Introduction
The PI3K/AKT signaling pathway regulates various
cellular functions including tumorigenesis by inhibiting apoptosis
and activating proliferation of cancer cells (1–5).
Phosphoinositide 3-kinases (PI3Ks) are lipid kinases that
phosphorylate phosphatidylinositol-4, 5-biphosphate
(PIP2) to phosphatidylinositol-3, 4, 5-triphosphate
(PIP3) (6). The
resulting PIP3 phosphorylates and activates protein
kinase B (AKT, also known as PKB) serine/threonine-specific protein
kinase, which plays a key role in multiple cellular processes.
Importantly, the activated AKT regulates many events related with
tumor malignancies such as metastasis, apoptosis, proliferation,
invasion, migration and motility (7–12).
Notably, this pathway is often found to be overactive in cancer
cells. Many researchers have accordingly tried to develop cancer
drugs to inhibit this crucial signaling pathway at some point. The
PI3K/AKT pathway is known to be negatively regulated by a
well-known tumor suppressor phosphatase and tensin homologue
(PTEN), in which PTEN dephosphorylates PIP3 to
PIP2 preventing activation of AKT (13–15).
The PI3K/AKT pathway can be hyper-activated in PTEN defective cells
(16–18) and PTEN is frequently found to be
mutated or deleted in various human cancers (16–21).
Therefore, suppressing the PTEN function is an alternative way for
cancer cells to obtain oncogenic activity. Neural precursor cell
expressed, developmentally downregulated 4-1 (NEDD4-1) is an
effective target in this regard. Wang et al suggested that
NEDD4-1 degrades PTEN protein by catalyzing PTEN
poly-ubiquitination (22). In
fact, an inverse relationship between PTEN and NEDD4-1 expression
levels is often found in human urinary lung cancer (23). Considering these findings,
elimination of NEDD4-1 function seems to be an effective means of
inhibiting tumorigenesis in a PTEN-dependent manner.
In an effort to identify the physiological mechanism
that triggers the induction of NEDD4-1-mediated PTEN
downregulation, we found the involvement of p34SEI-1 in
this process. p34SEI-1 is known to act as a
transcriptional regulator, cell cycle regulator, senescence
inhibitor and apoptosis inhibitor (22,24–29).
Especially, it plays a critical role in tumor pathogenesis acting
as an oncoprotein. We previously showed that the expression level
of p34SEI-1 significantly increased in breast cancer
patients relative to healthy subjects (26). Several different research groups
have suggested that p34SEI-1 exerts oncogenic effects by
deregulating several vital pathways. For example,
p34SEI-1 overexpression is associated with upregulation
of E2F-mediated transcription, transformation of NIH3T3
fibroblasts, promotion of tumor growth in athymic nude mice, and
chromosomal instability (22,24–29).
Most importantly, our previous study showed the p34SEI-1
protein maximizes oncogenic characteristics by providing an
anti-apoptotic function to cancer cells and increasing the survival
of tumor cells (26). According to
our data, the p34SEI-1 confers resistance to various
apoptotic stimuli on human breast cancer cells by stabilizing XIAP
(X-linked inhibitor of apoptosis protein) (26). In the process of elucidating the
mechanism of p34SEI-1 mediated tumorigenesis, we
suspected that p34SEI-1 might obtain its oncogenic
potential in part through activation of the PI3K/AKT pathway.
Therefore, we tested the involvement of p34SEI-1 in the
regulation of the PI3K/AKT signaling pathway via NEDD4-1 mediated
PTEN degradation.
In this report, we suggest that p34SEI-1
oncogenic protein promotes tumor progression by inducing NEDD4-1
mediated PTEN ubiquitination/degradation and activating the
PI3K/AKT pathway.
Materials and methods
Cell lines and cell culture
MCF7 breast cancer and HEK293 human epithelial
kidney cells were used for this study. Each cell line was cultured
in DMEM medium (WelGENE Inc., Daegu, Korea) supplemented with 10%
fetal bovine serum (Gibco-BRL, Carlsbad, CA, USA) and 1%
antibiotic-antimycotic (Gibco-BRL). All of the cells were cultured
at 37°C in a humidified atmosphere composed of 95% air and 5%
CO2.
Western blot analysis
Cells were washed in an ice-cold PBS buffer and
lased in RIPA lysis buffer (1% sodium deoxycholate, 0.1% SDS, 1%
Triton X-100, 10 mM Tris-HCl, pH 8.0, 140 mM NaCl, 0.025%
NaN3 and 1.0 mM protease inhibitor). The protein amount
was quantified using a protein assay kit (Bio-Rad, Seoul, Korea).
Each protein sample was subjected to SDS-PAGE and transferred to an
Immobilon Transfer Membranes (Millipore, cat. no. IPVH00010,
Billerica, MA, USA). The filter was blocked in 5% non-fat dry
milk/0.1% Tween/TBS followed by incubation with each corresponding
antibody. Immune-detection was done by using the Power Opti-ECL
Western blotting Detection reagent (Bionote, Hwaseong, Korea).
Antibodies used in this study were purchased as follows:
p34SEI-1 (Enzo Life Sciences, ALX-804-645, Farmingdale,
NY, USA), pAKT (Ser473) (Cell Signaling, cat. no. 9271, Danvers,
MA, USA), PTEN (Santa Cruz Biotechnology, sc-7974, Santa Cruz, CA,
USA), NEDD4-1 (Santa Cruz Biotechnology, sc-25508), and γ-tubulin
(Santa Cruz Biotechnology, sc-7396).
Overexpression and knockdown of
p34SEI-1 and NEDD4-1
For ectopic overexpression of p34SEI-1,
MCF7 or HEK293 cells were transfected for 12, 24 or 48 h with 6-8
μg of either C-terminal EGFP-tagged p34SEI-1 expression
vector (p34SEI-1-EGFP) or a control vector (pEGFP) by
using Lipofectamine 2000 (Invitrogen, Seoul, Korea). To knockdown
endogenous p34SEI-1 and NEDD4-1, MCF7 or HEK293 cells
were transiently transfected with p34SEI-1 or NEDD4-1
specific siRNA (20 pmol siRNA final concentration) with a control
of scrambled siRNA. The following target sequences were used to
generate p34SEI-1 or NEDD4-1 siRNA; p34SEI-1
siRNA (5-CCGAAUUGGACUAC CUCAUdTdT-3) and NEDD4-1 siRNA
(5-UUCCAUGAAUC UAGAAGAACATT-3 (30). p34SEI-1 siRNA was
obtained from Santa Cruz Biotechnology (sc-62988) and NEDD4-1
oligonucleotides were chemically synthesized by ST Pharm Co. Ltd
(Seoul, Korea).
Immunoprecipitation
To analyze the interaction between
p34SEI-1 and NEDD4-1, HEK293 cells were co-transfected
with EGFP-tagged p34SEI-1 (p34SEI-1-EGFP) and
HA-tagged NEDD4-1 (pHA-NEDD4-1) plasmids and cell lysates were
immuno precipitated with either anti-EGFP (Abcam, ab290, Cambridge,
MA, USA) or anti-HA (Sigma, H9658, St. Louis, MO, USA) antibodies.
IP was performed by lysing cells in IP buffer (50 mM of Tris-HCl pH
7.4, 150 mM of NaCl, 10 mM of NaF, 10 mM of
Na3VO4, 1 mM of PMSF, 1% of NP-40) with
protease inhibitors, followed by pre-clearing with protein A/G
Sepharose (Santa Cruz Biotechnology, sc-2003). Pre-cleared lysates
were incubated with each antibody for 16 h at 4°C with continuous
agitation, and then protein A/G Sepharose was added. After 4 h, the
lysate-antibody-agarose A/G bead complex was collected by
centrifugation at 10,000 x g for 5 min, the complex was then washed
three times with IP buffer, and proteins were eluted from the beads
by boiling them in SDS sample buffer and analyzed by using a
western blot with the indicated a ntibodies. Proteins were probed
with the corresponding antibodies.
In vivo PTEN-ubiquitination assay
HEK293 cells were transfected with
p34SEI-1-EGFP plasmid and/or NEDD4-1 siRNA in the
presence of HA-Ub. Forty-eight hours after transfection, cells were
treated with 10 μM proteasome inhibitor MG132 (A.G. Scientific Inc,
M-1157, San Diego, CA, USA) for 16 h. The cells were lysed with
RIPA buffer with protease inhibitors. The lysates were centrifuged
to obtain cytosolic proteins. Ubiquitinated PTEN was
immunoprecipitated by anti-PTEN antibody (Santa Cruz Biotechnology,
sc-7974), followed by immunoblotting with anti-Ub antibody (Santa
Cruz Biotechnology, sc-8017).
Immunohistochemistry analysis
Four-micrometer-thick sections were sliced onto
Silane Coated Micro Slides (Muto Pure Chemicals Corp., Tokyo,
Japan) and incubated at 60°C for 2 h. The slides were then
deparaffinised by application of xylene and incubation (5 min x 3)
at room temperature. Sections were hydrated by applying graded
alcohol and endogenous peroxidase activity was quenched by
incubating the sections in methanol with 0.3%
H2O2 for 30 min at room temperature. After
washing the slides in PBS (5 min x 2), antigen retrieval was
performed by heating the slides in citrate buffer (0.01 M, pH 6.0)
using a microwave in a pressure cooker for 15 min. After heating,
the samples were allowed to cool for 2 h at room temperature
followed by washing with PBS (5 min x 2). An immunohistochemical
analysis was performed using p34SEI-1 (Biorbyt,
Cambridge, UK) and NEDD4-1 (Proteintech, cat. no. 13690-1-AP,
Chicago, IL, USA) rabbit polyclonal antibodies with 1:50 dilution
each by PBS. The tissues were incubated with primary antibodies for
2 h at room temperature and washed three times with PBS followed by
incubation using an Ultra Vision Quanto Detection System HRP DAB
(Lab Vision Corp., Fremont, CA, USA) according to the
manufacturer’s instructions. The immunostained slides were examined
by two independent observers and a consensus score was determined
for each specimen. A positive reaction for both antibodies was
scored into 4 grades, according to the intensity of the staining:
0, 1+, 2+ and 3+. The percentages of positive cells were also
scored into 4 categories: 0, 0%; 1, 1–30%; 2, 31–60%; and 3,
61–100%. The final score, calculated as the product of the
intensity score multiplied by the percentage score, was classified
as follows: 0, negative; 1–3, weak; 4–6, moderate; and 7–9, strong.
Samples with a final score less than 3 were grouped together as
expression negative while those with a score greater than 4 were
grouped together as expression positive.
Reverse transcription (RT)-PCR
The total RNA was extracted from HEK293 cells after
transfection with either pEGFP or p34SEI-1-EGFP using an
RNeasy mini kit (Qiagen, Hilden, Germany). For reverse
transcription, 1 μg RNA of each sample was subjected to cDNA
synthesis using an oligo (dT) primer and the ImProm-II™ Reverse
Transcription System (Promega, A3800, Madison, WI, USA) following
the manufacturer’s instructions. Each gene product was amplified
using 10 ng cDNA, the corresponding pair of primers, and an
AccuPower PCR PreMix system (Bioneer, Daejeon, Korea), in which the
β-actin gene product was used as an internal control (Table I).
 | Table I.Oligonucleotide sequences and
conditions for RT-PCR analysis. |
Table I.
Oligonucleotide sequences and
conditions for RT-PCR analysis.
| Primer name | Primer sequence
(5′→3′)a | Amplicon | Conditions size
(bp)b | Source |
|---|
| pRT-SEI-1 | F:
AGGACCTCAGCCACATTGAG | 142 bp | 60°C | This study |
| R:
GGTGCCCAAAGTTCATTGTC | | 27 cycles | |
| pRT-NEDD4-1 | F:
GGAGTTGCCAGAGAATGGTT | 151 bp | 60°C | This study |
| R:
TTGCCATGATAAACTGCCAT | | 27 cycles | |
| pRT-NF-κB | F:
CCGCACCTCCACTCCATCC | 121 bp | 62°C | Sarma et al
(37) |
| R:
ACATCAGCACCCAAGGACACC | | 26 cycles | Du and Galan
(38) |
| pRT-ACTB | F:
AGGTCGGAGTCAACGGATTTG | 377 bp | 58°C | This study |
| R:
GTGATGGCATGGACTGTGGT | | 21 cycles | |
Reporter assay
Cells were co-transfected with
p34SEI-1-EGFP and pGL4-NF-κB plasmids using
Lipofectamine 2000 (Invitrogen) transfection reagent following the
manufacturer’s protocol, in which the NF-κB genes were subcloned
into the pGL4 basic luciferase reporter vector (pGL4.1; Promega).
The following day, 14–18 h later, cells were lysed and luciferase
assays were performed using Luciferase Assay System (Promega, cat.
no. E1501) and the luciferase activity levels were measured after
standardization against pGL4.1.
Results
Effect of p34SEI-1on the
alteration of pAKT, PTEN, and NEDD4-1 protein levels
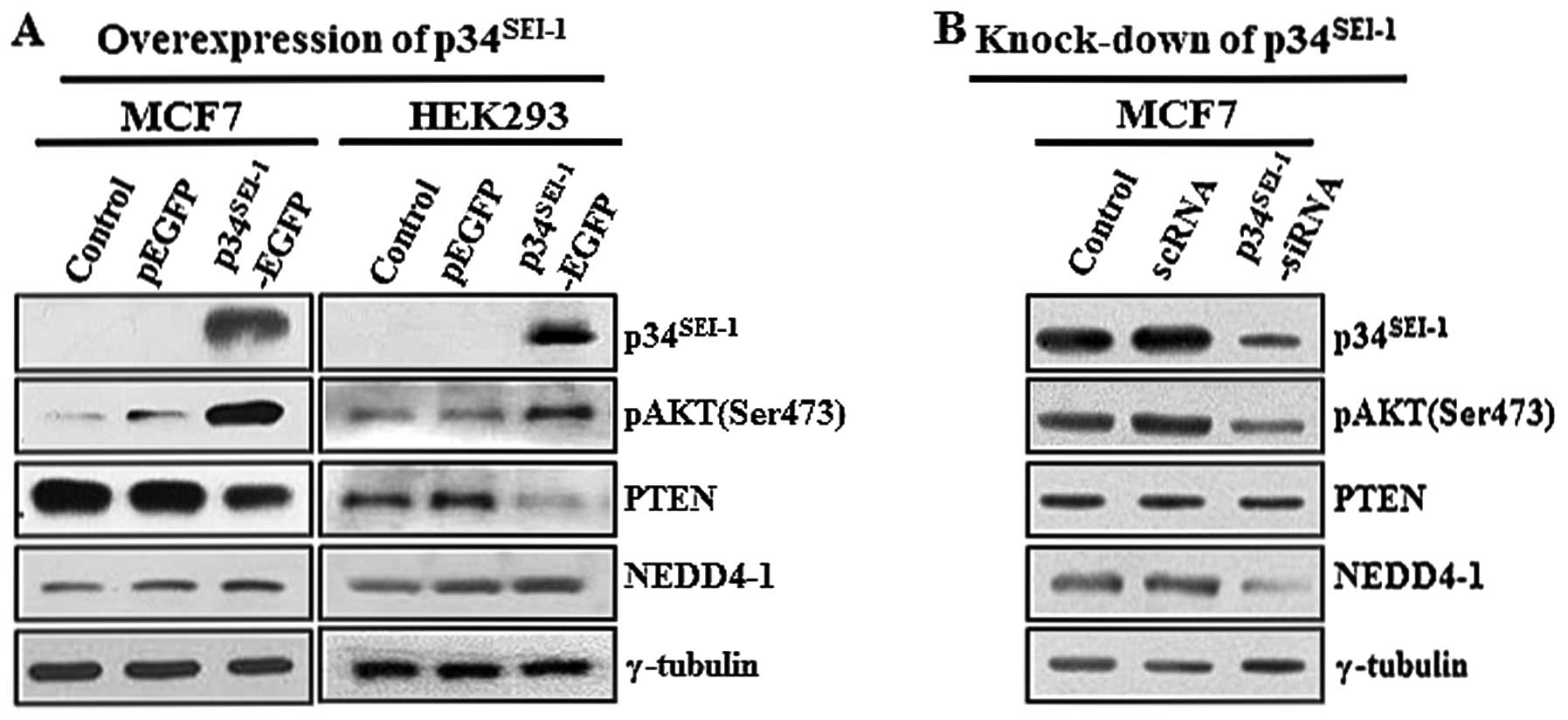
As part of an effort to identify how
p34SEI-1 promotes tumor progression in various cancer
cells, we initially tested the effect of p34SEI-1 on the
PI3K/AKT signaling pathway because of its vital roles in
tumorigenesis. After MCF7 and HEK293 cells were transfected with
the EGFP control vector (pEGFP) and C-terminal EGFP-tagged
p34SEI-1 (p34SEI-1-EGFP) plasmids, AKT
phosphorylation on serine 473 residue was examined because
phosphorylation on this residue is known to promote signal
transduction related with tumor malignancies. Our data showed that
p34SEI-1 overexpression significantly increased AKT
phosphorylation on this residue (Fig.
1A). In contrast, the AKT phosphorylation level was decreased
when MCF7 cells were treated with p34SEI-1-siRNA
compared to scrambled siRNA-treated control cells (Fig. 1B). This strongly suggests that
p34SEI-1 has a positive effect on AKT phosphorylation
and therefore activation of the PI3K/AKT signaling pathway. Next,
we tested the alteration of PTEN and NEDD4-1 expression levels in
response to ectopic expressed p34SEI-1 because they are
the main components affecting the PI3K/AKT signaling pathway.
Interestingly, the western blot analysis revealed that
p34SEI-1 overexpression slightly decreased PTEN but
increased NEDD4-1 at the protein levels (Fig. 1A). We then tested PTEN and NEDD4-1
protein levels in MCF7 cells transfected with
p34SEI-1-siRNA. As expected, NEDD4-1 expression
significantly decreased relative to the scrambled siRNA-treated
cells. However, no change in the PTEN protein level was detected,
likely due to a weak or even no effect of NEDD4-1 on downregulation
of PTEN (Fig. 1B). Taken together,
our results implicate that p34SEI-1 activates the
PI3K/AKT signaling pathway at least in part by regulating NEDD4-1
and PTEN.
p34SEI-1 induced
ubiquitination and degradation of PTEN in a NEDD4-1 dependent
way
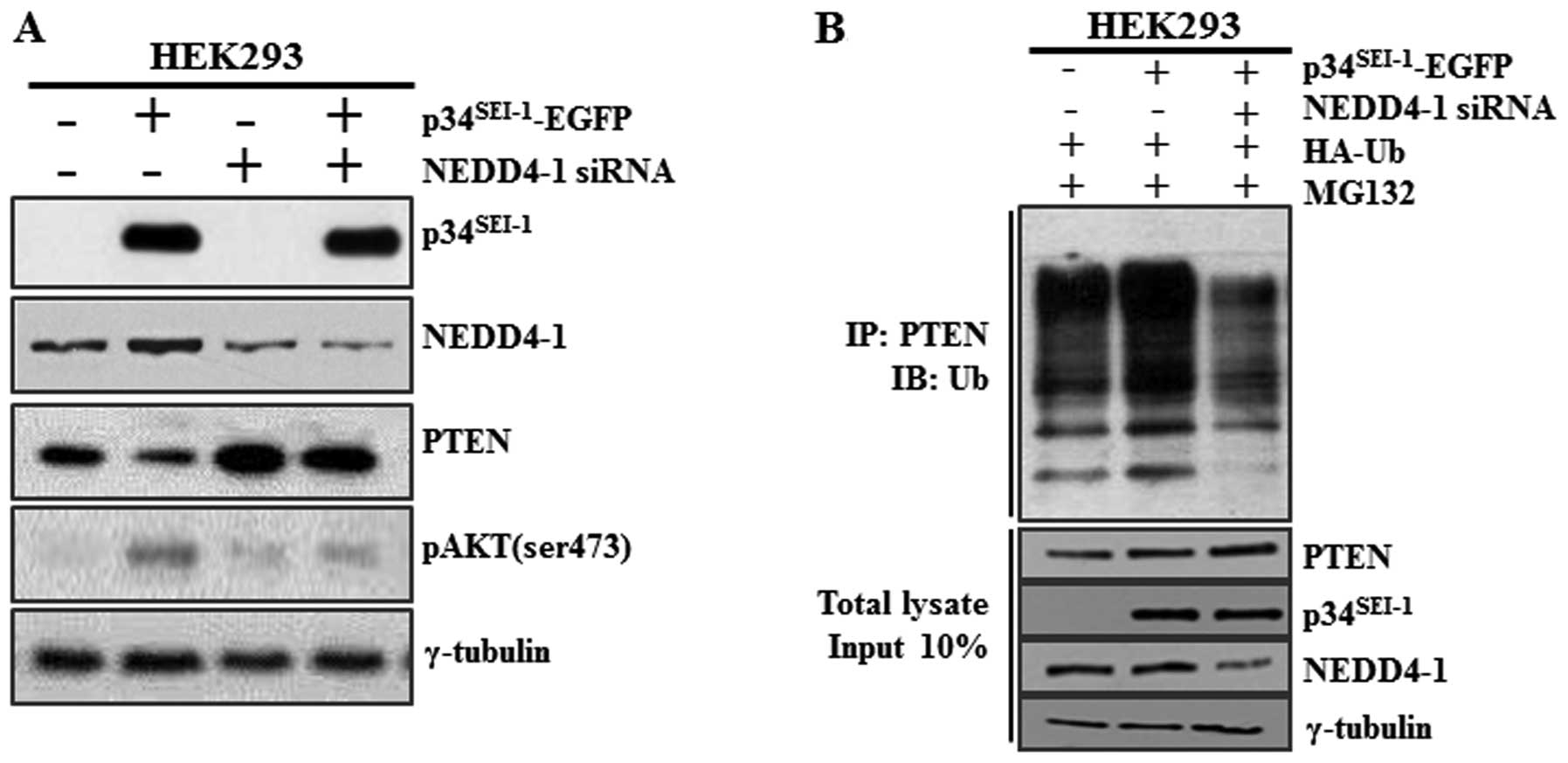
Our results imply that p34SEI-1 may
downregulate PTEN in a NEDD4-1-dependent manner. However, it is
possible that p34SEI-1 may affect other proteins or
pathways and indirectly cause PTEN downregulation. For example,
p34SEI-1 might activate different types of E3 ligases
rather than NEDD4-1 and downregulate PTEN. In fact, two different
research groups suggested apparently discrepant results. Wang et
al suggested that NEDD4-1 directly binds to PTEN and induces
its ubiquitination and degradation (31). However, Fouladkou et al
showed a contradictory result that NEDD4-1 is dispensable for the
regulation of PTEN stability (23,32).
To test whether PTEN downregulation by p34SEI-1 is
NEDD4-1 dependent, the PTEN protein level was checked after HEK293
cells were transfected with p34SEI-1-EGFP vector and/or
NEDD4-1 siRNA (Fig. 2A). Our
result showed that siRNA-induced NEDD4-1 silencing induced an
increase of endogenous PTEN protein level and a concurrent decrease
of AKT phosphorylation. Importantly, p34SEI-1
overexpression alone reduced the PTEN protein level but
co-transfection of p34SEI-1-EGFP and NEDD4-1 siRNA had
no effect on the PTEN protein level (Fig. 2A). This finding suggests that
p34SEI-1 decreases the PTEN protein level in a NEDD4-1
E3 ligase-dependent manner. The data strongly imply that
p34SEI-1 is responsible for the PTEN ubiquitination and
its subsequent degradation. Therefore, a ubiquitination assay was
performed to determine whether downregulation of PTEN stimulated by
p34SEI-1 is derived from ubiquitination-mediated protein
degradation. HEK293 cells were transfected with
p34SEI-1-EGFP vector and/or NEDD4-1 siRNA in the
presence of HA-Ub and MG132. The resulting cells were subjected to
immunoprecipitation (IP) and immunoblot (IB) analyses as mentioned
in Materials and methods. As shown in Fig. 2B, endogenous PTEN was significantly
ubiquitinated in p34SEI-1 overexpressing cells compared
to control cells. However, this did not occur in cells
co-transfected with p34SEI-1-EGFP and NEDD4-1 siRNA
indicating that NEDD4-1 is required for
p34SEI-1-mediated PTEN ubiquitination. Taken together,
these results strongly suggest that p34SEI-1 induces
PTEN ubiquitination in a NEDD4-1 dependent manner and thereby
targets PTEN for proteasomal degradation.
Positive effect of p34SEI-1 on
NEDD4-1 expression both at the transcriptional and protein
levels
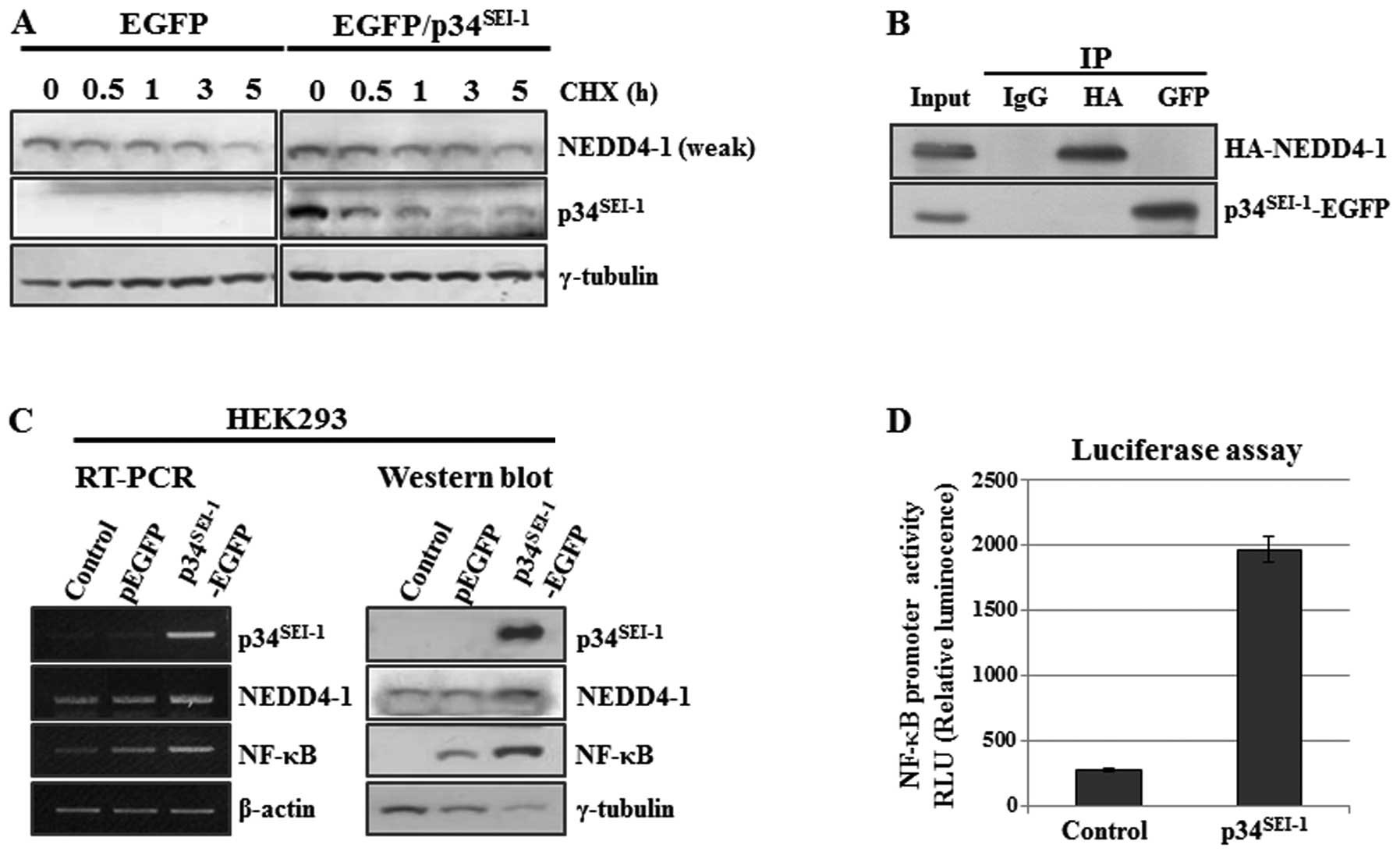
Our data indicate that p34SEI-1 might
exert positive effect on NEDD4-1 expression probably by controlling
NEDD4-1 turnover. This hypothesis was examined by employing
cycloheximide chase experiment. Briefly, HEK293 cells were
transfected with pEGFP and p34SEI-1-EGFP plasmids in the
presence of cycloheximide (CHX, 20 μg/ml), an inhibitor of
protein biosynthesis and NEDD4-1 protein levels were checked at the
indicated times of treatment as shown in Fig. 3A. NEDD4-1 expression slowly
decreased in p34SEI-1-EGFP transfected cells compared to
the control cells (Fig. 3A). This
strongly suggests that p34SEI-1 stabilizes NEDD4-1 by
preventing proteasome-dependent protein degradation. The next
question was how p34SEI-1 can stabilize NEDD4-1 protein.
One possibility is that p34SEI-1 may stabilize NEDD4-1
via direct interaction with it. To test this hypothesis, HEK293
cells were co-transfected with p34SEI-1 and NEDD4-1
overexpressing vectors (p34SEI-1-EGFP and HA-NEDD4-1)
and cell lysates were used for co-immunoprecipitation using
anti-EGFP (i.e., p34SEI-1-EGFP) or anti-HA (i.e.,
HA-NEDD4-1) antibodies. However, no direct interaction was detected
between p34SEI-1 and NEDD4-1 protein (Fig. 3B). Moreover, we also found the same
result from an examination of molecular interactions between
endogenous p34SEI-1 and NEDD4-1 (data not shown).
In addition, the positive effect of
p34SEI-1 on NEDD4-1 expression was also checked at the
transcriptional level because p34SEI-1 is a well-known
transcriptional co-activator. NEDD4-1 transcription was measured by
using RT-PCR after HEK293 cells were transfected with either pEGFP
or p34SEI-1-EGFP expression vector. Our data showed that
NEDD4-1 transcription was slightly increased by p34SEI-1
overexpression (Fig. 3C). However,
our luciferase assay showed no direct effect of p34SEI-1
on NEDD4-1 transcription (data not shown). Considering that
p34SEI-1 is a transcriptional co-activator, we could not
exclude the other possibility that p34SEI-1 might
indirectly activate NEDD4-1 transcription by working with other
transcriptional activator. To test this hypothesis, we first
searched transcription factor binding sites in the NEDD4-1 gene
promoter by using the TRANSFAC database (http://www.genome.ad.jp). Many putative transcription
factor binding sites were found in the NEDD4-1 gene promoter,
including NF-κB, FOXO3b, FOXO3a, FOXD1, FOXO1a, FOXO4 and Nkx2-2.
Among them, we initially focused on NF-κB because the PI3K/AKT
signaling pathway is known to be strongly related with NF-κB
activity (16,33,34).
More importantly, Judge et al suggested that NEDD4 is one of
the targets of NF-κB transcription factor (35). As shown in Fig. 3C, NF-κB expression was slightly or
significantly increased by p34SEI-1 overexpression both
at the transcriptional and protein levels, respectively. In a
further study, a luciferase assay was employed to examine the
direct effect of p34SEI-1 on the NF-κB transcriptional
activation. The luciferase assay was performed by transfecting
pGL4-NF-κB and p34SEI-1-EGFP into HEK293. As shown in
Fig. 3D, over-expressed
p34SEI-1 significantly activated NF-κB transcription.
Our data suggest that p34SEI-1 seems to indirectly
activate NEDD4-1 transcription through NF-κB transcription factor.
We showed that p34SEI-1 overexpression significantly
increased NF-κB transcription. Our further search of transcription
factor binding sites in the NF-κB gene promoter revealed the
putative binding site of p300, a known p34SEI-1
interacting protein. Therefore, it is possible that
p34SEI-1 may enhance transcriptional activation of NF-κB
via the interaction with p300. However, this needs to be tested
further. Considering all these data, we conclude that
p34SEI-1 exerts a positive effect on NEDD4-1 stability
but not through direct interaction with NEDD4-1, whereby
p34SEI-1 positively affects NEDD4-1 both at the
transcriptional and protein levels.
Correlation between p34SEI-1
and NEDD4-1 expression levels in breast tissues containing adjacent
histologically normal and invasive ductal carcinoma tissues
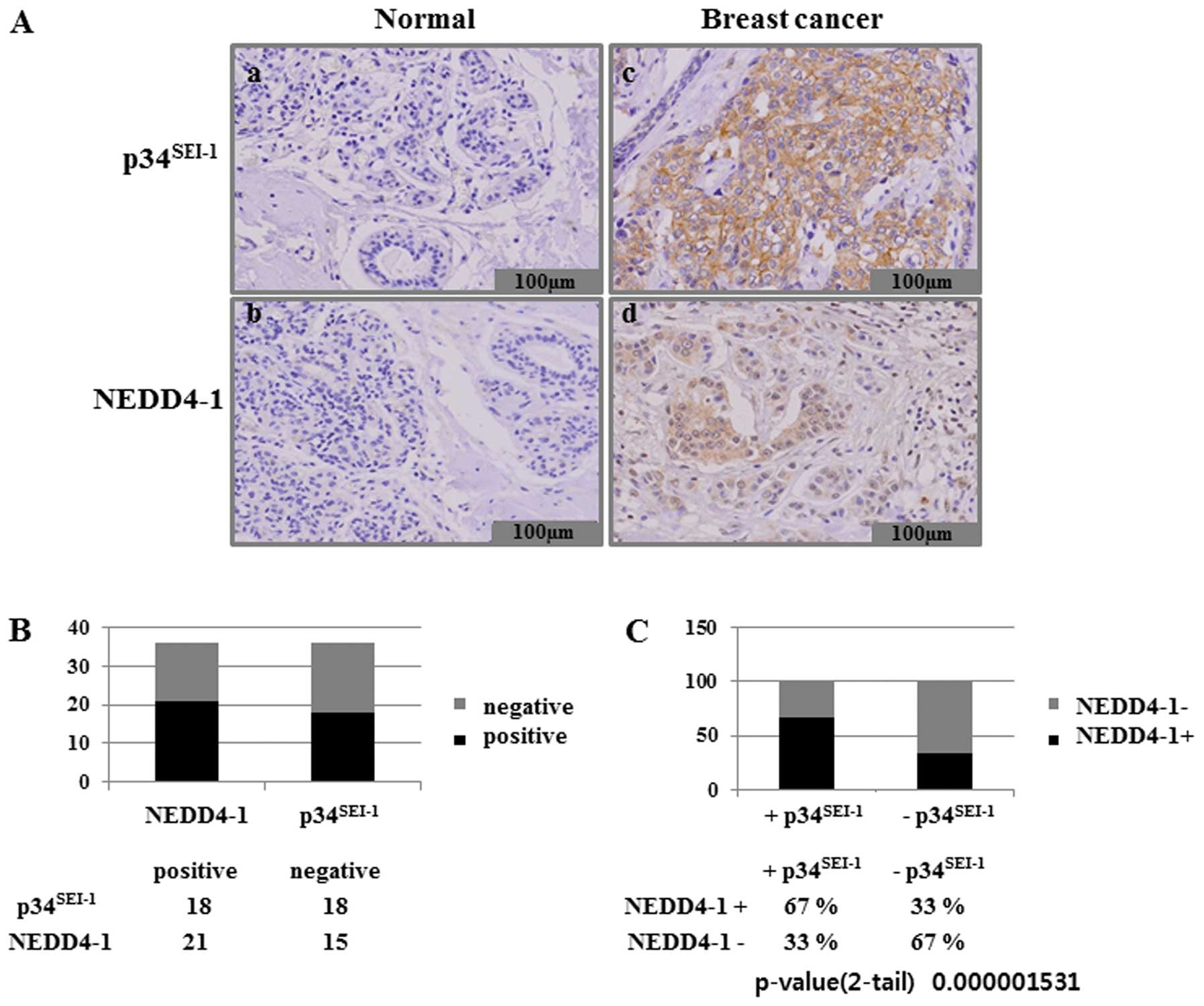
Considering the role of p34SEI-1 as a
positive regulator of NEDD4-1, we hypothesized that high level of
p34SEI-1 in cancer cells might contribute to similar
high levels of NEDD4-1 protein in cancer cells. We therefore
conducted an immunohistochemical analysis of both
p34SEI-1 and NEDD4-1 expression in 36 tissue samples
from patients with breast cancer. We assessed p34SEI-1
expression in formalin-fixed and paraffin-embedded samples obtained
from surgical resections of 36 patients with breast cancer. Normal
and cancer breast tissues showed immune-negative and strong
immune-positive staining, respectively, for both
p34SEI-1 and NEDD4-1 (Fig.
4A). This suggests that expression levels of
p34SEI-1 and NEDD4-1 are very low in normal breast
tissue but strong in breast cancer cells. The immunohistochemical
analysis also revealed that significant overexpression of
p34SEI-1 and NEDD4-1 was detected in 18 (50%) of the 36
and in 21 (58%) of the 36 breast cancer tissue samples,
respectively (Fig. 4B).
Interestingly, NEDD4-1 levels were found to be consistently higher
in the areas of high p34SEI-1 protein level. Our
examination of the co-expression of p34SEI-1 and NEDD4-1
in breast cancer cells showed that NEDD4-1 was expressed in 67% of
the p34SEI-1 positive samples (Fig. 4C). These data confirm that
p34SEI-1 has a positive effect on the NEDD4-1 expression
in cancer cells. This is consistent with the findings that both
p34SEI-1 and NEDD4-1 have oncogenic potential and their
expression levels are increased as tumor invasiveness progresses.
Taken together, our data strongly suggest that p34SEI-1
and NEDD4-1 are significantly over-expressed in breast cancer
tissues.
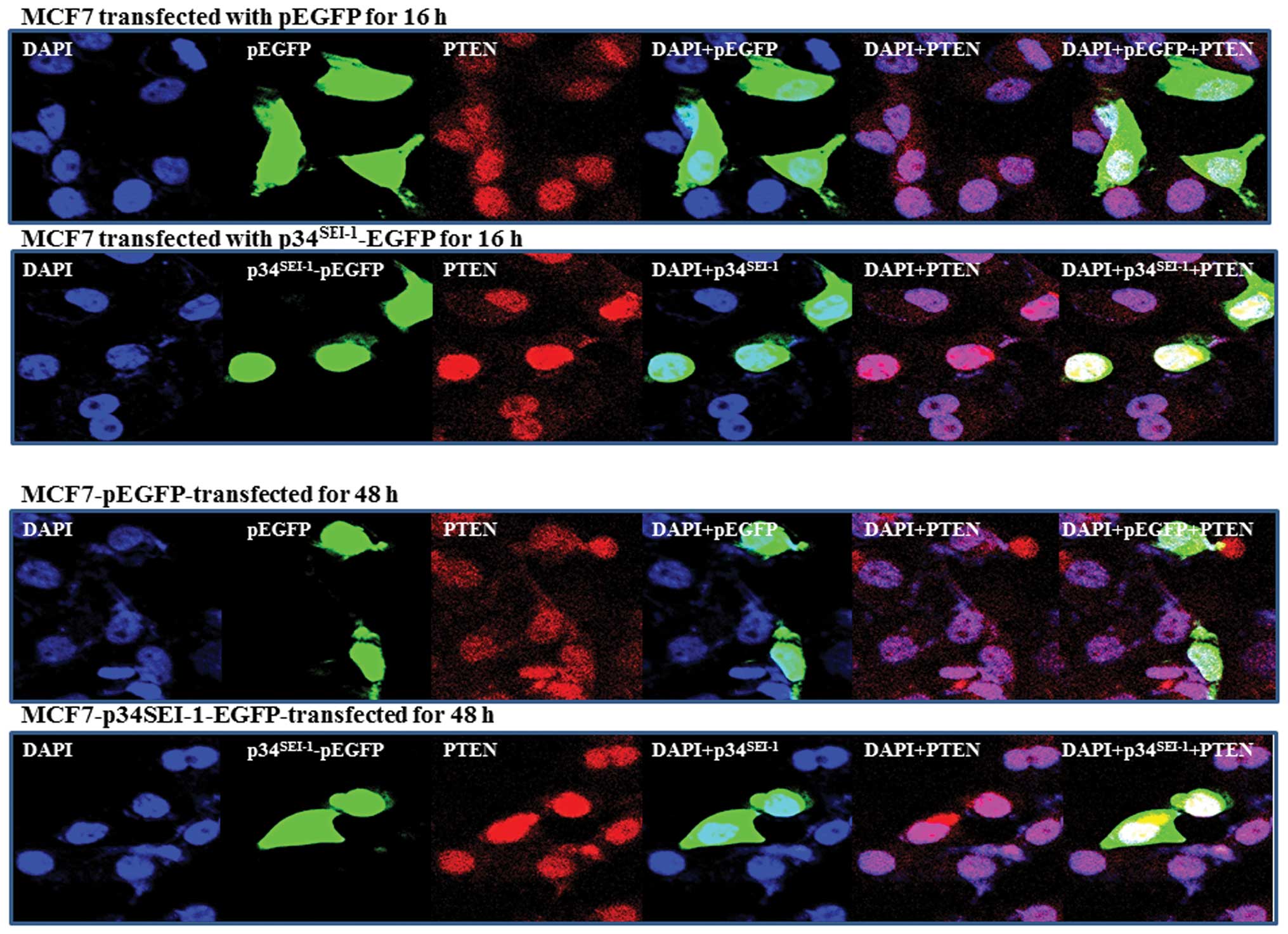
PTEN subcellular localization in response
to p34SEI-1
It has been suggested that PTEN is located in the
nucleus as well as the cytosol. Trotman et al suggested that
PTEN localization is affected by NEDD4-1, which can both mono- and
poly-ubiquitinate (31). PTEN
nuclear localization is mediated by its mono-ubiquitination. We
thus far have shown that p34SEI-1 induces
NEDD4-1-mediated PTEN poly-ubiquitination. Considering p34 has an
oncogenic potential working with NEDD4-1 E3 ligase, it was expected
that p34SEI-1 might affect PTEN nuclear localization. To
test this hypothesis, we analyzed PTEN nuclear localization in
response to p34SEI-1 by using immunofluorescence, as
mentioned in Materials and methods. In normal conditions,
endogenous PTEN is found both in the nucleus and cytoplasm of MCF7
cells. However, when p34SEI-1 is overexpressed as EGFP
fusion, our IF data revealed a different distribution of PTEN in
nucleus in p34SEI-1 overexpressing and control cells.
PTEN accumulated much more inside the nucleus after transfection
within 16 h and lasted until 48 h (Fig. 5). In contrast, the PTEN protein
level was slightly reduced in cytosolic accumulation compared to
non-transfected cells (Fig. 5).
This suggests that p34SEI-1 positively affects PTEN
nuclear import. Taken together, the results indicate that
p34SEI-1 affects subcellular localization of PTEN, in
which p34SEI-1 overexpression induces PTEN nuclear
localization.
Discussion
The PI3K signaling pathway plays important roles in
cells by controlling many different cellular functions, in which
AKT is the key regulator of the PI3K pathway to promote signal
transduction related with tumor malignancies. AKT is phosphorylated
and activated by either PI3K activation or PTEN inactivation. In
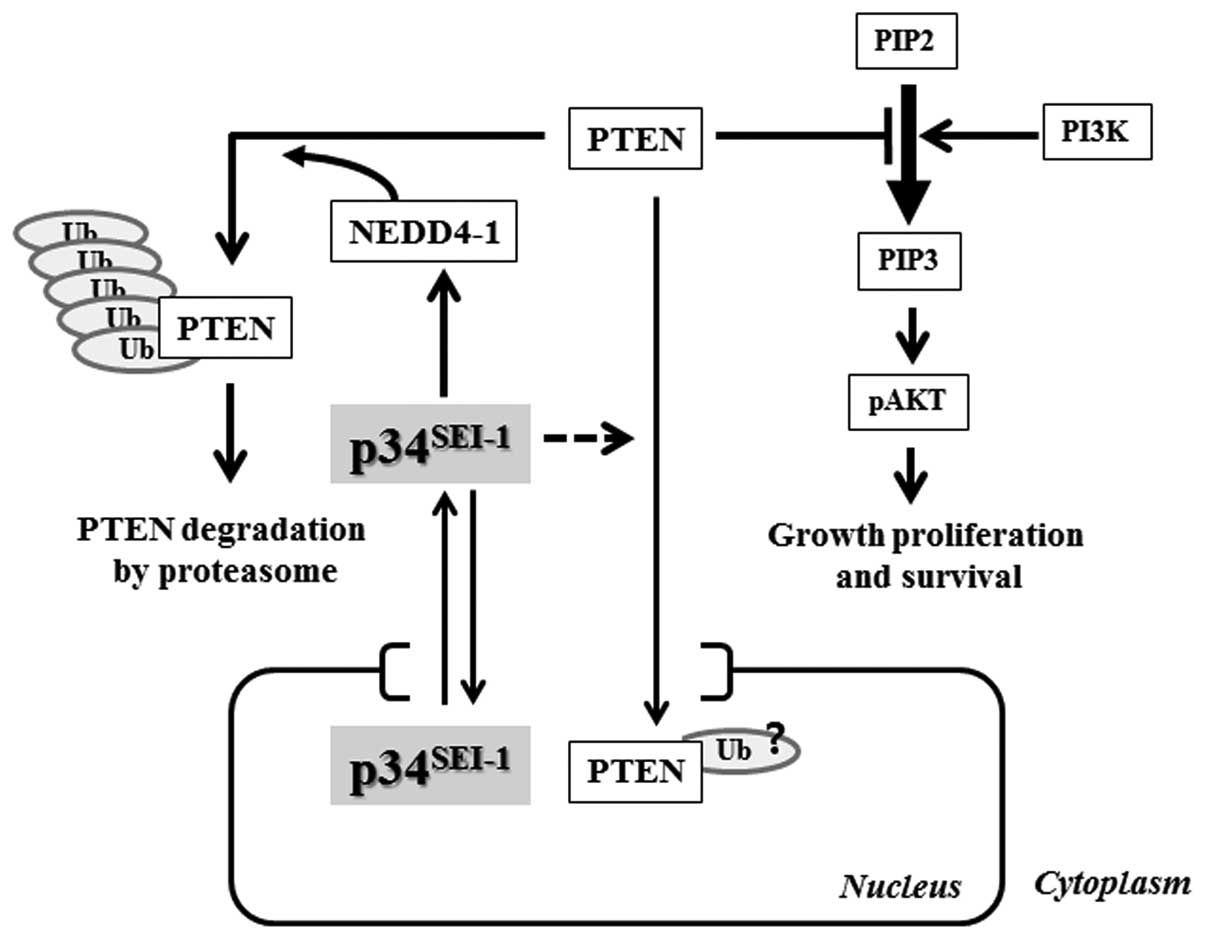
the present study, we suggest a mechanism of how
p34SEI-1 has oncogenic potential to promote
carcinogenesis. Our data show that p34SEI-1
overexpression stabilizes NEDD4-1 and in turn NEDD4-1 induces
poly-ubiquitination/degradation of a PTEN tumor suppressor, and
subsequently promotes tumorigenesis by positively regulating the
PI3K/AKT pathway as summarized in Fig.
6.
We previously showed that p34SEI-1
directly binds and stabilizes the X-linked inhibitor of apoptosis
protein (XIAP) leading to an anti-apoptotic effect (26). Interestingly, the group of Van
Themsche (36) suggested that XIAP
can act as an E3 ligase for PTEN and therefore directly induces
PTEN ubiquitination. Considering all these results, it is likely
that p34SEI-1 may have an indirect effect on PTEN
ubiquitination/degradation through XIAP. Thus, we also tested the
effect of p34SEI-1 on XIAP mediated PTEN
ubiquitination/degradation. However, our data revealed
XIAP-independent PTEN ubiquitination/degradation (data not shown).
Similarly, it needs to be considered that p34SEI-1 may
affect different types of PTEN ubiquitin ligases that are
responsible for PTEN ubiquitination/degradation.
Our data revealed that p34SEI-1 affects
PTEN subcellular localization as well as its protein expression. It
has been suggested that NEDD4-1 can mediate both mono- and
poly-ubiquitination of PTEN and it can cause PTEN nuclear transport
by catalyzing PTEN mono-ubiquitination (31). Considering the positive effect of
p34SEI-1 on NEDD4-1 expression, it is suspected that
p34SEI-1 may induce mono-ubiquitination as well as
poly-ubiquitination of PTEN. Therefore, p34SEI-1 seems
to positively control nuclear import of PTEN by upregulating
NEDD4-1 expression as shown in Fig.
5. However, our immunofluorescence data were unexpected in the
PTEN’s subcellular distribution in response to p34SEI-1
overexpression. The reason is that PTEN nuclear localization has
been suggested to be essential to suppress tumorigenesis and is
mediated by its mono-ubiquitination (31). Considering that p34SEI-1
is an oncoprotein, which normally activate tumorigenesis, it was
expected that p34SEI-1 over expression would inhibit
PTEN nuclear import. We thought that p34SEI-1 might
exert a negative effect on PTEN in the nucleus as in the case of
cytosol, even though p34SEI-1 induced PTEN nuclear
import. However, the mechanism by which p34SEI-1 exerts
a positive effect on PTEN nuclear localization remains unclear.
Further research is needed to determine the effects of
p34SEI-1 on PTEN nuclear localization.
In summary, our previous and current results suggest
that p34SEI-1 is highly expressed in human breast cancer
cells acting as an oncoprotein. In this process,
p34SEI-1 appears to cause tumorigenesis by inducing
NEDD4-1-mediated PTEN down-regulation and positively regulating the
PI3K/AKT pathway. Thus, therapeutic strategies that interfere with
the function of p34SEI-1 are expected to be promising
targets for new anticancer drugs for breast cancer treatment.
Abbreviations:
|
p34SEI-1
|
34-KD protein encoding SEI-1
(selected with Ink4a-1 as bait) gene;
|
|
PTEN
|
phosphatase and tensin homolog
deleted on chromosome ten;
|
|
NEDD4-1
|
neuronal precursor cell-expressed
developmentally downregulated 4-1;
|
|
PI3K
|
phosphoinositide-3 kinase
|
Acknowledgements
This study was supported by the
National Research Foundation of Korea (NRF) grant funded by the
Korean government (MEST) (nos. R11-2005-017-04001-0 and
2011-0030701) and Basic Science Research Program through the
National Research Foundation of Korea (NRF) funded by the Ministry
of Education, Science and Technology (2012R1A1A3012438).
References
|
1.
|
Morgensztern D and McLeod HL:
PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer
Drugs. 16:797–803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Cortot A, Armand JP and Soria JC:
PI3K-AKT-mTOR pathway inhibitors. Bull Cancer. 93:19–26. 2006.(In
French).
|
|
3.
|
Yap TA, Garrett MD, Walton MI, Raynaud F,
de Bono JS and Workman P: Targeting the PI3K-AKT-mTOR pathway:
progress, pitfalls, and promises. Curr Opin Pharmacol. 8:393–412.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
LoPiccolo J, Blumenthal GM, Bernstein WB
and Dennis PA: Targeting the PI3K/Akt/mTOR pathway: effective
combinations and clinical considerations. Drug Resist Updat.
11:32–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Yuan TL and Cantley LC: PI3K pathway
alterations in cancer: variations on a theme. Oncogene.
27:5497–5510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kong D and Yamori T: Advances in
development of phosphatidylinositol 3-kinase inhibitors. Curr Med
Chem. 16:2839–2854. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Qiao M, Sheng S and Pardee AB: Metastasis
and AKT activation. Cell Cycle. 7:2991–2996. 2008. View Article : Google Scholar
|
|
8.
|
Xue G, Restuccia DF, Lan Q, et al:
Akt/PKB-mediated phosphorylation of Twist1 promotes tumor
metastasis via mediating cross-talk between PI3K/Akt and TGF-beta
signaling axes. Cancer Discov. 2:248–259. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Brader S and Eccles SA: Phosphoinositide
3-kinase signalling pathways in tumor progression, invasion and
angiogenesis. Tumori. 90:2–8. 2004.PubMed/NCBI
|
|
10.
|
Grille SJ, Bellacosa A, Upson J, et al:
The protein kinase Akt induces epithelial mesenchymal transition
and promotes enhanced motility and invasiveness of squamous cell
carcinoma lines. Cancer Res. 63:2172–2178. 2003.PubMed/NCBI
|
|
11.
|
Toker A and Yoeli-Lerner M: Akt signaling
and cancer: surviving but not moving on. Cancer Res. 66:3963–3966.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Yoeli-Lerner M and Toker A: Akt/PKB
signaling in cancer: a function in cell motility and invasion. Cell
Cycle. 5:603–605. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Gagnon V, St-Germain ME, Parent S and
Asselin E: Akt activity in endometrial cancer cells: regulation of
cell survival through cIAP-1. Int J Oncol. 23:803–810.
2003.PubMed/NCBI
|
|
14.
|
Maehama T and Dixon JE: The tumor
suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem.
273:13375–13378. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Stambolic V, Suzuki A, de la Pompa JL, et
al: Negative regulation of PKB/Akt-dependent cell survival by the
tumor suppressor PTEN. Cell. 95:29–39. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Akca H, Demiray A, Tokgun O and Yokota J:
Invasiveness and anchorage independent growth ability augmented by
PTEN inactivation through the PI3K/AKT/NFκB pathway in lung cancer
cells. Lung Cancer. 73:302–309. 2011.PubMed/NCBI
|
|
17.
|
Carver BS, Chapinski C, Wongvipat J, et
al: Reciprocal feedback regulation of PI3K and androgen receptor
signaling in PTEN-deficient prostate cancer. Cancer Cell.
19:575–586. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Taylor CJ, Qiao J, Colon NC, Schlegel C,
Josifi E and Chung DH: Integrin-linked kinase regulates phosphatase
and tensin homologue activity to promote tumorigenesis in
neuroblastoma cells. Surgery. 150:162–168. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Tian XX, Pang JC, To SS and Ng HK:
Restoration of wild-type PTEN expression leads to apoptosis,
induces differentiation, and reduces telomerase activity in human
glioma cells. J Neuropathol Exp Neurol. 58:472–479. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
McMenamin ME, Soung P, Perera S, Kaplan I,
Loda M and Sellers WR: Loss of PTEN expression in paraffin-embedded
primary prostate cancer correlates with high Gleason score and
advanced stage. Cancer Res. 59:4291–4296. 1999.PubMed/NCBI
|
|
21.
|
Chang JG, Chen YJ, Perng LI, et al:
Mutation analysis of the PTEN/MMAC1 gene in cancers of the
digestive tract. Eur J Cancer. 35:647–651. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Wang X, Trotman LC, Koppie T, et al:
NEDD4-1 is a protooncogenic ubiquitin ligase for PTEN. Cell.
128:129–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Amodio N, Scrima M, Palaia L, et al:
Oncogenic role of the E3 ubiquitin ligase NEDD4-1, a PTEN negative
regulator, in non-small-cell lung carcinomas. Am J Pathol.
177:2622–2634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Hayashi R, Goto Y, Ikeda R, Yokoyama KK
and Yoshida K: CDCA4 is an E2F transcription factor family-induced
nuclear factor that regulates E2F-dependent transcriptional
activation and cell proliferation. J Biol Chem. 281:35633–35648.
2006. View Article : Google Scholar
|
|
25.
|
Hsu SI, Yang CM, Sim KG, Hentschel DM,
O’Leary E and Bonventre JV: TRIP-Br: a novel family of PHD zinc
finger-and bromodomain-interacting proteins that regulate the
transcriptional activity of E2F-1/DP-1. EMBO J. 20:2273–2285. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Hong SW, Kim CJ, Park WS, et al: p34SEI-1
inhibits apoptosis through the stabilization of the X-linked
inhibitor of apoptosis protein: p34SEI-1 as a novel target for
anti-breast cancer strategies. Cancer Res. 69:741–746. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Li Y, Nie CJ, Hu L, et al:
Characterization of a novel mechanism of genomic instability
involving the SEI1/SET/NM23H1 pathway in esophageal cancers. Cancer
Res. 70:5695–5705. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Tang DJ, Hu L, Xie D, et al: Oncogenic
transformation by SEI-1 is associated with chromosomal instability.
Cancer Res. 65:6504–6508. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Lee SL, Hong SW, Shin JS, et al: p34SEI-1
inhibits doxorubicin-induced senescence through a pathway mediated
by protein kinase C-delta and c-Jun-NH2-kinase 1 activation in
human breast cancer MCF7 cells. Mol Cancer Res. 7:1845–1853. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Lin Q, Wang J, Childress C, Sudol M, Carey
DJ and Yang W: HECT E3 ubiquitin ligase Nedd4-1 ubiquitinates ACK
and regulates epidermal growth factor (EGF)-induced degradation of
EGF receptor and ACK. Mol Cell Biol. 30:1541–1554. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Trotman LC, Wang X, Alimonti A, et al:
Ubiquitination regulates PTEN nuclear import and tumor suppression.
Cell. 128:141–156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Fouladkou F, Landry T, Kawabe H, et al:
The ubiquitin ligase Nedd4-1 is dispensable for the regulation of
PTEN stability and localization. Proc Natl Acad Sci USA.
105:8585–8590. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Burow ME, Weldon CB, Melnik LI, et al:
PI3-K/AKT regulation of NF-kappaB signaling events in suppression
of TNF-induced apoptosis. Biochem Biophys Res Commun. 271:342–345.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Holmes KM, Annala M, Chua CY, et al:
Insulin-like growth factor-binding protein 2-driven glioma
progression is prevented by blocking a clinically significant
integrin, integrin-linked kinase, and NF-kappaB network. Proc Natl
Acad Sci USA. 109:3475–3480. 2012. View Article : Google Scholar
|
|
35.
|
Judge AR, Koncarevic A, Hunter RB, Liou
HC, Jackman RW and Kandarian SC: Role for IkappaBalpha, but not
c-Rel, in skeletal muscle atrophy. Am J Physiol Cell Physiol.
292:C372–C382. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Van Themsche C, Leblanc V, Parent S and
Asselin E: X-linked inhibitor of apoptosis protein (XIAP) regulates
PTEN ubiquitination, content, and compartmentalization. J Biol
Chem. 284:20462–20466. 2009.PubMed/NCBI
|
|
37.
|
Sarma SN, Kim YJ and Ryu JC: Gene
expression profiles of human promyelocytic leukemia cell lines
exposed to volatile organic compounds. Toxicology. 271:122–130.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Du F and Galan JE: Selective inhibition of
type III secretion activated signaling by the Salmonella
effector AvrA. PLoS Pathog. 5:e10005952009. View Article : Google Scholar : PubMed/NCBI
|