomarker (21) |
| UBE2I |
Ubiquitin-conjugating enzyme E2I | Hs00163336_m1 |
Ubiquitin-activating protein | Protein
degradation | NSCLC prognostic
biomarker (19,20) |
| UBA6 | Ubiquitin-like
modifier activating enzyme-6 | Hs00414964_m1 |
Ubiquitin-conjugation for protein
degradation | Protein
degradation | NSCLC prognostic
biomarker (21) |
| Table IIGenes regulated by miR-200a, -200b
and -200c in H1299. |
Table II
Genes regulated by miR-200a, -200b
and -200c in H1299.
| Genes |
miR-200a |
miR-200b |
miR-200c |
|---|
|
|
|
|---|
| Predicted | Downregulated | Predicted | Downregulated | Predicted | Downregulated |
|---|
| mRNA | Protein | | mRNA | Protein | | mRNA | Protein |
|---|
| AHNAK | • | | | | | | | | |
| ATRX | | | ✓ | | | | • | ✓ | |
| DLC1 | • | ✓ | ✓ | • | ✓ | ✓ | • | | ✓ |
| E2F4 | • | | | | | | | | |
| HFE | | | ✓ | | | ✓ | • | ✓ | |
| HNRNPA3 | | | | • | ✓ | ✓ | | | |
| THBS1 | | | | | | | • | | |
| UBA6 | | | | • | | | | | |
| UBE2I | | | | • | | | | | |
| ZEB1 | • | ✓ | ✓ | • | ✓ | ✓ | • | ✓ | ✓ |
| ZEB2 | • | | ✓ | • | | ✓ | • | | ✓ |
Restoring miR-200 expression in H1299
cells
We analyzed the expression levels of miR-200a, -200b
-and 200c in a metastatic human NSCLC model, H1299 cells. Normal
human small airway epithelial cells (SAEC) were used as control
cells. The expression level of miR-200a, -200b and -200c in H1299
cells was at the detection limit (Fig.
2). In contrast, SAEC expressed higher levels of miR-200a,
-200b and -200c (data not shown; http://www.wvucancer.org/guoLab/Publications)
consistent with their normal epithelial phenotype. In order to
identify potential molecular targets of miR-200a, -200b, -200c,
H1299 cells were stably infected with lentiviral vectors expressing
miR-200a, -200b, -200c or negative control miRNAs. The infected
H1299 cells expressed high levels of exogenous miR-200a, -200b,
-200c, which were comparable with the levels exhibited in normal
lung epithelial SAEC cells (data not shown; http://www.wvucancer.org/guoLab/Publications).
miR-200 regulation on predicted molecular
targets
ZEB1 and ZEB2 genes are the most
extensively characterized targets of the miR-200 family (14,15)
and they were used as positive controls in the qRT-PCR experiments.
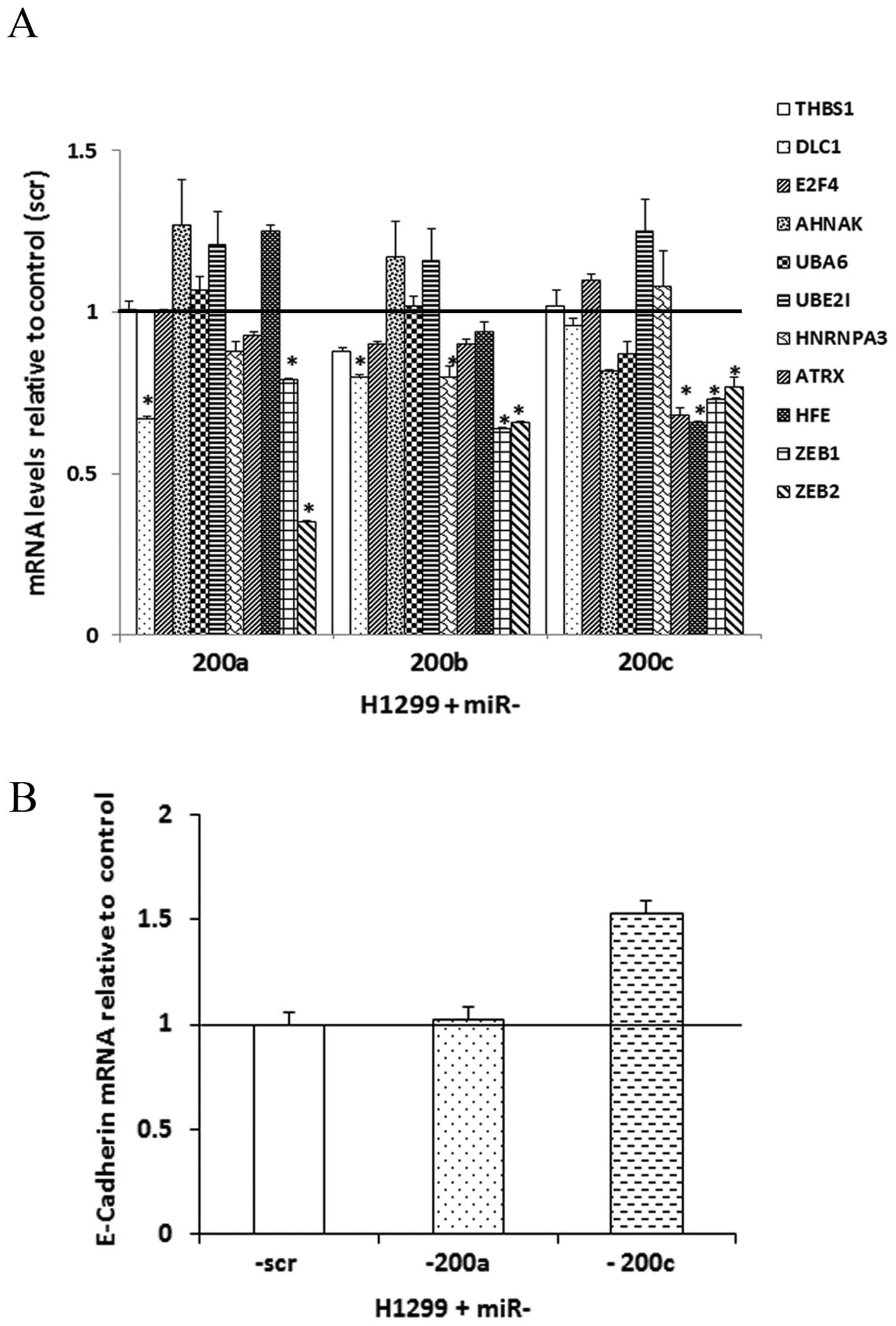
In H1299 cells over-expressing miR-200a, -200b and -200c,
ZEB1 and ZEB2 exhibited significant (p<0.05;
unpaired t-tests) downregulation at the mRNA expression level
(Fig. 2A). Following
overexpression of miR-200a, DLC1 showed significantly
decreased mRNA expression. Restoring miR-200b resulted in
significant downregulation of DLC1 and HNRNPA3 at the
mRNA level. The overexpression of miR-200c resulted in a
significant mRNA downregulation of ATRX and HFE
(Fig. 2A).
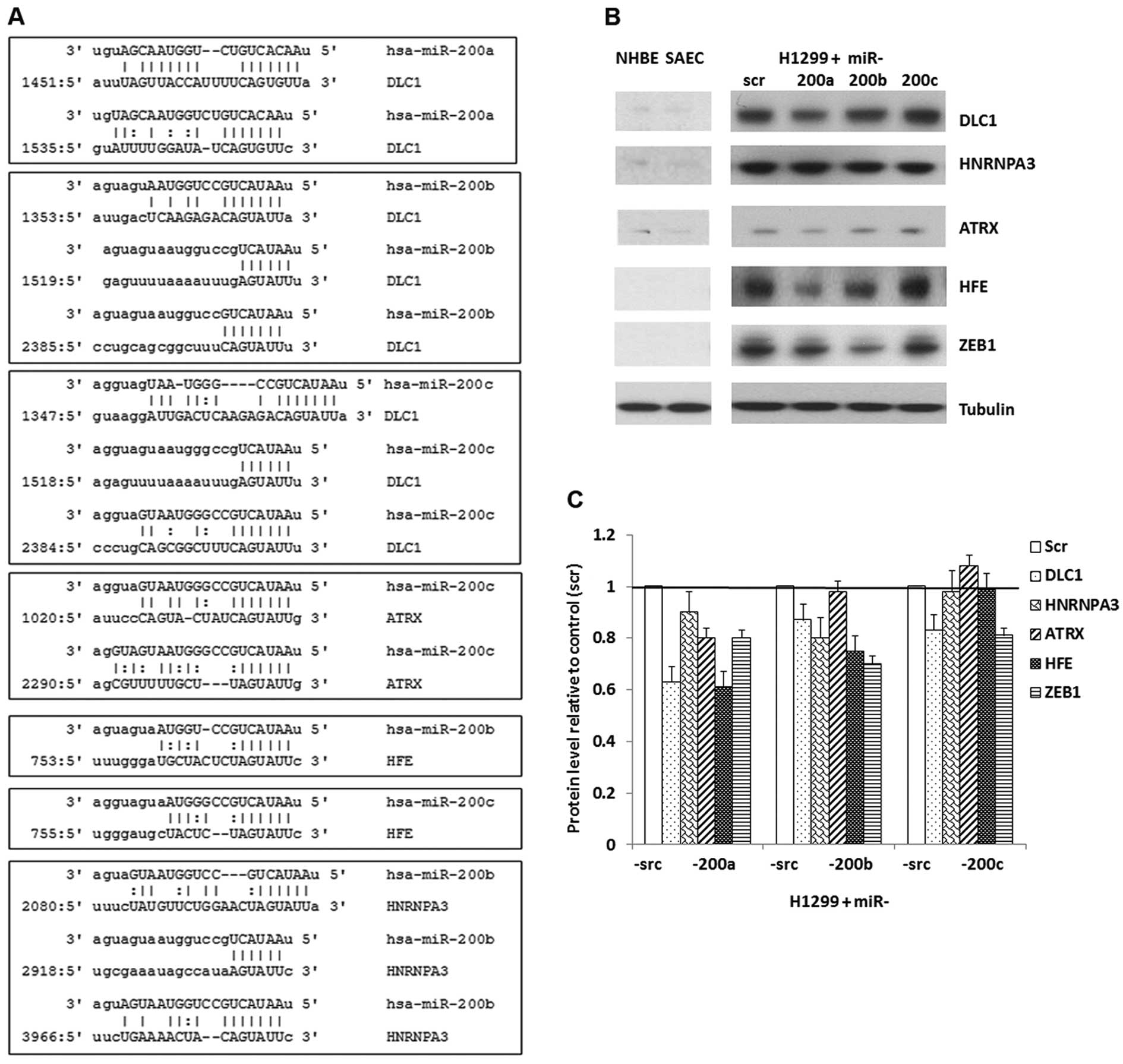
At the protein level, ZEB1 was reduced in
H1299 cells overexpressing miR-200a, -200b and -200c, with the most
downregulation (about 30%) in cells re-expressing miR-200b
(Fig. 3B and C). A considerable
downregulation of DLC1, ATRX and HFE was
observed in H1299 cells overexpressing miR-200a. It is noteworthy
that DLC1 and HFE had about 40% downregulation in
H1299 cells infected with miR-200a mimic, exhibiting a more
significant downregulation than ZEB1. In H1299 cells with
restored expression of miR-200b, the protein levels for
DLC1, HNRNPA3 and HFE were reduced by 15–30%.
The protein expression of DLC1 was decreased about 20% in H1299
cells that overexpressed miR-200c (Fig. 3B and C).
The results of the present study show that
miRNA-200a regulates ATRX, DLC1 and HFE;
miRNA-200b regulates DLC1, HFE and HNRNPA3;
miRNA-200c regulates ATRX, DLC1 and HFE
(Table II). The target prediction
results from each of the four bioinformatics algorithms are
provided on our website (http://www.wvucancer.org/guoLab/Publications). There
is a concordant downregulation of these potential targets at both
mRNA and protein expression levels in the corresponding H1299 cells
overexpressing the miR-200 family. All of the potential targets
contain binding sites for the corresponding miR-200 family members
according to the microRNA.org database (26), except for ATRX/miR-200a and
HFE/miR-200a. The 3′ UTR region of DLC1 contains 2
binding sites for miR-200a, 3 for miR-200b and 3 for miR-200c.
ATRX contains 1 binding site for miR-200c. HFE
contains 1 binding site for miR-200b and 1 for miR-200c.
HNRNPA3 contains 3 binding sites for miR-200b (Fig. 3A). HFE is a predicted target
of miR-200c, not miR-200a or -200b, in NSCLC. Although miR-200b and
-200c have the same seed sequence, the correlation between
HFE and miR-200b was not statistically significant (p=0.057)
in SCC patient samples. Therefore, it was not initially selected as
a predicted target of miR-200b in NSCLC. Nevertheless, HFE
was shown to be regulated by miR-200a, -200b and 200c in metastatic
H1299 cells. AHNAK and E2F4 are predicted targets of
miR-200a. However, miR-200a did not suppress the expression of
these two genes in H1299 cells (Fig.
2A). Similarly, UBA6 and UBE2I were not regulated
by miR-200b in metastatic lung cancer cells as they were predicted
to be. These results were summarized in Table II.
To further substantiate the regulatory effects of
miR-200 on these lung cancer prognostic markers, SCC patient
samples (n=57) (23,24) were screened to investigate the
correlation between the miR-200 family and the mRNA expression of
its predicted target genes as well as ZEB1 and ZEB2.
The results showed that all the downregulated genes had a
significant negative correlation with the corresponding miR-200
family member in SCC patient tumor tissues, except
ATRX/miR-200a and HFE/miR-200a (Table III). The results in the patient
samples further strengthened the in vitro findings.
| Table IIICorrelation between the expression of
miR-200 and its regulated genes in squamous cell lung cancer
patient tumors (n=57). |
Table III
Correlation between the expression of
miR-200 and its regulated genes in squamous cell lung cancer
patient tumors (n=57).
| Genes |
miR-200a |
miR-200b |
miR-200c |
|---|
| ATRX | −0.0629 | NA | −0.301a |
| DLC1 | −0.313a | −0.374a | −0.496a |
| HFE | −0.193 | −0.253b | −0.393a |
| HNRNPA3 | NA | 0.264a | NA |
| ZEB1 | −0.426a | −0.458a | −0.484a |
| ZEB2 | −0.395a | −0.379a | −0.382a |
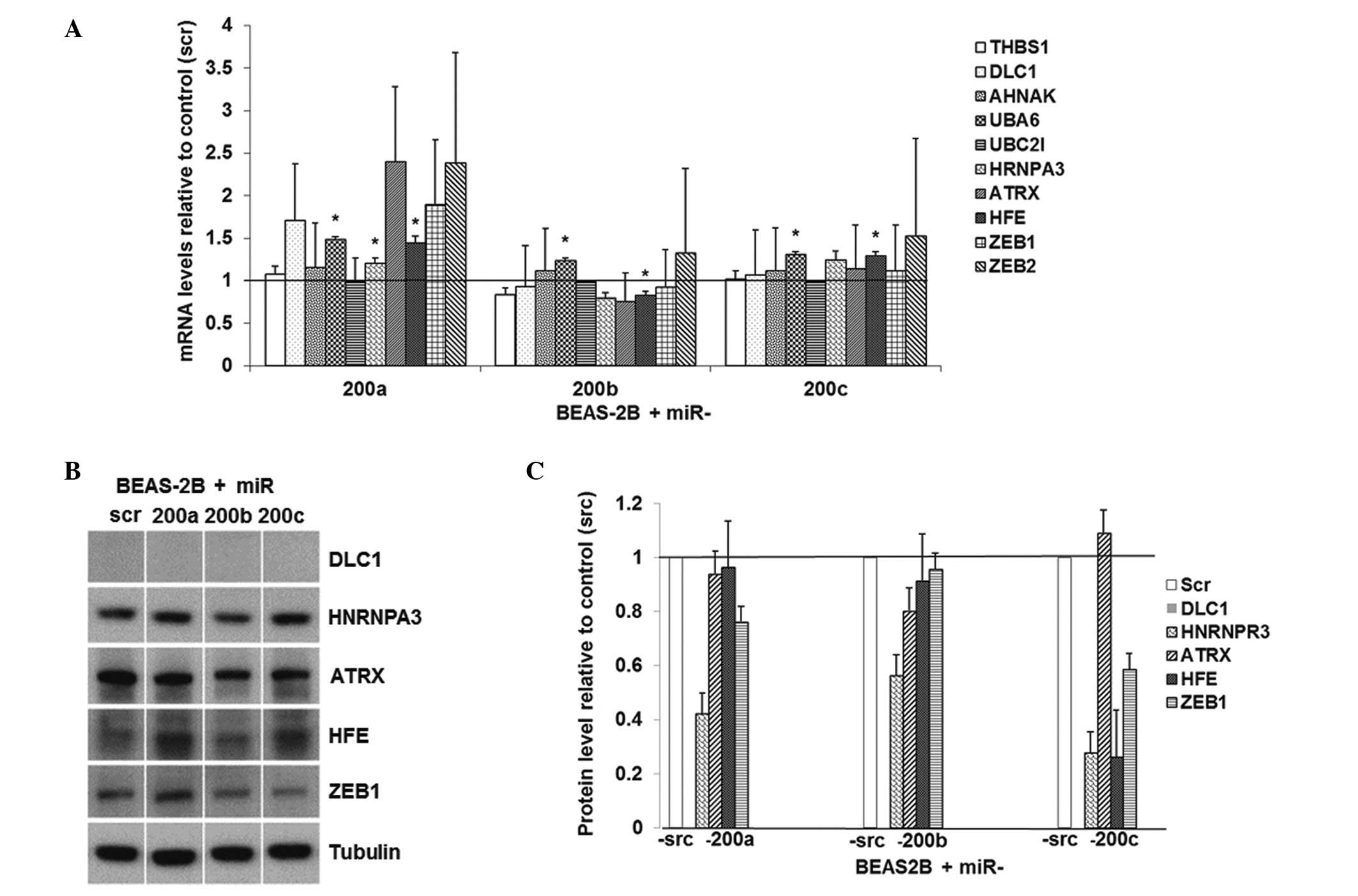
The regulation of miR-200 on these predicted target
genes was also evaluated in human immortalized lung epithelial
cells BEAS-2B. The overexpression of miR-200b in these cells
resulted in significantly downregulated mRNA level of HFE
(Fig. 4A). The overexpression of
miR-200a, -200b and -200c in BEAS-2B cells caused approximately 60,
40 and 70% downregulation of HNRNPR3 protein, respectively
(Fig. 4B and C). The
overexpression of miR-200b in BEAS-2B resulted in a 20%
downregulation of ATRX at the protein level and the
re-expression of miR-200c resulted in a 70% downregulation of
HFE at the protein level. These results indicate that
miR-200 family downregulates HNRNPR3, HFE and
ATRX in normal lung epithelial cells. Together, these
results substantiate the role of miR-200 family and its regulated
genes in lung cancer initiation and progression.
E-cadherin and miR-200
After the potential molecular targets of the miR-200
family were shown in the present study, we sought to explore the
effect of miR-200s on EMT in the metastatic NSCLC cells.
Re-expression of miR-200c induced a 1.53-fold upregulation of
E-cadherin (CDH1) through the downregulation of the
E-cadherin repressor transcriptional factors ZEB1 and
ZEB2, which is consistent with previous studies in breast
cancer or NSCLC cells (16,30,31)
(Fig. 2B). E-cadherin protein was
highly expressed in normal lung SAEC cells, but not in metastatic
NSCLC H1299 cells (data not shown; http://www.wvucancer.org/guoLab/Publications). These
results indicate that re-expression of miR-200 may reverse EMT
process.
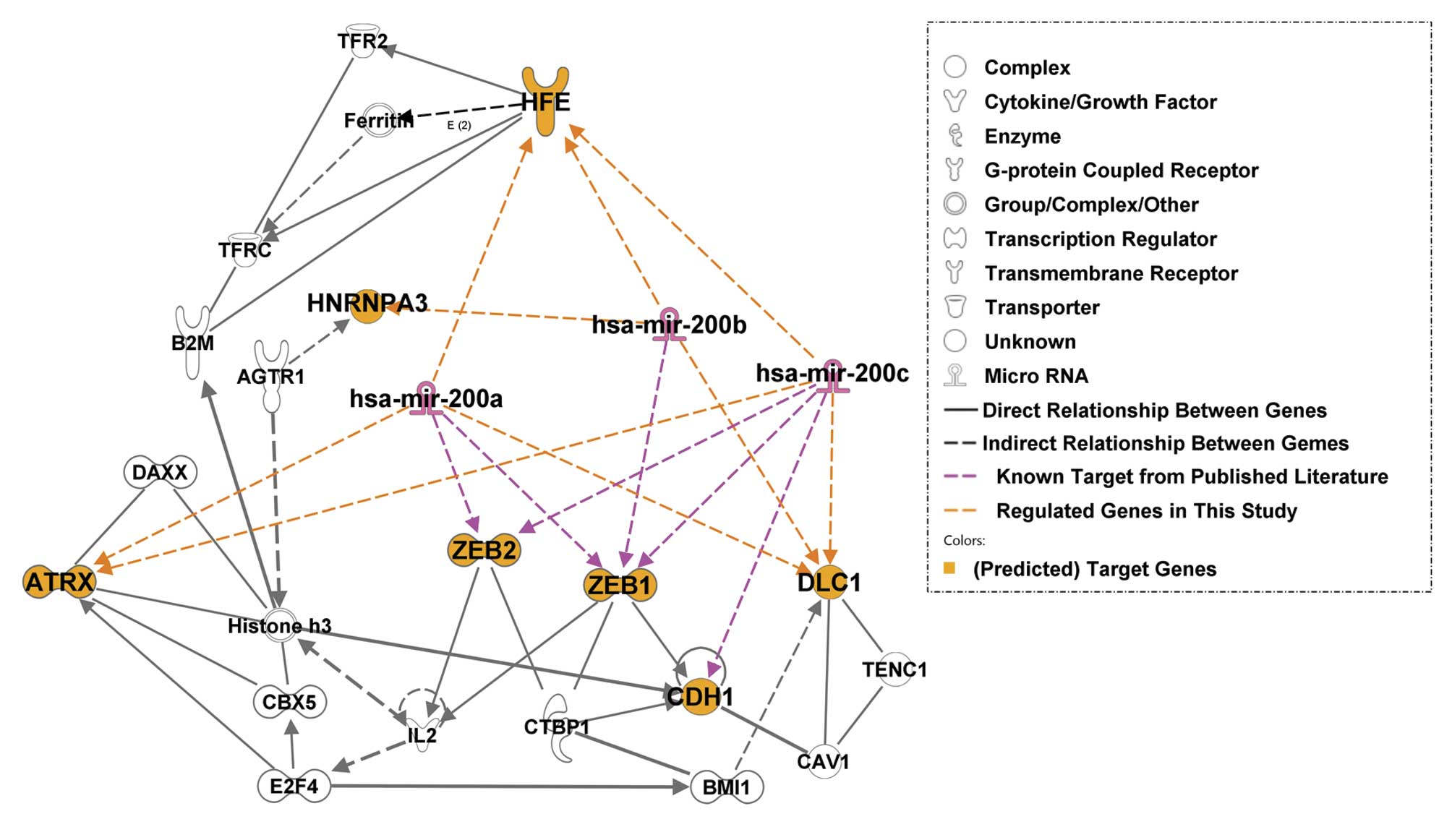
Molecular network analysis
Molecular network interactions and significant
canonical signaling pathways associated with miR-200s and their
predicted molecular targets were retrieved using IPA. The molecular
network map shows interactions between the miR-200s and their known
target genes, ZEB1 and ZEB2, as well as potential
targets identified from the present study (Fig. 5; the regulation identified in the
present study is shaded in orange font). Among the genes in the
molecular network, Histone H3 and E-cadherin (CDH1)
were major focal points in the miR-200 network. Histone H3
is a component of the nucleosome and CDH1 is a cell adhesion
protein and epithelial phenotype marker. These results suggest that
miR-200s and their potential target genes participate in molecular
interactions involved in gene transcription regulation, either
during the regulation of gene expression at the chromatin level or
in the regulation of cell-cell interactions as mediated by
E-cadherin. Increasing evidence indicates that chromatin remodeling
induced by DNA damage or epigenetic changes are responsible for
carcinogenesis.
The IPA functional analysis found a total of 69
canonical pathways associated with the miR-200 network, of which 13
canonical pathways were statistically significant (adjusted
p<0.05 with Benjamini-Hochberg tests; Table IV). The top signaling pathways
include virus entry via endocytic pathways, allograft rejection
signaling, OX40 signaling pathway, caveolar-mediated endocytosis
signaling, communication between innate and adaptive immune cells,
chronic myeloid leukemia signaling, molecular mechanisms of cancer
and DNA double-strand break repair by homologous recombination,
among others (Table IV).
Furthermore, the IPA functional analysis found 25 significant
diseases and disorders related to the miR-200 network. The top 3
diseases included genetic disorders, metabolic diseases and cancer
(Table V). At the molecular level,
beta-2-microglobulin (B2M), CDH1, ZEB1,
ZEB2, ATRX, HFE and the miR-200s are
involved in genetic disorders; B2M, transferrin receptor
2 (TFR2), HFE and angiotensin II receptor
(AGTR1) are involved in metabolic diseases; B2M,
E2F, miR-200s, ATRX, DLC1, ZEB1,
ZEB2, CDH1 and caveolin 1 (CAV1) are involved
in cancer (Table V). These results
indicate that miR-200 network involves complex signaling pathways
and mechanisms, and has implications in numerous human diseases and
disorders.
| Table IVTop 13 significant canonical pathways
related to the miR-200 molecular network in Ingenuity pathway
analysis. |
Table IV
Top 13 significant canonical pathways
related to the miR-200 molecular network in Ingenuity pathway
analysis.
| Canonical
pathways | P-value | Molecules |
|---|
| Virus entry via
endocytic pathways | 0.0002 | B2M, CAV1,
TFRC |
| Allograft rejection
signaling | 0.0019 | B2M, IL2 |
| OX40 signaling
pathway | 0.0025 | B2M, IL2 |
| Caveolar-mediated
endocytosis signaling | 0.0044 | B2M, CAV1 |
| Communication
between innate and adaptive immune cells | 0.0056 | B2M, IL2 |
| Chronic myeloid
leukemia signaling | 0.0071 | CTBP1, E2F4 |
| Molecular
mechanisms of cancer | 0.0102 | DAXX, CDH1,
E2F4 |
| DNA double-strand
break repair by homologous recombination | 0.0191 | ATRX |
| Lipid antigen
presentation by CD1 | 0.0257 | B2M |
| Antiproliferative
role of TOB in T cell signaling | 0.0355 | IL2 |
| Colorectal cancer
metastasis signaling | 0.0407 | CDH1, E2F4 |
| Role of CHK
proteins in cell cycle checkpoint control | 0.0447 | E2F4 |
| Cell cycle
regulation by BTG family proteins | 0.0468 | E2F4 |
| Table VTop 25 significant disease and
disorder functions related to the miR-200 molecular network in
Ingenuity pathway analysis. |
Table V
Top 25 significant disease and
disorder functions related to the miR-200 molecular network in
Ingenuity pathway analysis.
| Disease and
Disorders | P-value | Molecules |
|---|
| Genetic
disorder | 0.00004 | B2M, CDH1, IL2,
TFR2, ZEB2, ATRX, mir-200, CAV1, ZEB1, HFE, AGTR1 |
| Metabolic
disease | 0.00004 | B2M, TFR2, HFE,
AGTR1 |
| Cancer | 0.00005 | B2M, E2F4, mir-200,
ATRX, ZEB1, DLC1, CTBP1, CDH1, BMI1, IL2, ZEB2, CAV1, TFRC,
AGTR1 |
| Reproductive system
disease | 0.00005 | B2M, CDH1, BMI1,
IL2, ATRX, mir-200, CAV1, TFRC, DLC1, AGTR1 |
| Gastrointestinal
disease | 0.00021 | B2M, CDH1, BMI1,
IL2, mir-200, ZEB2, CAV1, TFRC, AGTR1 |
| Hepatic system
disease | 0.00021 | B2M, BMI1, IL2,
mir-200, DLC1, AGTR1 |
| Organismal injury
and abnormalities | 0.00021 | IL2, AGTR1 |
| Infection
mechanism | 0.00053 | CTBP1, E2F4, IL2,
CAV1, TFRC, ZEB1 |
| Infectious
disease | 0.00053 | CTBP1, B2M, IL2,
CAV1, TFRC, AGTR1 |
| Hematological
disease | 0.00122 | B2M, CTBP1, E2F4,
IL2, ATRX, CAV1, DLC1, AGTR1, HFE |
| Dermatological
diseases and conditions | 0.00138 | CAV1, ZEB1,
HFE |
| Inflammatory
response | 0.00138 | B2M, CDH1, IL2,
ZEB1, AGTR1 |
| Ophthalmic
disease | 0.00138 | ZEB1 |
| Respiratory
disease | 0.00186 | CTBP1, B2M, CDH1,
IL2, mir-200, CAV1, AGTR1 |
| Immunological
disease | 0.00247 | B2M, DAXX, E2F4,
CDH1, HNRNPA3, IL2, TFRC, DLC1, AGTR1 |
| Antimicrobial
response | 0.00276 | IL2 |
| Cardiovascular
disease | 0.00276 | B2M, IL2, CAV1,
AGTR1 |
| Inflammatory
disease | 0.00401 | B2M, DAXX, CDH1,
HNRNPA3, IL2, ZEB2, TFRC, ZEB1, DLC1, AGTR1 |
| Connective tissue
disorders | 0.00501 | B2M, DAXX, CDH1,
HNRNPA3, IL2, TFRC, DLC1, AGTR1 |
| Neurological
disease | 0.00550 | BMI1, ZEB2, CAV1,
AGTR1 |
| Renal and
urological disease | 0.00550 | B2M, CDH1,
AGTR1 |
| Skeletal and
muscular disorders | 0.00733 | B2M, DAXX, CDH1,
HNRNPA3, BMI1, IL2, TFRC, DLC1 |
| Developmental
disorder | 0.00961 | ATRX, ZEB2,
AGTR1 |
| Nutritional
disease | 0.01230 | IL2 |
| Endocrine system
disorders | 0.04720 | AGTR1 |
Discussion
Lung cancer is a dynamic and diverse disease and is
associated with numerous somatic mutations, deletions and
amplification events. Tumor recurrence and metastasis causes
lethality and failure in lung cancer treatment. About 35–50% of
stage I NSCLC patients will develop and die from tumor recurrence
within 5 years following surgery (32,33)
and adjuvant chemotherapy of stage II and stage III disease has
resulted in very modest survival benefits (34). Epithelial-mesenchymal transition
(EMT) is a key process in tumor metastasis. Novel therapeutic
approaches targeting EMT are needed to effectively prevent tumor
recurrence and metastasis.
miRNAs are small non-coding RNAs that regulate gene
expression via degradation or translational inhibition of target
mRNAs. Importantly, one miRNA can regulate the expression of
multiple genes because it can bind to its mRNA targets regardless
whether there is perfect seed sequence complementarity (5). Our previous studies identified
prognostic marker genes for NSCLC (19–21).
The expression of these prognostic biomarkers was associated with
metastatic potential in early stage NSCLC tumors. Identification of
miRNAs that regulate multiple prognostic biomarker genes could shed
light on the mechanisms underlying tumor metastasis and potentially
provide the basis for the development of novel therapeutic targets
to improve the clinical outcome.
Deregulated expression of miR-200 family members
has been observed in multiple cancer types (15,16,29,30).
Numerous studies showed that miR-200 family members regulate the
EMT and cancer cell invasion by suppressing the expression of
ZEB1 and ZEB2 genes (15,35).
ZEB1 and ZEB2 are key transcription factors
regulating EMT by binding to an E box upstream of their target
genes, which include E-cadherin, and repressing their expression.
Moreover, ZEB1 and ZEB2 can repress the transcription
of miR-200 genes via negative feed-back loop mechanism (15,36).
Absence of E-cadherin in cell junctions renders loss of cell-cell
communication, thereby allowing cancerous cells to acquire an
aggressive, invasive phenotype. Thus, downregulation of E-cadherin
is associated with increased lymph node metastasis and
poor-prognosis of NSCLC (37).
Numerous studies have reported a stringent control of E-cadherin
expression by the miR-200s via suppression of ZEB1 and
ZEB2(16,30,31).
The miR-200 family expression is lost in mesenchymal subtypes of
epithelial cancers and negatively correlates with cancer cell
invasion (15,16) and metastasis in NSCLC (38).
On the other hand, overexpression of miR-200 was
also found in cholangiocarcinoma malignant cells compared to
non-malignant cells (39),
melanoma cell lines (29), ovarian
cancer (40), colorectal cancer
(41) and NSCLC (Fig. 1B). These results indicate that
expression of miR-200 family during tumorigenesis is rather
complex, and may adopt a bimodal pattern with elevated levels in
primary tumors and dramatic downregulation in metastatic cells.
Alternatively, the role of miR-200 may differ depending on cancer
type and stage.
Despite strong evidence that miR-200s inhibit EMT
and suppress cancer cell invasion, several functional
overexpression studies have yielded conflicting results on the role
of miR-200s in metastasis, supporting both their anti-metastatic
(18,30,42–44)
and pro-metastatic (45,46) potential.
The present study sought to determine whether some
of our previously identified human lung cancer prognostic markers
are potential molecular targets of miR-200a, -200b and -200c
microRNAs. The study goal was to explore whether biomarkers
associated with NSCLC poor prognosis are functionally involved in
EMT and metastasis through miR-200 regulation. In order to identify
new molecular targets of the miR-200 family, we used the H1299
NSCLC cell line. This cell line is p53-deficient, has
metastatic characteristics and is devoid of miR-200 family and
E-cadherin expression. We chose this cell model in order to
identify new molecular targets following re-expression of miR-200a,
-200b and -200c, independent of p53 regulation (47–49).
p53 is a tumor suppressor protein that regulates the
expression of a myriad of genes and miRNAs including the miR-200
family (47–50). The present study shows an increased
expression of E-cadherin in H1299 cells re-expressing miR-200c,
consistent with other published results (16,30).
It has been shown that E-cadherin promoter is methylated in NSCLC
cells (51) and re-expression of
E-cadherin may not be solely dependent on suppression of
ZEB1 but also dependent on other processes such as promoter
demethylation (52). The modest
re-expression level of E-cadherin in the present study is in
agreement with other reports (16,18,52),
suggesting that more significant downregulation of ZEB1 and
ZEB2 might be required for full re-activation of
E-cadherin.
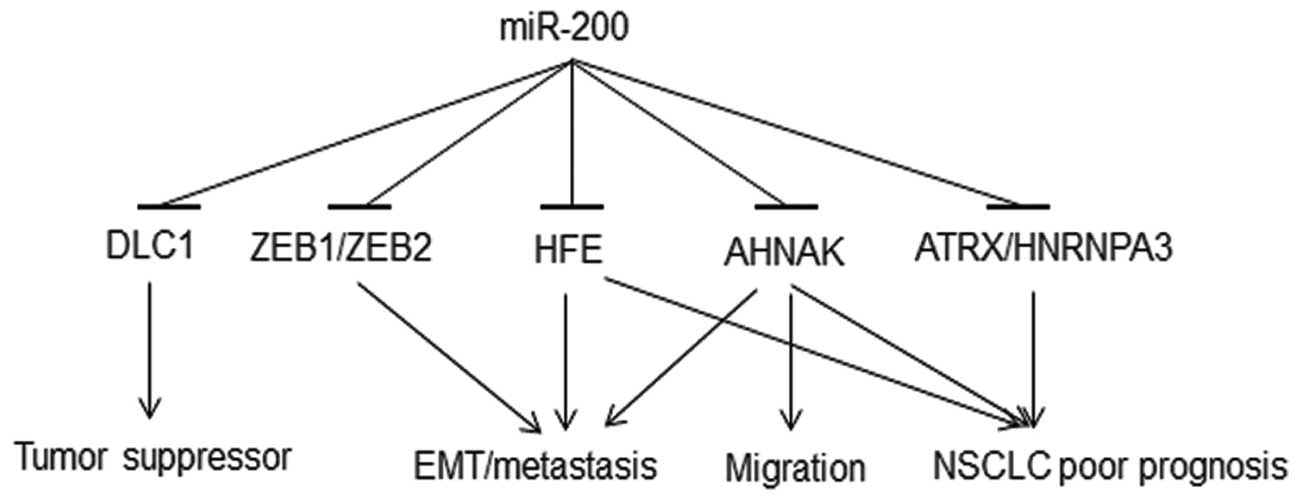
This study identified a regulation of miR-200
family on their potential novel molecular targets. The results show
that DLC1, ATRX and HFE genes are regulated by
miR-200a and miR-200c; DLC1, HFE and HNRNPA3
are regulated by miR-200b. Although, the changes in the expression
of these genes at the mRNA and protein levels after re-expression
of miR-200s in H1299 cells were relatively small (<2-fold), such
small changes in the expression of microRNA targets are very common
(16,53). Despite restoration of normal
miR-200 levels, some other components of the post-transcriptional
gene silencing pathway in H1299 cells might be in limiting amounts,
for example Dicer, which is commonly downregulated in cancer cells
(54). These findings are
consistent with the model that miR-200s regulate their targets
differentially either by targeting mRNA for degradation or/and by
inhibiting its translation. All of these potential novel molecular
targets of the miR-200 family are prognostic biomarkers for NSCLC
(19–21). These genes showed overexpression in
metastasis-prone NSCLC cells (H1299) compared with normal lung
epithelial cells (NHBE and SAEC; Fig.
4B). HFE is important in iron metabolism disorder and
oxidative stress (55). HFE
polymorphism is associated with multiple cancer types and
chemoresponse (56). The complex
of beta-2-microglobulin (B2M) and its receptor HFE activates
EMT and promotes metastases in human prostate, breast, lung and
renal cancer cells both in vivo and in vitro, through
the modulation of iron responsive pathways (57). Inhibition of either B2M or
HFE reverses EMT (57). Our
results show that HFE is regulated by all three of the
studied miR-200 family members, indicating new mechanisms in EMT
induction and lung cancer metastasis. AHNAK, a
pseudopod-specific protein, also controls EMT in metastatic cancer
cells (58). AHNAK
knockdown in metastatic cells causes reduced cell migration and
induces mesenchymal-epithelial transition (MET). Consistent results
were observed in clinical cohorts, in which overexpression of
AHNAK was associated with poor prognosis of NSCLC (19). Overexpression of
ATRX(19) and
HNRNPA3(59) was observed
in NSCLC tumors and poor prognosis patients. Collectively, loss of
miR-200 could lead to overexpression of HFE, AHNAK,
ATRX and HNRNPA3, which in turn is associated with
poor prognosis of NSCLC (Fig.
6).
DLC1, a tumor suppressor gene, is frequently
silenced in various types of human cancer (60). DLC1 was first identified in
primary human hepatocellular carcinoma, with an inhibitory effect
on the growth of breast and liver tumors (61). Downregulation of DLC1 by
miR-200a in primary human liver cells has been previously reported
(62). In the present study,
re-expression of miR-200a, -200b or -200c in metastatic human NSCLC
cells resulted in DLC1 downregulation at both mRNA and
protein expression levels, with miR-200a exerting the most
significant repression of DLC1. The overexpression of
miR-200 family observed in primary tumors (Fig. 1B) could downregulate DLC1,
which is involved in tumorigenesis (Fig. 6). Downregulation of DLC1, in
turn, is associated with poor prognosis of NSCLC (21). These results, again, indicate
potential pleiotropic regulatory mechanisms of miR-200 in lung
cancer development and progression. Together, these results
indicate that deregulation of miR-200 induces aberrant expression
of multiple genes involved in lung cancer carcinogenesis, EMT, cell
migration and metastasis, with significant implications on NSCLC
clinical outcome. The proposed mechanisms of miR-200 regulation in
carcinogenesis and metastasis are illustrated in Fig. 6.
IPA functional pathway analyses found that the
miR-200 molecular network involved canonical pathways of immune
response, molecular mechanisms of cancer, metastasis signaling
transduction, cell-cell communication, proliferation and DNA
repair. These results indicate that miR-200 is essential in
regulating signaling pathways responsible for many biological
functions and complex molecular mechanisms (Table IV). Moreover, the miR-200 related
molecular network is implicated in at least 25 human diseases and
abnormalities, including genetic disorders, metabolic disease,
cancer and reproductive system disease, among many others (Table V).
In conclusion, this study combined computational
predictions and quantitative experimental validations to
demonstrate that the miR-200 family regulates multiple NSCLC
prognostic marker genes. The identified regulation, direct or
indirect, provides important insights of possible microRNA
regulatory mechanisms in EMT and lung cancer metastasis and lays a
foundation for future functional analysis. These potential
molecular targets, each with significant prognostic value in NSCLC
patients, are involved in the regulation of gene transcription and
signal transduction pathways. The findings of the miR-200
downregulation of DLC1, ATRX, HNRPNA3,
AHNAK and HFE in metastatic human NSCLC cells and the
proposed regulatory mechanisms in tumorigenesis and metastasis
could provide the basis for the development of novel therapeutic
approaches for the treatment of this deadly disease. In our future
research, reporter luciferase assays with mutations at specific
seed sequences will be carried out to validate the direct
interaction of these genes downregulated by miR-200 family.
Acknowledgements
We thank Rebecca Raese for her help
in editing the manuscript. We thank Yuya Kudo for his assistance in
figure preparation. This project was supported by the American
Cancer Society (122300-IRG-09-061-04-IRG to A.V.I.) and Susan G.
Komen (KG110350 to A.V.I.). Software license for Ingenuity Pathway
Analysis was supported by NIH/NCRR P2016477. This project is
supported by the NIH R01LM009500 (PI: N.L.G.) and NCRR P20 RR16440
ARRA Supplement (PD: N.L.G.). The findings and conclusions in this
report are those of the author(s) and do not necessarily represent
the views of the National Institute for Occupational Safety and
Health.
References
|
1.
|
Mirshahidi HR and Hsueh CT: Updates in
non-small cell lung cancer - insights from the 2009 45th annual
meeting of the American Society of Clinical Oncology. J Hematol
Oncol. 3:182010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Monteiro J and Fodde R: Cancer stemness
and metastasis: therapeutic consequences and perspectives. Eur J
Cancer. 46:1198–1203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelialmesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Dohadwala M, Yang SC, Luo J, et al:
Cyclooxygenase-2-dependent regulation of E-cadherin: prostaglandin
E(2) induces transcriptional repressors ZEB1 and snail in non-small
cell lung cancer. Cancer Res. 66:5338–5345. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kasinski AL and Slack FJ: MicroRNAs en
route to the clinic: progress in validating and targeting microRNAs
for cancer therapy. Nat Rev Cancer. 11:849–864. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Edwards JK, Pasqualini R, Arap W and Calin
GA: MicroRNAs and ultraconserved genes as diagnostic markers and
therapeutic targets in cancer and cardiovascular diseases. J
Cardiovasc Transl Res. 3:271–279. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Fabbri M: miRNAs as molecular biomarkers
of cancer. Expert Rev Mol Diagn. 10:435–444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Krek A, Grun D, Poy MN, et al:
Combinatorial microRNA target predictions. Nat Genet. 37:495–500.
2005. View
Article : Google Scholar
|
|
10.
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Nicoloso MS, Spizzo R, Shimizu M, Rossi S
and Calin GA: MicroRNAs - the micro steering wheel of tumour
metastases. Nat Rev Cancer. 9:293–302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kozomara A and Griffiths-Jones S: miRBase:
integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39:D152–D157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Gregory PA, Bracken CP, Bert AG and
Goodall GJ: MicroRNAs as regulators of epithelial-mesenchymal
transition. Cell Cycle. 7:3112–3118. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Gregory PA, Bert AG, Paterson EL, et al:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Guarino M, Rubino B and Ballabio G: The
role of epithelial-mesenchymal transition in cancer pathology.
Pathology. 39:305–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Cochrane DR, Howe EN, Spoelstra NS and
Richer JK: Loss of miR-200c: a marker of aggressiveness and
chemoresistance in female reproductive cancers. J Oncol.
2010:8217172010. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Guo L, Ma Y, Ward R, et al: Constructing
molecular classifiers for the accurate prognosis of lung
adenocarcinoma. Clin Cancer Res. 12:3344–3354. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Guo NL, Wan YW, Tosun K, et al:
Confirmation of gene expression-based prediction of survival in
non-small cell lung cancer. Clin Cancer Res. 14:8213–8220. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Wan YW, Sabbagh E, Raese R, et al: Hybrid
models identified a 12-gene signature for lung cancer prognosis and
chemoresponse prediction. PLoS One. 5:e122222010. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Shedden K, Taylor JM, Enkemann SA, et al:
Gene expression-based survival prediction in lung adenocarcinoma: a
multi-site, blinded validation study. Nat Med. 14:822–827. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Raponi M, Zhang Y, Yu J, et al: Gene
expression signatures for predicting prognosis of squamous cell and
adenocarcinomas of the lung. Cancer Res. 66:7466–7472. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Raponi M, Dossey L, Jatkoe T, et al:
MicroRNA classifiers for predicting prognosis of squamous cell lung
cancer. Cancer Res. 69:5776–5783. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Chen K and Rajewsky N: Natural selection
on human microRNA binding sites inferred from SNP data. Nat Genet.
38:1452–1456. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Betel D, Wilson M, Gabow A, Marks DS and
Sander C: The microRNA.orgurisimplemicroRNA.org resource: targets
and expression. Nucleic Acids Res. 36:D149–D153. 2008.
|
|
27.
|
Papadopoulos GL, Reczko M, Simossis VA,
Sethupathy P and Hatzigeorgiou AG: The database of experimentally
supported targets: a functional update of TarBase. Nucleic Acids
Res. 37:D155–D158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Jan CH, Friedman RC, Ruby JG and Bartel
DP: Formation, regulation and evolution of Caenorhabditis
elegans 3′UTRs. Nature. 469:97–101. 2011. View Article : Google Scholar
|
|
29.
|
Elson-Schwab I, Lorentzen A and Marshall
CJ: MicroRNA-200 family members differentially regulate
morphological plasticity and mode of melanoma cell invasion. PLoS
One. 5:e131762010. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Ceppi P, Mudduluru G, Kumarswamy R, et al:
Loss of miR-200c expression induces an aggressive, invasive, and
chemoresistant phenotype in non-small cell lung cancer. Mol Cancer
Res. 8:1207–1216. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Takeyama Y, Sato M, Horio M, et al:
Knockdown of ZEB1, a master epithelial-to-mesenchymal transition
(EMT) gene, suppresses anchorage-independent cell growth of lung
cancer cells. Cancer Lett. 296:216–224. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Hoffman PC, Mauer AM and Vokes EE: Lung
cancer. Lancet. 355:479–485. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Naruke T, Goya T, Tsuchiya R and Suemasu
K: Prognosis and survival in resected lung carcinoma based on the
new international staging system. J Thorac Cardiovasc Surg.
96:440–447. 1988.PubMed/NCBI
|
|
34.
|
Shields TH, LoCicero J, Reed CE and Feins
RH: General Thoracic Surgery. Lippincott, Williams & Wilkins;
Philadelphia, PA: 2009
|
|
35.
|
Burk U, Schubert J, Wellner U, et al: A
reciprocal repression between ZEB1 and members of the miR-200
family promotes EMT and invasion in cancer cells. EMBO Rep.
9:582–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Bracken CP, Gregory PA, Kolesnikoff N, et
al: A double-negative feedback loop between ZEB1-SIP1 and the
microRNA-200 family regulates epithelial-mesenchymal transition.
Cancer Res. 68:7846–7854. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Sulzer MA, Leers MP, van Noord JA, Bollen
EC and Theunissen PH: Reduced E-cadherin expression is associated
with increased lymph node metastasis and unfavorable prognosis in
non-small cell lung cancer. Am J Respir Crit Care Med.
157:1319–1323. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Patnaik SK, Kannisto E, Knudsen S and
Yendamuri S: Evaluation of microRNA expression profiles that may
predict recurrence of localized stage I non-small cell lung cancer
after surgical resection. Cancer Res. 70:36–45. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Meng F, Henson R, Lang M, et al:
Involvement of human micro-RNA in growth and response to
chemotherapy in human cholangiocarcinoma cell lines.
Gastroenterology. 130:2113–2129. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Iorio MV, Visone R, Di LG, et al: MicroRNA
signatures in human ovarian cancer. Cancer Res. 67:8699–8707. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Xi Y, Formentini A, Chien M, et al:
Prognostic values of microRNAs in colorectal cancer. Biomark
Insights. 2:113–121. 2006.PubMed/NCBI
|
|
42.
|
Gibbons DL, Lin W, Creighton CJ, et al:
Contextual extracellular cues promote tumor cell EMT and metastasis
by regulating miR-200 family expression. Genes Dev. 23:2140–2151.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Olson P, Lu J, Zhang H, et al: MicroRNA
dynamics in the stages of tumorigenesis correlate with hallmark
capabilities of cancer. Genes Dev. 23:2152–2165. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Roybal JD, Zang Y, Ahn YH, et al: miR-200
inhibits lung adenocarcinoma cell invasion and metastasis by
targeting Flt1/VEGFR1. Mol Cancer Res. 9:25–35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Dykxhoorn DM, Wu Y, Xie H, et al: miR-200
enhances mouse breast cancer cell colonization to form distant
metastases. PLoS One. 4:e71812009. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Korpal M, Ell BJ, Buffa FM, et al: Direct
targeting of Sec23a by miR-200s influences cancer cell secretome
and promotes metastatic colonization. Nat Med. 17:1101–1108. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Chang CJ, Chao CH, Xia W, et al: p53
regulates epithelialmesenchymal transition and stem cell properties
through modulating miRNAs. Nat Cell Biol. 13:317–323. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Schubert J and Brabletz T: p53 spreads out
further: suppression of EMT and stemness by activating miR-200c
expression. Cell Res. 21:705–707. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Kim T, Veronese A, Pichiorri F, et al: p53
regulates epithelialmesenchymal transition through microRNAs
targeting ZEB1 and ZEB2. J Exp Med. 208:875–883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Hermeking H: p53 enters the microRNA
world. Cancer Cell. 12:414–418. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Wang G, Hu X, Lu C, et al:
Promoter-hypermethylation associated defective expression of
E-cadherin in primary non-small cell lung cancer. Lung Cancer.
62:162–172. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Suzuki M, Sunaga N, Shames DS, et al: RNA
interference-mediated knockdown of DNA methyltransferase 1 leads to
promoter demethylation and gene re-expression in human lung and
breast cancer cells. Cancer Res. 64:3137–3143. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Sansom SE, Nuovo GJ, Martin MM, et al:
miR-802 regulates human angiotensin II type 1 receptor expression
in intestinal epithelial C2BBe1 cells. Am J Physiol Gastrointest
Liver Physiol. 299:G632–G642. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Yan M, Huang HY, Wang T, et al:
Dysregulated expression of dicer and drosha in breast cancer.
Pathol Oncol Res. 18:343–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Martin PM, Gnana-Prakasam JP, Roon P, et
al: Expression and polarized localization of the hemochromatosis
gene product HFE in retinal pigment epithelium. Invest Ophthalmol
Vis Sci. 47:4238–4244. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Lee SY, Liu S, Mitchell RM, et al: HFE
polymorphisms influence the response to chemotherapeutic agents via
induction of p16INK4A. Int J Cancer. 129:2104–2114. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Josson S, Nomura T, Lin JT, et al:
β2-microglobulin induces epithelial to mesenchymal transition and
confers cancer lethality and bone metastasis in human cancer cells.
Cancer Res. 71:2600–2610. 2011.
|
|
58.
|
Shankar J, Messenberg A, Chan J, et al:
Pseudopodial actin dynamics control epithelial-mesenchymal
transition in meta-static cancer cells. Cancer Res. 70:3780–3790.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Boukakis G, Patrinou-Georgoula M,
Lekarakou M, Valavanis C and Guialis A: Deregulated expression of
hnRNP A/B proteins in human non-small cell lung cancer: parallel
assessment of protein and mRNA levels in paired tumour/non-tumour
tissues. BMC Cancer. 10:4342010. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Durkin ME, Yuan BZ, Zhou X, et al: DLC-1:
a Rho GTPase-activating protein and tumour suppressor. J Cell Mol
Med. 11:1185–1207. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Yuan BZ, Miller MJ, Keck CL, et al:
Cloning, characterization, and chromosomal localization of a gene
frequently deleted in human liver cancer (DLC-1) homologous to rat
RhoGAP. Cancer Res. 58:2196–2199. 1998.PubMed/NCBI
|
|
62.
|
Banaudha K, Kaliszewski M, Korolnek T, et
al: MicroRNA silencing of tumor suppressor DLC-1 promotes efficient
hepatitis C virus replication in primary human hepatocytes.
Hepatology. 53:53–61. 2011. View Article : Google Scholar : PubMed/NCBI
|